[level-membership-for-basic-science-category]
CHAPTER 25 Protein Hardware for Signaling
This chapter introduces proteins that transduce signals in the cytoplasm: protein kinases, protein phosphatases, guanosine triphosphatases (GTPases), and adapter proteins. Remarkably, kinases and GTPases use the same strategy to operate molecular switches that carry information through signaling pathways: the simple addition and removal of inorganic phosphate. Protein kinases add phosphate groups to specific protein targets, and phosphatases remove them. GTPases bind guanosine triphosphate (GTP) and hydrolyze it to guanosine diphosphate (GDP) and inorganic phosphate, which dissociates. In both cases, the presence or absence of a single phosphate group switches a protein between active and inactive conformations.
These molecular switches are often linked in series to form a signaling cascade that can both transmit and refine signals. Enzymes along signaling pathways (including kinases) often act as amplifiers. Turning on the binary switch of one enzyme molecule can produce many product molecules, each of which, in turn, may continue to propagate and amplify the original signal by activating downstream molecules. Other pathways involve negative feedback loops. Few signaling pathways are linear; instead, most branch and intersect, allowing cells to integrate information from multiple receptors and to control multiple effector systems simultaneously. Chapter 27 illustrates the functions of molecular switches in several signaling pathways.
Protein Phosphorylation
For historical and practical reasons, it has been easier to study protein kinases than protein phosphatases, so most research and accounts of regulation by phosphorylation emphasize kinases (witness the 361,167 PubMed hits for “protein kinase” compared with 127,267 for “protein phosphatase” in August 2006). Furthermore, many researchers assumed incorrectly that phosphatases are always active, leading to a lack of interest in their roles in signaling reactions. Readers should not forget that both directions are important on this two-way street.
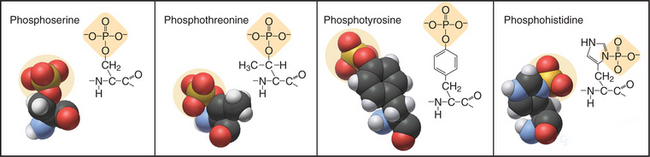
In eukaryotes, more than 99% of protein phosphorylation occurs on serine and threonine residues, but phosphorylation of tyrosine residues regulates many processes in animals (Fig. 25-1). Bacteria and Archaea use histidine and aspartate phosphorylation for signaling (see Fig. 27-11), but these modifications are little known in eukaryotes. Phosphohistidine and phosphoaspartate are more difficult to assay than are other phosphorylated residues, so pathways using these phosphoamino acids might have escaped detection.
Effects of Phosphorylation on Protein Structure and Function
Protein Kinases
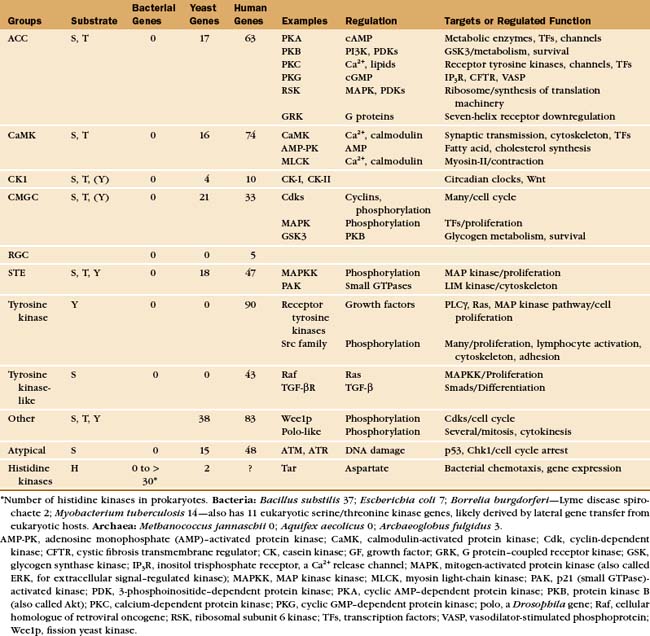
Protein kinases catalyze the transfer of the g-phosphate from adenosine triphosphate (or, rarely, guanosine triphosphate) to amino acid side chains of proteins. Protein kinases are important, as is evident from the remarkable number of genes: 116 in budding yeast (second only to transcription factor genes), 409 in nematode worms (second only to seven-helix receptor genes), and 518 in humans. Most protein kinases in eukaryotes are either serine/threonine kinases or tyrosine kinases (Appendix 25-1). The difference is that most serine/threonine kinases phosphorylate either serine and threonine but not tyrosine, while most tyrosine kinases phosphorylate tyrosine but not serine or threonine.
Most serine/threonine and tyrosine kinases had a common evolutionary origin and share similar structures and catalytic mechanisms, despite differences in substrate specificity. Tyrosine kinases emerged in animals after their divergence from fungi. Nevertheless, fungi have phosphotyrosine owing to two families of serine/threonine kinases that also phosphorylate tyrosine and three protein tyrosine phosphatases to reverse these reactions. A family of 40 “atypical” protein kinases had a separate origin from the major family. Lipid kinases have a catalytic domain related to typical protein kinases. They phosphorylate inositol phospholipids (see Fig. 26-7) or a few proteins.
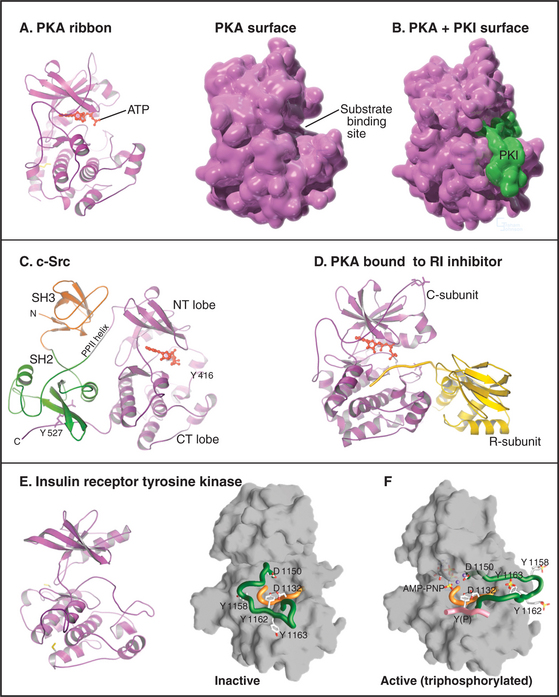
The catalytic domain of eukaryotic protein kinases consists of about 260 residues in two lobes surrounding the ATP-binding pocket (Fig. 25-3). Despite extensive sequence divergence, all of these kinases have a similar polypeptide fold with conserved residues at critical positions required for catalysis.
Each kinase has a restricted range of protein substrates, so activation of a particular protein kinase changes the phosphorylation and activity of a discrete set of target proteins. Substrate specificity is achieved by selective binding of substrates to a groove on the surface of the kinase (Fig. 25-3B). This groove recognizes amino acids that flank the phosphorylatable residue and position the acceptor amino acid side chain in the active site. Typically, all substrates that bind a particular kinase have similar residues surrounding the target serine, threonine, or tyrosine (a consensus target sequence). For example, the consensus sequence for protein kinase A (PKA) is Arg-Arg-Gly-Ser/Thr-Ile. The arginines and isoleucine flanking the target serine or threonine residue specify binding to PKA. Interactions outside the catalytic site may also contribute to specific binding.
In addition to the catalytic domain, most protein kinases have other domains for regulation or localization (Fig. 25-4). Adapter domains, such as SH2, SH3, and pleckstrin homology domains (Fig. 25-11), target kinases to specific sites in the cell. Such localization can either bring together a kinase and its substrates or limit their interaction. Transmembrane segments anchor receptor kinases to membranes. Receptor tyrosine kinases usually have additional residues inserted in the kinase domain and at the C-terminus. Phosphorylation of tyrosines in these inserts creates binding sites for effector proteins with SH2 domains (see Figs. 27-6, 27-7, and 27-8).
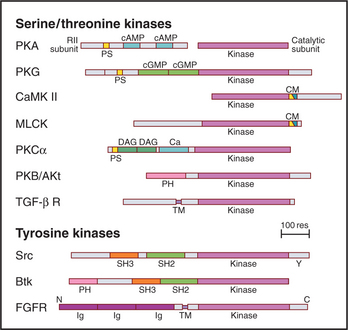
Figure 25-4 protein kinase domain architecture. See Appendix 25-1 for definitions of the kinase names. Btk, Bruton tyrosine kinase; Ca, calcium-binding site; cAMP and cGMP, cyclic nucleotide–binding sites; CM, overlapping pseudosubstrate/calmodulin-binding site; DAG, diacylglycerol-binding site; FGFR, fibroblast growth factor receptor; Ig, immunoglobulin domains; PH, pleckstrin homology domain; PS, pseudosubstrate sequences; SH2 and SH3, Src-homology domains; TM, transmembrane domain.
Prokaryotes generally lack serine/threonine/tyrosine kinases but use a large family of histidine kinases for signal transduction (see Fig. 27-11). These prokaryotic kinases differ in structure, mechanism and evolution-ary origin from eukaryotic kinases. A few bacteria have acquired eukaryotic kinases by lateral transfer of genes.
Regulation of Protein Kinases
Each kinase has its own regulatory mechanism, but most involve one or more of three strategies: (1) phosphorylation, (2) interactions with intrinsic peptides or extrinsic subunits that may themselves be targets for second messengers or regulatory proteins, and (3) targeting to specific cellular locations, such as the nucleus, plasma membrane, or cytoskeleton, enhancing interaction with specific substrates.
Phosphorylation
Phosphorylation can either activate or inhibit protein kinases. In some cases, another kinase molecule of the same type carries out the phosphorylation, but often another type of kinase is responsible. When linked in series, different types of kinases form signaling cascades that can amplify and sharpen the response to a stimulus (see Fig. 27-5):
Regulation of Substrate Binding
Peptides that are intrinsic to the kinase or part of a separate protein can inhibit kinases by competing with protein substrates for binding to the enzyme (Figs. 25-3B and D and 25-4):
Targeting
Protein Phosphatases
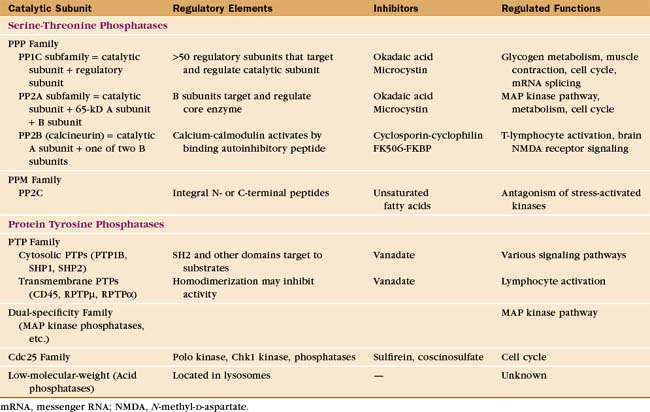
Eukaryotes have several families of protein phosphatases that remove phosphate from amino acid side chains (Table 25-1 and Fig. 25-5). Like protein kinases, most protein phosphatases are active toward either phosphoserine/threonine or phosphotyrosine, although several dual-specificity phosphatases can dephosphorylate all three residues. The 90 active protein tyrosine phosphatases far outnumber the 20 serine/threonine phosphatase genes in the human genome. Each tyrosine phosphatase is thought to act on a limited number of substrates. The small number of serine/threonine phosphatases achieve specificity by associating with an array of accessory subunits, which regulate enzyme activity and target catalytic subunits to particular substrates. Domains flanking the catalytic domains also regulate enzyme activity (Fig. 25-6).
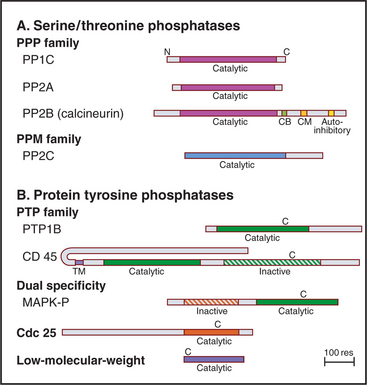
PPP Family of Serine/Threonine Phosphates
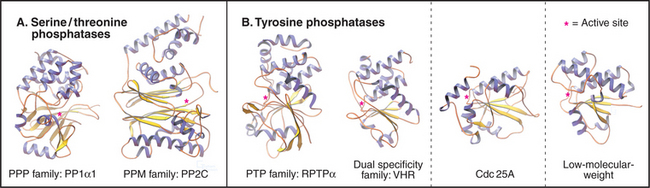
Members of the PPP family of serine/threonine phosphatases are found in Bacteria, Archaea, and all tissues of eukaryotes. PP1 and PP2A are two of the most evolutionarily conserved enzymes. All three PPP subfamilies share the same catalytic fold with a two-metal ion cluster (Fe2+ and Zn2+ in vivo) in the active site (Fig. 25-5A). Diverse regulatory subunits restrict the substrates for PP1 and PP2A by targeting catalytic subunits to specific sites in the cell, as illustrated by the following examples:
PP2B, also known as calcineurin, is the only cytoplasmic phosphatase regulated by Ca2+. PP2B consists of two subunits: an A subunit with the phosphatase active site and a B subunit that is similar to calmodulin (see Fig. 3-12C) but does not participate in the response to Ca2+. At low concentrations of Ca2+, a C-terminal autoinhibitory segment of the A subunit blocks its own active site. An increase in cytoplasmic Ca2+ activates PP2B by first binding to calmodulin. Calcium-calmodulin then binds the autoinhibitory segment and displaces it from the active site. The transcription factor NF-AT (nuclear factor–activated T cells) is the best known substrate (see Fig. 27-8). Activation of T-cell receptors on T lymphocytes releases Ca2+ in the cytoplasm and turns on PP2B. Dephosphorylation permits NF-AT to enter the nucleus, where it turns on the expression of several lymphocyte growth factors. PP2B is the indirect target of two potent drugs that inhibit the immune rejection of transplanted organs. These drugs—cyclosporin and FK506—bind two different small proteins: cyclophilin and FK-binding protein. Both drug-protein complexes bind PP2B and inhibit the phosphatase directly by blocking the active site. This prevents expression of genes regulated by NF-AT. Suppression of the immune response by cyclosporin revolutionized organ transplantation in humans.
Protein Tyrosine Phosphatases
The human genome contains 107 genes for protein tyrosine phosphatases (PTPs [Table 25-1]), matching the number of tyrosine kinase genes. PTPs participate in many processes, including lymphocyte activation and regulation of the cell cycle by reversing the actions of protein tyrosine kinases. In some cases, dephosphorylation of tyrosine activates the substrate, such as Src tyrosine kinase (Fig. 25-3C) and cyclin-dependent protein kinases (see Fig. 40-14). PTPs are tumor suppressors; somatic mutations that inactivate these enzymes are common in cancer cells. Eleven of these human PTP genes encode proteins that are missing key catalytic residues in the active site, so they do not hydrolyze phosphotyrosine. Perhaps they serve as adapters to bind proteins with phosphotyrosines.
The four families of PTPs represent a remarkable evolutionary tale. Sequence analysis and atomic structures (Fig. 25-5B) show that three of these families arose from different ancestors but have converged to have similar active sites. PTPs and dual-specificity phosphatases derived from a common ancestor and have similar three-dimensional structures, whereas Cdc25 and low-molecular-weight tyrosine phosphatases have different folds. Nevertheless, all bind phosphotyrosine in a narrow but deep pocket, transfer the phosphate to the sulfur atom of a cysteine in the sequence Cys-x-x-x-x-x-Arg, and release the phosphate in the rate-limiting step, when water attacks the phosphocysteine intermediate.
PTPs are said to have more restricted substrate specificity than serine/threonine phosphatases, but some well-characterized PTPs have multiple substrates. Localization contributes additional specificity. For example, a transmembrane segment that anchors a PTP such as CD45 (Fig. 25-6) to the plasma membrane can enhance its access to some substrates and restrict its access to other substrates.
PTP Subfamily
Many cytoplasmic PTPs are bound to cellular partners. Adapter domains, such as SH2 domains, bind the phosphatases SHP-1 and SHP-2 to phosphotyrosines. C-terminal hydrophobic residues bind the phosphatase PTP1B to the endoplasmic reticulum. Transient oxidation of the catalytic cysteine by hydrogen peroxide accompanies activation of some signaling pathways that depend on phosphorylation of tyrosines, such as the insulin pathway (see Fig. 27-7).
Other PTPs are transmembrane proteins with a single transmembrane segment linking a variety of extracellular domains to one or two PTP domains in the cytoplasm (Fig. 25-6). The membrane proximal PTP domain has phosphatase activity. In most cases, the second PTP domain is inactive, so it might have regulatory functions. Extracellular domains are attractive candidates as receptors, and some have been implicated in cellular adhesion, but no extracellular ligand has been shown to regulate phosphatase activity.
CD45, the best-characterized transmembrane PTP, constitutes a remarkable 10% of the plasma membrane protein of human white blood cells. CD45 is required for antigens to activate B and T lymphocytes (see Fig. 27-8). Lymphocytes that lack CD45 fail to release intracellular Ca2+, secrete lymphokines, or proliferate in response to antigen stimulation. It is thought that the CD45 phosphatase activates one or more Src-family tyrosine kinases associated with the T-cell receptor by dephosphorylating inhibitory phosphotyrosine residues (Fig. 25-3C).
Dual-Specificity Subfamily
The dual-specificity family of phosphatases prefers phosphotyrosine as a substrate, but owing to the fact that its substrate-binding site is shallower than that of a PTP, it can also dephosphorylate serine and threonine at about 1% of that rate. The most interesting members of this group, the MKPs, inactivate MAP kinases by dephosphorylating both phosphotyrosine and phosphothreonine residues (see Fig. 29-5B). Some members of this family, such as the five PTEN phosphatases, remove phosphate from lipids, specifically the D-3 position of polyphosphoinositides (see Fig. 26-7).
Cdc25 Subfamily
Cdc25 removes inhibitory phosphates from adjacent threonine and tyrosine residues on the master cell cycle kinases Cdk1 and Cdk2, releasing these enzymes to promote cell cycle progression (see Fig. 43-4). This is an example of a phosphatase having a positive effect on a biological process. Cdc25 itself is activated by serine/threonine phosphorylation during the cell cycle.
Cooperation between Kinases and Phosphatases
Some protein phosphatases are stably associated with their substrate proteins. One example is the dual-specificity MAP kinase phosphatase-3 (MKP-3) bound to MAP kinase (see Fig. 27-6). Following activation by upstream kinases, this MAP kinase is active only transiently, owing to dephosphorylation by the associated phosphatase. These have been called self-correcting signal complexes, but more broadly speaking, this is an example of a biological timer.
Pharmacological Agents for Studying Protein Kinases and Phosphatases
Inhibitors of protein kinases and protein phosphatases (Table 25-1) are widely used to explore the biological functions of these enzymes. Few, if any, of these inhibitors are entirely specific for one protein kinase or phosphatase. Given that these protein families are so large, caution is required in interpreting experiments with these agents. Nevertheless, some inhibitors of tyrosine kinases are successful anticancer drugs. Development of specific inhibitors of protein phosphatases is challenging, owing to the chemistry of the dephosphorylation reactions and the fact that the enzymes have similar active sites.
Guanosine Triphosphate–Binding Proteins
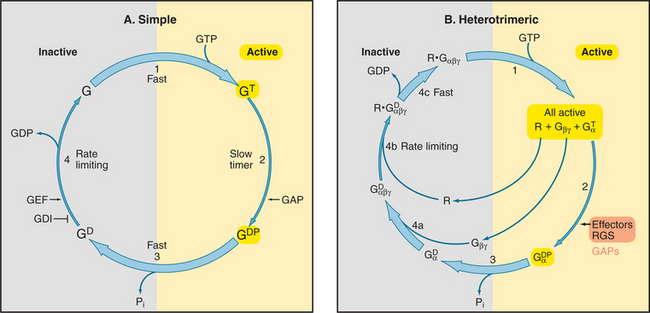
Cells use GTP-binding proteins (called GTPases or G-proteins) to regulate a host of functions, including protein synthesis, signal transduction from plasma membrane receptors, regulation of the cytoskeleton, membrane traffic, and nuclear transport (Appendix 25-2). These proteins had a common ancestor, share a homologous core domain that binds a guanine nucleotide (Fig. 25-7), and use a common enzymatic cycle of GTP binding, hydrolysis, and product dissociation to switch the protein on and off (Fig. 25-8).
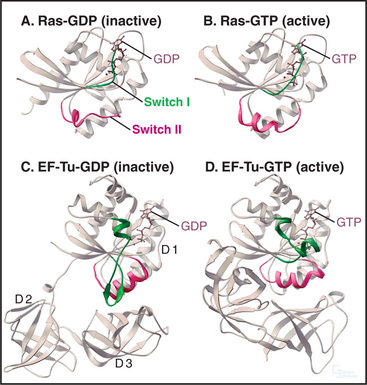
Genes for GTPases are ancient, as all forms of life use GTPases to regulate protein synthesis. Gene duplication and divergence created 10 families of GTPases in eukaryotes. Further molecular evolution produced multiple isoforms within these families to provide more specificity. Tubulin, the microtubule subunit (see Fig. 34-4), also binds and hydrolyzes GTP but has a completely different fold than do the GTPases considered here.
GTPases share a core GTP-binding domain of about 200 residues folded into a b-sheet of six strands sandwiched between five a-helices (Fig. 25-7). The architecture of this domain was maintained during the evolutionary divergence of the GTPases, despite the fact that about 80% of the residues in this core differ between the major classes. GTP binds in a shallow groove formed largely by loops at the ends of elements of secondary structure. A network of hydrogen bonds between the protein and guanine base, ribose, triphosphate, and Mg2+ anchor the nucleotide.
The four main classes of GTPases are elongation factors, small GTPases related to Ras, trimeric G-proteins, and dynamin-related GTPases. Small 20-kD GTPases such as Ras (Fig. 25-7A-B) consist simply of a GTP-binding core domain. The 39- to 45-kD Ga subunits of trimeric G-proteins have an additional domain of a-helices hinged to the core domain by two strands (Fig. 25-9). This helical domain covers the GTP-binding site. Elongation factor EF-Tu (Fig. 25-7C-D) and dynamin (see Fig. 22-11) have additional domains that are required for intermolecular interactions.
All GTPases use the same enzyme cycle. The conformation of a GTPase depends on whether GTP or GDP is bound. The active GTP-bound conformation interacts with effector proteins. The GDP conformation is inactive, because it does not bind effectors. Chapter 4 explains methods that are used to analyze the mechanism (see Figs. 4-6 and 4-7).
The GTPase cycle (Fig. 25-8) consists of four steps: (1) Rapid binding of GTP is coupled to changes in the conformations of three segments of the polypeptide called switch-I, -II, and -III. In the active GTP state, these switch loops form a binding site for downstream target proteins. (2) GTP hydrolysis is slow and irreversible. (3) Dissociation of the g-phosphate is fast and coupled to the return of the switch loops to the inactive conformation. (4) GTPases tend to accumulate in the inactive GDP-state, because GDP dissociates slowly and GTP cannot bind until GDP dissociates.
GTPases use diverse intrinsic or extrinsic protein modules to regulate the GTPase cycle. Most GTPases depend on other proteins, called guanine nucleotide exchange factors, to accelerate dissociation of GDP (Appendix 25-2). Although unrelated in structure, guanine nucleotide exchange factors have similar mechanisms. They distort the P loop, the part of the nucleotide binding site that interacts with the b phosphate, allowing GDP to escape, and then bind tightly to the nucleotide-free GTPase. Most small GTPases require extrinsic GTPase-activating proteins (GAPs) to stimulate the GTP hydrolysis that turns them off.
Elongation Factors
These GTPases act as timers to ensure the fidelity of protein synthesis (see Fig. 17-10). Elongation factor Tu (EF-Tu) has two accessory domains hinged to the GTPase core (Fig. 25-7C-D; see also Fig. 17-10). GTP EF-Tu binds and delivers aminoacyl-tRNAs (transfer RNAs) to the A site of a ribosome. If the tRNA anticodon matches the mRNA codon at the A site, it remains bound long enough for GTP to be hydrolyzed. This releases EF-Tu from the ribosome and allows the correct amino acid to be added to the growing polypeptide chain. The accessory factor EF-Ts is the GDP exchange factor for this system.
Small Guanosine Triphosphatases
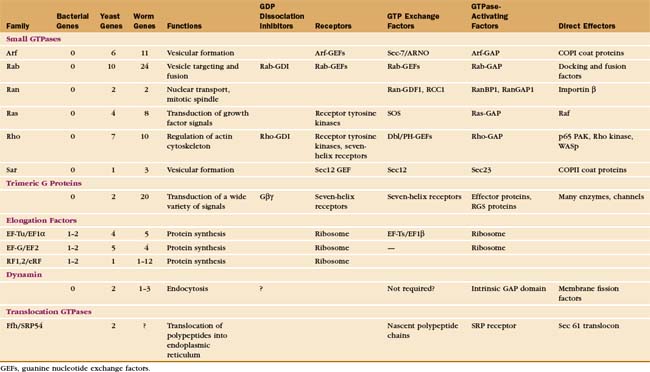
The six families of small GTPases consist of a single domain (Fig. 25-7A-B). Three types of small GTPases—Arf, Rab, and Sar—act as switches for intracellular traffic of membrane vesicles (see Fig. 21-6). The Ran family regulates nuclear transport (see Fig. 14-17) and assembly of the mitotic spindle (see Fig. 44-8). Other chapters explain these processes. This chapter introduces two families of small GTPases, Ras and Rho, that transduce signals from cell surface receptors.
Covalently attached lipids anchor many small GTPases to membrane bilayers. These hydrophobic chains are required for activity. Ras and some Rho and Rab proteins are modified with a C-15 or C-20 prenyl chain on a C-terminal cysteine. Arfs are myristoylated, but other small GTPases, such as Ran, are not modified and are soluble in the cytoplasm. Even membrane-associated GTPases may cycle into the cytoplasm when their lipid tails turn over or when they bind cytoplasmic regulatory proteins such as Rho-GDI (Rho-guanine nucleotide dissociation inhibitor [see Fig. 21-11]), Rab, Arf, and Sar GTPases also recycle through the cytoplasm as they direct vesicular traffic from one compartment to another. Membrane association can be regulated either by interactions with accessory proteins (in the case of Rab) or through guanine nucleotide–dependent conformational changes (in the case of Arf and Sar).
Ras is the prototypical small GTPase. Normally, Ras transmits signals from growth factor receptor tyrosine kinases to transcription factors that control genes required for cellular proliferation (see Figs. 27-6 and 27-7). In quiescent cells, Ras accumulates in the inactive GDP form. Stimulation of growth factor receptors attracts SOS, the Ras nucleotide exchange factor, to the membrane, where it activates Ras by exchanging GDP for GTP. Ras-GTP then activates a cascade of protein kinases that ultimately controls gene expression. Ras has low intrinsic GTPase activity (rate = 0.005s−1; half-time = 140 seconds) that is not stimulated by binding effector proteins. An accessory protein called Ras-GAP stimulates GTPase activity 105-fold by providing a crucial arginine for the active site. This inactivates Ras until SOS again stimulates dissociation of GDP. Mutations that inhibit GTP hydrolysis predispose to cancer, because without GTP hydrolysis, Ras is continually active and stimulates cellular growth.
The 16 isoforms in the human Rho family include Rho itself, Rac, and Cdc42. They regulate the actin and microtubule cytoskeletons, cellular growth, and cellular polarity. Stimulation of certain seven-helix receptors and receptor tyrosine kinases activates Rho family-GTPases by activating exchange factors that catalyze the exchange of GDP for GTP. Activated Rho-family proteins stimulate kinases (such as p21-activated kinase and Rho-kinase), which mediate downstream effects on the actin cytoskeleton. For example, Rho-kinase activates myosin-II by phosphorylating the regulatory light chain and inhibiting the light chain phosphatase (see Fig. 39-21). Activated Cdc42 binds Wiskott-Aldrich syndrome protein (WASp), a protein that regulates actin filament nucleation (see Figs. 33-17 and 38-8). WASp is defective in Wiskott-Aldrich syndrome, an inherited human disease characterized by a deficiency in blood cell function. Rac stimulates another protein related to WASp that regulates actin filament nucleation.
Trimeric G-Proteins
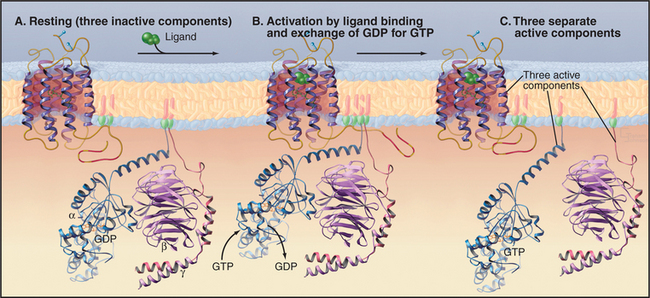
Trimeric G-proteins have three subunits (Fig. 25-9). Gα subunits have a GTP-binding domain similar to small GTPases plus a helical domain that helps to trap the bound nucleotide. Gβ and Gγ subunits bind tightly to each other and reversibly to Gα. Seven-helix receptors activate trimeric G-proteins by promoting dissociation of GDP bound to an inactive trimeric protein. GTP binding changes the conformation of Gα and releases Gβγ. This generates two signals, as both Gα and Gβγ can engage downstream effector proteins.
Subunit Diversity
Given more than 1000 genes for seven-helix receptors, many receptors use the same G-proteins to transduce signals. For example, the hundreds of different odorant receptors in the nose all activate Golfa (see Fig. 27-1). Trimeric G-proteins act on a limited variety of downstream effectors, including ion channels, kinases, and enzymes that produce second messengers (Table 25-2).
| Family (No. of Human Members) | Receptors | Effectors |
|---|---|---|
| Giα (7) | α-Adrenergic amines, acetylcholine, chemokines, various neurotransmitters, tastants | Inhibit adenylyl cyclase, open potassium channels, close calcium channels |
| Gqα (5) | α-Adrenergic amines, acetylcholine, various neurotransmitters | Activate phospholipase Cβ to produce IP3, which releases Ca2+ |
| Gsα (3) | β-Adrenergic amines, hormones (corticotropin, glucagon, parathyroid, thyrotropin, others) | Stimulate adenylyl cyclase to produce cAMP, receptor kinase |
| Gtα (2) | Rhodopsin, which absorbs light | Activate cGMP phosphodiesterase to break down cGMP, receptor kinase |
| G13α (2) | Thrombin and others | Rho and others |
| Golfα | Odorant receptors | Activate adenylyl cyclase, receptor kinase |
Structure of Trimeric G-Proteins
The GTPase core of Gα is linked to a helical domain that covers the GTP-binding site. Gβ is a torus-shaped molecule composed of seven modules, each folded into an antiparallel β-sheet (Fig. 25-9). These so-called WD repeats are found in many proteins but are best characterized in Gβ. Loops on one face of the torus interact with Gα, whereas those on the other face interact with Gγ. The small Gγ subunit associates surprisingly tightly with Gβ through an N-terminal helical coiled-coil and other interactions. A C-20 prenyl group on a C-terminal cysteine of most Gγ subunits anchors Gβγ to membrane bilayers. An N-terminal fatty acid, usually myristate, anchors Gα independently to lipid bilayers. Some Gα subunits have an additional palmitic acid anchor on a reactive cysteine. Tight interaction of Gβγ with Gα-GDP blocks effector interaction sites on both partners.
Guanosine Triphosphatase Cycle
Seven-helix receptors activate trimeric G-proteins by catalyzing the exchange of GDP for GTP on the Gα subunit. This dissociates the Gα from Gβγ, allowing each to activate effector proteins (Fig. 25-9). It is convenient to think about this process as two cycles: a GTPase cycle coupled to a subunit cycle (Fig. 25-8B).
The rate of GDP dissociation from trimeric G-proteins is near zero (half-life about 10 to 60 minutes) unless they bind an activated seven-helix receptor. Activated seven-helix receptors are guanine nucleotide exchange factors that increase the rate of GDP dissociation by many orders of magnitude, allowing GTP to exchange for GDP on a millisecond-to-second time scale, fast enough for vision (see Fig. 27-2). Interaction of the receptor with Gα and Gβγ triggers a conformational change that weakens the bonds of GDP to Gα. The cleft between the Gα domains must open transiently to release GDP and accept a GTP from solution.
Both effector proteins and extrinsic GTPase activators, called RGS proteins (regulators of G-protein signaling), stimulate GTP hydrolysis and terminate the signal. For example, Gqa-GTP binds and stimulates phospholipase Cb, which produces two second messengers: inositol 1,4,5-triphosphate (IP3) and diacylglycerol (see Fig. 26-12). At the same time, phospholipase Cb accelerates the GTPase of Gqa, providing negative feedback to turn off the signal. G-proteins regulate some physiological processes that require a rapid response, such as the heart rate (Gia) and vision (Gta). In these cases, a family of 20 or more RGS proteins stimulates GTPase activity of G-proteins about 100-fold, yielding half-times of less than 1 second. These RGS proteins work by stabilizing the transition state between GTP and GDP-Pi. Some RGS proteins also have a Rho–guanine nucleotide exchange factor domain, so they may connect seven-helix receptors and trimeric G-proteins to the Rho family of small GTPases.
Subunit Cycle
GTP binding, hydrolysis, and dissociation drive not only the GTPase cycle but also a linked subunit cycle (Fig. 25-8B). Ga cycles on and off both Gβγ and the receptor as it traverses its GTPase cycle. The conformational change in Ga that is induced by GTP binding affects all of its molecular interactions. GTP binding reduces the affinity of Ga for both its receptor and associated Gβγ subunits, so all three molecular partners separate on the cytoplasmic face of the membrane (Fig. 25-9). Once dissociated, the receptor, Ga, and Gβγ are each free to interact with other partners (Table 25-2). The conformation of Gβγ is the same whether bound to Ga or free, so its activation on dissociation from Gβγ is attributable simply to unmasking of effector binding sites. Gβγ is not a passive partner in these linked cycles, as it must be bound to Ga-GDP before activated membrane receptors can trigger dissociation of GDP. In addition to activating membrane targets, such as ion channels, Gβγ provides a membrane anchor to enhance the interaction of cytoplasmic effectors (such as kinases that phosphorylate seven-helix receptors) with membrane targets.
The properties of the linked GTPase and subunit cycles allow activation of a single receptor to generate a large signal. Although most seven-helix receptors are active only briefly (owing to rapid ligand dissociation and rapid inactivation [see Figs. 27-1 and 27-2]), they can turn on multiple G-proteins, each with a longer lifetime. Slow GTP hydrolysis by Ga subunits allows ample time for these G-proteins to activate downstream effector proteins. Because these effectors are typically enzymes or channels, they amplify the signal in a short time.
Mechanisms of Effector Activation
Both Gα and Gβγ subunits participate in signaling by interacting with downstream effector proteins. In some cases, Gα and Gβγ subunits act individually; in other cases, they may act synergistically and even antagonistically. The following two examples, elaborated on in Chapter 27, illustrate how G-proteins activate effector proteins.
In the eye, when the seven-helix photoreceptor rhodopsin absorbs a photon, the G-protein Gta (also called transducin) relays and amplifies a signal (see Fig. 27-2). Each activated rhodopsin generates about 500 Gta-GTPs, which bind an inhibitory subunit of the enzyme cGMP phosphodiesterase. This stimulates the activity of the phosphodiesterase, which lowers the cytoplasmic concentration of cGMP and closes an ion channel. The signal is transient, because both an RGS protein and the inhibitory subunit itself promote the hydrolysis of GTP bound to Gta. Inactive Gta-GDP dissociates from the inhibitory subunit, terminating the signal that flowed through Gta to the enzyme.
In the heart, the β-adrenergic receptor activates Gsa, releasing both Gsa-GTP and Gβγ to bind effector proteins (see Fig. 27-3). During its 10-second lifetime, Gsa-GTP binds and stimulates the enzyme adenylyl cyclase to produce the second messenger cAMP. Gβγ assists in receptor inactivation by binding β-ARK, the kinase that phosphorylates and turns off the β-adrenergic receptor, terminating the signal.
GTPases in Disease
Both abnormal activation or inactivation of G-proteins can cause disease (Table 25-3). Mutations that interfere with GTP hydrolysis cause Ga to accumulate in the GTP state and persistently activate downstream effectors. For example, mutations in arginine or glutamine residues of Ga that are crucial for GTP hydrolysis can cause tumors by prolonging the activation of pathways responsible for cell proliferation. Common variants in the sequence of other G-proteins are associated with high blood pressure and other common diseases.
| Disease | GTPase | Mechanism |
|---|---|---|
| Excess Signal | ||
| Cholera | Gsα | Cholera toxin ADP-ribosylation of R201 inhibits GTP hydrolysis in intestinal epithelium. |
| Pituitary and thyroid adenomas | Gsα | Somatic point mutations of R201 or Q227 inhibit GTP hydrolysis; constitutive activity mimics signal from hormones that stimulate proliferation and secretion by these glands. |
| Various cancers | Ras | Point mutations inhibit GTP hydrolysis, generating persistent stimulation of signals for cell proliferation. |
| Deficient Signal | ||
| Whooping cough | Giα | Pertussis toxin ADP-ribosylation of Giα C347 in the bronchial epithelium blocks receptor activation; connection to coughing not established. |
| Night blindness | Gtα | Germ line point mutation in G38. |
| Pseudohypoparathyroidism type Ia | Gsα | Point mutations result in loss of Gsα or may block its activation by receptors. |
C, cysteine; G, glycine; Q, glutamine; R, arginine.
Adapted from Farfel Z, Bourne HR, Iiri T: The expanding spectrum of G protein diseases. N Engl J Med 340:1012–1020, 1999.
Dynamin-Related GTPases
These large GTPases have an N-terminal GTP-binding domain and a C-terminal GTPase-activating domain that allows self-assembly into spiral polymers (see Fig. 22-11). Dynamin, the prototypical family member, also contains domains that target it to the plasma membrane, where it participates in endocytosis. Other dynamin family members regulate vacuolar trafficking in yeast and the division of mitochondria.
Experimental Tools
Mutations, especially those that constitutively acti-vate GTPases (by inhibiting GTP hydrolysis) or inactivate GTPases, have been powerful tools for investigating GTPase functions in live cells. For biochemical experiments, slowly hydrolyzed analogs of GTP, such as GTPγS (with a sulfur substituted for one of the γ-phosphate oxygens), are used to activate GTPases. Similarly, aluminum fluoride and beryllium fluoride bind very tightly in place of the hydrolyzed γ phosphate, keeping Ga in an active GDP-Pi state similar to GTP. The fungal metabolite brefeldin A blocks nucleotide exchange on some Arfs. This disrupts membrane traffic between the Golgi complex and the endoplasmic reticulum (see Fig. 21-14).
Molecular Recognition by Adapter Domains
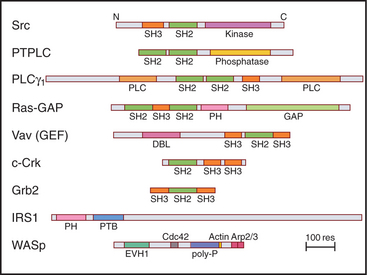
During the characterization of signaling pathways, several patterns of amino acid sequence appeared repeatedly in different proteins, such as Src (Figs. 25-3C and 25-4B). These turned out to be compactly folded domains (Fig. 25-10) that are incorporated into a variety of proteins (Fig. 25-11), including many unrelated to signaling. These domains mediate interactions of proteins with each other and with membrane lipids (Table 25-4).
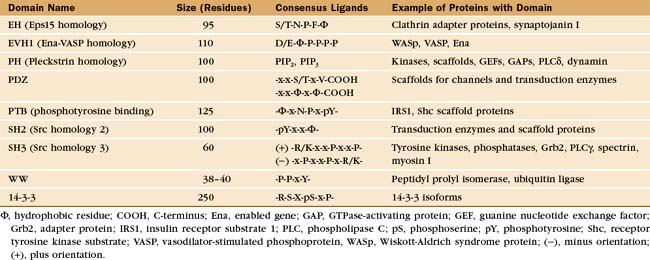
The names of adapter domains generally came from the proteins where they were originally recognized. For example, Src homology (SH) domains were first recognized in the Src tyrosine kinase (Fig. 25-3E). SH1 is the tyrosine kinase domain; SH2 domain binds phosphotyrosine peptides; and SH3 binds polyproline type II helices. Chapter 27 provides detailed examples of how adapter domains function. This section provides an overview of their structure and ligand-binding properties. The following points apply to adapter proteins in general.
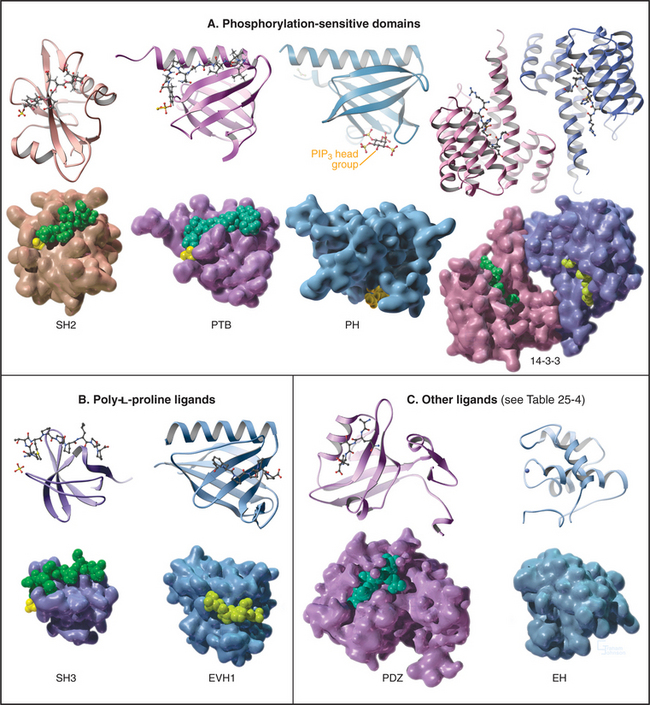
All members of each family of adapters have similar structures (and common evolutionary origins) but differ in their affinity for a range of similar ligands. For example, all SH2 domains bind peptides with a sequence phosphotyrosine-X-X-hydrophobic residue. All require phosphotyrosine but differ in their affinity for peptides depending on the hydrophobic residue and the intervening residues. This lock-and-key strategy creates specificity with lots of particular combinations, as in real locks and keys. Although lacking in sequence similarity, three of these domains—PH, PTB, and EVH1—have similar folds (Fig. 25-10) and might have had a common ancestor. Nevertheless, their ligands are quite distinct from each other and bind to different sites on the commonly folded domains.
Phosphorylation-Sensitive Adapters
SH2 Domains
SH2 domains bind short peptide sequences that begin with a phosphotyrosine. Like a two-pronged plug, these peptides insert into two cavities in the SH2 socket. Phosphotyrosine is one prong. It is the key residue, as it provides most of the binding energy by virtue of an extensive network of hydrogen bonds between the phosphate and its deep binding pocket. A phosphate on the tyrosine increases the affinity of a peptide for its partner SH2 domain by orders of magnitude. This allows reversible phosphorylation to control interactions between SH2 domains and their ligands. This switching mechanism is used in growth factor signaling and lymphocyte activation (see Figs. 27-6 to 27-9). The second prong is a hydrophobic side chain of the third residue C-terminal from phosphotyrosine. It inserts into a hydrophobic cavity on the surface of the SH2 domain. The size of this side chain is a major determinant of binding specificity. Two residues between these plug residues straddle the β-sheet of the SH2 with their side chains exposed to solvent.
SH2 interactions with target peptides and proteins are relatively weak, with Kds in the range of 0.1 to 1μM. This allows for rapid exchange of partners and dephosphorylation of the phosphotyrosine. Nevertheless, these interactions appear to be specific, the 115 human proteins with SH2 domains engaging a limited number of target phosphoproteins. SH2 domains target several signal transduction enzymes to receptor tyrosine kinases (see Fig. 27-6) as the first step in propagating signals to effectors, including second messengers and transcription factors. Intramolecular binding of the Src SH2 to a C-terminal phosphotyrosine regulates the catalytic activity of the enzyme (Fig. 25-3C).
Phosphotyrosine-Binding Domains
Most PTB domains require a phosphotyrosine at the C-terminal end of the peptide ligand. Hydrophobic residues preceding phosphotyrosine in the sequence contribute to binding specificity. The bound peptide is hydrogen-bonded onto the edge of a β-sheet, and phosphotyrosine interacts with basic residues. PTB domains target adapter proteins to phosphotyrosines on receptor tyrosine kinases, such as insulin receptor (see Fig. 27-7). In a few cases, PTB domains bind unphosphorylated peptide ligands. Note that the fold of PTB domains and their mode of interaction with ligand peptides have nothing in common with SH2 domains.
14-3-3 Proteins
Vertebrates have at least seven genes for 14-3-3 subunits that assemble into homodimers or heterodimers. Protein ligands with an appropriate sequence and a central phosphoserine bind each subunit with submicromolar affinity. Peptides with two appropriate sequences bind much more tightly. 14-3-3 proteins regulate protein kinases, including the Ras-activated kinase Raf (see Fig. 27-6) and cellular death pathways (see Chapter 46). During interphase or after DNA damage, a 14-3-3 protein inhibits the cell cycle phosphatase Cdc25, when it is phosphorylated on a serine (see Fig. 43-4).
WW Domains
These tiny adapter domains are found in more than 100 proteins. They bind certain phosphoserine or phosphothreonine peptides. These phosphorylation-dependent interactions regulate Cdc25 and ubiquitin-mediated protein destruction (see Chapter 23).
PH Domains
These compact domains of about 100 residues are named for the PH domain that was first recognized in pleckstrin, the major substrate for protein kinase C in platelets. PH domains bind polyphosphoinositides: PH domains of dynamin and phospholipase Cd prefer phosphatidylinositol (PI) (4,5) P2 (PIP2), whereas the PH domain of Bruton’s tyrosine kinase (Btk) favors PI(3,4,5)P3 (PIP3). These interactions target proteins with PH domains to membrane bilayers that are rich in PIP2 and PIP3 and make these membrane interactions responsive to the activities of phosphoinositide kinases (see Figs. 26-7 and 38-12). PH-domain proteins include kinases (PKB/Akt, PDK1), signaling scaffolding proteins (insulin receptor substrate 1 [IRS1]; see Fig. 27-7), enzymes (phospholipase Cγ1; see Figs. 26-4 and 26-12), and guanine nucleotide exchange factors. Mutations in the PH domain of the tyrosine kinase, Btk, reduce affinity for PI(3,4,5)P3 and cause a failure of β-lymphocyte development (see Fig. 28-9), resulting in immunodeficiency due to a lack of antibodies.
Adapters with Proline-Rich Ligands
SH3 Domains
SH3 domains found in 253 different human proteins bind to proline-rich sequences of numerous target proteins. These ligands form left-handed, type II polyproline helices (see Fig. 29-1) that make hydrophobic interactions with aromatic residues in a shallow groove on the SH3 domain, as well as hydrogen bonds contributed by ligand peptide carbonyl oxygens. Depending on the SH3 domain, the peptide can be oriented in either direction. Residues flanking the central proline helix contribute to binding specificity. Even optimal peptide ligands bind with relatively low affinity (Kds in the micromolar range), so they exchange rapidly. When incorporated into proteins, type II poly-l-proline helices with appropriate sequences bind somewhat more tightly, owing to secondary interactions.
SH3 domains of the adapter protein Grb2 (Fig. 25-11) link activated growth factor receptors to the nucleotide exchange protein for Ras (see Fig. 27-6). SH3 domains are also found in tyrosine kinases and cytoskeletal proteins, including myosin-I, spectrin, and cortactin.
EVH1 Domains
EVH1 domains (Fig. 25-11) are found in WASp (see Fig. 33-17) and other signaling proteins that regulate actin polymerization. EVH1 domains are folded like PH and PTB domains, but they bind type II proline-rich helices of target protein in a groove. The corresponding groove is occupied by an intrinsic a-helix in some PH domains. This site also differs from that for phosphotyrosine peptides on PTB domains. Thus, a common scaffold has diverged to form three completely different binding sites.
ACKNOWLEDGMENTS
Thanks go to Brad Nolen and Tony Hunter for their suggestions on revisions to this chapter.
Alonso A, Sasin J, Bottini N, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699-711.
Aramburu J, Heitman J, Crabtree GR. Calcineurin: A central controller of signalling in eukaryotes. EMBO Rep. 2004;5:343-348.
Barford D, Das AK, Egloff M-P. Structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133-164.
Berwick DC, Tavaré JM. Identifying protein kinase substrates: Hunting for the organ-grinder’s monkeys. Trends Biochem Sci. 2004;29:227-232.
Bhattacharyya RP, Reményi A, Yeh BJ, Lim WA. Domains, motifs, and scaffolds: The role of modular interactions in the evolution and wiring of cell signaling circuits. Annu Rev Biochem. 2006;75:655-680.
Bockoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743-781.
Bourdeau A, Dube N, Tremblay ML. Cytoplasmic protein tyrosine phosphatases, regulation and function: The roles of PTP1B and TC-PTP. Curr Opin Cell Biol. 2005;17:203-209.
Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 2004;84:1-39.
Cherfils J, Chardin P. GEFs: Structural basis of their activation of small GTP-binding proteins. Trends Biochem Sci. 1999;24:306-311.
Drause DS, VanEtten RA. Tyrosine kinases as targets for cancer therapy. New Engl J Med. 2005;353:172-187.
Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629-635.
Farfel Z, Bourne HR, Iiri T. The expanding spectrum of G protein diseases. N Engl J Med. 1999;340:1012-1020.
Ferrell JE. How responses get more switch-like as you move down a protein kinase cascade. Trends Biochem Sci. 1997;22:288-289.
Gallego M, Virshup DM. Protein serine/threonine phosphatases: Life, death, and sleeping. Curr Opin Cell Biol. 2005;17:197-202.
Hampoelz B, Knoblich JA. Heterotrimeric G proteins: New tricks for an old dog. Cell. 2004;119:453-456.
Herrmann C. Ras-effector interactions: After one decade. Curr Opin Struct Biol. 2003;13:122-129.
Hofmann F. The biology of cyclic GMP-dependent protein kinases. J Biol Chem. 2005;280:1-4.
Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373-398.
Iiri T, Farfel Z, Bourne HR. G-protein diseases furnish a model for the turn-on switch. Nature. 1998;394:35-38.
Ishii M, Kurachi Y. Physiological actions of regulators of G-protein signaling (RGS) proteins. Life Sci. 2003;74:163-171.
Kim C, Xuong N-H, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIa) subunits of PKA. Science. 2005;307:690-696.
Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002;27:514-520.
Manning G, Whyte DB, Marinez R, et al. The protein kinase complement of the human genome. Science. 2002;298:1912-1934.
Nourry C, Grant SG, Borg JP. PDZ domain proteins: Plug and play!. Sci STKE. 2003;2003(179):RE7.
Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906-7909.
Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445-452.
Pawson T, Scott JD. Protein phosphorylation in signaling: 50 years and counting. Trends Biochem Sci. 2005;30:286-290.
Rossman KL, Der CJ, Sondek J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167-180.
Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1:51-66.
Sprang SR. G-protein mechanisms: Insights from structural analysis. Annu Rev Biochem. 1997;66:639-678.
Wong W, Scott JD. AKAP signalling complexes: Focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959-970.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 25 Protein Hardware for Signaling
This chapter introduces proteins that transduce signals in the cytoplasm: protein kinases, protein phosphatases, guanosine triphosphatases (GTPases), and adapter proteins. Remarkably, kinases and GTPases use the same strategy to operate molecular switches that carry information through signaling pathways: the simple addition and removal of inorganic phosphate. Protein kinases add phosphate groups to specific protein targets, and phosphatases remove them. GTPases bind guanosine triphosphate (GTP) and hydrolyze it to guanosine diphosphate (GDP) and inorganic phosphate, which dissociates. In both cases, the presence or absence of a single phosphate group switches a protein between active and inactive conformations.
These molecular switches are often linked in series to form a signaling cascade that can both transmit and refine signals. Enzymes along signaling pathways (including kinases) often act as amplifiers. Turning on the binary switch of one enzyme molecule can produce many product molecules, each of which, in turn, may continue to propagate and amplify the original signal by activating downstream molecules. Other pathways involve negative feedback loops. Few signaling pathways are linear; instead, most branch and intersect, allowing cells to integrate information from multiple receptors and to control multiple effector systems simultaneously. Chapter 27 illustrates the functions of molecular switches in several signaling pathways.
Protein Phosphorylation
For historical and practical reasons, it has been easier to study protein kinases than protein phosphatases, so most research and accounts of regulation by phosphorylation emphasize kinases (witness the 361,167 PubMed hits for “protein kinase” compared with 127,267 for “protein phosphatase” in August 2006). Furthermore, many researchers assumed incorrectly that phosphatases are always active, leading to a lack of interest in their roles in signaling reactions. Readers should not forget that both directions are important on this two-way street.
In eukaryotes, more than 99% of protein phosphorylation occurs on serine and threonine residues, but phosphorylation of tyrosine residues regulates many processes in animals (Fig. 25-1). Bacteria and Archaea use histidine and aspartate phosphorylation for signaling (see Fig. 27-11), but these modifications are little known in eukaryotes. Phosphohistidine and phosphoaspartate are more difficult to assay than are other phosphorylated residues, so pathways using these phosphoamino acids might have escaped detection.
Effects of Phosphorylation on Protein Structure and Function
Protein Kinases
Protein kinases catalyze the transfer of the g-phosphate from adenosine triphosphate (or, rarely, guanosine triphosphate) to amino acid side chains of proteins. Protein kinases are important, as is evident from the remarkable number of genes: 116 in budding yeast (second only to transcription factor genes), 409 in nematode worms (second only to seven-helix receptor genes), and 518 in humans. Most protein kinases in eukaryotes are either serine/threonine kinases or tyrosine kinases (Appendix 25-1). The difference is that most serine/threonine kinases phosphorylate either serine and threonine but not tyrosine, while most tyrosine kinases phosphorylate tyrosine but not serine or threonine.
Most serine/threonine and tyrosine kinases had a common evolutionary origin and share similar structures and catalytic mechanisms, despite differences in substrate specificity. Tyrosine kinases emerged in animals after their divergence from fungi. Nevertheless, fungi have phosphotyrosine owing to two families of serine/threonine kinases that also phosphorylate tyrosine and three protein tyrosine phosphatases to reverse these reactions. A family of 40 “atypical” protein kinases had a separate origin from the major family. Lipid kinases have a catalytic domain related to typical protein kinases. They phosphorylate inositol phospholipids (see Fig. 26-7) or a few proteins.
The catalytic domain of eukaryotic protein kinases consists of about 260 residues in two lobes surrounding the ATP-binding pocket (Fig. 25-3). Despite extensive sequence divergence, all of these kinases have a similar polypeptide fold with conserved residues at critical positions required for catalysis.
Each kinase has a restricted range of protein substrates, so activation of a particular protein kinase changes the phosphorylation and activity of a discrete set of target proteins. Substrate specificity is achieved by selective binding of substrates to a groove on the surface of the kinase (Fig. 25-3B). This groove recognizes amino acids that flank the phosphorylatable residue and position the acceptor amino acid side chain in the active site. Typically, all substrates that bind a particular kinase have similar residues surrounding the target serine, threonine, or tyrosine (a consensus target sequence). For example, the consensus sequence for protein kinase A (PKA) is Arg-Arg-Gly-Ser/Thr-Ile. The arginines and isoleucine flanking the target serine or threonine residue specify binding to PKA. Interactions outside the catalytic site may also contribute to specific binding.
In addition to the catalytic domain, most protein kinases have other domains for regulation or localization (Fig. 25-4). Adapter domains, such as SH2, SH3, and pleckstrin homology domains (Fig. 25-11), target kinases to specific sites in the cell. Such localization can either bring together a kinase and its substrates or limit their interaction. Transmembrane segments anchor receptor kinases to membranes. Receptor tyrosine kinases usually have additional residues inserted in the kinase domain and at the C-terminus. Phosphorylation of tyrosines in these inserts creates binding sites for effector proteins with SH2 domains (see Figs. 27-6, 27-7, and 27-8).
Figure 25-4 protein kinase domain architecture. See Appendix 25-1 for definitions of the kinase names. Btk, Bruton tyrosine kinase; Ca, calcium-binding site; cAMP and cGMP, cyclic nucleotide–binding sites; CM, overlapping pseudosubstrate/calmodulin-binding site; DAG, diacylglycerol-binding site; FGFR, fibroblast growth factor receptor; Ig, immunoglobulin domains; PH, pleckstrin homology domain; PS, pseudosubstrate sequences; SH2 and SH3, Src-homology domains; TM, transmembrane domain.
Prokaryotes generally lack serine/threonine/tyrosine kinases but use a large family of histidine kinases for signal transduction (see Fig. 27-11). These prokaryotic kinases differ in structure, mechanism and evolution-ary origin from eukaryotic kinases. A few bacteria have acquired eukaryotic kinases by lateral transfer of genes.
Regulation of Protein Kinases
Each kinase has its own regulatory mechanism, but most involve one or more of three strategies: (1) phosphorylation, (2) interactions with intrinsic peptides or extrinsic subunits that may themselves be targets for second messengers or regulatory proteins, and (3) targeting to specific cellular locations, such as the nucleus, plasma membrane, or cytoskeleton, enhancing interaction with specific substrates.
Phosphorylation
Phosphorylation can either activate or inhibit protein kinases. In some cases, another kinase molecule of the same type carries out the phosphorylation, but often another type of kinase is responsible. When linked in series, different types of kinases form signaling cascades that can amplify and sharpen the response to a stimulus (see Fig. 27-5):
Regulation of Substrate Binding
Peptides that are intrinsic to the kinase or part of a separate protein can inhibit kinases by competing with protein substrates for binding to the enzyme (Figs. 25-3B and D and 25-4):
Targeting
Protein Phosphatases
Eukaryotes have several families of protein phosphatases that remove phosphate from amino acid side chains (Table 25-1 and Fig. 25-5). Like protein kinases, most protein phosphatases are active toward either phosphoserine/threonine or phosphotyrosine, although several dual-specificity phosphatases can dephosphorylate all three residues. The 90 active protein tyrosine phosphatases far outnumber the 20 serine/threonine phosphatase genes in the human genome. Each tyrosine phosphatase is thought to act on a limited number of substrates. The small number of serine/threonine phosphatases achieve specificity by associating with an array of accessory subunits, which regulate enzyme activity and target catalytic subunits to particular substrates. Domains flanking the catalytic domains also regulate enzyme activity (Fig. 25-6).
PPP Family of Serine/Threonine Phosphates
Members of the PPP family of serine/threonine phosphatases are found in Bacteria, Archaea, and all tissues of eukaryotes. PP1 and PP2A are two of the most evolutionarily conserved enzymes. All three PPP subfamilies share the same catalytic fold with a two-metal ion cluster (Fe2+ and Zn2+ in vivo) in the active site (Fig. 25-5A). Diverse regulatory subunits restrict the substrates for PP1 and PP2A by targeting catalytic subunits to specific sites in the cell, as illustrated by the following examples:
[/not-level-membership-for-basic-science-category]
