Chapter 12 Principles of Clinical Pharmacology
Basic Concepts in Pharmacokinetics
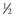 ). From this concept, the following equation is derived:
). From this concept, the following equation is derived: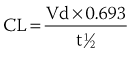
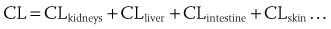
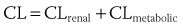
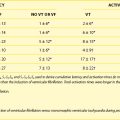
Absorption of drugs can vary with timing of dose in relation to a meal. For example, concentrations of dronedarone (a currently investigational antiarrhythmic drug) are twofold to threefold higher when taken with a meal compared with peak concentrations when taken on an empty stomach.1
Intersubject Variability in Drug Action
Several factors can modulate the response obtained following the administration of a particular drug to a particular patient at a particular time. This statement argues against the “one size fits all” concept and clearly defines the need for individualized drug therapy. To fully integrate the basic principles underlying clinical pharmacology, the prescriber needs to understand the principles of pharmacokinetics, pharmacodynamics, and drug efficacy. Figure 12-1 depicts the three major principles that define the relationship between drug dose and clinical outcome.
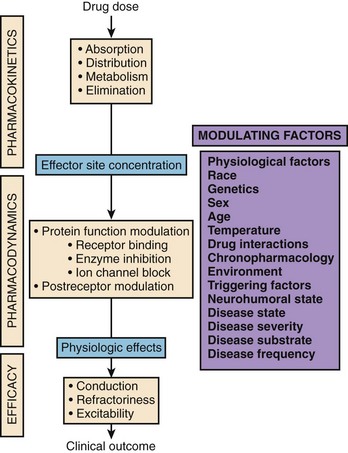
As discussed earlier, pharmacokinetics describes the relationship between the dose administered and the observed concentrations of a drug or its metabolites in selected biologic fluids. Concentrations of active or toxic substances at their effector or toxic sites are often of the greatest interest. Pharmacodynamics describes the relationship between the concentration of an active substance at its effector site and the physiological effects observed. Currently, most drugs are aimed at either direct or indirect modulation of a protein function. For most of them, there is a range of concentrations for which changes in protein function are linearly related to drug concentration. Finally, drug efficacy links the physiological effects observed following the administration of a drug to clinical outcome. Several major clinical trials in recent years, such as the Cardiac Arrhythmia Suppression Trial, have demonstrated that achievement of expected pharmacodynamic response is not necessarily related to a desirable clinical outcome (i.e., drug effectiveness).2,3
Drugs with a Narrow Therapeutic Index: Antiarrhythmic Agents
The notion that monitoring plasma drug concentrations could provide a method for adjusting doses to reduce inter-individual variability in response arose during the development of new antimalarial drugs during World War II. Shortly thereafter, this notion was applied to quinidine therapeutics.4 This concept was derived from the well-recognized relationships between “normal” plasma ion concentrations or hormonal levels and a “normal” physiological state. Using such a framework, it was observed in initial trials that plasma concentrations of quinidine below 3 µg/mL were rarely associated with an antiarrhythmic response, whereas concentrations above 8 µg/mL were frequently associated with QRS widening, cinchonism, and hypotension.5 Thus, a tentative therapeutic range of 3 to 8 µg/mL was defined.
Using the same approach, relatively well-defined therapeutic ranges were also established for lidocaine (4 to 8 µg/mL), mexiletine (500 to 1000 ng/mL), and procainamide (4 to 8 µg/mL) for patients presenting with ventricular arrhythmias.6–9 However, as drug assays developed further and experience accumulated, it became evident that the therapeutic concentration window was very wide with these antiarrhythmic agents and that wide intersubject variability existed. Therapeutic ranges, such as the one for quinidine (2 to 5 µg/mL), had to be redefined because of impurities and metabolites interfering with early fluorometric methods.10 Also, the overlap between effective and toxic concentrations (narrow therapeutic/toxic window) in different patients was significant, and it became almost impossible to predict, for a specific patient, plasma levels associated with efficacy or toxicity.
Subsequently, another important source of intersubject variability was identified in patients treated with the potent class Ic antiarrhythmic agent encainide.11 In a small clinical study, 10 of 11 patients with ventricular arrhythmias responded to the drug (encainide) with arrhythmia suppression and QRS widening, and the eleventh had no response. In the 10 responders, peak plasma encainide ranged from 3 to 200 ng/mL. In the single nonresponder, peak plasma encainide was the highest (300 ng/mL). Further studies demonstrated the importance of active metabolites (O-demethyl encainide [ODE] and 3 methoxy-O-demethyl encainide [MODE]) in accounting for encainide action, but a simple therapeutic range—based solely on the plasma concentrations of the parent compound or in combination with the metabolites—could not be defined.12
Propafenone is another class Ic antiarrhythmic agent that shows wide intersubject variability in its response and in the formation of active metabolites.13 In addition, the drug exhibits varying electrophysiological (sodium, calcium, and potassium channel block) and pharmacologic (β-blocking) effects depending on the route of administration, the metabolism status, and the plasma concentrations of its enantiomers.13,14 Several investigators have tried to derive combined therapeutic ranges for the metabolites—the enantiomers—and for the combinations of the parent drug plus metabolites, without success.
The situation with antiarrhythmic agents is not unique and is observed with other drugs that have a narrow therapeutic index. For example, doses and plasma concentrations of warfarin that were required to maintain the international normalized ratio (INR) within acceptable limits (2 to 3) vary widely among individuals.15–17 There is no rationale to use the plasma concentrations of each warfarin enantiomer, rather than INR values, to adjust warfarin doses.
Pharmacogenetics
Genetically Determined Pharmacokinetic Factors
Genetically determined abnormalities in the ability to biotransform drugs range from apparently benign conditions such as Gilbert’s syndrome (a deficiency in glucuronyl transferase activity) to the rare but potentially fatal syndrome of pseudo-cholinesterase deficiency. This most widely studied polymorphic drug oxidation trait is a deficiency in the cytochrome P450 isozyme (CYP2D6) responsible, among others, for the biotransformation of the antihypertensive drug debrisoquine to its inactive 4-hydroxy metabolite.18,19 Following the oral administration of a single 10-mg dose of debrisoquine, a metabolic ratio (debrisoquine/4-hydroxydebrisoquine), established from an 8-hour urinary excretion profile, can discriminate between two distinct phenotypes.20 Individuals with a ratio greater than 12.6 are defined as poor metabolizers (PMs), whereas a value less than this antimode reflects the ability to extensively metabolize (EM) the probe drug. Family studies indicated that the deficient trait is inherited as an autosomal recessive character.18 Regardless of geographic location, about 5% to 10% of whites are PMs. At the other end of the spectrum, 2% to 5% are known as ultra-rapid metabolizers (UM), since they exhibit very high expression levels and activity of CYP2D6.
The CYP2D6 gene is located on the long arm of chromosome 22 (q11.2-qter).21 Deletion or transition mutations in the gene lead to splicing errors during messenger ribonucleic acid (mRNA) processing and result in unstable proteins.22,23 Therefore, the CYP2D6 protein is functionally absent in PMs. Deoxyribonucleic acid (DNA) assays based on allele-specific amplification with the polymerase chain reaction (PCR) allow identification of approximately 95% of all PMs.23–25
CYP2D6 activity can also be inhibited by drugs, including quinidine, some tricyclic antidepressants, and some selective serotonin reuptake inhibitors (SSRIs; fluoxetine and paroxetine).26
CYP2D6 can metabolize substances via various C-oxidations, including aromatic, alicyclic, and aliphatic hydroxylation; N- and S-oxidation; as well as O-dealkylation. For example, the metabolism of several classes of cardiovascular drugs such as β-blockers and class I antiarrhythmic drugs, as well as the metabolism of neuroleptics and antidepressants, co-segregates with the debrisoquine 4-hydroxylase polymorphism.27 The clinical consequences of genetically determined polymorphic drug metabolism depend on the pharmacologic activity or toxicity of the parent compound compared with that of the metabolites formed by CYP2D6. Clinically important variations can be encountered in the following four situations:
Pharmacologic Effects Are Mediated by the Parent Compound Alone
Mexiletine is a class Ib antiarrhythmic agent that undergoes stereoselective disposition because of an extensive metabolism; less than 10% of an administered oral dose is recovered unchanged in urine.28,29 The major metabolites formed by carbon and nitrogen oxidation are hydroxymethylmexiletine, p-hydroxymexiletine, m-hydroxymexiletine, and N-hydroxymexiletine.28–31 Antiarrhythmic activity resides solely in mexiletine, and all metabolites are inactive. The formation of hydroxymethylmexiletine, p-hydroxymexiletine, and m-hydroxymexiletine is genetically determined and co-segregates with polymorphic debrisoquine 4-hydroxylase (CYP2D6) activity.32 Hence, subjects with the EM phenotype form large amounts of these metabolites. Conversely, clearance of mexiletine is twofold smaller and elimination half-life is longer in subjects with the PM phenotype. Consequently, at the same dose, mean plasma concentrations of mexiletine are higher, and drug accumulation is expected to occur in PM patients during chronic therapy.32 This may lead to side effects such as ataxia and muscle weakness because of the increased block of sodium channels in peripheral nerves.
Combined administration of low-dose quinidine, which is a selective and potent inhibitor of CYP2D6, inhibits mexiletine metabolism through its three CYP2D6 major oxidative pathways and alters mexiletine disposition to such an extent that the pharmacokinetic parameters of the drug are no longer different between EMs and PMs.32 Mexiletine and quinidine have been used in combination to improve antiarrhythmic efficacy and to decrease the incidence of gastrointestinal side effects.33 Because of decreased clearance and increased elimination half-life during quinidine coadministration, EM patients undergoing combined therapy should exhibit higher trough concentrations and lesser peak-to-trough fluctuations in mexiletine plasma concentrations. Drug accumulation and long-term side effects remain a risk if dosage adjustments are not made.
Specific genotypes are associated with metabolic bio-inactivation and, hence, the dose requirement or efficacy of certain drugs. For example, a specific genetic profile (activity of CYP2C9) is associated with higher or lower than average doses required to maintain the INR in the desired range for patients receiving warfarin therapy, and dose prediction based on a pharmacogenetic algorithm is superior to empiric dosing in rapidly achieving the desired target INR.34
A Metabolite Is More Active than the Parent Compound
Initial clinical trials with encainide reported a series of observations that led to important conclusions about the potential role of active metabolites in mediating drug effects. In the study of encainide effects as related to metabolite concentrations, among the 11 subjects, ODE and MODE were found in the urine of the 10 responders with respect to clinical effects but were not detected in the single nonresponder.11 Electrophysiological studies demonstrated that ODE is approximately 10-fold more potent a sodium channel blocker than the parent drug, whereas MODE is approximately threefold more potent; the metabolites had refractoriness-prolonging properties, whereas the parent drug had only minor effects.12,35–37
Drug metabolism studies clearly demonstrated that CYP2D6 is involved in the sequential metabolism of encainide into ODE and into MODE.12 Patients unable to form ODE or MODE are therefore PMs with low CYP2D6 activity. In normal volunteers with the EM phenotype, pretreatment with low-dose quinidine decreased encainide systemic clearance fivefold and decreased the partial metabolic clearance of encainide to ODE + MODE 13-fold.38 These data are compatible with the finding of inhibition of encainide biotransformation by quinidine (inhibition of CYP2D6). Coadministration of quinidine to volunteers having EM properties blunted encainide-induced QRS prolongation.38
Clopidogrel, useful when combined with acetylsalicylic acid (ASA) in stroke prevention in patients with atrial fibrillation who are not eligible for warfarin therapy, is metabolized from a prodrug to active drug by cytochrome P450 2C19. Patients with loss of function-variant alleles in the gene encoding this enzyme have apparent failure of clopidogrel efficacy (as demonstrated in studies in patients with vascular disease).26,39
The Parent Compound and the Metabolite Have Different Pharmacologic Effects
Systematic evaluation of the dose-response and concentration-response relationships for propafenone demonstrated substantial inter-individual variability in the extent of QRS prolongation and in minimal effective plasma concentrations required for arrhythmia suppression. Follow-up studies have shown that propafenone biotransformation to 5-hydroxy propafenone is catalyzed by CYP2D6 and that 5-hydroxy propafenone exerts sodium channel blocking action in vitro similar to those of the parent drug; however, a second metabolite, N-desalkyl propafenone, is somewhat less potent.40–42 Administration of low-dose quinidine for a short period to a group of patients receiving chronic propafenone therapy resulted in a 2.5-fold increase in plasma propafenone with a commensurate decrease in 5-hydroxy propafenone concentrations.43
Although propafenone and 5-hydroxy propafenone are roughly equipotent as sodium channel blockers, the parent drug is substantially more potent as a β-blocker.14 High concentrations of propafenone that can be observed in PMs can produce clinically detectable β-blockade similar to approximately 20 mg of propranolol every 8 hours. Propafenone metabolism is known to be saturable in EMs; that is, doubling the daily dosage from 450 to 900 mg/day results in a disproportionate sixfold increase in mean plasma propafenone concentrations.44 Thus β-blocking effects are expected in patients with the PM phenotype or in EMs receiving high dosages of the drug.44
Combined administration of propafenone and quinidine was also tested over a 1-year period in patients with atrial fibrillation in the Combined Administration of Quinidine and Propafenone for Atrial Fibrillation (CAQ-PAF) study.45 The objective of the study was to demonstrate that combined administration of propafenone and quinidine would be superior to propafenone alone to prevent the recurrence of atrial fibrillation. The rationale was that increased plasma propafenone concentrations caused by combined quinidine administration would be associated with additional electrophysiological (sodium, potassium, and calcium channel blocks) and pharmacologic (β-blocking) effects that are mediated mostly by propafenone itself compared with the effects that can be observed from propafenone and its 5-hydroxy metabolite. The results demonstrated that chronic administration of quinidine was able to inhibit CYP2D6 and propafenone metabolism over a 1-year period. Recurrence of atrial fibrillation was very low in genetically determined PMs (1 of 11) and in patients with propafenone plasma levels greater than 1500 ng/mL but very high in patients with propafenone plasma concentrations lower than 1000 ng/mL. This example illustrates that combined drug administration to alter patient phenotype can be associated with improved efficacy of a drug.
Venlafaxine is another example of a drug and its metabolite having different pharmacologic effects between EMs and PMs. Venlafaxine is a new-generation drug considered a first-line agent for the treatment of depressive disorders. It strongly inhibits presynaptic reuptake of noradrenaline and serotonin and weakly inhibits the presynaptic reuptake of noradrenaline and serotonin. It also weakly inhibits dopamine reuptake.46 Following oral administration, venlafaxine undergoes extensive first-pass metabolism.47,48 It is metabolized to several metabolites, including O-desmethyl venlafaxine, a pharmacologically active metabolite that inhibits noradrenaline and serotonin reuptake with potencies similar to those of venlafaxine.49 The disposition of venlafaxine is genetically determined and co-segregates with CYP2D6 activity in humans.50 Subjects with the PM phenotype have fourfold to eightfold higher plasma concentrations of venlafaxine and a 20-fold lower capability to form the O-desmethyl metabolite. Since the O-desmethyl metabolite and venlafaxine have a similar potency for serotonin reuptake, no difference in antidepressant activity is expected between EMs and PMs of CYP2D6. However, case studies suggested that higher plasma concentrations of venlafaxine caused by low CYP2D6 activity could increase the risk of cardiovascular toxicity, since venlafaxine (and possibly not the metabolite) is a potent blocker of the cardiac sodium channel.51 Venlafaxine has weak affinity for CYP2D6 and low propensity for causing drug interaction. However, several other CYP2D6 substrates such as the first-generation H1 antagonist diphenhydramine, can inhibit the metabolism of venlafaxine, increase the plasma concentrations of the parent compound up to fourfold, and potentially predispose patients to increased risk of cardiac toxicity.52
Toxicity Resides Within the Metabolite
A major form of toxicity-limiting chronic procainamide therapy is the drug-induced lupus syndrome.53 The exact mechanism whereby procainamide is capable of initiating this autoimmune syndrome is unclear. In preliminary metabolic studies, incubation of procainamide with mouse hepatic microsomes produced a reactive metabolite.54 Comparison with microsomal incubations of compounds modified at the site of the aromatic amine (N-acetyl procainamide [NAPA], p-hydroxyprocainamide, or desaminoprocainamide) led to the conclusion that oxidation of the primary aromatic amine of procainamide is involved in the production of such a reactive metabolite.53,55 The formation of N-hydroxyprocainamide was confirmed in both rat and human hepatic microsomes, and characterization of the reaction showed that it was mediated by cytochrome P450.56,57 Moreover, in vitro studies with genetically engineered microsomes expressing high levels of CYP2D6 exhibited the highest activity for the formation of N-hydroxyprocainamide.58 In vitro results were corroborated by clinical observations that the formation of nitroprocainamide, the potentially stable end product of N-hydroxyprocainamide, was absent in PMs of CYP2D6 but present in subjects with high CYP2D6 activity.50 Finally, formation of N-hydroxy procainamide was prevented in EMs during the combined administration of quinidine, a potent CYP2D6 inhibitor.50 These results indicate that CYP2D6 becomes the key enzyme in the formation of the toxic metabolite. Subjects with functionally deficient CYP2D6 activity (PMs) may therefore be at lower risk of procainamide-induced lupus erythematosus.
Genetically Determined Pharmacodynamic Factors
Over the past decade, great advances in the field of molecular biology have made it possible to elucidate the genetic causes of the inherited forms of the long QT syndrome (LQTS).59–61 These exciting discoveries have important implications for the understanding and therapy of this condition and have led to a better understanding of cardiac repolarization and arrhythmias in general. However, the prevalence of inherited LQTS is low. It is increasingly recognized that the concomitant use of older and recently introduced agents whether from new therapeutic classes or from those once believed to be safe (such that they were made available over the counter) put patients at increased risk for cardiac toxicity. Indeed, the list of drugs associated with the acquired form of LQTS is still growing. Genetic markers associated with an increased risk of drug-induced LQTS have also been identified.63 That is, mutations in genes encoding for specific ion channel proteins predispose patients, who are otherwise apparently “normal,” to excessive responses to drugs causing prolongation of cardiac repolarization and increased risk of torsades de pointes. In 1998, Priori et al demonstrated for the first time that a recessive variant of the Romano-Ward LQTS is present in the population.64 A homozygous missense mutation in the pore region of KvLQT1 was found in a 9-year-old boy with normal hearing, a prolonged Q-T interval, and syncopal episodes during exercise. However, the parents of the proband were heterozygous for the mutation and had a normal Q-T interval. In 1997, Donger et al identified a missense mutation in the C-terminal domain of KvLQT1 that was not associated with significant prolongation of the Q-T interval but the administration of QT-prolonging drugs predisposed patients to torsades de pointes.65 These recent observations suggested that mutations in cardiac potassium channel genes (and possibly other genes encoding for proteins involved in cardiac repolarization) may predispose patients with normal Q-T intervals to the acquired LQTS during treatment with drugs modulating cardiac repolarization.
Drug Interactions
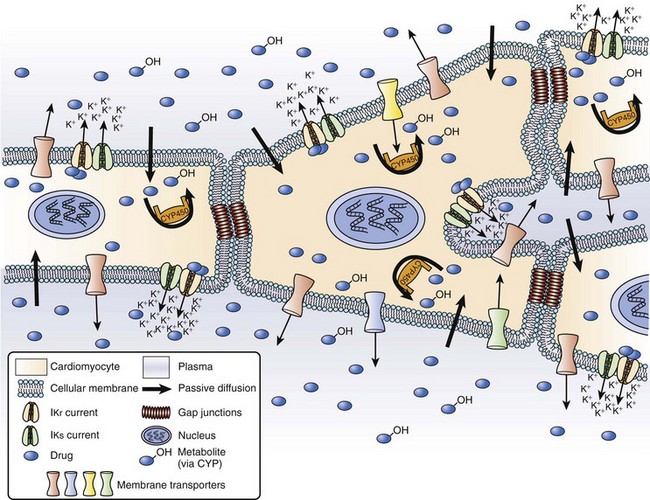
Clinicians and regulatory agencies have recently been concerned about the risk of prolongation of cardiac repolarization caused by drugs other than antiarrhythmic drugs. This concern is justified, since electrocardiogram (ECG) monitoring is not routinely employed in therapy with several of these agents. Such undesirable drug actions were first reported, as proarrhythmic events following the administration of the H1 antagonist terfenadine.66,67 The underlying mechanism of Q-T interval prolongation and torsades de pointes during terfenadine therapy was shown to be related to IKr block.68,69 Block of IKr was also demonstrated for several other agents, such as astemizole, cisapride, pimozide, thioridazine, droperidol, domperidone, macrolide antibiotics (erythromycin, clarithromycin), imidazole anti-fungals, and sildenafil, which have all been associated with proarrhythmic events and deaths in some patients.70–78
Proarrhythmia with these drugs is almost always observed during combined drug administration. Therefore, some authors have concluded that concomitant treatment with IKr blockers may predispose patients to proarrhythmia. However, this hypothesis has not been proven. Competitive antagonism at the receptor level would predict that combined use of IKr blockers should lead to a decrease in drug effects rather than synergistic activity. Indeed, combined use of dofetilide and NAPA, or NAPA and diphenhydramine, is associated with a decrease in action potential prolongation when the drugs are used together compared with when the drugs are used alone. Similarly, concomitant administration of dofetilide and erythromycin was associated with a decrease in overall action potential prolongation compared with dofetilide alone.79 Thus, proarrhythmia observed during the concomitant administration of IKr blockers in patients cannot be related solely to their electrophysiological properties on IKr.
Proarrhythmia with combined use of IKr blockers is usually observed under conditions of decreased metabolic capacity. For example, the induction of torsades de pointes during concomitant therapy with terfenadine and erythromycin or ketoconazole has been explained mainly on the basis of a specific cytochrome P450 enzyme inhibition.80,81 Terfenadine is known to be metabolized by CYP3A4.82 Erythromycin and imidazole (oral anti-fungals) are known inhibitors of CYP3A4; in subjects receiving the combination of terfenadine and erythromycin, erythromycin causes a decrease in the formation of the inactive acid metabolite as well as accumulation of terfenadine, which may lead to prolongation of cardiac repolarization (QT) and torsades de pointes. A similar mechanism can be described for other agents. Thus, combined administration of CYP3A4 substrates leads to the accumulation of one of these drugs; if the drug exhibits potent IKr blocking properties, proarrhythmia (torsades de pointes) caused by prolonged repolarization may be observed.
A third factor may also play a major role in drug-induced LQTS. P-glycoprotein (P-gp) is a versatile transporter that is able to pump a wide variety of xenobiotics outside a cell.83 P-gp is located primarily in the villous columnar epithelial cells of the small intestine and in hepatocytes, but it can also be found in cardiac myocytes.84 CYP3As and P-gp can function together by preventing cellular entry of lipophilic toxic compounds or by decreasing intracellular concentration of drugs. P-gp and CYP3As share tremendous substrate or inhibitor specificity, or both, so that the substrates or inhibitors of CYP3A4 can also simultaneously inhibit P-gp. Under conditions of combined treatment with IKr–CYP3A4–P-gp substrates, not only plasma concentrations but also intracellular cardiac concentrations of IKr blockers can be increased. Some frequently used drugs can also inhibit P-gp, including amiodarone, verapamil, and itraconazole. These drugs can lead to digoxin toxicity by reducing digoxin excretion.
Finally, as with CYP3A4, significant inter-individual variation exists in the expression of P-gp, and genetic polymorphisms have been described for both CYP3As and MDR1 (P-gp).85,86 The authors of this chapter have found that 29% of Canadians of French origin possess two mutated alleles (exon 26) of MDR1, which have recently been associated with altered drug concentrations.86 Thus, some patients may be at increased of proarrhythmia caused by mutations in these genes as well as mutations in genes associated with LQTS.
Key References
The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406-410.
Couture L, Nash JA, Turgeon J. The ATP-binding cassette transporters and their implication in drug disposition: A special look at the heart. Pharmacol Rev. 2006;58:244-258.
Drolet B, Zhang S, Deschênes D, et al. Droperidol lengthens cardiac repolarization due to block of the rapid component of the delayed rectifier potassium current. J Cardiovasc Electrophysiol. 1999;10:1597-1604.
Funck-Brentano C, Kroemer HK, Lee JT, Roden DM. Propafenone. N Engl J Med. 1990;322:518-525.
Funck-Brentano C, Turgeon J, Woosley RL, Roden DM. Effect of low dose quinidine on encainide pharmacokinetics and pharmacodynamics: Influence of genetic polymorphism. J Pharmacol Exp Ther. 1989;249:134-142.
Idle JR, Mahgoub A, Angelo MM, et al. The metabolism of [14C]-debrisoquine in man. Br J Clin Pharmacol. 1979;7:257-266.
Koch-Weser J. Correlation of serum concentrations and pharmacologic effects of antiarrhythmic drugs. In: Acheson GH, Maxwell RA, editors. Pharmacology and the future of man. Basel: Karger, 1973.
Lessard E, Fortin A, Bélanger PM, et al. Role of CYP2D6 in the N-hydroxylation of procainamide. Pharmacogenetics. 1997;7:381-390.
Lessard E, Yessine MA, Hamelin BA, et al. Diphenhydramine alters the disposition of venlafaxine trough inhibition of CYP2D6 activity in humans. J Clin Psychopharmacol. 2001;21:175-184.
Michaud V, Vanier MC, Brouillette D, et al. Combination of phenotype assessments and CYP2C9-VKORC1 polymorphisms in the determination of warfarin dose requirements in heavily medicated patients. Clin Pharmacol Ther. 2008;83(5):740-748.
Mitcheson JS, Chen J, Lin M, et al. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329-12333.
Muth EA, Moyer JA, Haskins JT, et al. Biochemical, neurophysiological, and behavioral effects of Wy-45,030, an ethyl cyclohexanol derivative. Drug Dev Res. 1991;23:191-193.
Paulussen A, Lavrijsen K, Bohets H, et al. Two linked mutations in transcriptional regulatory elements of the CYP3A5 gene constitute the major genetic determinant of polymorphic activity in human. Pharmacogenetics. 2000;10:415-424.
Price Evans DA, Mahgoub A, Sloan TP, et al. A family and population study of the genetic polymorphism of debrisoquine oxidation in a white British population. J Med Genet. 1980;17:102-105.
Roden DM, Stein CM. Clopidogrel and the concept of high-risk pharmacokinetics. Circulation. 2009;119(16):2127-2130.
Salata JJ, Jurkiewicz NK, Wallace AA, et al. Cardiac electrophysiological actions of the histamine H1-receptor antagonists astemizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ Res. 1995;76:110-119.
Thompson KA, Iansmith DHS, Siddoway LA, et al. Potent electrophysiologic effects of the major metabolites of propaferone in canine Purkinje fibers. J Pharmacol Exp Ther. 1988;244:950-955.
Turgeon J, Fiset C, Giguère R, et al. Influence of debrisoquine phenotype and of quinidine on mexiletine disposition in man. J Pharmacol Exp Ther. 1991;259:789-798.
Woodcock J, Lesko LJ. Pharmacogenetics-tailoring treatment for the outliers. N Engl J Med. 2009;360(8):811-813.
1 Zareba KM. Dronedarone: A new antiarrhythmic agent. Drugs Today (Barc). 2006;42(2):75-86.
2 Ruskin JN. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321:386-388.
3 The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406-410.
4 Edgar AL, Sokolow M. Experiences with the photofluorometric determination of quinidine in blood. J Lab Clin Med. 1950;36:478-484.
5 Sokolow M, Edgar AL. Blood quinidine concentrations as a guide in the treatment of cardiac arrhythmias. Circulation. 1950;1:576-592.
6 Gianelly R, von der Gruben JO, et al. Effect of lidocaine on ventricular arrhythmias in patients with coronary heart disease. N Engl J Med. 1967;277:1215-1219.
7 Campbell NPS, Kelly JG, Shanks RG, et al. Mexiletine (Ko 1173) in the management of ventricular dysrhythmias. Lancet. 1973;2(7826):404-407.
8 Koch-Weser J. Serum drug concentrations as therapeutic guides. N Engl J Med. 1972;287:227-231.
9 Koch-Weser J. Correlation of serum concentrations and pharmacologic effects of antiarrhythmic drugs. In: Acheson GH, Maxwell RA, editors. Pharmacology and the future of man. Basel: Karger, 1973.
10 Kessler KM, Lowenthal DT, Warner H, et al. Quinidine elimination in patients with congestive heart failure or poor renal function. N Engl J Med. 1974;290:706-709.
11 Roden DM, Reele SB, Higgins SB, et al. Total suppression of ventricular arrhythmias by encainide. Pharmacokinetic and electrocardiographic characteristics. N Engl J Med. 1980;302:877-882.
12 Barbey JT, Thompson KA, Echt DS, et al. Antiarrhythmic activity, electrocardiographic effects and pharmacokinetics of the encainide metabolites O-desmethyl encainide and 3-methoxy-O-desmethyl encainide in man. Circulation. 1988;77:380-391.
13 Funck-Brentano C, Kroemer HK, Lee JT, Roden DM. Propafenone. N Engl J Med. 1990;322:518-525.
14 Kroemer HK, Funck-Brentano C, Silberstein DJ, et al. Stereoselective disposition and pharmacologic activity of propafenone enantiomers. Circulation. 1989;79:1068-1076.
15 Breckenridge A, Orme M, Wesseling H, et al. Pharmacokinetics and pharmacodynamics of the enantiomers of warfarin in man. Clin Pharmacol Ther. 1974;15:424-430.
16 Chan E, McLachlan A, O’Reilly R, Rowland M. Stereo-chemical aspects of warfarin drug interactions: Use of combined pharmacokinetic-pharmacodynamic model. Clin Pharmacol Ther. 1994;56:286-294.
17 Furuya H, Fernandez-Salguero P, Gregory W, et al. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenetics. 1995;5:389-392.
18 Price Evans DA, Mahgoub A, Sloan TP, et al. A family and population study of the genetic polymorphism of debrisoquine oxidation in a white British population. J Med Genet. 1980;17:102-105.
19 Idle JR, Mahgoub A, Angelo MM, et al. The metabolism of [14C]-debrisoquine in man. Br J Clin Pharmacol. 1979;7:257-266.
20 Daily AK, Armstrong M, Monkman SC, et al. Genetic and metabolic criteria for the assignment of debrisoquine 4-hydroxylation (cytochrome P4502D6) phenotypes. Pharmacogenetics. 1991;1:33-41.
21 Kimura S, Umeno M, Skoda RC, et al. The human debrisoquine 4-hydroxylase (CYP2D) locus: Sequence and identification of the polymorphic CYP2D6 gene, a related gene, and a pseudogene. Am J Hum Genet. 1989;45:889-904.
22 Gonzalez FJ, Skoda RC, Kimura S, et al. Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature. 1988;331:442-446.
23 Gonzalez FJ, Meyer UA. Molecular genetics of the debrisoquin-sparteine polymorphism. Clin Pharmacol Ther. 1991;50:233-238.
24 Broly F, Gaedigk A, Heim M, et al. Debrisoquine/sparteine hydroxylation genotype and phenotype: Analysis of common mutations and alleles of CYP2D6 in a European population. DNA Cell Biol. 1991;10:545-558.
25 Broly F, Marez D, Sabbagh N, et al. An efficient strategy for detection of known and new mutations of the CYP2D6 gene using single strand conformation polymorphism analysis. Pharmacogenetics. 1995;5:373-384.
26 Roden DM, Stein CM. Clopidogrel and the concept of high-risk pharmacokinetics. Circulation. 2009;119(16):2127-2130.
27 Eichelbaum M, Gross AS. The genetic polymorphism of debrisoquine/sparteine metabolism. Clinical aspects. Pharmacol Ther. 1990;46:377-394.
28 Beckett AH, Chidomere EC. The identification and analysis of mexiletine and its metabolic products in man. J Pharm Pharmacol. 1977;29:281-285.
29 Beckett AH, Chidomere EC. The distribution, metabolism and excretion of mexiletine in man. Postgrad Med J. 1977;53(Suppl 1):60-66.
30 Grech-Bélanger O, Turgeon J, Lalande M, Bélanger PM. Meta-hydroxymexiletine, a new metabolite of mexiletine. Isolation, characterization, and species differences in its formation. Drug Metab Dispos. 1991;19:458-461.
31 Turgeon J, Paré JRJ, Lalande M, et al. Isolation and structural characterization by spectroscopic methods of two glucuronide metabolites of mexiletine after N-oxidation and deamination. Drug Metab Dispos. 1992;20:762-769.
32 Turgeon J, Fiset C, Giguère R, et al. Influence of debrisoquine phenotype and of quinidine on mexiletine disposition in man. J Pharmacol Exp Ther. 1991;259:789-798.
33 Duff HJ, Roden DM, Primm RK, et al. Mexiletine in the treatment of resistant ventricular tachycardia: Enhancement of efficacy and reduction of dose-related side effects by combination with quinidine. Circulation. 1983;67:1124-1128.
34 Woodcock J, Lesko LJ. Pharmacogenetics-tailoring treatment for the outliers. N Engl J Med. 2009;360(8):811-813.
35 Carey ELJr, Duff HJ, Roden DM, et al. Encainide and its metabolites. Comparative effects in man on ventricular arrhythmia and electrocardiographic intervals. J Clin Invest. 1984;73:539-547.
36 Dawson AK, Roden DM, Duff HJ, et al. Differential effects of O-dimethyl encainide on induced and spontaneous arrhythmias in the conscious dog. Am J Cardiol. 1984;54:654-658.
37 Duff HJ, Dawson AK, Carey EL, et al. The electrophysiologic actions of O-demethyl encainide: An active metabolite [abstract]. Clin Res. 1981;29:270A.
38 Funck-Brentano C, Turgeon J, Woosley RL, Roden DM. Effect of low dose quinidine on encainide pharmacokinetics and pharmacodynamics: Influence of genetic polymorphism. J Pharmacol Exp Ther. 1989;249:134-142.
39 Michaud V, Vanier MC, Brouillette D, et al. Combination of phenotype assessments and CYP2C9-VKORC1 polymorphisms in the determination of warfarin dose requirements in heavily medicated patients. Clin Pharmacol Ther. 2008;83(5):740-748.
40 Siddoway LA, Roden DM, Woosley RL. Clinical pharmacology of propafenone: Pharmacokinetics, metabolism and concentration-response relations. Am J Cardiol. 1984;54:9D-12D.
41 Malfatto G, Zaza A, Forster M, et al. Electrophysiologic, inotropic and antiarrhythmic effects of propafenone, 5-hydroxypropafenone and N-depropylpropafenone. J Pharmacol Exp Ther. 1988;246:419-426.
42 Thompson KA, Iansmith DHS, Siddoway LA, et al. Potent electrophysiologic effects of the major metabolites of propaferone in canine Purkinje fibers. J Pharmacol Exp Ther. 1988;244:950-955.
43 Funck-Brentano C, Kroemer HK, Pavlou H, et al. Genetically-determined interaction between propafenone and low dose quinidine: Role of active metabolites in modulating net drug effect. Br J Clin Pharmacol. 1989;27:435-444.
44 Lee JT, Kroemer HK, Silberstein DJ, et al. The role of genetically determined polymorphic drug metabolism in the beta-blockade produced by propafenone. N Engl J Med. 1990;322:1764-1768.
45 O’Hara GE, Philippon F, Gilbert M, et al. Combined administration of quinidine and propafenone for atrial fibrillation: The CAQ-PAF pilot study [abstract]. Eur Heart J. 1999;20:104.
46 Bolden-Watson C, Richelson E. Blockade by newly-developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sci. 1993;52:1023-1029.
47 Howell SR, Husbands GEM, Scatina JA, Sisenwine SF. Metabolic disposition of 14C-venlafaxine in mouse, rat, dog, rhesus monkey and man. Xenobiotica. 1993;23:349-359.
48 Wang CP, Howell SR, Scantina J, Sisenwine SF. The disposition of venlafaxine enantiomers in dogs, rats, and human receiving venlafaxine. Chirality. 1992;4:84-90.
49 Muth EA, Moyer JA, Haskins JT, et al. Biochemical, neurophysiological, and behavioral effects of Wy-45,030, an ethyl cyclohexanol derivative. Drug Dev Res. 1991;23:191-193.
50 Lessard E, Yessine MA, Hamelin BA, et al. Influence of CYP2D6 activity on the disposition of the antidepressant agent venlafaxine in humans. Pharmacogenetics. 1999;9:435-443.
51 Khalifa M, Daleau P, Turgeon J. Mechanism of sodium channel block by venlafaxine in guinea pig ventricular myocytes. J Pharmacol Exp Ther. 1999;291:280-284.
52 Lessard E, Yessine MA, Hamelin BA, et al. Diphenhydramine alters the disposition of venlafaxine trough inhibition of CYP2D6 activity in humans. J Clin Psychopharmacol. 2001;21:175-184.
53 Uetrecht JP, Freeman RW, Woosley RL. The implications of procainamide metabolism to its induction of lupus. Arthritis Rheum. 1981;24:994-999.
54 Freeman RW, Uetrecht JP, Woosley RL, et al. Covalent binding of procainamide in vitro and in vivo to hepatic protein in mice. Drug Metab Dispos. 1981;9:188-192.
55 Freeman RW, Woosley RL, Oates JA, Harbison RL. Evidence for the biotransformation of procainamide to a reactive metabolite. Toxicol Appl Pharmacol. 1979;50:9-16.
56 Uetrecht JP, Sweetman BJ, Woosley RL, Oates JA. Metabolism of procainamide to a hydroxylamine by rat and human hepatic microsomes. Drug Metab Dispos. 1984;12:77-81.
57 Budinsky RA, Roberts SM, Coats EA, et al. The formation of procainamide hydroxylamine by rat and human liver microsomes. Drug Metab Dispos. 1987;15:37-43.
58 Lessard E, Fortin A, Bélanger PM, et al. Role of CYP2D6 in the N-hydroxylation of procainamide. Pharmacogenetics. 1997;7:381-390.
59 Roden DM, Spooner PM. Inherited long QT syndromes: A paradigm for understanding arrhythmogenesis. J Cardiovasc Electrophysiol. 1999;10:1664-1683.
60 Keating MT. The long QT syndrome. A review of recent molecular genetic and physiologic discoveries. Medicine. 1996;75:1-5.
61 Splawski I, Shen J, Timothy KW, et al. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics. 1998;51:86-97.
62 Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805-811.
63 Mitcheson JS, Chen J, Lin M, et al. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329-12333.
64 Priori SG, Schwartz PJ, Napolitano C, et al. A recessive variant of the Romano-Ward long-QT syndrome? Circulation. 1998;97:24202425.
65 Donger C, Denjoy I, Berthet M, et al. KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation. 1997;96:2778-2781.
66 MacConnell TJ, Stanners AJ. Torsades de pointes complicating treatment with terfenadine. BMJ. 1991;302:1469.
67 Mathews DR, McNutt B, Okerholm R, et al. Torsades de pointes occurring in association with terfenadine use. JAMA. 1991;266:2375-2376.
68 Rampe D, Wible B, Brown AM, Dage RC. Effects of terfenadine and its metabolites on a delayed rectifier K+ channel cloned from human heart. Mol Pharmacol. 1993;44:1240-1245.
69 Roy ML, Dumaine R, Brown AM. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94:817-823.
70 Salata JJ, Jurkiewicz NK, Wallace AA, et al. Cardiac electrophysiological actions of the histamine H1-receptor antagonists astemizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ Res. 1995;76:110-119.
71 Drolet B, Khalifa M, Daleau P, et al. Block of the rapid component of the delayed rectifier potassium current by the prokinetic agent cisapride underlies drug-related lengthening of the QT interval. Circulation. 1998;97:204-210.
72 Kang J, Wang L, Cai F, Rampe D. High affinity blockade of the HERG cardiac K(+) channel by the neuroleptic pimozide. Eur J Pharmacol. 2000;392:137-140.
73 Drolet B, Vincent F, Rail J, et al. Thioridazine lengthens repolarization of cardiac ventricular myocytes by block of the delayed rectifier potassium current. J Pharmacol Exp Ther. 1999;288:1261-1268.
74 Drolet B, Zhang S, Deschênes D, et al. Droperidol lengthens cardiac repolarization due to block of the rapid component of the delayed rectifier potassium current. J Cardiovasc Electrophysiol. 1999;10:1597-1604.
75 Drolet B, Rousseau G, Daleau P, et al. Domperidone should not be considered a no-risk alternative to cisapride in the treatment of gastrointestinal motility disorders. Circulation. 2000;102:1883-1885.
76 Daleau P, Lessard E, Groleau MF, Turgeon J. Erythromycin blocks the rapid component of the delayed rectifier potassium current and lengthens repolarization of guinea pig ventricular myocytes. Circulation. 1995;91:3010-3016.
77 Dumaine R, Roy ML, Brown AM. Blockade of HERG and Kv1.5 by ketoconazole. J Pharmacol Exp Ther. 1998;286:727-735.
78 Geelen P, Drolet B, Rail J, et al. Sildenafil (Viagra) prolongs cardiac repolarization by blocking the rapid component of the delayed rectifier potassium current. Circulation. 2000;102:275-277.
79 Antzelevitch C, Sun ZQ, Zhang ZQ, Yan GX. Cellular and ionic mechanisms underlying erythromycin-induced long QT intervals and torsades de pointes. J Am Coll Cardiol. 1996;28:1836-1848.
80 Honig PK, Wortham DC, Zamani K, et al. Terfenadine-ketoconazole interaction. Pharmacokinetic and electrocardiographic consequences. JAMA. 1993;269:1513-1518.
81 Honig PK, Woosley RL, Zamani K, et al. Changes in the pharmacokinetics and electrocardiographic pharmacodynamics of terfenadine with concomitant administration of erythromycin. Clin Pharmacol Ther. 1992;52:231-238.
82 Kuang TY, Morgan A, Lazarev A, Cantilena LR. Human CYP3A4 as a potential in vitro screening system for terfenadine drug interactions [abstract]. Clin Pharmacol Ther. 1994;55:139.
83 Bellamy WT. P-glycoproteins and multidrug resistance. Annu Rev Pharmacol Toxicol. 1996;36:161-183.
84 Couture L, Nash JA, Turgeon J. The ATP-binding cassette transporters and their implication in drug disposition: A special look at the heart. Pharmacol Rev. 2006;58:244-258.
85 Paulussen A, Lavrijsen K, Bohets H, et al. Two linked mutations in transcriptional regulatory elements of the CYP3A5 gene constitute the major genetic determinant of polymorphic activity in human. Pharmacogenetics. 2000;10:415-424.
86 Kim RB, Leake BF, Choo EF, et al. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70:189-199.