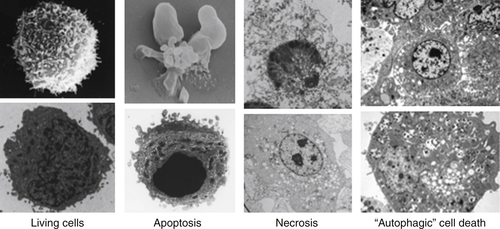
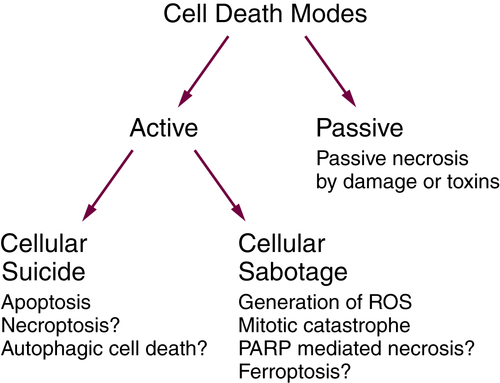
Apoptosis
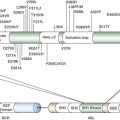
Caspase Activation
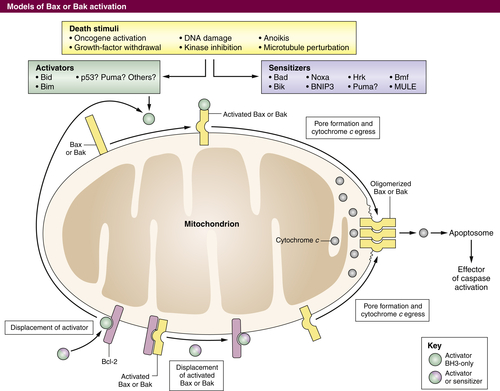
The Mitochondrial Pathway of Apoptosis
The BCL-2 Protein Family
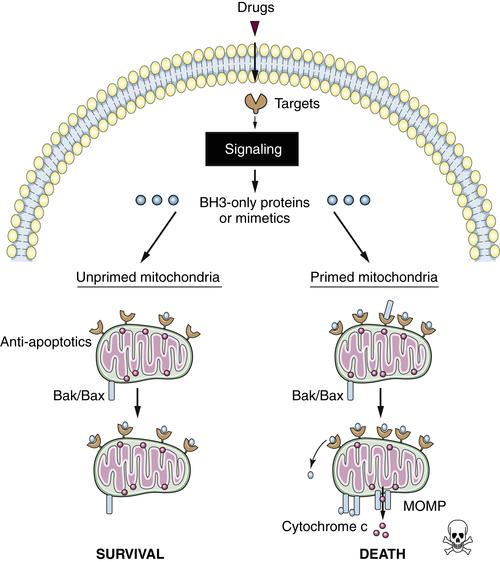
Mitochondrial Priming and Chemotherapy Response
Targeted Therapies and Apoptosis
Directly Targeting the BCL-2 Family in Cancer Treatment
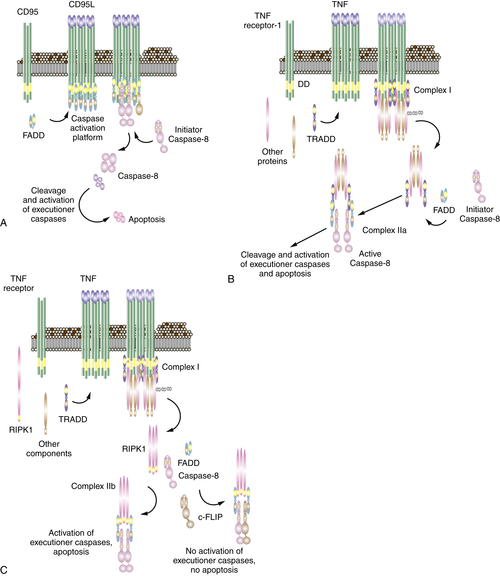
The Death Receptor Pathway of Apoptosis
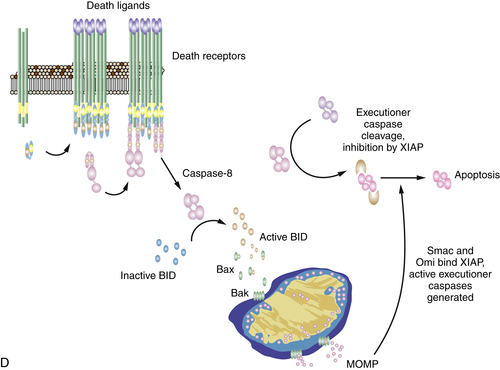
Mitochondrial Pathway Activation by Caspase-8
Necrosis
Passive and Active Necrosis
Necrosis as an Adjunct to Other Cell Death Modalities
Ischemia and Necrosis
Necroptosis Is a Form of Active Necrosis
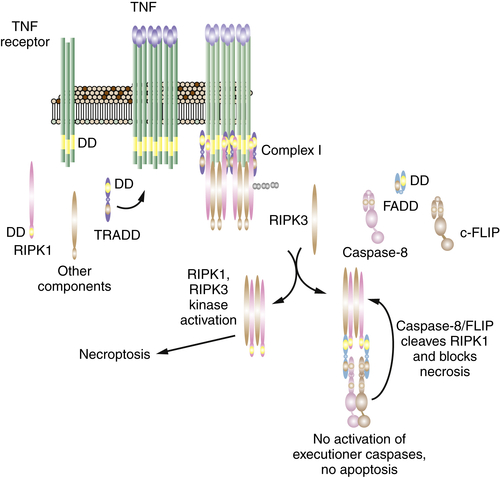
Necroptosis, RIP Kinases, and Cancer Therapy
Autophagy
Protein and Organelle Quality Control
Cell Catabolism
Quality Control and Catabolism Overlap
Dual and Context-Specific Role for Autophagy in Cancer
Process of Autophagy
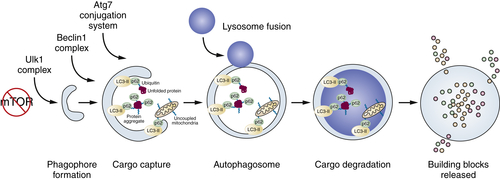
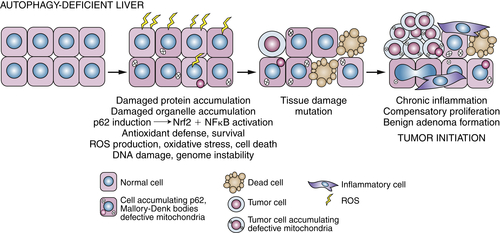
Autophagy-Mediated Tumor Suppression
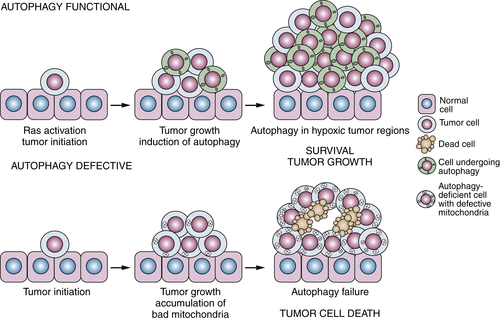
Tumor Promotion by Autophagy
Role of Autophagy in Cancer Therapy
1. A matter of life and death. Cancer Cell . 2002 ; 1 : 19 – 30 .
2. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ . 2009 ; 16 : 3 – 11 .
3. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ . 2012 ; 19 : 107 – 120 .
4. Distinct death mechanisms in Drosophila development. Curr Opin Cell Biol . 2010 ; 22 : 889 – 895 .
5. The pantheon of the fallen: why are there so many forms of cell death? Trends Cell Biol . 2012 ; 22 : 555 – 556 .
6. Human caspases: activation, specificity, and regulation. J Biol Chem . 2009 ; 284 : 21777 – 21781 .
7. Means to an End: Apoptosis and Other Cell Death Mechanisms . Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press ; 2011 .
8. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer . 1972 ; 26 : 239 – 257 .
9. Cell death: critical control points. Cell . 2004 ; 116 : 205 – 219 .
10. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem . 1999 ; 274 : 20049 – 20052 .
11. Lysosomes and autophagy in cell death control. Nat Rev Cancer . 2005 ; 5 : 886 – 897 .
12. Caspase-independent cell death: leaving the set without the final cut. Oncogene . 2008 ; 27 : 6452 – 6461 .
13. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol Cell . 2012 ; 46 : 387 – 397 .
14. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell . 1985 ; 41 : 899 – 906 .
15. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A . 1985 ; 82 : 7439 – 7443 .
16. Involvement of the bcl-2 gene in human follicular lymphoma. Science . 1985 ; 228 : 1440 – 1443 .
17. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci . 2009 ; 122 : 437 – 441 .
18. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer . 2008 ; 8 : 121 – 132 .
19. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev. Mol Cell. Biol . 2010 ; 11 : 621 – 632 .
20. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science . 2001 ; 292 : 727 – 730 .
21. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell . 2002 ; 2 : 183 – 192 .
22. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev . 2000 ; 14 : 2060 – 2071 .
23. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science . 2007 ; 315 : 856 – 859 .
24. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell . 2006 ; 9 : 351 – 365 .
25. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell . 2001 ; 8 : 705 – 711 .
26. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J . 1995 ; 14 : 5589 – 5596 .
27. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell . 2005 ; 17 : 393 – 403 .
28. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell . 2005 ; 17 : 525 – 535 .
29. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature . 2003 ; 426 : 671 – 676 .
30. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol . 2012 ; 30 ( 25 ) : 3127 – 3135 .
31. The hallmarks of cancer. Cell . 2000 ; 100 : 57 – 70 .
32. Intrinsic tumour suppression. Nature . 2004 ; 432 : 307 – 315 .
33. A role for proapoptotic Bax and Bak in T-cell differentiation and transformation. Blood . 2010 ; 116 : 5237 – 5246 .
34. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature . 1991 ; 349 : 254 – 256 .
35. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell . 1994 ; 78 : 703 – 711 .
36. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature . 2011 ; 471 : 110 – 114 .
37. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell . 2005 ; 7 : 227 – 238 .
38. Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel-induced apoptosis in breast cancer cells. Cell Death Differ . 2010 ; 17 : 1624 – 1635 .
39. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J Cell Biol . 2009 ; 187 : 429 – 442 .
40. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell . 1993 ; 74 : 957 – 967 .
41. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell . 2001 ; 7 : 683 – 694 .
42. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science . 2000 ; 288 : 1053 – 1058 .
43. Induction of bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene . 1994 ; 9 : 3743 – 3751 .
44. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science . 2004 ; 303 : 1010 – 1014 .
45. p53 has a direct apoptogenic role at the mitochondria. Mol Cell . 2003 ; 11 : 577 – 590 .
46. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science . 2011 ; 334 : 1129 – 1133 .
47. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell . 2012 ; 151 : 344 – 355 .
48. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest . 2007 ; 117 : 112 – 121 .
49. Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell . 2007 ; 12 : 171 – 185 .
50. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol . 2012 ; 30 : 488 – 496 .
51. An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood . 2011 ; 118 : 3579 – 3590 .
52. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood . 2002 ; 99 : 3530 – 3539 .
53. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med . 2004 ; 350 : 2129 – 2139 .
54. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science . 2004 ; 304 : 1497 – 1500 .
55. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A . 2004 ; 101 : 13306 – 13311 .
56. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A . 2006 ; 103 : 14907 – 14912 .
57. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med . 2001 ; 344 : 1052 – 1056 .
58. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med . 2007 ; 4 : e294 .
59. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res . 2007 ; 67 : 11867 – 11875 .
60. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res . 2011 ; 17 : 2140 – 2148 .
61. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res . 2010 ; 70 : 6670 – 6681 .
62. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med . 2012 ; 18 : 521 – 528 .
63. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res . 2005 ; 65 : 6282 – 6293 .
64. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell . 2011 ; 43 : 432 – 448 .
65. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell . 2011 ; 43 : 449 – 463 .
66. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene . 2013 ; 32 : 1341 – 1350 .
67. Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cell Biol . 2005 ; 6 : 268 – 275 .
68. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell . 2009 ; 20 : 4871 – 4884 .
69. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell . 2007 ; 129 : 983 – 997 .
70. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell . 2010 ; 18 : 802 – 813 .
71. RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol Cell . 2011 ; 44 : 9 – 16 .
72. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature . 2011 ; 471 : 363 – 367 .
73. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol . 2011 ; 13 : 1437 – 1442 .
74. Autophagy: renovation of cells and tissues. Cell . 2011 ; 147 : 728 – 741 .
75. A unified nomenclature for yeast autophagy-related genes. Dev Cell . 2003 ; 5 : 539 – 545 .
76. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol . 2009 ; 10 : 458 – 467 .
77. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell . 2004 ; 15 : 1101 – 1111 .
78. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol . 2008 ; 9 : 1004 – 1010 .
79. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol . 2012 2012:736905 .
80. Mechanisms of mitophagy. Nat Rev Mol Cell Biol . 2010 ; 12 : 9 – 14 .
81. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature . 2006 ; 441 : 880 – 884 .
82. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature . 2006 ; 441 : 885 – 889 .
83. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol . 2010 ; 12 : 213 – 223 .
84. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell . 2007 ; 131 : 1149 – 1163 .
85. Autophagy suppresses tumorigenesis through elimination of p62. Cell . 2009 ; 137 : 1062 – 1075 .
86. Autophagy-deficient mice develop multiple liver tumors. Genes Dev . 2011 ; 25 : 795 – 800 .
87. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany, NY) . 2009 ; 1 : 425 – 437 .
88. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab . 2008 ; 8 : 318 – 324 .
89. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab . 2008 ; 8 : 325 – 332 .
90. Autophagy and lymphocyte homeostasis. Curr Top Microbiol Immunol . 2009 ; 335 : 85 – 105 .
91. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol . 2005 ; 169 : 425 – 434 .
92. The role of autophagy during the early neonatal starvation period. Nature . 2004 ; 432 : 1032 – 1036 .
93. Autophagy and metabolism. Science . 2010 ; 330 : 1344 – 1348 .
94. Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS One . 2011 ; 6 : e17412 .
95. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol . 2005 ; 25 : 1025 – 1040 .
96. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell . 2006 ; 10 : 51 – 64 .
97. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer . 2012 ; 12 : 401 – 410 .
98. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res . 2009 ; 15 : 5308 – 5316 .
99. mTOR signaling in growth control and disease. Cell . 2012 ; 149 : 274 – 293 .
100. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science . 2011 ; 331 : 456 – 461 .
101. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol . 2011 ; 13 : 1016 – 1023 .
102. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev . 2010 ; 20 : 51 – 56 .
103. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A . 2011 ; 108 : 11121 – 11126 .
104. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal . 2010 ; 3 ( 119 ) ra31 .
105. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev . 2011 ; 25 : 460 – 470 .
106. Pancreatic cancers require autophagy for tumor growth. Genes Dev . 2011 ; 25 : 717 – 729 .
107. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol . 2008 ; 10 : 676 – 687 .
108. IKK connects autophagy to major stress pathways. Autophagy . 2010 ; 6 : 189 – 191 .
109. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics . 1999 ; 59 : 59 – 65 .
110. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature . 1999 ; 402 : 672 – 676 .
111. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest . 2003 ; 112 : 1809 – 1820 .
112. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A . 2003 ; 100 : 15077 – 15082 .
113. p62: a versatile multitasker takes on cancer. Trends Biochem Sci . 2012 ; 37 : 230 – 236 .
114. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol . 2010 ; 30 : 3275 – 3285 .
115. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol . 2011 ; 193 : 275 – 284 .
116. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene . 2008 ; 27 : 6228 – 6244 .
117. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature . 2008 ; 456 : 259 – 263 .
118. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev . 2007 ; 21 : 1621 – 1635 .
119. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science . 2012 ; 336 : 225 – 228 .
120. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev . 2007 ; 21 : 1367 – 1381 .
121. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell . 2012 ; 21 : 297 – 308 .
122. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell . 2010 ; 22 : 165 – 178 .
123. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res . 2011 ; 17 : 3478 – 3489 .
124. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest . 2007 ; 117 : 326 – 336 .
125. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature . 2011 ; 481 : 385 – 388 .
126. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A . 2010 ; 107 : 8788 – 8793 .
127. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res . 2011 ; 17 : 654 – 666 .
128. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One . 2012 ; 7 e41831 .
129. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A . 2012 ; 109 : 8253 – 8258 .
130. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell . 2011 ; 19 : 613 – 628 .