html xmlns=”http://www.w3.org/1999/xhtml”>
22
Protein Biomarkers for Detecting Cancer
Molecular Screening
and
No matter how many mistakes you make or how slow you progress, you are still way ahead of everyone who isn’t trying.
(http://www.marcandangel.com/2012/06/08/60-quotes-change-the-way-you-think/)
Biomarkers of early cancer detection, specific markers of a malignancy type, and predictive markers of response to treatment will aid in the early diagnosis and selection of the most efficient therapies. An exponential growth in technologies has been achieved toward this goal in the past decade. However, it is safe to say that the field of disease biomarkers produced many more publications on the subject than actual “clinically actionable” targets. By no means a criticism, this statement reflects on the status quo of a discipline that has been “trying really hard” but finding the goal more elusive with each step forward. 1 Why has the progress been slow? Is it just the technology that is not on par with the complexity of human biology? Or perhaps, by focusing mainly on a paradigm of “DNA-mRNA-protein” as a fundamental driving force that defines a phenotype, are we oversimplifying and hence misinterpreting a system? 2,3 We bring these very general questions to the attention of the reader for two reasons: first to spark a debate on the fundamental issues that biomarker discovery entails and second to put our detailed proteomics discussion into the perspective within the larger context of biomarker discovery.
Establishing a panel of biomarkers for early diagnosis of cancer holds tremendous promise but also faces daunting obstacles. The major challenge stems from the very nature of a biological system, its complexity, dynamics, variability, and versatility all making it difficult to draw clear lines between a state that would be considered normal and one that appears to be slightly different and in danger of becoming abnormal. Then come barriers of logistics, such as a dichotomy between a need to mainly rely on population studies while facing tremendous intra- and interpersonal phenotypic variability, defining adequate controls, availability of specimens, and the danger of their potential adulteration before analysis (i.e., in the course of collection, processing, and storage). Last but not least is the requirement for sensitive technologies capable of measuring target compounds directly from a biological milieu with a level of specificity and selectivity that allows differentiation among discrete cohorts of individuals/patients that is reliable enough to justify the risk of acting on specific clinical modalities. There is a growing recognition that achieving true breakthroughs in the use of biomarkers to improve human conditions, while delivering health care in an economically sustainable way, requires concerted and coordinated efforts of stakeholders across disciplines and across borders. A large consortium of authors has recently published an in-depth analysis of the status of implementation of proteomics biomarkers of disease with a focus on postdiscovery/postvalidation barriers and outlined a need for a roadmap to biomarker implementation. 4 In our review, we concentrate on the preimplementation stages of the biomarker pipeline from the perspective of technologies that are currently available for biomarker discovery and validation, with a focus on protein biomarkers for which mass spectrometry (MS) plays a primary role.
The discipline of MS-based proteomics 5 emerged through a serendipitous convergence of major technological developments in DNA sequencing, MS analysis of proteins/peptides and bioinformatics. In the enthusiasm that followed the success of the Human Genome Project, it appeared that understanding how the functional phenotype of a cell/tissue/organism relates to its protein/peptide repertoire was imminent, leading to exaggerated expectations that the new technology of proteomics would deliver meaningful results in a short time. A rush for quick success resulted in controversies and disappointments, and unavoidably triggered questions as to the fundamental validity and practical usefulness of proteomics approaches. 6–8 The involvement of funding agencies (e.g., the National Cancer Institute [NCI]), which funded programs aimed at addressing various stumbling blocks of proteomics technologies (e.g., the Clinical Proteomics Technologies for Cancer), 9–15 and of professional organizations and consortia (e.g., the Human Proteome Organization [HUPO] 16–20 and the Proteomics Specification in Time and Space [PROSPECTS] Network 21 ) has been crucial, providing funding and building foundations and collaborations among various disciplines to ensure future success in developing protein-based biomarkers of disease. Now, after more than a decade of intense effort, the promise of proteomics remains valid. Accumulated experience, however, required reassessment of the prospects of achieving quick payoffs—especially in the context of translational research, as illustrated by a recent breast cancer biomarker study using an animal model that is reviewed at the end of this chapter. Nevertheless, the field is progressing steadily, and the lessons learned in the early days of the proteomics bonanza are informing the directions of new developments. 22–30 Thousands of papers on the subject have been published to date. Here we focus on selected aspects of method development that, in our opinion, will play a significant role in moving this effort toward its eventual success.
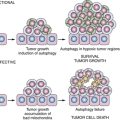
Before discussing different aspects of technology for protein biomarker discovery, it is important to ponder the nature of the objects of inquiry, that is, a protein and a proteome, in the context of the goal of finding biomarkers of physiological processes leading to tumorigenesis. At first approximation, a protein can be viewed as a gene that has been realized in a dimension defined by the available amino acid building blocks. With this simple though simplistic concept of a protein-gene equivalence in mind, the proteome can be reduced to a catalogue of gene products present in a cell. From this perspective, experiments aimed at monitoring gross changes in the repertoire of “protein parts”—that is, the presence or absence of a specific gene product, or significant differences in protein levels—would lead to the detection of robust biomarkers. Most protein biomarker discovery studies to date were based on these operational definitions of a protein and a proteome. The adopted strategies represented a natural extension of familiar approaches used in functional genomics and took advantage of the availability of the required, albeit imperfect, experimental tools. However, although proteins start their lives as strings of amino acid residues arranged in an order prescribed by DNA, they further acquire a multitude of attributes that endow them with functional competence: they fold into specific shapes; they might need to be “cut to order” by proteolytic enzymes; they are decorated with modifications, many of which are transient by design; they are localized to the proper compartment of a cell; they interact with other cell components, including other proteins, to form “molecular machines”; and, once their mission has been fulfilled, they need to be disposed of in an orderly fashion. Defects in any of these processes could lead to, or result from, disease and hence could be used as valuable biomarkers. 31–36 However, because of the very nature of experimental design and confounding factors of system complexity and limitations of technology, these types of biomarker candidates are likely to be missed in studies geared solely toward detecting changes in peptide/protein concentration. Although technically more challenging, approaches that target specific attributes of protein structure and function, including but not limited to the examples listed earlier, are gaining importance in the biomarker discovery field. Of note, systems biology–based predictions are still far from being able to model all aspects of protein “life.” Thus the analysis of a final protein product is the only approach currently available for characterization of protein-driven cell processes. As we discuss in this chapter, MS-based proteomics approaches enable exploration of biomarkers in various contexts.
Defining “Normal”
“Always remember that you are absolutely unique. Just like everyone else.” This maxim, attributed to Margaret Mead, is also an excellent comment on the challenges intrinsic to biomarker discovery. What is normal for one person might be outside the norm for another. Hence, the “perfect world” scenario of early detection of malignancy, or any other malady, would use “self” as a control and rely on serial analyses of specimens collected at different time points, thus focusing on changes in, rather than absolute levels of, putative markers of health or disease. For a variety of reasons, longitudinal collection of specimens for the population at large is not feasible. Therefore, we are limited to epidemiological approaches. In this context, assessing the level of variability within a population is vital to provide a baseline/reference point for evaluating disease consequences in terms of biomarkers. 37 To this end, a number of tools have been proposed and/or are being developed. A protein equivalent to the HapMap, 38 and repositories of human DNA sequences, 39–41 is the foundation on which disease-related changes in protein repertoire will need to be built. Databases of MS-identified peptides are being generated for human 42 and model organisms (e.g., mouse 43 ). In addition, the normal range of posttranslational modifications (PTMs) should be considered. 44 For example, population proteomics proposes using targeted affinity capture approaches combined with high-throughput MS to screen large numbers of samples for a broad array of modifications. 45 Antibodies or lectins could be used for enrichment. In this regard, the Human Protein Atlas is a valuable resource with respect to antibodies, 46,47 and it will be important to generate the lectin equivalent. 48,49 Examples of these datasets include publications on the normal urine, 50 breast, 51 oral epithelium, 52 liver, 53,54 and brain 55 proteomes and PTMs in various settings. 56 These catalogues of normal proteomes and PTMs will be crucial for identifying disease-related changes—that is, candidate biomarkers.