[level-membership-for-pediatrics-category]
Chapter 84 Progeria
Clinical Manifestations
Ophthalmological changes seen in HGPS include dry eye syndrome, hyperopia, and keratitis.
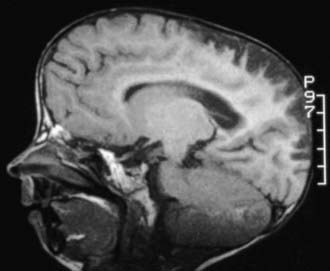
Progressive vascular changes are a major feature of HGPS. Medial smooth muscle cells are lost, vascular remodeling is defective resulting in intimal thickening, disrupted elastin, and deposition of extracellular matrix. Sclerotic plaques deposited in the aorta, coronary, and cerebral blood vessels lead to stenosis (Fig. 84-1). Neurologic abnormalities include transient ischemic attacks, stroke, and seizures, but the major cause of death is cardiovascular compromise. In the absence of neurologic events, motor and mental developments are usually normal.
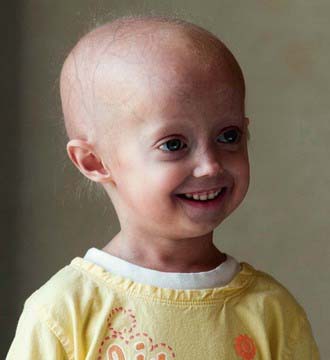
The constellation of bone, hair, subcutaneous fat, and skin changes results in the marked physical resemblance among patients with HGPS (Fig. 84-2).
Molecular Pathogenesis
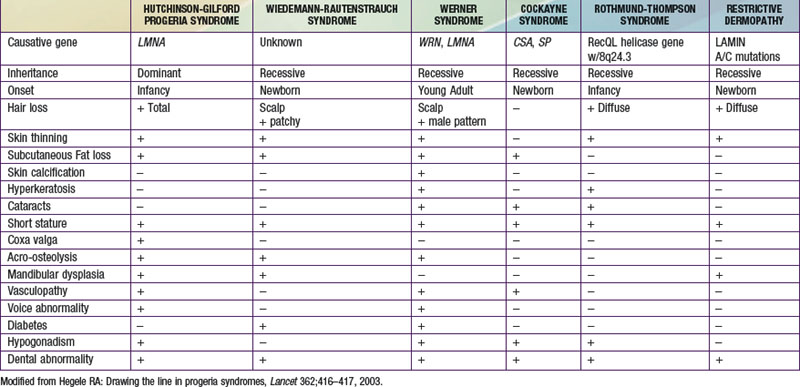
Prognosis and Treatment
The Progeria Foundation (www.progeriaresearch.org) maintains a progeria registry to help with diagnosis and to clearly define the incidence and molecular basis of the disorder. The Foundation website is an excellent source of current information on HGPS for families of children with the disorder.
Brown WT, Gordon LB, Collis FS. Hutchinson-Gilford progeria syndrome. In GeneReviews at GeneTests: Medical Genetics Information Rescurce (website) http://www.ncbi.nlm.nih.gov/sites/GeneTests/review/disease/progeria?db=genetests&search_param=contains Accessed February 17, 2011
Capell BC, Erdos MR, Madigan JP, et al. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879-12884.
Gordon L, McCarten K, Giobbie-Hurder A, et al. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120:824-833.
Meridith M, Gordon L, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592-604.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 84 Progeria
