[level-membership-for-basic-science-category]
Chapter 8 Principles of Toxicology
| Abbreviations | |
|---|---|
| ATP | Adenosine triphosphate |
| CNS | Central nervous system |
| CO | Carbon monoxide |
| DNA | Deoxyribonucleic acid |
| EDTA | Ethylenediamine tetraacetic acid |
| GI | Gastrointestinal |
Therapeutic Overview
Mechanisms of Action
Many toxic substances are known primarily to affect a specific target organ, such as the liver, nervous system, lungs, or kidneys, whereas others target the immune system or may affect normal fetal and cell growth and development. It is important to note, however, that although certain anatomical or functional characteristics may predispose organs to injury, designation of toxicants as “organ specific” (e.g., neurotoxic, cardiotoxic) is somewhat misleading, for several reasons. First, toxic substances may have adverse effects throughout the body, and although a chemical may primarily affect one organ, other organs may also incur less notable injury. Second, a toxic response in one tissue will undoubtedly have serious consequences for other tissues depending on the characteristics of the chemical (physical state, site of exposure, dose, and duration of exposure) and of the patient (general health, nutritional state, age, sex, enzyme induction, immune response, antioxidant concentrations). Thus a minor insult to a compromised organ may cause unexpected injury to that organ and may also affect other organs.
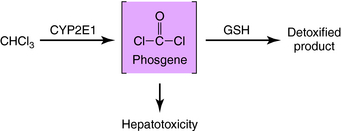
Because of a dual blood supply from the hepatic artery and the portal vein, the liver may be exposed to toxicants entering from the systemic circulation (e.g., via inhalation) or from the splanchnic circulation (absorbed from the gastrointestinal [GI] tract). The leaky capillary system of the hepatic sinusoids promotes the extraction of toxicants from the blood into the liver. The liver contains enzymes that metabolize many endogenous and exogenous chemicals (see Chapter 2). In some cases chemical modification produced by biotransformation results in bioactivation (production of toxic-reactive metabolites). This is exemplified by the bioactivation of chloroform to phosgene (Fig. 8-1). Compounds that enhance the activity of CYP2E1 (enzyme-inducing agents) or reduce the hepatic concentration of glutathione can exacerbate chloroform-induced liver injury. Other compounds that can cause liver injury after metabolism include several solvents (carbon tetrachloride, halogenated benzenes) and carcinogens (aflatoxin B, aromatic amines).
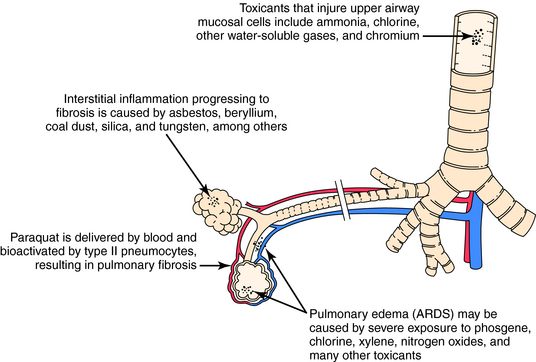
The blood can serve as the route of exposure of the lung in the case of paraquat, a herbicide taken up by type II pneumocytes. Biotransformation in the lung yields a reactive intermediate that undergoes redox cycling, which produces reactive oxygen species that injure the cell. Although exposure to paraquat usually occurs after ingestion or skin contamination, death results from pulmonary injury. Similarly, certain pyrrolidine alkaloids are metabolized in the liver to compounds that circulate in the blood and produce toxicity in the lung. The actions of toxicants on pulmonary tissue are shown in Figure 8-2.
The kidney is also susceptible to toxicants. The mechanisms of renal injury are similar to those in other organs but also include unique mechanisms. Delivery of blood-borne toxicants to the kidney is high, because the kidney receives 25% of cardiac output, and its functions include filtering, concentrating, and eliminating toxicants. As water is reabsorbed, the concentration of chemicals in the tubule can increase to toxic levels. In some cases the concentration may exceed the solubility of a chemical and lead to precipitation and obstruction of the affected area (Fig. 8-3).
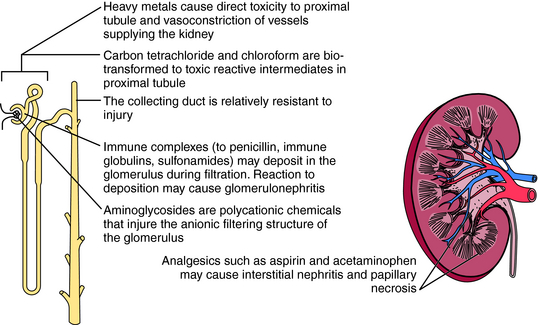
The kidney can compensate for excessive chemical exposure because, like many organs, it has a reserve mass. Thus tissue injury equal to one entire kidney must occur before loss of function is clinically apparent. The kidney also replaces lost functional capacity by hypertrophy. In addition, the kidney has developed binding proteins to remove heavy metals. For example, metallothioneins avidly bind cadmium, protecting the kidney and other organs from toxicity. However, renal toxicity will develop if exposure exceeds the binding capacity or if a large amount of the cadmium-metallothionein complex accumulates.
In addition to being a target for chemical-mediated injury, the immune system may mediate injury by producing hypersensitivity reactions—adverse events caused by an immune response to foreign antigens (see Chapter 6). Indeed, cell-mediated hypersensitivity initiated by T lymphocytes may lead to contact dermatitis as a consequence of exposure to nickel and several industrial chemicals.
Pharmacokinetics
Generally the means by which a toxicant reaches its target organ is governed by the same pharmacokinetic principles that govern the actions of therapeutic drugs (see Chapter 2). These principles also provide the basis for methods to reduce, reverse, or prevent toxicity of many substances.
It is generally too late to prevent cell or organ damage after the toxicant reaches its target, although the effects of many toxicants may still be minimized at this stage. Compounds metabolized to reactive intermediates are good examples. An antidote that prevents biotransformation can minimize injury produced by a reactive intermediate. The competitive antagonism of methanol or ethylene glycol by ethanol is based on this principle (see Chapter 32). Similarly, drugs that inhibit certain cytochrome P450 isozymes can reduce biotransformation of some toxicants, either reducing or enhancing their effects.
Relationship of Mechanisms of Action to Chemical Toxicity
Treatment is focused on preventing cyanide from reaching its target, cytochrome oxidase. Because cyanide has a high affinity for ferric iron, ferric iron in the form of oxidized hemoglobin can be provided by administration of sodium nitrite. This converts hemoglobin to methemoglobin, the ferric form, which effectively competes with cyanide for cytochrome a3, forming cyanmethemoglobin. Cyanide is removed from the body after being converted to thiocyanate by the enzyme rhodanese and is ultimately excreted in the urine. Sodium thiosulfate is administered to facilitate thiocyanate formation. An alternative is to administer hydroxocobalamin, which reacts with cyanide to produce cyanocobalamin. Because hydroxocobalamin and cyanocobalamin have little toxicity, they hold promise for the treatment of cyanide poisoning.
Mechanisms involved in heavy metal toxicity are poorly understood. Many metals function as essential enzyme cofactors. Substitution by a similar but toxic metal may produce enzymatic dysfunction. Metals are generally very reactive and may bind key sulfhydryl groups in active centers of enzymes. Besides causing direct metal toxicity, metals can also produce hypersensitivity reactions. Nickel, chromium, gold, and others cause cell-mediated (type IV) hypersensitivity reactions (Table 8-1).
| Metal | Mechanism | Target Organs |
|---|---|---|
| Arsenic | Reacts with sulfhydryl groups | Peripheral neurons |
| GI tract | ||
| Interferes with oxidative phosphorylation | Liver | |
| Cardiovascular system | ||
| Lead | Reacts with sulfhydryl groups | Hematopoietic system |
| Central and peripheral nervous systems | ||
| Interferes with heme synthesis | Kidney | |
| Direct toxic effect | Central nervous system | |
| Mercury | Reacts with sulfhydryl groups | Central and peripheral nervous systems |
| Some forms have direct cytotoxic effects | Kidney | |
| GI tract | ||
| Respiratory tract |
Treatment for metal toxicity focuses on increasing excretion of the metal from the body. Chelator drugs bind the metal between two or more functional groups to form a complex that is excreted in urine. Specific chelators work best in the removal of certain metals. The uses of chelators are summarized in Table 8-2.
TABLE 8–2 Metals Chelated by Therapeutic Agents
| Chelator | Metal |
|---|---|
| Succimer | Lead, arsenic, mercury |
| Deferoxamine | Iron |
| EDTA | Lead |
| Penicillamine | Copper, lead |
| British anti-Lewisite | Lead, arsenic, mercury |
EDTA, Ethylenediamine tetraacetic acid.
Low-concentration lead exposure may pose special risks for children. Subtle neurological injuries including depressed IQ scores and learning disorders have been reported. This may be due to enhanced accumulation in the immature nervous system. Lead may cross the blood-brain barrier more easily in children, and their CNS may also be less capable of removing it. Children with blood lead concentrations exceeding 10 µg/dL are considered to be at risk.
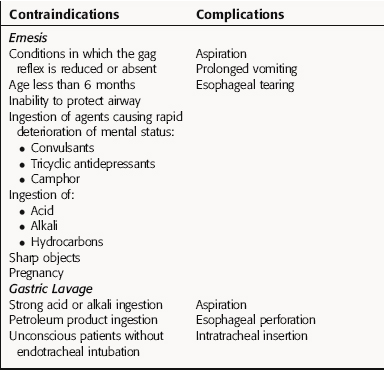
A summary of some common antidotes for several types of chemical toxicants is provided in Table 8-3. In addition, because emesis and gastric lavage are often used for the treatment of toxicity, the main contraindications and problems associated with these procedures are presented in Table 8-4.
| Toxicant | Antidote | Mechanism |
|---|---|---|
| Cyanide | Cyanide kit (sodium nitrite, sodium thiosulfate) | Induction of methemoglobinemia; cyanide binds preferentially to methemoglobin |
| Metals | Chelators | Binding of metal with subsequent urinary excretion |
| Methanol, ethylene glycol | Ethanol | Competition for alcohol dehydrogenase |
| CO | O2 | Displaces CO molecules from carboxyhemoglobin |
TABLE 8–4 Contraindications and Complications of Emesis and Gastric Lavage
Chemical Exposure and Health Outcomes
Exposure to environmental toxicants may occur in several ways and under different circumstances. Catastrophic accidents producing immediate morbidity and mortality are infrequent. In 1984 an explosion in a pesticide factory produced more than 2000 deaths and 10,000 injuries in Bhopal, India. High concentrations of methylisocyanate, a chemically reactive gas, were released in an urban setting and caused immediate lung damage and death in both workers and residents of the city.
Dart RC, editor. Medical Toxicology, ed 3, Philadelphia, Lippincott: Williams and Wilkins, 2004.
Klaassen CD, editor. Casarett and Doull’s Toxicology: The basic science of poisons, ed 6, McGraw Hill: New York, 2001.
Younglai EV, Wu YJ, Foster WG. Reproductive toxicology of environmental toxicants: Emerging issues and concerns. Curr Pharm Des. 2007;13:3005-3019.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 8 Principles of Toxicology
| Abbreviations | |
|---|---|
| ATP | Adenosine triphosphate |
| CNS | Central nervous system |
| CO | Carbon monoxide |
| DNA | Deoxyribonucleic acid |
| EDTA | Ethylenediamine tetraacetic acid |
| GI | Gastrointestinal |
Therapeutic Overview
Mechanisms of Action
Many toxic substances are known primarily to affect a specific target organ, such as the liver, nervous system, lungs, or kidneys, whereas others target the immune system or may affect normal fetal and cell growth and development. It is important to note, however, that although certain anatomical or functional characteristics may predispose organs to injury, designation of toxicants as “organ specific” (e.g., neurotoxic, cardiotoxic) is somewhat misleading, for several reasons. First, toxic substances may have adverse effects throughout the body, and although a chemical may primarily affect one organ, other organs may also incur less notable injury. Second, a toxic response in one tissue will undoubtedly have serious consequences for other tissues depending on the characteristics of the chemical (physical state, site of exposure, dose, and duration of exposure) and of the patient (general health, nutritional state, age, sex, enzyme induction, immune response, antioxidant concentrations). Thus a minor insult to a compromised organ may cause unexpected injury to that organ and may also affect other organs.
Because of a dual blood supply from the hepatic artery and the portal vein, the liver may be exposed to toxicants entering from the systemic circulation (e.g., via inhalation) or from the splanchnic circulation (absorbed from the gastrointestinal [GI] tract). The leaky capillary system of the hepatic sinusoids promotes the extraction of toxicants from the blood into the liver. The liver contains enzymes that metabolize many endogenous and exogenous chemicals (see Chapter 2). In some cases chemical modification produced by biotransformation results in bioactivation (production of toxic-reactive metabolites). This is exemplified by the bioactivation of chloroform to phosgene (Fig. 8-1). Compounds that enhance the activity of CYP2E1 (enzyme-inducing agents) or reduce the hepatic concentration of glutathione can exacerbate chloroform-induced liver injury. Other compounds that can cause liver injury after metabolism include several solvents (carbon tetrachloride, halogenated benzenes) and carcinogens (aflatoxin B, aromatic amines).
The blood can serve as the route of exposure of the lung in the case of paraquat, a herbicide taken up by type II pneumocytes. Biotransformation in the lung yields a reactive intermediate that undergoes redox cycling, which produces reactive oxygen species that injure the cell. Although exposure to paraquat usually occurs after ingestion or skin contamination, death results from pulmonary injury. Similarly, certain pyrrolidine alkaloids are metabolized in the liver to compounds that circulate in the blood and produce toxicity in the lung. The actions of toxicants on pulmonary tissue are shown in Figure 8-2.
The kidney is also susceptible to toxicants. The mechanisms of renal injury are similar to those in other organs but also include unique mechanisms. Delivery of blood-borne toxicants to the kidney is high, because the kidney receives 25% of cardiac output, and its functions include filtering, concentrating, and eliminating toxicants. As water is reabsorbed, the concentration of chemicals in the tubule can increase to toxic levels. In some cases the concentration may exceed the solubility of a chemical and lead to precipitation and obstruction of the affected area (Fig. 8-3).
The kidney can compensate for excessive chemical exposure because, like many organs, it has a reserve mass. Thus tissue injury equal to one entire kidney must occur before loss of function is clinically apparent. The kidney also replaces lost functional capacity by hypertrophy. In addition, the kidney has developed binding proteins to remove heavy metals. For example, metallothioneins avidly bind cadmium, protecting the kidney and other organs from toxicity. However, renal toxicity will develop if exposure exceeds the binding capacity or if a large amount of the cadmium-metallothionein complex accumulates.
In addition to being a target for chemical-mediated injury, the immune system may mediate injury by producing hypersensitivity reactions—adverse events caused by an immune response to foreign antigens (see Chapter 6). Indeed, cell-mediated hypersensitivity initiated by T lymphocytes may lead to contact dermatitis as a consequence of exposure to nickel and several industrial chemicals.
[/not-level-membership-for-basic-science-category]
