[level-membership-for-basic-science-category]
Product stability and stability testing
Michael E. Aulton
Chapter contents
The stability of pharmaceutical products
Stabilization of pharmaceuticals
Key points
• Pharmaceutical products tend to deteriorate due to chemical, physical and microbiological causes.
• Careful formulation of the product is necessary to ensure adequate stability.
The stability of pharmaceutical products
Pharmaceutical products tend to deteriorate on storage. The shelf-life of a pharmaceutical product is the period of time during which, if stored correctly, it is expected to retain acceptable chemical, physical and microbiological stability. The expiry date, or expiration date, is the date given on the product’s primary and secondary packaging which represents the end of the shelf-life.
Stabilization of pharmaceuticals
Pharmaceuticals should be formulated and stored in a way that minimizes degradation. The following factors are relevant to most mechanisms of degradation. Oxidation and photodegradation are also important degradation mechanisms and these are dealt with separately later in this chapter.
Temperature
The rate of degradation reactions is markedly influenced by temperature. Storage of the product in a refrigerator (at 2–8 °C) is an option if the product is unstable at room temperature.
Using a freezer (at less than −15 °C) to store unstable formulations is sometimes adopted, however, this makes storage and distribution of the product inconvenient. Also, because a liquid product needs to be thawed before administration, there is the risk of degradation occurring if heat is used to achieve this. Moreover, some drugs, for instance amoxicillin are less stable in solution when frozen than when stored at refrigerator temperature (McDonald et al 1989). Freezing can cause degradation of biopharmaceuticals (see Chapter 46) and live vaccines, though the inclusion of a cryoprotectant, such as trehalose, in the formulation may protect against this.
Finished products are most at risk of exposure to unacceptable temperatures during transportation or storage in vehicles, such as in ambulances (Helm et al 2003, Lucas et al 2004, Priston et al 2005).
Solvent
Replacing an aqueous solvent in a formulation with a non-aqueous one is a potential means of avoiding hydrolysis.
The dielectric constant of a solvent is related to its polarity, more polar solvents having higher values. The dielectric constant can influence the rate at which charged species react. However, the practical considerations of choosing a solvent for a formulation, such as its toxicity and compatibility with the drug, usually outweigh consideration of any effect due to the solvent’s dielectric constant.
Solid dosage forms of a drug, such as tablets or capsules, are usually more stable than liquid ones. However, reactions can occur in water adsorbed onto the surface of a drug particle or other dosage form component. This is why poorly stored aspirin tablets may smell of acetic acid, formed by the hydrolysis of aspirin. Injection formulations of unstable drugs, such as penicillins, can be formulated as freeze-dried powders, which are reconstituted with water or saline (0.9% w/v sodium chloride solution) immediately before administration.
Suspension formulations are often more stable than a solution formulation of the same drug because much of the drug is protected within the insoluble particles.
Acid and base catalysis
A catalyst is a species that accelerates the rate of a reaction without itself being consumed in the reaction. Hydrolysis is often catalysed by hydrogen ions (which exist as H3O+ in solution) or hydroxyl ions. The pH of an aqueous formulation is therefore a critical factor which determines its stability. Specific acid catalysis is catalysis by hydrogen ions and specific base catalysis is catalysis by hydroxyl ions.
Investigation of the relationship between pH and the degradation rate of a drug is performed during the preformulation phase of drug development (Chapter 23). Plotting the logarithm to base 10 of the first-order reaction rate constant against pH may yield useful information about the degradation mechanism. A typical curve is shown in Figure 49.1a. The increase in degradation rate at low pH is due to specific acid catalysis. The increase in rate seen at high pH is due to specific base catalysis. Straight lines with gradients of −1 in the acidic region and +1 in the basic region are characteristic of specific acid and specific base catalysed hydrolysis. The flat region of the curve is largely due to uncatalysed hydrolysis. The product should be maintained at a pH within this region for optimal stability. Cefuroxime and other cephalosporin antibiotics typically show this shape of graph.
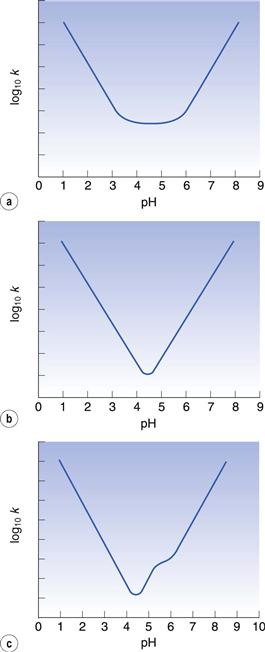
For some drugs, such as many penicillins, the uncatalysed reaction with water is relatively less important than that shown in Figure 49.1a, so there is no flat base to the curve and a V-shaped graph results (Fig. 49.1b). In this case, the pH needs to be more precisely controlled than in the previous example because there is a narrower region of optimal stability.
Many drug molecules undergo ionization, to an extent that often depends on the pH. These typically give a curve as shown in Figure 49.1c. The ionized and unionized forms of the drug degrade at different rates. Therefore, the rate of reaction changes as the pH influences the relative proportion of ionized drug present and this gives an inflection point on the graph. Aspirin is an example of a drug which shows this characteristic.
Other species in a formulation besides H3O+ or OH− may act as acids and bases and thus catalyse degradation reactions. This is known as general acid catalysis and general base catalysis respectively. Buffer ions are a common cause of this, so careful selection of buffer for use in a formulation is needed. Hydrolysis of the amide bond of the antimicrobial drug chloramphenicol is catalysed by several common buffers, including phosphate and acetate. Borate buffer, however, does not catalyse degradation and is used, for example, to buffer eye drops.
Ionic strength
The ionic strength of a medium is related to the concentration of ionic species in it. Changing the ionic strength by adding electrolyte to a solution has some influence on the rate of many degradation reactions. This effect is not high enough to be of importance in the formulation of drug solutions. However, it can be important in laboratory experiments to investigate the influence of pH on degradation rate. In this case, care should be taken to ensure the ionic strength of the various buffer solutions used is kept the same in order to avoid interference with the results.
Light
Containers made from tinted glass protect the product from light to some extent because they allow less ultraviolet light to penetrate than those which are untinted (Chapter 47). Placing the product in an opaque outer container such as a cardboard box is also an option, but it must be borne in mind that the patient might not return the product to its secondary packaging following use.
Oxygen
Oxidation reactions are less influenced by temperature than most other degradation reactions, so low-temperature storage may be less successful as a stabilization option. Flushing containers with an inert gas such as nitrogen before they are sealed will reduce the amount of oxygen in the product. However this technique will not remove all oxygen and is best suited for single-use containers such as ampoules.
Oxidation reactions are generally promoted by high pH, so aqueous products which are susceptible to oxidation should be formulated at as low a pH as possible.
Heavy metal ions, such as Cu2+ and Fe3+, catalyse oxidation reactions, acting at the initiation and propagation stages. These ions are present at trace levels in all formulations. A chelating agent such as ethylenediamine tetraacetic acid (EDTA) or citric acid has a stabilizing effect by binding to the heavy metal ions and preventing them from acting as catalysts.
Chelating agents are usually used in combination with an antioxidant. Antioxidants act in one of two ways. They may act as oxygen scavengers – oxygen is removed from the formulation by reacting preferentially with the antioxidant. The other mechanism by which antioxidants act is to terminate free radical reactions. The antioxidant forms a free radical which is relatively unreactive and so cannot contribute to free radical propagation. Sodium metabisulphite and ascorbic acid are examples of water-soluble antioxidants used in aqueous formulations. Ascorbyl palmitate, butylated hydroxytoluene and α-tocopherol are oil-soluble antioxidants used in oily formulations.
Physical stability
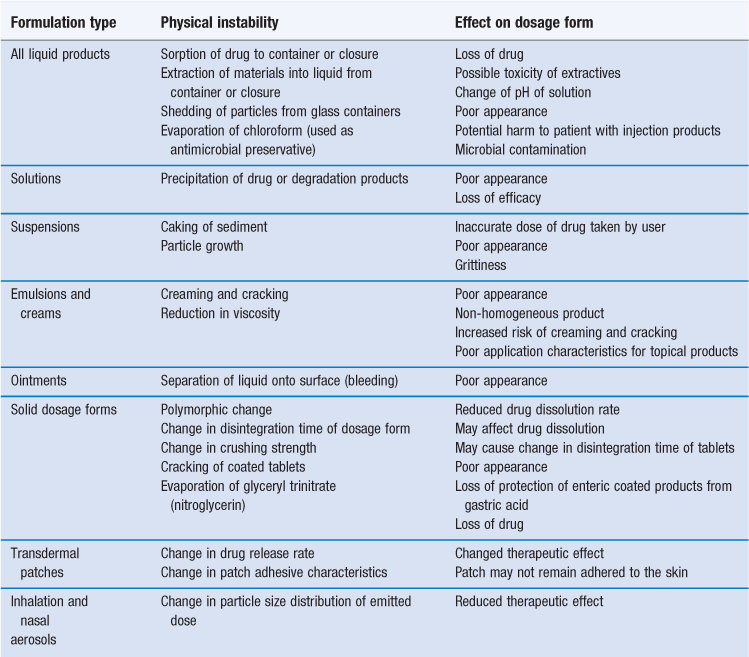
Common physical causes of instability are summarized in Table 49.1. Most of these are due to changes in the physical properties of the dosage form, either spoiling the product’s appearance or reducing its effectiveness. The other potential problems are loss of drug due to sorption or evaporation, or contamination of the product by extractables from the container.
Molecules of the drug or other formulation component may be lost from a formulation by adsorption onto the surface of the container or closure or by absorption of molecules into plastic or rubber containers or closures. Adsorption and absorption often operate together and are collectively known as sorption. Non-polar molecules are susceptible to sorption to plastics and rubber (Chapter 47). For instance, diazepam is lost from solutions in contact with plastic packaging. Loss of antimicrobial preservative to rubber closures is a problem with injection dosage forms (Chapter 36). Sorption is enhanced where the drug (or preservative) is present at low concentration. If the drug molecule ionizes, the pH of the solution may influence the extent of sorption because the unionized form of the molecule, being less polar than the ionized form, may undergo more sorption.
Glyceryl trinitrate (nitroglycerin) evaporates from tablets, where it can then be lost by sorption to plastic packaging. To avoid this, glyceryl trinitrate tablets need to be packaged in glass bottles with aluminium-lined closures.
Plastic packaging materials may be permeable to water vapour. Aqueous products packaged in plastic containers may therefore lose water on storage and the drug content becomes more concentrated.
The term extractable or extractive is used to describe any material which is released from packaging materials into the dosage form. The common packaging material polyvinyl chloride (PVC) is rendered flexible by the addition of a plasticizer (e.g. diethylhexylphthalate, DEHP) which may migrate into injection solutions. This is a particular problem where the solution contains a non-aqueous solvent or surfactant. Paclitaxel, a cytotoxic drug, needs to be administered in dilute solution by slow intravenous infusion. The injection also contains polyoxyethylated castor oil (a material used to solubilize the drug) with ethanol as a cosolvent. Paclitaxel injection therefore cannot be added to infusion fluids contained in PVC because it induces DEHP extraction. Glass or polyethylene infusion containers should therefore be used instead (Allwood and Martin 1996).
Glass containers can release hydroxyl ions into an aqueous product, changing its pH (Chapter 47). This may especially occur during heating in an autoclave, and surface-treated glass is available to minimize hydroxyl ion extraction for formulations where this is of concern.
Microbiological stability
Deterioration due to the presence of microorganisms can either render the product harmful to the patient or have an adverse effect on the product’s properties (Chapter 50). Microbiological deterioration is a critical factor in the stability of sterile products, once the container is opened. Injection products generally need to be used immediately the container is opened and products for use in the eye have a short in-use life once opened.
Stability testing
The purpose of stability testing is to quantify the kinetics of degradation of a product so that the shelf-life of a formulated pharmaceutical product can be defined. Testing must be performed on the exact product that will be marketed and ultimately be used by the patient. However, prior knowledge of the drug’s stability is needed early in the development process in order to assist with its formulation. Therefore, different approaches to the evaluation of stability are needed at the various stages of the development of a product. Information gained during the stability assessment of the active pharmaceutical ingredient alone (Chapter 48) may influence some of the test conditions at this stage.
Accelerated stability testing (stress testing)
Preformulation assessment
In the early stages of the development of a new drug, during the preformulation phase, qualitative assessment of the drug’s likely susceptibility to hydrolysis, oxidation and light degradation is performed (Chapter 23). At this stage, potential degradation mechanisms and degradation products are identified and the development of stability-indicating assays is commenced. Susceptibility of the drug to hydrolysis is assessed by heating solutions of it in water, dilute acid and dilute base. Oxidation is investigated by comparing solutions heated with, and without, flushing with oxygen. The solid-state stability is also studied by storing drug at high temperature, either on its own or combined with potential excipients which may be used in a solid dosage form. Determination of the effect of pH on the drug’s stability is studied by heating solutions that are buffered at a range of pH values. The effect of light on the stability of the drug is discussed later in this chapter.
Prediction of shelf-life by stress testing
Determination of the shelf-life of a formulated product must eventually be performed on the actual product at realistic storage temperatures and humidities. However, the shelf-lives of commercial pharmaceutical products are typically of several years. Testing over such an extended period would be impractical early in product development in order to decide if a particular formulation was of sufficient stability. The Arrhenius equation (Chapter 7) allows the prediction of reaction rates at proposed storage temperatures, from data obtained at high temperatures. For example, if it is desired to formulate a new drug as a solution dosage form, simple buffered solutions of the drug are stored at a range of elevated temperatures for time periods ranging from minutes to days, depending on the relative stability of the drug (Chapter 48). In general, the rate of degradation will increase as the temperature of the test is increased (Chapter 7). Accelerated reaction rates are calculated at each temperature and the Arrhenius equation is used to predict the reaction rate at room temperature, which is then used to calculate the estimated shelf-life. Later in the development process, once the drug molecule’s degradation characteristics have been assessed, prototype formulations must be subjected to the same process to allow optimization of the stability of the finished product.
There are a number of pitfalls in the use of this type of testing. The precision of the shelf-life estimate is poor, so studies typically yield estimates with a wide range of uncertainty. At high temperatures, other degradation reactions may take place that are not significant at normal storage temperatures. Alternatively, at high temperatures, the degradation products which are initially formed from the drug (the primary degradation products) may rapidly react to form further degradation products (the secondary degradation products) and so will not accumulate. Therefore, later in the development process, when the product undergoes stability testing at normal storage conditions, the primary degradation products may accumulate. If these degradation products are not foreseen, they could cause interference with chromatographic analysis of the product, or raise questions about the toxicity profile of the formulation.
The kinetics of drug degradation may also change at different temperatures. A reduction in the concentration of dissolved oxygen will also tend to occur at high temperatures, so use of the Arrhenius equation may not provide a reliable estimate of shelf-life in liquid products which degrade by oxidation. In the case of solid dosage forms, high temperatures often reduce moisture levels associated with drug or excipients, also leading to poor stability prediction. Semisolid dosage forms are often unsuitable for this type of stress testing due to melting of ingredients (particularly excipient bases) at elevated temperature.
Nevertheless, despite these drawbacks, the use of the Arrhenius equation to predict room temperature stability does allow the decision to be made concerning whether a particular formulation type is likely to be sufficiently stable to allow commercial production. It also allows the relative stability of formulations to be studied at an early stage of the development process.
There is however, no substitute for properly-conducted, long-term stability tests carried out under the normal storage conditions that are likely to be experienced by the product.
Temperature cycling
Temperature cycling studies involve storing the product at alternating high and low temperatures. These are often designed to subject liquid products to repeated freezing and thawing, for example. This may reveal stability problems because it potentially accelerates physical deterioration of the product. Temperature fluctuations encourage particle growth in suspensions (Chapter 26), the cracking of emulsions (Chapter 27) and precipitation of dissolved drug from solutions. Such studies also allow the effects of extreme temperature variations during distribution of the product to be evaluated (Helm et al 2003, Lucas et al 2004, Priston et al 2005).
Photostability testing
Photostability studies are carried out at various stages of the product development process and involve investigation of the effect of light on drugs and formulated products. Drug or product is exposed to light, provided by artificial-daylight fluorescent lamps that emit long-wavelength ultraviolet and visible light to simulate indirect, indoor sunlight. The study is performed within a cabinet which has a controlled temperature, typically of 25 °C. Initial photostability studies are carried out using pure drug, spread over the base of shallow containers and directly exposed to the light. After a period of storage, the material is chemically analysed to assess the degree of photodegradation. The drug’s photosensitivity is shown by a reduction in assay of the drug molecule, the formation of photodegradation products or a colour change.
Following this, the formulated pharmaceutical product is tested. The product must be stored in exactly the same packaging that will be used for marketing the product. If light exposure causes an unacceptable amount of change in the product, redesign of the packaging is required to increase light protection.
A light dose of 1.2 million lux hours is typically used in photostability studies; this corresponds to an extended period of exposure to indoor light. The light dose received by the samples under test will also depend on the distance from the light source. The light output from the lamps may vary as the lamps age. Frequent measurements of light energy are therefore required at various positions within the light stability cabinet throughout the lifetime of the lamps.
Long-term stability testing
Stress testing, as described above, gives useful information about the likely stability of a formulated product in a relatively short timescale. However, before it can be marketed, a product must undergo long-term stability testing at conditions representing realistic storage conditions. This involves storing the product under predicted worst-case conditions of temperature and humidity in controlled-temperature cabinets or rooms. Samples are removed at intervals and tested over a minimum period of 12 months. The tests performed will include assays of the drug and other formulation components, such as the antimicrobial preservative and determination of drug degradation products. Other tests may be required, such as pH determination, evaluation of the product’s physical characteristics and possibly also microbiological tests. Furthermore, specific tests are needed for specific dosage forms.
Any protocol should be designed to ensure that the product remains of adequate quality throughout its proposed shelf-life at the proposed storage conditions when it is marketed.
It is advisable at this stage to perform, in parallel, storage of product at slightly higher than normal temperature/humidity combinations. These are known as accelerated degradation conditions. Reference to accelerated testing in this context should be distinguished from stress testing (discussed above), where more extreme temperatures are used. Accelerated test conditions represent a moderately stressful environment which will allow stability problems to be detected more quickly. Significant degradation at these temperatures gives some early warning of stability problems that are likely to develop on prolonged storage at ambient temperatures.
Climatic zones
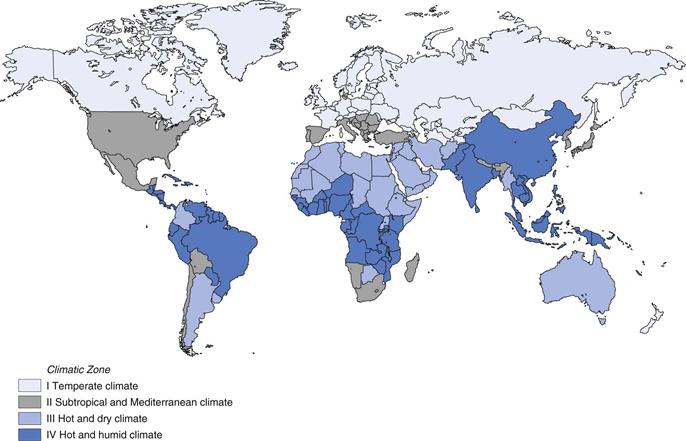
The shelf-life of a product depends on its storage temperature and, for susceptible products, also on the humidity. Environmental conditions of temperature and humidity vary between countries around the world. A pharmaceutical manufacturer may market a product in several countries, or indeed continents. In order to avoid having to carry out long-term stability testing under different conditions for each country, and to simplify the long-term stability testing of a product for the global market, four climatic zones have been defined. The worldwide climatic zones recognized by World Health Organization (2009) are mapped in Figure 49.2.
Climatic Zone I.
Temperate climate, includes Canada, New Zealand, northern Europe, Russia, United Kingdom
Climatic Zone II.
Subtropical and Mediterranean climate, includes Japan, southern Europe, USA, southern Africa, parts of South America
Climatic Zone III.
Hot and dry climate, includes Argentina, Australia, Botswana, Middle East, northern Africa
Climatic Zone IV.
Hot and humid climate, includes Brazil, much of central Africa including Ghana and Nigeria, Indonesia, Nicaragua, the Philippines, Malaysia
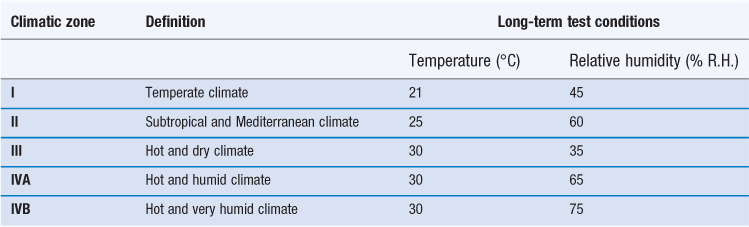
These are based on observed temperatures and relative humidities, both outside and inside buildings, from which mean temperatures and average humidity values are calculated. The storage conditions used for long-term stability testing simulate the worst-case average indoor temperature and humidity experienced in that geographical zone (Dietz et al 1993). The values of temperature and humidity set by WHO for long-term stability testing are shown in Table 49.2. Note that for this purpose, WHO has split Climatic Zone IV into two: IVA ‘Hot and humid climate’ and IVB ‘Hot and very humid climate’.
Testing protocols
Because the packaging of a product may affect its stability, products which are undergoing long-term stability testing should be stored in the exact packaging that is intended to be used when the product is marketed. Liquid products packaged in containers with closures need to be stored inverted, to allow any interaction of the product with the closure to be detected, for instance sorption of preservative into rubber vial closures or into bottle caps. This is not necessary where the product has no separate closure, such as for an ampoule.
At least three separate batches of a finished pharmaceutical product should be placed on stability testing. This is to allow any batch-to-batch differences in stability to be detected. A difference might arise, for example, in a liquid product having differing pH between batches. In a solid dosage form, there may be differences in moisture content, say, following wet granulation. Stability testing should be performed on each individual strength, dosage form, container type and container size of the finished product.
Testing should be performed at least every three months in the first year, every six months in the second year and annually thereafter. Long-term testing should continue at least for the period of the proposed shelf-life.
During the development process, smaller batch sizes are made than will be necessary for full-scale production purposes. However batch size can have an effect on the stability of the final product. For example, any differences in residual moisture content in solid dosage forms or differing viscosities in semisolid dosage forms may result in stability differences. Therefore batches of, at least, pilot-scale should be used for stability testing, i.e. batches that, although smaller than the projected manufacturing batch size for the marketed product, use essentially the same production equipment.
During storage of stability test samples, humidity is controlled because of the potentially harmful effects of moisture on products such as solid dosage forms. However, no humidity control is required for products which are unaffected by humidity, such as aqueous products in glass containers.
Storage under low humidity conditions provides a significant stress for aqueous products packaged in plastic containers that may lose water vapour from the product thorough the plastic, resulting in a more concentrated product. This is especially important for products likely to be stored within Climatic Zone III (hot and dry). Water loss from plastic containers stored at 30 °C is twice as rapid at 35% relative humidity, representative of a dry climate, compared to 65%, representative of a humid one. An additional stability testing requirement for such products is that they are also stored at very low humidity to detect excessive moisture loss. For instance, for Climatic Zones III and IV, additional storage at 35% relative humidity at 30 °C is required.
Representative protocol
An example of a recommended testing schedule that combines the above requirements for climatic Zones II, IVA and IVB is shown in Table 49.3. To reduce the amount of routine testing, it is common practice in industry to perform Climatic Zone I stability testing using the same conditions as required for Climatic Zone II testing (i.e. under slightly harsher conditions than are actually required for Zone I).
Table 49.3
Examples of recommended minimum stability testing schedules for pharmaceutical products
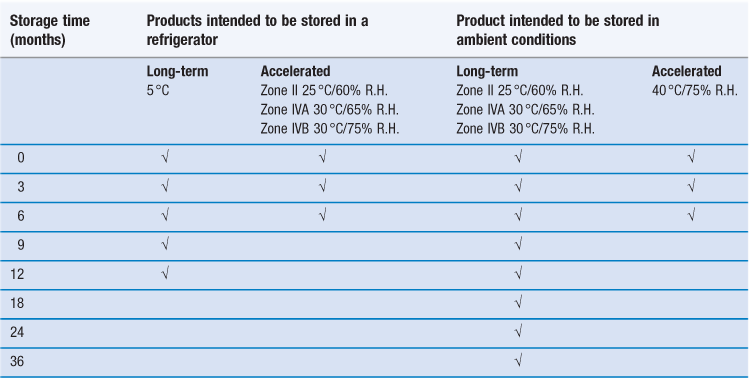
(Assessments should be made at least at the time points indicated by a tick.)
For products where refrigerated storage is envisaged, the 5 °C storage condition (actually, 5±3 °C is allowed, hence a range of 2–8 °C) represents realistic storage temperatures experienced in a refrigerator. In this case, the higher temperature/humidity accelerated degradation conditions are actually the appropriate ambient conditions, thus in effect simulating a non-functioning refrigerator.
Stability assessments for specific dosage forms
A comprehensive list of possible stability assessments (both chemical and physical) for a wide range of dosage forms is given in Table 49.4. It is not necessary to perform all possible assessments at each time point. Judgement can be made on this issue following preliminary results.
Table 49.4
Recommended stability assessments (physical and chemical) for a range of dosage forms
| Finished pharmaceutical product (chapter in this book where more information can be found) | Stability assessment |
| Tablets (Chapter 30) | Dissolution (or disintegration, if relevant), water content and hardness/friability. |
| Capsules (Chapters 33 and 34) | Hard capsules: brittleness, dissolution (or disintegration, if relevant), water content and level of microbial contamination. Soft capsules: dissolution (or disintegration, if relevant), level of microbial contamination, pH, leakage and pellicle formation. |
| Oral solutions, suspensions and emulsions (Chapters 24, 26 and 27) | Formation of precipitate, clarity (for solutions), pH, viscosity, extractables, level of microbial contamination. Additionally for suspensions: dispersibility, rheological properties, mean size and size distribution of particles. Also polymorphic conversion, if applicable. Additionally for emulsions: phase separation, mean size and distribution of dispersed globules. |
| Powders and granules for oral solution or suspension (Chapter 28) | Water content and reconstitution time. Reconstituted products (solutions and suspensions) should be evaluated as described under ‘Oral solutions suspensions and emulsions’, after preparation according to the recommended labelling, through the maximum intended use period. |
| Pressurized metered-dose inhalers and nasal aerosols (Chapters 37 and 38) | Dose content uniformity, labelled number of medication actuations per container meeting dose content uniformity, aerodynamic particle size distribution, microscopic evaluation, water content, leak rate, level of microbial contamination, valve delivery (shot weight), extractables/leachables from plastic and elastomeric components, weight loss, pump delivery, foreign particulate matter and extractables/leachables from plastic and elastomeric components of the container, closure and pump. Samples should be stored in upright, inverted and on-the-side orientations. For suspension-type aerosols: microscopic examination of appearance of the valve components and container’s contents for large particles, changes in morphology of the drug particles, extent of agglomerates, crystal growth, foreign particulate matter, corrosion of the inside of the container or deterioration of the gaskets. |
| Nasal sprays: solutions and suspensions (Chapter 38) | Clarity (for solution), level of microbial contamination, pH, particulate matter, unit spray medication content uniformity, number of actuations meeting unit spray content uniformity per container, droplet and/or particle size distribution, weight loss, pump delivery, microscopic evaluation (for suspensions), foreign particulate matter and extractables/leachables from plastic and elastomeric components of the container, closure and pump. |
| Topical, ophthalmic and otic (ear) preparations (Chapters 27, 39 and 41) | Includes ointments, creams, lotions, pastes, gels, solutions, eye drops and cutaneous sprays. For topical preparations: clarity, homogeneity, pH, suspendability (for lotions), consistency, viscosity, particle size distribution (for suspensions, when feasible), level of microbial contamination/sterility and weight loss (when appropriate). For ophthalmic or otic products (e.g. creams, ointments, solutions and suspensions): sterility, particulate matter and extractable volume. For cutaneous sprays: pressure, weight loss, net weight dispensed, delivery rate, level of microbial contamination, spray pattern, water content and particle size distribution (for suspensions). |
| Transdermal patches (Chapter 39) | In vitro release rates, leakage, level of microbial contamination/sterility, peel and adhesive forces. |
| Suppositories and pessaries (Chapter 42) | Softening range, disintegration and dissolution (at 37 °C). |
| Small volume parenterals (Chapter 36) | Colour, clarity (for solutions), particulate matter, pH, sterility, endotoxins. For powders for injection solution include in addition: reconstitution time, water content, clarity, colour, pH, sterility, pyrogen/endotoxin and particulate matter. It may be appropriate to consider monitoring of sterility after reconstitution into a product. For suspension for injection include in addition: particle size distribution, dispersibility and rheological properties. For emulsion for injection include in addition: phase separation, viscosity, mean size and size distribution of dispersed phase globules. |
| Large volume parenterals (Chapter 36) | Colour, clarity, particulate matter, pH, sterility, pyrogen/endotoxin and volume. |
(Adapted from recommendations of World Health Organization, 2009.)
Evaluation of results
The results of tests on samples subject to stability studies should remain within the specification set for the product. For instance, degradation of drug should be within allowable limits and the product’s appearance, smell and if relevant, taste should be acceptable.
It is common for the lower acceptable limit for the drug content of a product to be set at 90% of the labelled content of drug. This is not the same as 90% of the original concentration, because this will vary due to manufacturing variation and whether an overage was included during manufacturing.
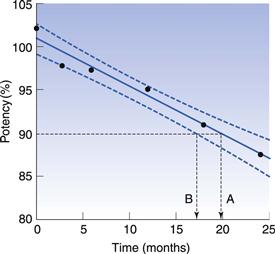
Various statistical tests are applied to ensure that the amount of drug remaining at the expiry date is above the lower acceptable limit. Figure 49.3 shows hypothetical stability results for a tablet product which, for simplicity, is assumed to degrade by zero-order kinetics (Chapter 7). Some scatter is seen in the results due to variability of the analytical technique and non-homogeneity within the product. Linear regression analysis enables the average rate of degradation to be determined, and this gives an estimated time of 20 months to reach 90% of the labelled potency. However, linear regression calculates average values, both of the slope of the regression line and of the intercept at the y-axis. Thus it is possible that the true shelf-life is less than this estimate. To avoid this possibility, the 95% confidence bounds for the regression are determined. These define the region within which there is a 95% probability that the true regression line lies (Fig. 49.3). The shelf-life is determined as the time at which the lower 95% confidence boundary crosses the lower specification limit, which in this example is at 17 months. This method of calculating shelf-life incorporates a safety margin to allow for the uncertainty in both the degradation rate and the initial drug content (the y-axis intercept value).
In cases where the degradation does not follow zero-order kinetics, appropriate mathematical transformation of the data is required before linear regression is performed. For example, for a drug degrading by first-order kinetics (Chapter 7), logarithmic transformation of the assay data is first performed. Knowledge of the order of the degradation reaction is obtained from studies performed at higher (stress) temperatures, which would have been performed earlier, probably at the preformulation stage (Chapter 23). This is because during long-term ambient testing, insufficient degradation would occur to allow accurate determination of the order of reaction.
As a minimum of three batches are subject to stability testing, the shelf-life is calculated for each batch separately and the shortest calculated value is selected as the product’s shelf-life. Increasing the number of data points available may have the effect of decreasing the width of the confidence boundary, making it closer to the average regression line. This would improve the estimate of the shelf-life. Pooling the data from all of the batches when calculating the 95% confidence limits therefore potentially gives a longer shelf-life. This is permitted if the slopes and intercepts of the regression lines for each batch are shown to be statistically equivalent by the statistical technique of analysis of covariance. Extrapolation of the data beyond the period for which data are available is not advisable (or allowable by regulatory authorities) because of the possibility that the degradation mechanism and kinetics may change.
Concluding comments
Pharmaceutical products tend to deteriorate due to chemical, physical and microbiological causes. Chemical degradation of the drug in a product is influenced by factors such as the presence of water, oxygen, light or incompatible ingredients in the product. Deterioration can be reduced in a number of ways. Careful formulation of the product is necessary to ensure adequate stability. This may be achieved through attention to general factors, such as appropriate selection of the solvent or pH, or by including specific stabilizing additives, such as an antioxidant or antimicrobial preservative. Packaging plays an important role in the prevention of product deterioration, for instance in protecting the contents from moisture. However, the packaging must avoid interactions such as sorption or the release of extractives. Storage conditions of the manufactured product (temperature, humidity and light exposure) must be appropriate. Adequate stability of the product needs to be demonstrated during its development before it can be assigned a shelf-life. Stress testing is valuable in predicting the likely stability of a product, but long-term testing is needed before a product can be marketed. In conclusion, stability is an important factor which may affect the quality and efficacy of all pharmaceutical products.
References
1. Allwood MC, Martin H. The extraction of diethylhexylphthalate (DEHP) from polyvinyl chloride components of intravenous infusion containers and administration sets by paclitaxel injection. International Journal of Pharmaceutics. 1996;127(1):65–71.
2. Dietz R, Feilner K, Gerst F, Grimm W. Drug stability testing: classification of countries according to climatic zone. Drugs Made in Germany. 1993;36(3):99–103.
3. Helm M, Castner T, Lampl L. Environmental temperature stress on drugs in prehospital emergency medical service. Acta Anaesthesiologica Scandinavica. 2003;47(4):425–429.
4. Lucas TI, Bishara RH, Seevers RH. A stability program for the distribution of drug products. Pharmaceutical Technology, July 2004;68–73.
5. McDonald C, Sunderland VB, Lau H, Shija R. The stability of amoxicillin sodium in normal saline and glucose (5%) solution in the liquid and frozen states. Journal of Clinical Pharmacy and Therapeutics. 1989;14(1):45–52.
6. Priston MJ, Grant IC, Hughes JM, Marquis PT. Pilot study on potential degradation of drug efficacy resulting from Antarctic storage, transport and field conditions. International Journal of Circumpolar Health. 2005;64(2):184–186.
7. World Health Organization. Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. Annex 2 of WHO Technical Report Series No. 953, 43rd Report of WHO Expert Committee on Specifications for Pharmaceutical Preparations Geneva, Switzerland: WHO; 2009.
Bibliography
1. Barnes AR, Nash S. Near-patient stability studies. Journal of Clinical Pharmacy and Therapeutics. 1996;21:49–55.
2. Byrn SR, Xu W, Newman AW. Chemical reactivity in solid-state pharmaceuticals: formulation implications. Advanced Drug Delivery Reviews. 2001;48:115–136.
3. Connors KA, Amidon GL, Stella VJ. Chemical Stability of Pharmaceuticals: a Handbook for Pharmacists. 2nd edn New York: Wiley; 1986.
4. Duarte Favaro RM, Iha MH, Pires Bianchi M. Liquid chromatographic determination of geometrical retinol isomers and carotene in enteral feeding formulas. de L Journal of Chromatography A. 2003;1021:125–132.
5. International Conference on Harmonisation of Technical Requirements for registration of pharmaceuticals for human use, Geneva, Switzerland. ICH Harmonised Tripartite Guideline (known as ‘ICH Guidelines’).
6. Q1A 2003 Stability testing of new drug substances and products.
7. Q1B 1996 Stability testing: Photostability testing of new drug substances and products.
8. Q1C 1996 Stability testing for new dosage forms.
9. Q1E 2003 Evaluation of stability data.
10. Q1C 1995 Stability testing of biotechnological/biological products.
11. Ma M, Lee T, Kwong E. Interaction of methylparaben preservative with selected sugars and sugar alcohols. Journal of Pharmaceutical Sciences. 2002;91:1715–1723.
12. Tashima T, Kawakami U, Harada M, et al. Isolation and identification of new oligomers in aqueous solution of glutaraldehyde. Chemical and Pharmaceutical Bulletin. 1987;35:4169–4180.
13. Vermes A, van der Sijs H, Guchelaar HJ. An accelerated stability study of 5–flucytosine in intravenous solution. Pharmacy World and Science. 1999;21:35–39.
14. Zahn M. A risk-based approach to establish stability testing conditions for tropical countries. Journal of Pharmaceutical Sciences. 2006;95:946–965.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Product stability and stability testing
Michael E. Aulton
Chapter contents
The stability of pharmaceutical products
Stabilization of pharmaceuticals
Key points
• Pharmaceutical products tend to deteriorate due to chemical, physical and microbiological causes.
• Careful formulation of the product is necessary to ensure adequate stability.
The stability of pharmaceutical products
Pharmaceutical products tend to deteriorate on storage. The shelf-life of a pharmaceutical product is the period of time during which, if stored correctly, it is expected to retain acceptable chemical, physical and microbiological stability. The expiry date, or expiration date, is the date given on the product’s primary and secondary packaging which represents the end of the shelf-life.
Stabilization of pharmaceuticals
Pharmaceuticals should be formulated and stored in a way that minimizes degradation. The following factors are relevant to most mechanisms of degradation. Oxidation and photodegradation are also important degradation mechanisms and these are dealt with separately later in this chapter.
Temperature
The rate of degradation reactions is markedly influenced by temperature. Storage of the product in a refrigerator (at 2–8 °C) is an option if the product is unstable at room temperature.
Using a freezer (at less than −15 °C) to store unstable formulations is sometimes adopted, however, this makes storage and distribution of the product inconvenient. Also, because a liquid product needs to be thawed before administration, there is the risk of degradation occurring if heat is used to achieve this. Moreover, some drugs, for instance amoxicillin are less stable in solution when frozen than when stored at refrigerator temperature (McDonald et al 1989). Freezing can cause degradation of biopharmaceuticals (see Chapter 46) and live vaccines, though the inclusion of a cryoprotectant, such as trehalose, in the formulation may protect against this.
Finished products are most at risk of exposure to unacceptable temperatures during transportation or storage in vehicles, such as in ambulances (Helm et al 2003, Lucas et al 2004, Priston et al 2005).
Solvent
Replacing an aqueous solvent in a formulation with a non-aqueous one is a potential means of avoiding hydrolysis.
The dielectric constant of a solvent is related to its polarity, more polar solvents having higher values. The dielectric constant can influence the rate at which charged species react. However, the practical considerations of choosing a solvent for a formulation, such as its toxicity and compatibility with the drug, usually outweigh consideration of any effect due to the solvent’s dielectric constant.
Solid dosage forms of a drug, such as tablets or capsules, are usually more stable than liquid ones. However, reactions can occur in water adsorbed onto the surface of a drug particle or other dosage form component. This is why poorly stored aspirin tablets may smell of acetic acid, formed by the hydrolysis of aspirin. Injection formulations of unstable drugs, such as penicillins, can be formulated as freeze-dried powders, which are reconstituted with water or saline (0.9% w/v sodium chloride solution) immediately before administration.
Suspension formulations are often more stable than a solution formulation of the same drug because much of the drug is protected within the insoluble particles.
Acid and base catalysis
A catalyst is a species that accelerates the rate of a reaction without itself being consumed in the reaction. Hydrolysis is often catalysed by hydrogen ions (which exist as H3O+ in solution) or hydroxyl ions. The pH of an aqueous formulation is therefore a critical factor which determines its stability. Specific acid catalysis is catalysis by hydrogen ions and specific base catalysis is catalysis by hydroxyl ions.
Investigation of the relationship between pH and the degradation rate of a drug is performed during the preformulation phase of drug development (Chapter 23). Plotting the logarithm to base 10 of the first-order reaction rate constant against pH may yield useful information about the degradation mechanism. A typical curve is shown in Figure 49.1a. The increase in degradation rate at low pH is due to specific acid catalysis. The increase in rate seen at high pH is due to specific base catalysis. Straight lines with gradients of −1 in the acidic region and +1 in the basic region are characteristic of specific acid and specific base catalysed hydrolysis. The flat region of the curve is largely due to uncatalysed hydrolysis. The product should be maintained at a pH within this region for optimal stability. Cefuroxime and other cephalosporin antibiotics typically show this shape of graph.
For some drugs, such as many penicillins, the uncatalysed reaction with water is relatively less important than that shown in Figure 49.1a, so there is no flat base to the curve and a V-shaped graph results (Fig. 49.1b). In this case, the pH needs to be more precisely controlled than in the previous example because there is a narrower region of optimal stability.
Many drug molecules undergo ionization, to an extent that often depends on the pH. These typically give a curve as shown in Figure 49.1c. The ionized and unionized forms of the drug degrade at different rates. Therefore, the rate of reaction changes as the pH influences the relative proportion of ionized drug present and this gives an inflection point on the graph. Aspirin is an example of a drug which shows this characteristic.
Other species in a formulation besides H3O+ or OH− may act as acids and bases and thus catalyse degradation reactions. This is known as general acid catalysis and general base catalysis respectively. Buffer ions are a common cause of this, so careful selection of buffer for use in a formulation is needed. Hydrolysis of the amide bond of the antimicrobial drug chloramphenicol is catalysed by several common buffers, including phosphate and acetate. Borate buffer, however, does not catalyse degradation and is used, for example, to buffer eye drops.
Ionic strength
The ionic strength of a medium is related to the concentration of ionic species in it. Changing the ionic strength by adding electrolyte to a solution has some influence on the rate of many degradation reactions. This effect is not high enough to be of importance in the formulation of drug solutions. However, it can be important in laboratory experiments to investigate the influence of pH on degradation rate. In this case, care should be taken to ensure the ionic strength of the various buffer solutions used is kept the same in order to avoid interference with the results.
Light
Containers made from tinted glass protect the product from light to some extent because they allow less ultraviolet light to penetrate than those which are untinted (Chapter 47). Placing the product in an opaque outer container such as a cardboard box is also an option, but it must be borne in mind that the patient might not return the product to its secondary packaging following use.
Oxygen
Oxidation reactions are less influenced by temperature than most other degradation reactions, so low-temperature storage may be less successful as a stabilization option. Flushing containers with an inert gas such as nitrogen before they are sealed will reduce the amount of oxygen in the product. However this technique will not remove all oxygen and is best suited for single-use containers such as ampoules.
Oxidation reactions are generally promoted by high pH, so aqueous products which are susceptible to oxidation should be formulated at as low a pH as possible.
Heavy metal ions, such as Cu2+ and Fe3+, catalyse oxidation reactions, acting at the initiation and propagation stages. These ions are present at trace levels in all formulations. A chelating agent such as ethylenediamine tetraacetic acid (EDTA) or citric acid has a stabilizing effect by binding to the heavy metal ions and preventing them from acting as catalysts.
Chelating agents are usually used in combination with an antioxidant. Antioxidants act in one of two ways. They may act as oxygen scavengers – oxygen is removed from the formulation by reacting preferentially with the antioxidant. The other mechanism by which antioxidants act is to terminate free radical reactions. The antioxidant forms a free radical which is relatively unreactive and so cannot contribute to free radical propagation. Sodium metabisulphite and ascorbic acid are examples of water-soluble antioxidants used in aqueous formulations. Ascorbyl palmitate, butylated hydroxytoluene and α-tocopherol are oil-soluble antioxidants used in oily formulations.
Physical stability
Common physical causes of instability are summarized in Table 49.1. Most of these are due to changes in the physical properties of the dosage form, either spoiling the product’s appearance or reducing its effectiveness. The other potential problems are loss of drug due to sorption or evaporation, or contamination of the product by extractables from the container.
Molecules of the drug or other formulation component may be lost from a formulation by adsorption onto the surface of the container or closure or by absorption of molecules into plastic or rubber containers or closures. Adsorption and absorption often operate together and are collectively known as sorption. Non-polar molecules are susceptible to sorption to plastics and rubber (Chapter 47). For instance, diazepam is lost from solutions in contact with plastic packaging. Loss of antimicrobial preservative to rubber closures is a problem with injection dosage forms (Chapter 36). Sorption is enhanced where the drug (or preservative) is present at low concentration. If the drug molecule ionizes, the pH of the solution may influence the extent of sorption because the unionized form of the molecule, being less polar than the ionized form, may undergo more sorption.
Glyceryl trinitrate (nitroglycerin) evaporates from tablets, where it can then be lost by sorption to plastic packaging. To avoid this, glyceryl trinitrate tablets need to be packaged in glass bottles with aluminium-lined closures.
Plastic packaging materials may be permeable to water vapour. Aqueous products packaged in plastic containers may therefore lose water on storage and the drug content becomes more concentrated.
The term extractable or extractive is used to describe any material which is released from packaging materials into the dosage form. The common packaging material polyvinyl chloride (PVC) is rendered flexible by the addition of a plasticizer (e.g. diethylhexylphthalate, DEHP) which may migrate into injection solutions. This is a particular problem where the solution contains a non-aqueous solvent or surfactant. Paclitaxel, a cytotoxic drug, needs to be administered in dilute solution by slow intravenous infusion. The injection also contains polyoxyethylated castor oil (a material used to solubilize the drug) with ethanol as a cosolvent. Paclitaxel injection therefore cannot be added to infusion fluids contained in PVC because it induces DEHP extraction. Glass or polyethylene infusion containers should therefore be used instead (Allwood and Martin 1996).
Glass containers can release hydroxyl ions into an aqueous product, changing its pH (Chapter 47). This may especially occur during heating in an autoclave, and surface-treated glass is available to minimize hydroxyl ion extraction for formulations where this is of concern.
Microbiological stability
Deterioration due to the presence of microorganisms can either render the product harmful to the patient or have an adverse effect on the product’s properties (Chapter 50
[/not-level-membership-for-basic-science-category]
