Chapter 52 Proarrhythmia Syndromes
Proarrhythmia usually is defined as provocation of a new arrhythmia or aggravation of an existing arrhythmia during therapy with a drug at concentrations usually not considered toxic.1–9 However, drug-drug interactions that may lead to unrecognized elevations in drug concentrations, for example, an IKr (delayed rectifier potassium [K+] current)–blocking drug and a macrolide antibiotic, that can precipitate proarrhythmia. Other therapies (e.g., devices or ablation procedures) or pathophysiological conditions may also cause proarrhythmia.2,10,11 The various proarrhythmia syndromes are summarized in Box 52-1, and the drugs and conditions most frequently associated with proarrhythmia are listed in Table 52-1. Most types of proarrhythmia occur in the setting of structural heart disease, but proarrhythmia may also occur in individuals without apparent heart disease. Antiarrhythmic drugs have a higher risk of causing ventricular proarrhythmia, but drugs that are considered generally safe may cause proarrhythmia in susceptible individuals.2,4–6
Table 52-1 Drugs and Conditions Associated with Torsades de Pointes Ventricular Tachycardia
| DRUG CLASS | SPECIFIC DRUGS |
|---|---|
| ANTIARRHYTHMIC | |
| Class Ia | Quinidine |
| Procainamide | |
| Disopyramide | |
| Class Ic | Propafenone |
| Class III | Amiodarone |
| d, l-Sotalol | |
| d-Sotalol | |
| Dofetilide | |
| Ibutilide | |
| Azimalide | |
| Psychotropic | Haloperidol |
| Phenothiazines | |
| Risperidone | |
| Tricyclic antidepressants | |
| Tetracyclic antidepressants | |
| Diuretics | Furosemide |
| Hydrochlorothiazide | |
| Indapamide | |
| Metolazone | |
| Antimicrobial | Erythromycin |
| Clarithromycin | |
| Ciprofloxacin | |
| Levofloxacin | |
| Moxifloxacin | |
| Ofloxacin | |
| Trimethoprim-sulfamethoxazole | |
| Pentamidine | |
| Antifungal | Ketoconazole |
| Fluconazole | |
| Itraconazole | |
| Antihistamines | Diphenhydramine |
| Terfenadine* | |
| Astemizole* | |
| Cholinergic antagonists | Cisapride* |
| Narcotics | Methadone |
| DRUGS FOR OTHER CONDITIONS | |
| Bradycardia | Complete heart block or significant bradycardia |
| Electrolyte abnormalities | Hypokalemia |
| Hypomagnesemia | |
| Hypocalcemia | |
| Nervous system injury | Subarachnoid hemorrhage |
* No longer commercially available.
Drug-Induced Long QT Syndrome and Torsades De Pointes Ventricular Tachycardia
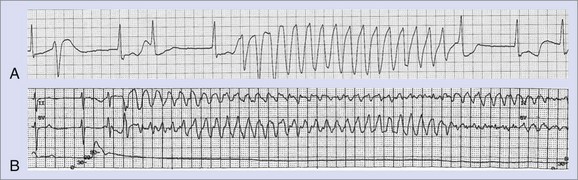
The greatest risk of ventricular proarrhythmia has been identified with drugs that prolong the Q-T interval on the electrocardiogram (ECG), which can predispose to a potentially life-threatening form of polymorphic ventricular tachycardia (VT), termed torsades de pointes VT (Figure 52-1).1–9 Other pathophysiological conditions that result in excessive prolongation of the Q-T interval may also cause this proarrhythmia (see Table 52-1).1–9 Syncope occurring early after the initiation of quinidine therapy was recognized as early as the 1920s, but it was not until the advent of continuous electrocardiographic monitoring that “quinidine syncope” was recognized to be caused by this pause-dependent polymorphic VT.12 The term torsades de pointes was initially coined by Dessertenne in 1966 to describe this polymorphic tachyarrhythmia that is often characterized by beat-to-beat changes in the QRS axis.13 Torsades de pointes VT often terminates spontaneously and may cause syncope, but it may transition into a sustained polymorphic VT or ventricular fibrillation (VF) and cause sudden cardiac death (SCD) (see Figure 52-1).
The most common drugs and conditions associated with excessive Q-T interval prolongation and torsades de pointes VT are summarized in Table 52-1. An up-to-date list of drugs associated with torsades de pointes VT is maintained at www.longqt.org. Antiarrhythmic drugs that prolong the ventricular action potential duration (APD), including class Ia drugs (quinidine, procainamide, disopyramide) and class III drugs (amiodarone, sotalol, dofetilide, ibutilide, azimilide), have all been reported to cause torsades de pointes VT.2–9 Rarely, the class Ic drug propafenone has been reported to cause torsades de pointes VT. Associated bradycardia and electrolyte abnormalities (hypokalemia, hypomagnesemia, or both), often caused by diuretic use, increase the probability of torsades de pointes VT in the setting of class Ia or III antiarrhythmic drug use. Although amiodarone significantly prolongs the Q-T interval, the incidence of torsades de pointes VT associated with amiodarone use is relatively low.2,14 Diuretics, by virtue of causing profound hypokalemia or hypomagnesemia, may be associated with torsades de pointes VT when other drugs are not used. The diuretic indapamide, which blocks the slowly activating component of the slow delayed rectifier K+ current (Iks), may cause excessive Q-T interval prolongation and torsades de pointes VT.15
Noncardiovascular drugs associated with torsades de pointes VT include tricyclic antidepressants (e.g., imipramine), antipsychotic drugs (e.g., haloperidol, risperidone, phenothiazine), antihistamines (e.g., diphenhydramine, terfenadine, astemizole), macrolide antibiotics (e.g., erythromycin, clarithromycin), quinolones (e.g., ciprofloxacin, levofloxacin, moxifloxacin, ofloxacin), pentamadine, anti-fungal agents, and cisapride.1–9 A number of drugs, including cisapride, terfenadine, and astemizole, have been withdrawn from the market because of an unacceptably high incidence of torsades de points VT. The risk of torsades de pointes VT caused by terfenadine or cisapride is attributed to the accumulation of these drugs in plasma as a consequence of coadministration of another drug (e.g., erythromycin or ketoconazole) that inhibits their cytochrome P450 3A4–mediated biotransformation to noncardioactive metabolites.5,6,8
Isolated profound bradycardia (most often in the setting of complete heart block) may be associated with profound Q-T interval prolongation and torsades de pointes VT, particularly in the setting of drugs or other factors that prolong the Q-T interval.13 Neurologic events such as subarachnoid hemorrhage have been associated with marked repolarization abnormalities, Q-T interval prolongation, and torsades de pointes VT. Adrenergic stimuli in the setting of dobutamine infusion have also been reported to cause torsades de pointes VT. Case reports have also implicated anesthetic agents, including halothane, as a cause for torsades de pointes VT.
Cellular Mechanisms of Torsades De Pointes Ventricular Tachycardia
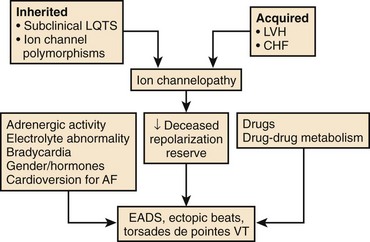
An increase in inward currents, a reduction of outward currents, or a combination of both during phase 2 and 3 of the ventricular action potential leads to prolongation of the ventricular action potential, which manifests as Q-T interval prolongation on the ECG (Figure 52-2).1–7 Mutations of genes encoding K+, sodium (Na+), and calcium (Ca2+) channels have been linked to the congenital long QT syndrome (LQTS). Disease states such as left ventricular hypertrophy and heart failure are associated with a reduction in outward currents caused by the downregulation of transient outward K+ current (Ito), IKr, the inward rectifier K+ current (IK1), or all, which occurs in a spatially heterogeneous manner.16,17 A reduction in net outward currents, an increase in inward currents, or a combination of both, facilitates the development of early afterdepolarizations (EADs) that develop in M cells or Purkinje cells because of the activation or reactivation of arrhythmogenic inward currents, including Ca2+ channels or the Na+-Ca2+ exchange current.1–718 These EADs may initiate torsades de pointes VT, which is then maintained by re-entry (see Figure 52-2).1,6 Intracellular calcium overload, as in the setting of heart failure, may facilitate the development of EADs in the setting of acquired LQTS.17
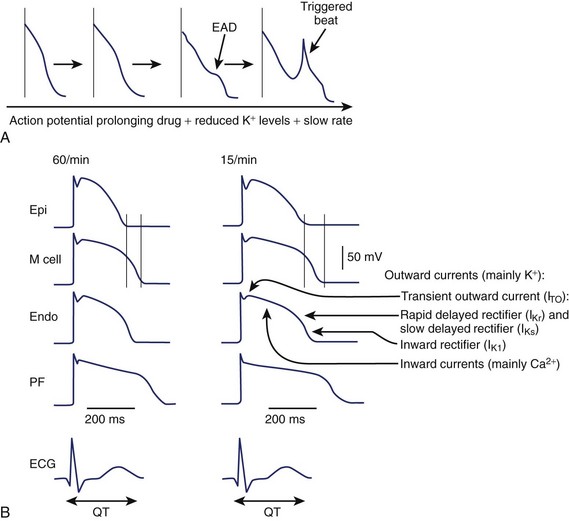
In the ventricular myocardium, the APD varies in a spatially heterogeneous manner because of differences in current densities in specific cell types (Purkinje fibers and endocardial, mid-myocardial, or epicardial cells).1,6,16,18 The ventricular APD is longest in the mid-myocardial layer (M cells) and shorter in the epicardial and endocardial regions. This dispersion of ventricular repolarization is increased in disease states, including ventricular hypertrophy, and may be further exaggerated by the administration of a drug that prolongs the Q-T interval.16 Marked spatial dispersion of ventricular repolarization appears to be a prerequisite for torsades de pointes VT because it provides the functional substrate for re-entry.19 Functional block may vary on a beat-to-beat basis, contributing to spiral re-entrant waves, which explains the polymorphic nature of the arrhythmia.1,6,20
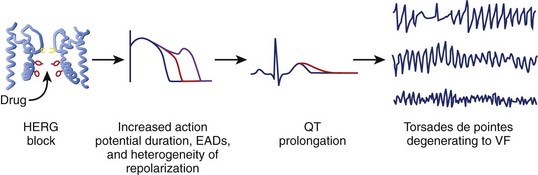
Although multiple ionic mechanisms may contribute to the development of torsades de pointes VT, the vast majority of drugs associated with torsades de pointes VT are thought to act by blockade of the rapid component of IKr (Figure 52-3).1,3,7 The HERG gene (also known as KCNH2) regulates the expression of IKr. The molecular structure of the drug-binding domain in the HERG channel makes it more vulnerable to blockade by a variety of drugs compared with other K+ channels. A small proportion of individuals who develop drug-induced torsades de pointes may have subclinical forms of congenital LQTS.3–621 Some polymorphisms of genes responsible for the expression or regulation of the ion channels involved in congenital LQTS have also been implicated in drug-induced torsades de pointes.3,4,21 Mutations in HERG may enhance blockade of the channel by certain drugs.
A normal QT at the start of antiarrhythmic drug therapy does not exclude the risk for proarrhythmia.2,5,7 The concept of reduced repolarization reserve and the risk of proarrhythmia was initially introduced by Roden.5,22 Cardiac repolarization is determined by IKr and IKs as well as other inward and outward currents during the plateau of the action potential. A defect in one of these currents may not be apparent if other currents that contribute to repolarization are intact. Roden has hypothesized that “physiologic processes such as drug metabolism or cardiac repolarization include multiple redundancies” and that “loss of these redundancies due to congenital or acquired conditions may enhance susceptibility to proarrhythmic responses even in the absence of a baseline phenotype.”5 For example, decreased expression of IKs because of a mutation in one of the genes responsible for expression of IKs, or because of a pathophysiological state such as heart failure or atrial fibrillation (AF), may not become apparent until the individual is treated with a drug that blocks IKr, which results in marked prolongation of the Q-T interval and proarrhythmia in this setting of reduced repolarization reserve (Figure 52-4).
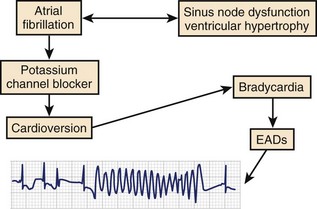
The period immediately after cardioversion to sinus rhythm from AF appears to be a time of great risk for torsades de pointes VT.9,23,24 The potential mechanism underlying this event is shown in Figure 52-5. The sustained tachycardia and the neurohumoral changes associated with AF may alter some electrophysiological properties, including downregulation of repolarizing currents. Restoration of sinus rhythm is often associated with bradycardia and action potential prolongation that could be further exaggerated by the downregulation of K+ channels. The action potential prolongation also facilitates increased sarcoplasmic calcium, predisposing to triggered activity. A similar situation may occur after atrioventricular (AV) junction ablation for the management of persistent AF. The early period after ablation is a particularly vulnerable period for SCD, which is believed to be caused by bradycardia-dependent polymorphic VT.25 Successful AV junction ablation is associated with abrupt slowing of the heart rate. Sudden slowing of the heart rate and the associated prolongation of the ventricular APD after ablation could increase the likelihood of EADs. Consistent with this hypothesis, we have observed exaggerated bradycardia-dependent prolongation of the Q-T interval and increased Q-T interval dispersion in patients with significant left ventricular dysfunction after total AV junction ablation.26
Risk Factors for Torsades de Pointes Ventricular Tachycardia
The risk factors for torsades de pointes VT are summarized in Box 52-2.1–9,27,28 The presence of multiple risk factors (e.g., female gender, cardiac hypertrophy, electrolyte abnormalities, and prior history of VT) dramatically increase the risk of developing torsades de pointes VT. Diuretic use in the setting of normal serum potassium concentrations has been reported to be a risk factor for torsades de pointes VT.29 This may be caused by total body potassium depletion that may not be reflected by serum potassium concentrations or the direct action potential–prolonging effects of some diuretic drugs.
Women have a twofold to threefold greater risk of developing torsades de pointes VT during treatment with action potential–prolonging drugs independent of other risk factors.27,30 In the general population, women have longer Q-T intervals compared with men. In experimental models, females have longer ventricular APDs and increased transmural dispersion of repolarization compared with males, which may be explained, in part, by reduced expression of IKr as well as other K+ currents.28 Testosterone shortens the ventricular APD, whereas estrogen prolongs the APD. Gender-specific differences in drug transport and metabolism may also contribute to the modulation of the pharmacokinetics of IKr-blocking drugs.28
The risk of torsades de pointes VT is generally related to drug dose.6,31 However, torsades de pointes VT has been described as an idiosyncratic reaction that occurs during quinidine therapy, and syncope secondary to torsades de pointes occurs within hours of initiation of this drug therapy.6,12 The development of torsades de pointes VT at low quinidine concentrations may be secondary to the IKr-blocking effects in conjunction with augmentation of the late Na+ current resulting in a decrease in net repolarizing currents; however, at higher quinidine concentrations, the more dominant Na+ channel–blocking effects balance the IKr block and restore the repolarization reserve.6 The risk of torsades de pointes VT associated with sotalol or dofetilide therapy clearly increases significantly with dose, which is further exacerbated by other risk factors, including renal failure.31 The incidence of ventricular proarrhythmia during sotalol initiation at doses up to 320 mg/day was 1.8%; this incidence increased to 4.5% at doses up to 480 mg/day and to 6.8% at doses greater than 640 mg/day.
Some cardiac disease processes such as ventricular hypertrophy or significant left ventricular dysfunction are associated with significant prolongation of the APD.32–34 These electrophysiological changes are caused by changes in repolarizing current densities, including decreases in Ito, IK1, IKr, and IKs.16,35 Changes in these repolarizing currents in their densities, balance, or both may alter the response of specific channel blockers, resulting in exaggerated prolongation of the APD and increased dispersion of ventricular repolarization that provides the substrate for torsades de pointes VT.
Electrocardiographic Harbingers of Torsades de Pointes Ventricular Tachycardia
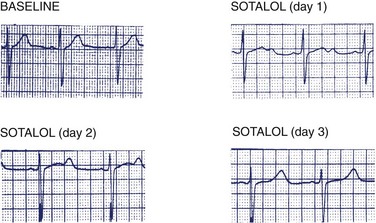
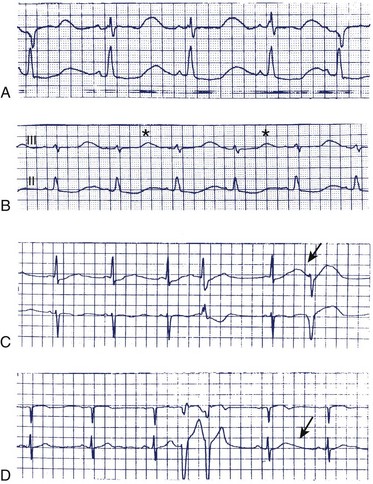
Excessive Q-T interval prolongation and morphologic changes in the T wave usually precede the development of torsades de pointes VT.6,29,34 Figure 52-6 illustrates the changes in the Q-T interval and T-wave morphology during sotalol initiation that precede the development of torsades de pointes VT. Note the development of Q-T interval prolongation and a U wave, both of which become progressively more prominent over the 48 to 72 hours after the initiation of sotalol administration. The magnitude of the Q-T interval prolongation may be exaggerated by abrupt slowing of the heart rate (e.g., after a compensatory pause). The prominence of the U waves may vary on a beat-to-beat basis and may be modified by abrupt changes in heart rate. Figure 52-7 illustrates and Box 52-3 summarizes some of the electrocardiographic harbingers of torsades de pointes VT. Note the excessive Q-T interval prolongation, the development of prominent U waves, T-wave alternans, post-extrasystolic pause exaggeration of the Q-T interval prolongation, and T-wave morphologic changes and clustering of repetitive polymorphic ventricular premature beats.
Dispersion of Ventricular Repolarization and Torsades de Pointes Ventricular Tachycardia
Abrupt changes in the Q-T interval and in the relationship between the Q-T interval and heart rate may develop immediately before the onset of torsades de pointes VT.7,36 Increased Q-T interval dispersion, defined as the difference between the shortest and the longest Q-T intervals measured on the 12-lead ECG, may precede the development of torsades de pointes VT. Class Ia antiarrhythmic drugs were shown to significantly increase Q-T interval dispersion compared with the drug-free state in patients who developed torsades de pointes VT during drug therapy.19,34 In contrast, patients who did not develop this proarrhythmia during drug therapy did not manifest a significant increase in Q-T interval dispersion compared with those in the drug-free state. Amiodarone did not increase Q-T interval dispersion in the patients who developed torsades de pointes VT on class Ia drugs. The low incidence of torsades de pointes VT observed during amiodarone therapy likely reflects its homogeneous effects on ventricular repolarization, which may, in part, be caused by calcium channel–blocking and β-adrenergic–blocking effects.19,34 The time from the peak of the T wave to its return to baseline has been shown to correlate directly with the development of torsades de points VT.7 Amiodarone therapy causes less bradycardia-induced prolongation of the APD of M cells isolated from patients with heart failure compared with M cells isolated from patients with heart failure who were not treated with amiodarone.37 The reduced dispersion of ventricular repolarization likely explains the low incidence of torsades de pointes VT occurring in association with amiodarone compared with class I or other class III antiarrhythmic drugs.
Treatment
The initial treatment for torsades de pointes VT is magnesium sulfate (1 to 2 g IV bolus).38 Magnesium should be administered even if the serum magnesium level is normal. The drug implicated should be discontinued, and other reversible causes should be corrected (e.g., electrolyte abnormalities, bradycardia). Overdrive atrial or ventricular pacing is effective when the arrhythmia recurs despite magnesium administration.39 Isoproterenol infusion may also be used to increase heart rate and shorten the Q-T interval. Atropine has been reported to eliminate torsades de pointes VT, presumably via the increase in heart rate reversing the Q-T interval prolongation.40 Serum K+ should be maintained in the high normal range (4.5 to 5 mEq/L). Elevation of the extracellular K+ concentration increases outward K+ currents and reduces the magnitude of drug block of IKr, thus shortening ventricular repolarization.41 Modest increases in serum K+ may reverse the repolarization abnormalities associated with some variants of LQTS or abnormalities associated with antiarrhythmic drug use and cause Q-T interval shortening as well as reduced Q-T interval dispersion.42 The mechanism by which magnesium prevents torsades de pointes VT is uncertain. Magnesium does not shorten the APD or the Q-T interval and likely exerts its effect by blocking Ca2+ channels. In experimental models, Na+ channel blockers have been reported to antagonize the effects of class Ia and III drugs on APD and to prevent proarrhythmia.43,44 K+ channel–activating compounds have also been reported to prevent EAD and proarrhythmia in animal models.45 However, these agents have not become mainstream therapies for torsades de pointes VT.
Should Antiarrhythmic Drug Therapy Be Initiated in the Hospital?
The safety of initiating antiarrhythmic drug therapy in the outpatient setting is still being debated.31 Approximately 50% of episodes of ventricular proarrhythmia occur within 72 hours of initiation of antiarrhythmic drug therapy for AF. It has been estimated that 1200 patients would have to be monitored for 3 days to prevent one torsades de pointes VT–related death. The Symptomatic Atrial Fibrillation Investigative Research on Dofetilide (SAFIRE-D) investigators reported a low incidence (0.8%) of torsades de pointes VT during the initial 3 days of dofetilide therapy in patients with AF.46 Nevertheless, the guidelines for dofetilide initiation approved by the U.S. Food and Drug Administration (FDA) require that this drug be initiated in the hospital. Similar recommendations can be found in the product monograph for sotalol approved for the treatment of AF. ECG monitoring during drug initiation should be undertaken for high-risk patients but does not appear to be cost effective for low-risk patients. Moreover, efforts at the prevention of proarrhythmia must be continued throughout the course of antiarrhythmic drug therapy.
Proarrhythmia Caused by Sodium Channel Blocking Drugs
Sudden Cardiac Death After Myocardial Infarction
Cardiac Arrhythmia Suppression Trials
The Cardiac Arrhythmia Suppression Trial (CAST) investigators reported that patients with frequent ventricular premature beats after a myocardial infarction (MI) who were treated with flecainide or encainide had a higher mortality rate compared with patients treated with placebo.47 The proarrhythmic risk was not just limited to the early phase of drug therapy but increased over the course of the trial. Subgroup analyses suggested that recurrent myocardial ischemia played an important role in the increased risk of arrhythmic death.48–50 Total mortality rate and risk of cardiac arrest were higher in patients who had sustained a non–Q-wave MI and in those who had not received thrombolytic therapy. The relative risk (RR) of death or cardiac arrest was 10.6 in patients sustaining a non–Q-wave MI versus 1.45 in those patients sustaining a Q-wave MI.49 This observation led to the hypothesis that recurrent ischemia in the setting of class Ic drug therapy altered the electrophysiological milieu in the infarct region, predisposing to a proarrhythmic response. Consistent with this hypothesis, concomitant β-blocker and flecainide or encainide use was not associated with an increased RR of death.49
CAST-II demonstrated the increased proarrhythmic potential of the class I antiarrhythmic drug moricizine during early dosage titration.51 Death or nonfatal cardiac arrest during the drug initiation phase was significantly greater in patients treated with moricizine compared with those treated with placebo. The RR of death or nonfatal cardiac arrest during the initial 2 weeks of moricizine therapy was 5.6. Although survival during long-term follow-up was similar in the moricizine and placebo groups, CAST-II was terminated early because it was deemed unlikely that a survival benefit from moricizine would be observed if the trial were completed.
CAST and meta-analyses of class I anti-arrhythmic drugs in patients with ischemic heart disease, left ventricular dysfunction, or both indicate that these drugs increase the risk of lethal cardiovascular events.52,53 Therefore such drugs are contraindicated in patients with a history of prior MI. By extension, many clinicians also believe that these drugs are relatively contraindicated in any patient with ischemic heart disease or risk factors for ischemic heart disease, as well as in patients with significant left ventricular dysfunction.
Mechanisms of Ventricular Proarrhythmia in Ischemic Heart Disease
Na+ channel–blocking agents slow conduction in atrial and ventricular muscles in a use-dependent manner (i.e., greater conduction block occurs at faster heart rates). Some Na+ channel–blocking agents (encainide and flecainide) produce extensive block at slow physiological heart rates, which increases the risk of proarrhythmia.2,54 By causing excessive slowing of conduction that exceeds changes in refractoriness in diseased tissue, class Ic antiarrhythmic drugs may stabilize a re-entrant circuit that has previously been unable to sustain re-entry and thus establish the electrophysiological substrate for sustained VT or convert nonsustained VT to sustained monomorphic VT.2,54 This model of re-entry assumes that the drug alters only conduction velocity. In reality, many Na+ channel–blocking drugs alter conduction velocity and refractoriness in a heterogeneous manner that depends on the physiological milieu. Increased dispersion of tissue refractoriness may also promote ventricular re-entry.
Experimental studies demonstrated that encainide and flecainide enhanced induction of sustained VT in dogs with prior MI without VT inducible at baseline.55–58 These drugs cause preferential conduction slowing in the peri-infarct zone, which results in unidirectional block or marked slowing of conduction in the direction transverse to fiber orientation that facilitates ventricular re-entry. In experimental models of acute ischemia, the presence of a Na+ channel–blocking agent significantly increased the incidence of spontaneous ventricular fibrillation (VF).54 Class I drugs, including lidocaine and flecainide, produce exaggerated effects on conduction velocity and repolarization in ischemic tissue, contributing to the substrate for VF.58–61 Ranger et al evaluated the concentration dependence of flecainide proarrhythmia in dogs with acute or chronic MI.56 Proarrhythmia was observed in 79% of dogs with chronic MI versus 55% of dogs with acute ischemia. However, the concentration dependence of flecainide proarrhythmia differed substantially between the two groups: proarrhythmia occurred at therapeutic concentrations in the setting of acute MI and most likely manifested as VF, whereas 20-fold higher concentrations of flecainide were required to induce proarrhythmia in the dogs with chronic MI, and proarrhythmia manifested as induced sustained monomorphic VT.56
Studies in experimental models have demonstrated that β-blockers, in the setting of acute ischemia, reduce the proarrhythmic effects of Na+ channel blockers.62,63 Under normal conditions, isoproterenol reverses flecainide’s effect on Na+ channel block. However, under conditions of membrane depolarization, as would be expected in ischemia, isoproterenol amplifies flecainide’s effects on sodium channel block.63 Thus an enhanced sympathetic tone in the setting of acute ischemia may further modulate the interaction between a class I drug and ischemic tissue to promote proarrhythmia.
Other Cardiac Disease States
Information about the proarrhythmic potential of class I Na+ channel–blocking drugs in cardiac disease models without ischemic heart disease is limited. However, cardiac hypertrophy and ventricular dysfunction in the setting of volume overload are associated with heterogeneous abnormalities of ventricular conduction and repolarization.17 In the setting of cardiac disease, the resting membrane potential is likely elevated in some cells, which might predispose them to greater antiarrhythmic drug-induced Na+ channel conduction block. In addition, frequent ventricular premature beats, which may occur in the setting of left ventricular dysfunction, may be associated with greater conduction delays in the presence of class I drugs and thus predispose to ventricular re-entry. Some Na+ channel–blocking drugs produce exaggerated slowing of conduction in models of disease states.54,64 Thus any Na+ channel–blocking drug may likely produce exaggerated electrophysiological responses in diseased ventricular tissue with responses occurring in a heterogeneous manner, thereby predisposing to ventricular proarrhythmia. Therefore selective Na+ channel–blocking drugs are relatively contraindicated in all patients with significant left ventricular dysfunction.
Incessant Ventricular Tachycardia Secondary to Sodium Channel–Blocking Drugs
Some patients treated with Na+ channel–blocking drugs, particularly class Ic drugs such as flecainide or propafenone, may develop slow, incessant VT.2,65 This usually occurs in the setting of structural heart disease (e.g., prior MI) and marked conduction slowing because the circulating wavefront is less likely to encounter refractory tissue and be abolished. Incessant VT may be hemodynamically tolerated because of its slow rate. However, the arrhythmia may degenerate into a hemodynamically significant VT or VF and be lethal.65 This arrhythmia is usually observed with higher or toxic drug doses or in the setting of other pathophysiological conditions that exaggerate the effects of the drug on conduction (e.g., acidosis, hyperkalemia, or concomitant Na+ channel blockers, including phenytoin). Incessant monomorphic VT may also be observed with overdose of tricyclic antidepressants. The anticholinergic effects of tricyclic antidepressants cause an increase in sinus rate, which results in exaggerated Na+ channel block.
Treatment
The antiarrhythmic drug should be discontinued, and electrolyte abnormalities or acidosis should be corrected. Hypertonic saline or sodium bicarbonate may reverse the conduction slowing and terminate the arrhythmia.66,67 However, Na+ may exacerbate heart failure, so caution is required in patients with significant left ventricular dysfunction. β-blockers may be beneficial because the magnitude of conduction slowing is less at slower heart rates. Na+ channel–blocking drugs should be avoided.
Ventricular Proarrhythmia in Wolff-Parkinson-White Syndrome
In the setting of ventricular pre-excitation, adenosine, digoxin, β-blockers, verapamil, and diltiazem may promote conduction of AF in an antegrade fashion across the accessory pathway. If the antegrade refractory period of the accessory pathway is short, this may provoke VF. Accordingly, these drugs are contraindicated in this setting.68
Atrial Flutter with 1:1 Atrioventricular Conduction
In patients with atrial flutter, Na+ channel–blocking drugs may significantly slow the atrial flutter cycle length such that 1:1 AV conduction develops and the ventricular rate increases (Figure 52-8).69,70 This phenomenon has been reported with quinidine and has been attributed to quinidine’s vagolytic effects. Slowing of the atrial flutter cycle length also occurs in association with flecainide and propafenone, particularly when rate-controlling (AV node–blocking) antiarrhythmic drugs are not prescribed. During atrial flutter, the atrial rate typically is approximately 300 beats/min. In most individuals not on AV node blocking drugs, 2:1 AV conduction is present and the ventricular response is approximately 150 beats/min. Flecainide or propafenone may slow the atrial flutter rate to approximately 200 to 220 beats/min. This may allow the ventricular response to increase from 150 to 200 to 220 beats/min with 1:1 AV conduction. An intraventricular conduction delay pattern is often observed because of the marked rate-dependent conduction block associated with these drugs at higher heart rates. In fact, this arrhythmia may be misdiagnosed as VT. This form of proarrhythmia may occur in patients receiving class I antiarrhythmic drugs for the treatment of AF. Such patients may have intermittent atrial flutter, or the antiarrhythmic drug may alter the electrophysiological substrate in the atria to predispose to atrial flutter.
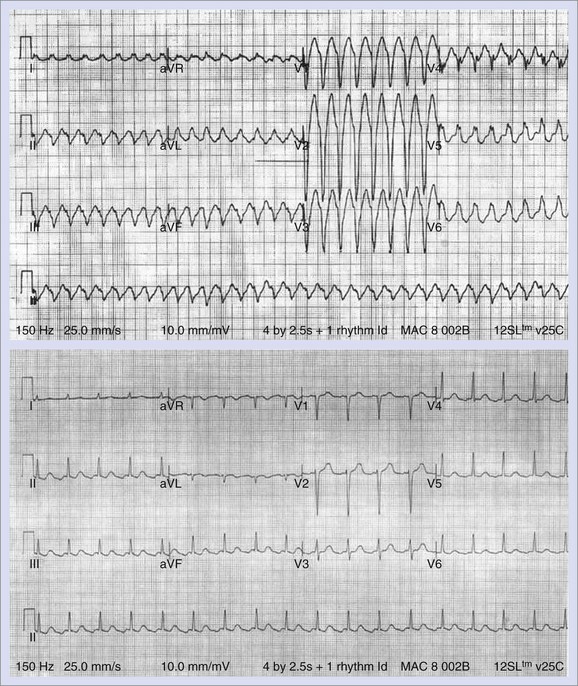
Increased Cardiac Mortality Associated with Class III Drugs
The Survival with Oral D-Sotalol (SWORD) trial investigators reported increased cardiovascular mortality with d-sotalol compared with placebo in patients with left ventricular dysfunction.71,72 As in CAST, excess mortality rates on active drug continued to be observed through the course of follow-up. The mechanism or mechanisms of the increased mortality rate are uncertain. Torsades de pointes VT is certainly a possibility. However, VF in the setting of acute ischemia precipitated by ischemia-mediated exaggeration of antiarrhythmic drug effects (similar to the mechanism proposed in CAST) is also another plausible cause. In contrast, the Danish Investigation of Arrhythmia and Mortality on Dofetilide (DIAMOND) did not report an increased mortality rate associated with dofetilide compared with placebo in patients with left ventricular dysfunction.73 Differences in trial design likely explain these apparently divergent outcomes. In DIAMOND, drug therapy was initiated in the hospital during ECG monitoring, and therapy was stopped if excessive Q-T interval prolongation was observed. In contrast, the SWORD trial design favored drug doses associated with Q-T interval prolongation. Furthermore, patients in the SWORD trial were generally healthy and hence less likely to benefit substantially from drug therapy.
Induced Proarrhythmia and Device Therapy
Anti-tachycardia pacing therapies for the termination of sustained VT are not always effective and may, at times, accelerate VT to VF or to a more hemodynamically unstable form of VT (Figure 52-9).10,74 The risk of acceleration of VT is relatively small (4%) and is similar for ramp and burst pacing modalities. Syncope in the setting of an implantable cardioverter-defibrillator (ICD) may be caused by ventricular proarrhythmia provoked by anti-tachycardia pacing therapies; the risk may be reduced by careful programming of the ICD. Sometimes, concomitant antiarrhythmic drug therapy may increase the efficacy of anti-tachycardia pacing therapies by slowing the VT rate and may minimize the risk of anti-tachycardia pacing–induced proarrhythmia. The risk of ventricular arrhythmia associated with implantable atrial defibrillators has been reported to be extremely low when the cardioversion shock is synchronized to the R wave and the shock is delivered at a slow heart rate.75 False-positive detection of AF caused by far-field R-wave oversensing can lead to inappropriate therapies, which may rarely initiate VF.76 Atrial anti-tachycardia pacing for treatment of atrial flutter or tachycardia may initiate sustained AF, although the risk is generally low.
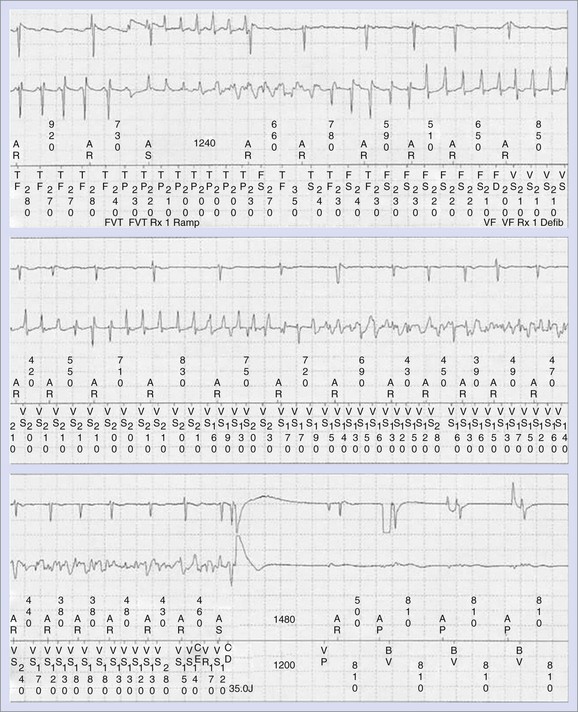
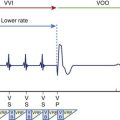
Normal operation of the pacing system may, at times, promote ventricular proarrhythmia. Sweeney et al analyzed the intracardiac initiation sequence of more than 1300 VT or VF episodes from two recent clinical studies.77 They found pacing-associated short-long-short sequences at the onset of 21% to 35% of all VT or VF episodes. The short-long-short sequences were further classified as pacing-permitted or pacing-facilitated onsets. Pacing-permitted onset of VT or VF was observed more frequently with the managed ventricular pacing algorithm and the VVI mode, whereas pacing-facilitated onset of VT or VF was more commonly observed with the DDD pacing mode. An example of VT initiated after a short-long-short sequence during pacing associated with the managed ventricular pacing algorithm is shown in Figure 52-10.
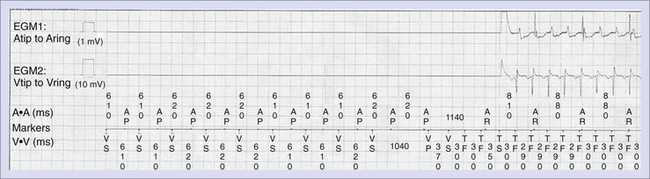
Precipitation of ventricular arrhythmias, including cases of VT storm immediately after cardiac resynchronization therapy (CRT), has been reported.10,78,79 Left ventricular pacing may need to be discontinued to prevent this. Left ventricular pacing via epicardial coronary sinus lead implantation reverses the transmural activation sequence, delaying endocardial depolarization and repolarization. The resulting increased dispersion of ventricular repolarization may contribute to the ventricular proarrhythmia that is occasionally observed with cardiac resynchronization therapy. Although some clinical trial data have suggested that the risk of SCD may be increased in patients treated with CRT versus an ICD, a recent meta-analysis of 14 trials did not suggest any excess risk of SCD from CRT (RR, 1.07; 95% confidence interval [CI], 0.79 to 1.46).80,81
Deaths related to lead failure–induced proarrhythmia have been reported in patients with ICDs.10
Antiarrhythmic Drug Effects on Defibrillation Thresholds
Some antiarrhythmic drugs, by virtue of their effects on passive and active membrane properties, may alter defibrillation thresholds. Na+ channel–blocking drugs have been reported to have no effect on defibrillation thresholds and do not increase them. These divergent effects appear, in part, to depend on the experimental model or the type of anesthesia used.82–84 In patients with left ventricular dysfunction, lidocaine has been reported to increase defibrillation thresholds.84 Reports suggest that drugs that prolong the APD and exert predominantly class III antiarrhythmic effects (e.g., procainamide, sotalol, azimilide, dofetilide) do not have any significant effect on defibrillation energy requirements, or they reduce them.84–89 The effects of amiodarone on defibrillation thresholds are being debated. Some studies have reported no change in defibrillation thresholds during long-term amiodarone therapy, whereas other investigators have reported an increase in defibrillation thresholds.89 This ongoing debate concerns reassessment of marginal defibrillation thresholds at initiation of an antiarrhythmic drug known to increase the defibrillation threshold.
Proarrhythmia After Ablation Procedures for Atrial Fibrillation
Left atrial tachycardia or flutter is a recognized complication of ablation techniques aimed at resolving AF.11,90,91 Ostial ablation of the of the pulmonary veins may result in focal atrial tachycardias in 1% of patients, whereas circumferential ablation approaches are associated with an 18% to 25% risk of focal or re-entrant left atrial tachycardias. Many of these arrhythmias resolve during follow-up; therefore repeat ablation may not be required.
Key References
The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406-412.
The Cardiac Arrhythmia Suppression Trial II (CAST) Investigators. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:227-233.
Dessertenne F. La tachycardie ventriculaire a deux foyers opposes variables. Arch Mal Coeur Vaiss. 1966;59:263-272.
Gillis AM. Effects of antiarrhythmic drugs on QT interval dispersion—relationship to antiarrhythmic action and proarrhythmia. Prog Cardiovasc Dis. 2000;42:385-396.
Hreiche R, Morissette P, Turgeon J. Drug-induced long QT syndrome in women: Review of current evidence and remaining gaps. Gend Med. 2008;5:124-135.
Roden DM. Cellular basis of drug-induced torsades de pointes. Br J Pharmacol. 2008;154:1502-1507.
Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005;115:2025-2032.
Tung R, Zimetbaum P, Josephson ME. A critical appraisal of implantable cardioverter-defibrillator therapy for the prevention of sudden cardiac death. J Am Coll Cardiol. 2008;52:1111-1121.
Tzivoni D, Banai S, Schuger C, et al. Treatment of torsade de pointes with magnesium sulfate. Circulation. 1988;77:392-397.
Waldo AL, Camm AJ, de Ruyter H, et al. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet. 1996;348:7-12.
1 Roden DM. Cellular basis of drug-induced torsades de pointes. Br J Pharmacol. 2008;154:1502-1507.
2 Roden DM. Mechanisms and management of proarrhythmia. Am J Cardiol. 1998;82:49I-57I.
3 Roden DM. Long QT syndrome: Reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59-69.
4 Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005;115:2025-2032.
5 Roden DM. Proarrhythmia as a pharmacogenomic entity: A critical review and formulation of a unifying hypothesis. Cardiovasc Res. 2005;67:419-425.
6 Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013-1022.
7 Ahmad K, Dorian P. Drug-induced QT prolongation and proarrhythmia: An inevitable link? Europace. 2007;9(Suppl 4):iv16-iv22.
8 Heist EK, Ruskin JN. Drug-induced proarrhythmia and use of QTc-prolonging agents: Clues for clinicians. Heart Rhythm. 2005;2(2 Suppl):S1-S8.
9 Shantsila E, Watson T, Lip GY. Drug-induced QT-interval prolongation and proarrhythmic risk in the treatment of atrial arrhythmias. Europace. 2007;9(Suppl 4):iv37-iv44.
10 Tung R, Zimetbaum P, Josephson ME. A critical appraisal of implantable cardioverter-defibrillator therapy for the prevention of sudden cardiac death. J Am Coll Cardiol. 2008;52:1111-1121.
11 Daoud EG, Weiss R, Augostini R, et al. Proarrhythmia of circumferential left atrial lesions for management of atrial fibrillation. J Cardiovasc Electrophysiol. 2006;17:157-165.
12 Selzer A, Wray HW. Quinidine syncope—paroxysmal ventricular fibrillation occurring during treatment of chronic atrial arrhythmias. Circulation. 1964;30:17-26.
13 Dessertenne F. La tachycardie ventriculaire a deux foyers opposes variables. Arch Mal Coeur Vaiss. 1966;59:263-272.
14 Middlekauff HR, Stevenson WG, Saxon LA, et al. Amiodarone and torsades de pointes in patients with advanced heart failure. Am J Cardiol. 1995;76:499-502.
15 Fiset C, Drolet B, Hamelin BA, et al. Block of IKs by the diuretic agent indapamide modulates cardiac electrophysiological effects of the class III antiarrhythmic drug dl-sotalol. J Pharmacol Exp Ther. 1997;283:148-156.
16 Gillis AM, Mathison HJ, Kulicz E, et al. Dispersion of ventricular repolarization and ventricular fibrillation in left ventricular hypertrophy: Influence of selective potassium channel blockers. J Pharmacol Exp Ther. 2000;292:381-386.
17 Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270-278.
18 Antzelevitch C, Sicouri S. Clinical relevance of cardiac arrhythmias generated by afterdepolarizations. Role of M cells in the generation of U waves, triggered activity and torsade de pointes. J Am Coll Cardiol. 1994;23:259-277.
19 Hii JTY, Wyse DG, Gillis AM, et al. Precordial QT interval dispersion as a marker of torsade de pointes: Disparate effects of class la antiarrhythmic drugs and amiodarone. Circulation. 1992;86:1376-1382.
20 Asano Y, Davidenko JM, Baxter WT, et al. Optical mapping of drug-induced polymorphic arrhythmias and torsade de pointes in the isolated rabbit heart. J Am Coll Cardiol. 1997;29:831-842.
21 Lehtonen A, Fodstad H, Laitinen-Forsblom P, et al. Further evidence of inherited long QT syndrome gene mutations in antiarrhythmic drug-associated torsades de pointes. Heart Rhythm. 2007;4:603-607.
22 Roden DM. Taking the “idio” out of “idiosyncratic”: Predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;20:1029-1034.
23 Choy AM, Darbar D, Dell’Orto S, et al. Exaggerated QT prolongation after cardioversion of atrial fibrillation. J Am Coll Cardiol. 1999;34:396-401.
24 Lafuente-Lafuente C, Mouly S, Longás-Tejero MA, et al. Antiarrhythmic drugs for maintaining sinus rhythm after cardioversion of atrial fibrillation: A systematic review of randomized controlled trials. Arch Intern Med. 2006;166:719-728.
25 Conti JB, Mills RMJr., Woodard DA, et al. QT dispersion is a marker for life-threatening ventricular arrhythmias after atrioventricular nodal ablation using radiofrequency energy. Am J Cardiol. 1997;79:1412-1414.
26 Raj SR, Gillis AM, Mitchell B, et al. Paced QT dispersion and QT morphology after radiofrequency atrioventricular junction ablation: impact of left ventricular function. Pacing Clin Electrophysiol. 2003;26:662-668.
27 Houltz B, Darpo B, Swedberg K, eWolbrette DL. Risk of proarrhythmia with class III antiarrhythmic agents: Sex-based differences and other issues. Am J Cardiol. 2003;91:39D-44D.
28 Hreiche R, Morissette P, Turgeon J. Drug-induced long QT syndrome in women: Review of current evidence and remaining gaps. Gend Med. 2008;5:124-135.
29 Houltz B, Darpö B, Swedberg K, et al. Effects of the IKr-blocker almokalant and predictors of conversion of chronic atrial tachyarrhythmias to sinus rhythm. A prospective study. Cardiovasc Drugs Ther. 1999;13:329-338.
30 Lehmann MH, Hardy S, Archibald D, et al. Sex difference in risk of torsade de pointes with d, l-Sotalol. Circulation. 1996;94:2534-2541.
31 Thibault B, Nattel S. Optimal management with class I and class III antiarrhythmic drugs should be done in the outpatient setting: Protagonist. J Cardiovasc Electrophysiol. 1999;10:472-481.
32 Volders PGA, Sipido KR, Vos MA, et al. Cellular basis of biventricular hypertrophy and arrhythmogenesis in dogs with chronic complete atrioventricular block and acquired torsade de pointes. Circulation. 1998;98:1136-1147.
33 Vos MA, de Groot SHM, Verduyn SC, et al. Enhanced susceptibility for acquired torsade de pointes arrhythmias in the dog with chronic, complete AV block is related to cardiac hypertrophy and electrical remodeling. Circulation. 1998;98:1125-1135.
34 Gillis AM. Effects of antiarrhythmic drugs on QT interval dispersion—relationship to antiarrhythmic action and proarrhythmia. Prog Cardiovasc Dis. 2000;42:385-396.
35 Gillis AM, Geonzon RA, Mathison HJ, et al. The effects of barium, dofetilide and 4-aminopyridine (4-AP) on ventricular repolarization in normal and hypertrophied rabbit heart. J Pharmacol Exp Ther. 1998;289:262-270.
36 Hinterseer M, Thomsen MB, Beckmann BM, et al. Beat-to-beat variability of QT intervals is increased in patients with drug-induced long-QT syndrome: A case control pilot study. Eur Heart J. 2008;29:185-190.
37 Drouin E, Charpentier F, Gauthier C, et al. Electrophysiological characteristics of cells spanning the left ventricular wall of human heart: Evidence for the presence of M cells. J Am Coll Cardiol. 1995;26:185-192.
38 Tzivoni D, Banai S, Schuger C, et al. Treatment of torsade de pointes with magnesium sulfate. Circulation. 1988;77:392-397.
39 Viskin S, Fish R, Roth A, et al. Prevention of torsade de pointes in the congenital long QT syndrome: Use of a pause prevention pacing algorithm. Heart. 1998;79:417-419.
40 Tan HL, Wilde AAM, Peters RJ. Suppression of torsades de pointes by atropine. Heart. 1998;79:99-100.
41 Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:407-411.
42 Tan HL, Alings M, Van Olden RW, et al. Long-term (subacute) potassium treatment in congenital HERG-related long QT syndrome (LQTS2). J Cardiovasc Electrophysiol. 1999;10:229-233.
43 Chezalviel-Guilbert F, Davy J-M, Poirier J-M, et al. Mexiletine antagonizes effects of sotalol on QT interval duration and its proarrhythmic effects in a canine model of torsade de pointes. J Am Coll Cardiol. 1995;26:787-792.
44 Sicouri S, Antzelevitch D, Heilmann C, et al. Effects of sodium channel block with mexiletine to reverse action potential prolongation in in vitro models of the long QT syndrome. J Cardiovasc Electrophysiol. 1997;8:1280-1290.
45 Brosch SF, Studenik C, Heistracher P. Abolition of drug-induced early afterdepolarizations by potassium channel activators in guinea-pig Purkinje fibers. Clin Exp Pharmacol Physiol. 1998;25:225-230.
46 Singh S, Zoble RG, Yellen L, et al. Efficacy and safety of oral dofetilide in converting to and maintaining sinus rhythm in patients with chronic atrial fibrillation or atrial flutter: The symptomatic atrial fibrillation investigative research on dofetilide (SAFIRE-D) study. Circulation. 2000;102:2385-2390.
47 The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406-412.
48 Anderson J, Platia E, Hallstrom A, et al. Interaction of baseline characteristics with the hazard of encainide, flecainide and moricizine therapy in patients with myocardial infarction: A possible explanation for increased mortality in the cardiac arrhythmia suppression trial (CAST). Circulation. 1994;90:2843-2852.
49 Greenberg HM, Dwyer EMJr., Hochman JS, et al. Interaction of ischemia and encainide/flecainide treatment: A proposed mechanism for the increased mortality in CAST I. Br Heart J. 1995;74:631-635.
50 Kennedy HL. Beta-blocker prevention of proarrhythmia and proischemia: Clues from CAST, CAMIAT, and EMIAT. Am J Cardiol. 1997;80:1208-1211.
51 The Cardiac Arrhythmia Suppression Trial II (CAST) Investigators. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:227-233.
52 Coplen SE, Antman EM, Berlin JA, et al. Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion. A meta-analysis of randomized control trials. Circulation. 1990;82:1106-1116.
53 Flaker GC, Blackshear JL, McBride R, et al. Antiarrhythmic drug therapy and cardiac mortality in atrial fibrillation. J Am Coll Cardiol. 1992;20:527-532.
54 Nattel S. Experimental evidence for proarrhythmic mechanisms of antiarrhythmic drugs. Cardiovasc Res. 1998;37:567-577.
55 Brugada J, Boersma L, Kirchhof C, et al. Proarrhythmic effects of flecainide. Experimental evidence for increased susceptibility to reentrant arrhythmias. Circulation. 1991;84:1808-1818.
56 Ranger S, Nattel S. Determinants and mechanisms of flecainide-induced promotion of ventricular tachycardia in anesthetized dogs. Circulation. 1995;92:1300-1311.
57 Coromilas J, Saltman AE, Waldecker B, et al. Electrophysiological effects of flecainide on anisotropic conduction and reentry in infarcted canine hearts. Circulation. 1995;91:2245-2263.
58 Restivo M, Yin H, Caref EB, et al. Reentrant arrhythmias in the subacute infarction period. The proarrhythmic effect of flecainide acetate on functional reentrant circuits. Circulation. 1995;91:1236-1246.
59 Carson DL, Cardinal R, Savard P, et al. Relationship between an arrhythmogenic action of lidocaine and its effects on excitation patterns in acutely ischemic porcine myocardium. J Cardiovasc Pharmacol. 1996;8:126-136.
60 Aupetit JF, Timour Q, Loufoua-Moundanga J, et al. Profibrillatory effects of lidocaine in the acutely ischemic porcine heart. J Cardiovasc Pharmacol. 1995;25:810-816.
61 Kou WH, Nelson SD, Lynch JJ, et al. Effect of flecainide acetate on prevention of electrical induction of ventricular tachycardia and occurrence of ischemic ventricular fibrillation during the early postmyocardial infarction period: Evaluation in a conscious canine model of sudden death. J Am Coll Cardiol. 1987;9:359-365.
62 Myerberg RJ, Kessler KM, Cox MM, et al. Reversal of proarrhythmic effects of flecainide acetate and encainide hydrochloride by propranolol. Circulation. 1989;80:1571-1579.
63 Cragun KT, Johnson SB, Packer DL. β-Adrenergic augmentation of flecainide-induced conduction slowing in canine Purkinje fibers. Circulation. 1997;96:2701-2708.
64 Yuan F, Pinto JM, Li Q, et al. Characteristics of I(K) and its response to quinidine in experimental healed myocardial infarction. J Cardiovasc Electrophysiol. 1999;10:844-854.
65 Winkle RA, Mason JW, Griffin JC, et al. Malignant ventricular tachyarrhythmias associated with the use of encainide. Am Heart J. 1981;102:857-863.
66 Bellet S, Hamden G, Somlyo A, et al. A reversal of the cardiotoxic effects of procainamide by molar sodium lactate. Am J Med Sci. 1959;237:177-189.
67 Bajaj AK, Woosley RL, Roden DM. Acute electrophysiologic effects of sodium administration in dogs treated with O-desmethyl encainide. Circulation. 1989;80:994-1002.
68 Exner DV, Muzyka T, Gillis AM. Proarrhythmia in patients with the Wolff-Parkinson-White syndrome after standard doses of intravenous adenosine. Ann Intern Med. 1995;122:351-352.
69 Crijns HJ, van Gelder IC, Lie KI. Supraventricular tachycardia mimicking ventricular tachycardia during flecainide treatment. Am J Cardiol. 1988;62:1303-1306.
70 Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117:141-150.
71 Waldo AL, Camm AJ, de Ruyter H, et al. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet. 1996;348:7-12.
72 Pratt CM, Camm AJ, Cooper W, et al. Mortality in the survival with oral d-sotalol (SWORD) trial: Why did patients die? Am J Cardiol. 1998;81:869-876.
73 Torp-Pedersen C, Moller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med. 1999;341:857-865.
74 Vlay LC, Vlay SC. Pacing induced ventricular fibrillation in internal cardioverter defibrillator patients: A new form of proarrhythmia. Pacing Clin Electrophysiol. 1997;20:132-133.
75 Tse HF, Lau CP, Sra JS, et al. Atrial fibrillation detection and R-wave synchronization by Metrix implantable atrial defibrillator: Implications for long-term efficacy and safety. The Metrix Investigators. Circulation. 1999;99:1446-1451.
76 Gillis AM, Unterberg-Buchwald C, Schmidinger H, Massimo S, et al. GEM III AT Worldwide Investigators. Safety and efficacy of advanced atrial pacing therapies for atrial tachyarrhythmias in patients with a new implantable dual chamber cardioverter-defibrillator. J Am Coll Cardiol. 2002;40:1653-1659.
77 Sweeney MO, Ruetz LL, Belk P, et al. Bradycardia pacing-induced short-long-short sequences at the onset of ventricular tachyarrhythmias: A possible mechanism of proarrhythmia? J Am Coll Cardiol. 2007;50:614-622.
78 Basu Ray I, Fendelander L, Singh JP. Cardiac resynchronization therapy and its potential proarrhythmic effect. Clin Cardiol. 2007;30:498-502.
79 Kantharia BK, Patel JA, Nagra BS, et al. Electrical storm of monomorphic ventricular tachycardia after a cardiac-resynchronization-therapy-defibrillator upgrade. Europace. 2006;8:625-628.
80 McAlister FA, Wiebe N, Ezekowitz JA, et al. Meta-analysis: Beta-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784-794.
81 Catanchin A, Anderson L, Jones S, et al. When life-saving devices terminate life. J Cardiovasc Electrophysiol. 2008;19(3):316-318.
82 Natale A, Jones DL, Kim YH, et al. Effects of lidocaine on defibrillation threshold in the pig: Evidence of anesthesia related increase. Pacing Clin Electrophysiol. 1991;14:1239-1244.
83 Stevens SK, Haffajee CI, Naccarelli GV, et al. Effects of oral propafenone on defibrillation and pacing thresholds in patients receiving implantable cardioverter-defibrillators. Propafenone Defibrillation Threshold Investigators. J Am Coll Cardiol. 1996;28:418-422.
84 Echt DS, Gremillion ST, Lee JT, et al. Effects of procainamide and lidocaine on defibrillation energy requirements in patients receiving implantable cardioverter defibrillator devices. J Cardiovasc Electrophysiol. 1994;5:752-760.
85 Dorian P, Newman D, Sheahan R, et al. d-Sotalol decreases defibrillation energy requirements in humans: A novel indication for drug therapy. J Cardiovasc Electrophysiol. 1996;7:952-961.
86 Qi XQ, Newman D, Dorian P. Azimilide decreases defibrillation voltage requirements and increases spatial organization during ventricular fibrillation. J Interv Card Electrophysiol. 1999;3:61-67.
87 Bhatia A, Nangia V, Solis J, et al. Biventricular pacing and QT interval prolongation. J Cardiovasc Electrophysiol. 2007;18:623-627.
88 Simon RD, Sturdivant JL, Leman RB, et al. The effect of dofetilide on ventricular defibrillation thresholds. Pacing Clin Electrophysiol. 2009;32(1):24-28.
89 Hohnloser SH, Dorian P, Roberts R, et al. Effect of amiodarone and sotalol on ventricular defibrillation threshold: The optimal pharmacological therapy in cardioverter defibrillator patients (OPTIC) trial. Circulation. 2006;114:104-109.
90 Katritsis D, Wood MA, Shepard RK, et al. Atrial arrhythmias following ostial or circumferential pulmonary vein ablation. J Interv Card Electrophysiol. 2006;16:123-130.
91 Chugh A, Oral H, Lemola K, Hall B, et al. Prevalence, mechanisms, and clinical significance of macroreentrant atrial tachycardia during and following left atrial ablation for atrial fibrillation. Heart Rhythm. 2005;2(5):464-471.