Principles of Antimicrobial Action and Resistance
1. List the five general categories of antimicrobial actions.
2. Define antibiotic and antimicrobial.
3. Define and differentiate bactericidal and bacteriostatic agents.
4. Compare and contrast the following terms: biologic versus clinical resistance, environmentally mediated versus microorganism-mediated resistance and intrinsic versus acquired resistance.
5. Describe the basic structure and chemical principle for the mechanism of beta-lactam antimicrobials.
6. List common β-lactam antibiotics and provide an example of a common pathogen susceptible to these agents
7. Describe the chemical principle for the mechanism of resistance to β-lactam antibiotics.
8. Describe the chemical principle for the mechanisms of glycopeptide agents.
9. List common glycopeptides and provide an example of a common pathogen susceptible to these agents.
10. List examples of cell membrane inhibitors, inhibitors of protein synthesis, inhibitors of deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) synthesis, and metabolic inhibitors. Provide an example of a common pathogen susceptible to each of the agents listed.
11. List five general mechanisms for antimicrobial resistance and provide an example for each.
12. Describe how the dissemination of antimicrobial resistance affects diagnostic microbiology, including effects on sensitivity testing, therapeutics, and organism identification.
Because antimicrobial agents play a central role in the control and management of infectious diseases, understanding their mode of action and the mechanisms of microorganisms to circumvent antimicrobial activity is important, especially because diagnostic laboratories are expected to design and implement tests that measure a pathogen’s response to antimicrobial activity (see Chapter 12). Much of what is discussed here regarding antimicrobial action and resistance is based on antibacterial agents, but the principles generally apply to almost all antiinfective agents. More information about antiparasitic, antifungal, and antiviral agents can be found in Parts IV, V, and VI, respectively.
Antimicrobial Action
Principles
Several key steps must be completed for an antimicrobial agent to successfully inhibit or kill an infecting microorganism (Figure 11-1). First, the agent must be in an active form. This is ensured through the pharmacodynamic design of the drug, which takes into account the route by which the patient receives the agent (e.g., orally, intramuscularly, intravenously). Second, the antibiotic must also be able to achieve sufficient levels or concentrations at the site of infection so that it has a chance to exert an antibacterial effect (i.e., it must be in anatomic approximation with the infecting bacteria). The ability to achieve adequate levels depends on the pharmacokinetic properties of the agent, such as rate of absorption, distribution, metabolism, and excretion of the agent’s metabolites. Table 11-1 provides examples of various anatomic limitations characteristic of a few commonly used antibacterial agents. Some agents, such as ampicillin and ceftriaxone, achieve therapeutically effective levels in several body sites, whereas others, such as nitrofurantoin and norfloxacin, are limited to the urinary tract. Therefore, a knowledge of the site of infection can substantially affect the selection of the antimicrobial agent for therapeutic use.
TABLE 11-1
Anatomic Distribution of Some Common Antibacterial Agents
| Serum-Blood* | Cerebrospinal Fluid | Urine | |
| Ampicillin | + | + | + |
| Ceftriaxone | + | + | + |
| Vancomycin | + | ± | + |
| Ciprofloxacin | + | ± | + |
| Gentamicin | + | − | + |
| Clindamycin | + | − | − |
| Norfloxacin | − | − | + |
| Nitrofurantoin | − | − | + |
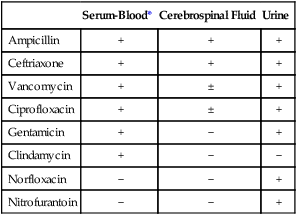
+, Therapeutic levels generally achievable at that site; ±, therapeutic achievable levels moderate to poor; −, therapeutic levels generally not achievable at that site.
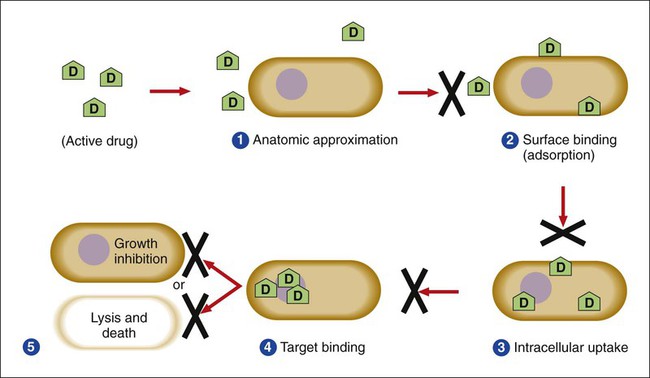
The remaining steps in antimicrobial action relate to direct interactions between the antibacterial agent and the bacterial cell. The antibiotic is attracted to and maintains contact with the cell surface. Because most targets of antibacterial agents are intracellular, uptake of the antibiotic to some location inside the bacterial cell is required. Once the antibiotic has achieved sufficient intracellular concentration, binding to a specific target occurs. This binding involves molecular interactions between the antimicrobial agent and one or more biochemical components that play an important role in the microorganism’s cellular metabolism. Adequate binding of the target results in disruption of cellular processes, leading to cessation of bacterial cell growth and, depending on the antimicrobial agent’s mode of action, cell death. Antimicrobial agents that inhibit bacterial growth but generally do not kill the organism are known as bacteriostatic agents. Effectively reducing the growth rate of an organism provides adequate protection in individuals whose immune system is capable of removing the agent of infection. Agents that usually kill target organisms are said to be bactericidal (Box 11-1). Bacteriocidal agents are more effective against organisms that are more difficult to control in combination with the host’s immune system.
The primary goal in the development and design of antimicrobial agents is to optimize a drug’s ability to efficiently achieve all steps outlined in Figure 11-1 while minimizing toxic effects on human cells and physiology. Different antibacterial agents exhibit substantial specificity in terms of their bacterial cell targets, that is, their mode of action. For this reason, antimicrobial agents are frequently categorized according to their mode of action.
Mode of Action of Antibacterial Agents
The interior of the bacterial cell has several potential antimicrobial targets. However, the processes or structures most frequently targeted are cell wall (peptidoglycan) synthesis, the cell membrane, protein synthesis, metabolic pathways, and DNA and RNA synthesis (Table 11-2).
TABLE 11-2
Summary of Mechanisms of Action for Commonly Used Antibacterial Agents
| Antimicrobial Class | Mechanism of Action | Spectrum of Activity |
| Aminoglycosides (e.g., gentamicin, tobramycin, amikacin, streptomycin, kanamycin) | Inhibit protein synthesis by binding to 30S ribosomal subunit | Gram-positive and gram-negative bacteria; not anaerobic bacteria |
| β-lactams (e.g., penicillin, ampicillin, mezlocillin, piperacillin, cefazolin, cefotetan, ceftriaxone, cefotaxime, ceftazidime, aztreonam, imipenem) | Inhibit cell wall synthesis by binding enzymes involved in peptidoglycan production (i.e., penicillin-binding proteins [PBPs]) | Both gram-positive and gram-negative bacteria, but spectrum may vary with the individual antibiotic. |
| Chloramphenicol | Inhibits protein synthesis by binding 50S ribosomal subunit | Gram-positive and gram-negative bacteria |
| Fluoroquinolones (e.g., ciprofloxacin, ofloxacin, norfloxacin) | Inhibit DNA synthesis by binding DNA gyrase and topoisomerase IV | Gram-positive and gram-negative bacteria, but spectrum may vary with individual antibiotic |
| Glycylglycines (e.g., tigecycline) | Inhibition of protein synthesis by binding to 30S ribosomal subunit | Wide spectrum of gram-positive and gram-negative species including those resistant to tetracycline |
| Ketolides (e.g., telithromycin) | Inhibition of protein synthesis by binding to 50S ribosomal subunit | Gram-positive cocci including certain macrolide-resistant strains and some fastidious gram-negatives (e.g., H. influenzae and M. catarrhalis) |
| Lipopeptides (e.g., daptomycin) | Binding and disruption of cell membrane | Gram-positive bacteria including those resistant to beta-lactams and glycopeptides |
| Nitrofurantoin | Exact mechanism uncertain; may have several bacterial enzyme targets and directly damage DNA | Gram-positive and gram-negative bacteria |
| Oxazolidinones (e.g., linezolid) | Bind to 50S ribosomal subunit to interfere with initiation of protein synthesis | Wide variety of gram-positive bacteria, including those resistant to other antimicrobial classes |
| Polymyxins (e.g., polymyxin B and colistin) | Disruption of cell membrane | Gram-negative bacteria, poor activity against most gram-positive bacteria |
| Rifampin | Inhibits RNA synthesis by binding DNA-dependent, RNA polymerase | Gram-positive and certain gram-negative (e.g., N. meningitidis) bacteria |
| Streptogramins (e.g., quinupristin/dalfopristin) | Inhibit protein synthesis by binding to two separate sites on the 50S ribosomal subunit | Primarily gram-positive bacteria |
| Sulfonamides | Interfere with folic acid pathway by binding the enzyme dihydropteroate synthase | Gram-positive and many gram-negative bacteria |
| Tetracycline | Inhibits protein synthesis by binding 30S ribosomal subunit | Gram-positive and gram-negative bacteria, and several intracellular bacterial pathogens (e.g., chlamydia) |
| Trimethoprim | Interferes with folic acid pathway by binding the enzyme dihydrofolate reductase | Gram-positive and many gram-negative bacteria |
Inhibitors of Cell Wall Synthesis
β-Lactam (Beta-Lactam) Antimicrobial Agents.
β-lactam antibiotics have a four-member, nitrogen-containing, β-lactam ring at the core of their structure (Figure 11-2). The antibiotics differ in ring structure and attached chemical groups. This drug class comprises the largest group of antibacterial agents, and dozens of derivatives are available for clinical use. Types of β-lactam agents include penicillins, cephalosporins, carbapenems, and monobactams. The popularity of these agents results from their bactericidal action and lack of toxicity to humans; also, their molecular structures can be manipulated to achieve greater activity for wider therapeutic applications.
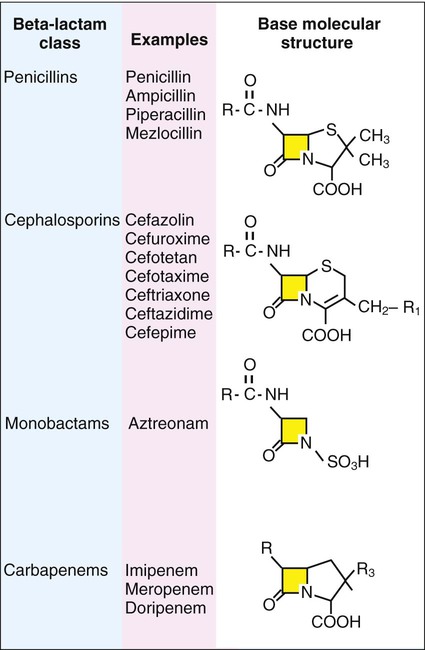
To circumvent the development of antimicrobial resistance, β-lactam combinations comprised of a β-lactam with antimicrobial activity (e.g., ampicillin, amoxicillin, piperacillin) and a beta-lactam without activity capable of binding and inhibiting β-lactamases (e.g., sulbactam, clavulanate, tazobactam) have been developed. The binding β-lactam “ties up” the β-lactamases produced by the bacteria and allows the other β-lactam in the combination to exert its antimicrobial effect. Examples of these β-lactam/β-lactamase inhibitor combinations include ampicillin/sulbactam, amoxicillin/clavulanate, and piperacillin/tazobactam. Such combinations are effective only against organisms that produce β-lactamases that are bound by the inhibitor; they have little effect on resistance that is mediated by altered PBPs (see Mechanisms of Antibiotic Resistance later in this chapter).
Glycopeptides and Lipopeptides.
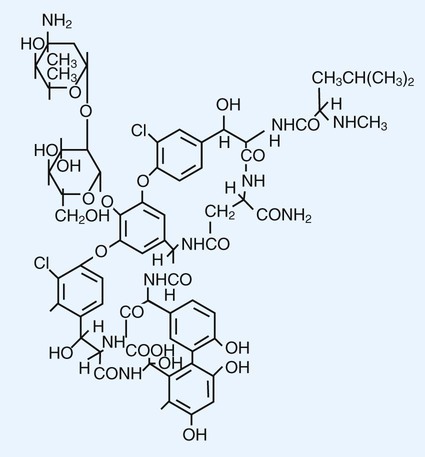
Glycopeptides are the other major class of antibiotics that inhibit bacterial cell wall synthesis by binding to the end of the peptidoglycan, interfering with transpeptidation. This is a different mechanism from that of the β-lactams, which bind directly to the enzyme. Two such antibiotics, vancomycin and teicoplanin, are large molecules and function differently from β-lactam antibiotics (Figure 11-3). With glycopeptides, the binding interferes with the ability of the PBP enzymes, such as transpeptidases and transglycosylases, to incorporate the precursors into the growing cell wall. With the cessation of cell wall synthesis, cell growth stops and death often follows. Because glycopeptides have a different mode of action, the resistance to β-lactam agents by gram-positive bacteria does not generally hinder their activity. However, because of their relatively large size, they cannot penetrate the outer membrane of most gram-negative bacteria to reach their cell wall precursor targets. Therefore, this agent is usually ineffective against gram-negative bacteria. Teicoplanin is approved for use throughout the world but is not currently available in the United States. When vancomycin is used, its levels should be monitored because the potential for toxicity.
Inhibitors of Protein Synthesis
Aminoglycosides and Aminocyclitols.
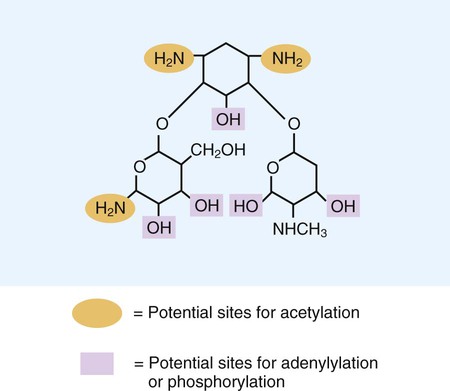
Aminoglycosides (aminoglycosidic aminocyclitol) inhibit bacterial protein synthesis by irreversibly binding to protein receptors on the organism’s 30S ribosomal subunit. This process interrupts several steps, including initial formation of the protein synthesis complex, accurate reading of the messenger RNA (mRNA) code, and formation of the ribosomal-mRNA complex. The structure of a commonly used aminoglycoside, gentamicin, is shown in Figure 11-4. Other aminoglycosides include tobramycin, amikacin, streptomycin, and kanamycin. The spectrum of activity of aminoglycosides includes a wide variety of aerobic gram-negative and certain gram-positive bacteria, such as S. aureus. Bacterial uptake of the aminoglycosides is accomplished by using them in combination with cell wall–active antibiotics, such as β-lactams or vancomycin. Anaerobic bacteria are unable to uptake these agents intracellularly and therefore are typically not inhibited by aminoglycosides. Aminoglycosides are associated with toxicity, and blood levels should be monitored during therapy. The major toxicities are nephrotoxicity and auditory or vestibular toxicity.
Inhibitors of DNA and RNA Synthesis
Fluoroquinolones.
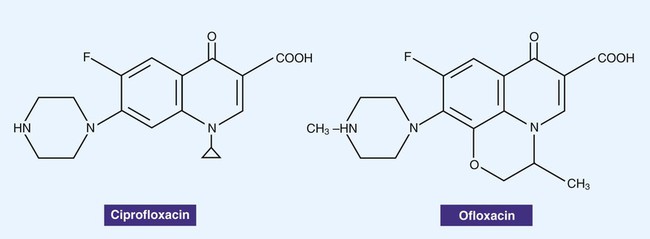
Fluoroquinolones, also often simply referred to as quinolones, are derivatives of nalidixic acid, an older antibacterial agent. The structures of two quinolones, ciprofloxacin and ofloxacin, are shown in Figure 11-5. These agents bind to and interfere with DNA gyrase enzymes involved in the regulation of bacterial DNA supercoiling, a process essential for DNA replication, recombination, and repair. The newer fluoroquinolones also inhibit topoisomerase IV. Topoisomerase IV functions very similarly to DNA gyrase, unlinking DNA after replication. The fluoroquinolones are potent bactericidal agents and have a broad spectrum of activity that includes gram-negative and gram-positive organisms. The fluoroquinolones target the DNA gyrase in gram-negative organisms and topoisomerase IV in gram-positive organisms. Because these agents interfere with DNA replication and therefore cell division, the drugs are bacteriocidal. However, the spectrum of activity varies with the individual quinolone agent. Toxicity varies with a variety of factors. Tendinitis and rupture of the Achilles tendon have been associated with fluoroquinolone treatment in the general population, and the risk is greater in older patients.
Inhibitors of Other Metabolic Processes
Sulfonamides.
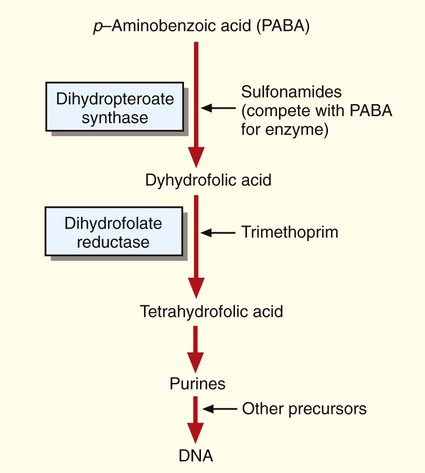
The bacterial folic acid pathway produces precursors required for DNA synthesis (Figure 11-6). Sulfonamides target and bind to one of the enzymes, dihydropteroate synthase, and disrupt the folic acid pathway. Several different sulfonamide derivatives are available for clinical use. These agents are active against a wide variety of bacteria, including the gram-positive and gram-negative (except P. aeruginosa) species. Sulfonamides are moderately toxic, causing vomiting, nausea, and hypersensitivity reactions. Sulfonamides are also antagonistic for several other medications, including warfarin, phenytoin, and oral hypoglycemic agents.
Trimethoprim.
Like the sulfonamides, trimethoprim targets the folic acid pathway. However, it inhibits a different enzyme, dihydrofolate reductase (see Figure 11-6). Trimethoprim is active against several gram-positive and gram-negative species. Frequently, trimethoprim is combined with a sulfonamide (usually sulfamethoxazole) into a single formulation to produce an antibacterial agent that can simultaneously attack two targets on the same folic acid metabolic pathway. This drug combination can enhance activity against various bacteria and may help prevent the emergence of bacterial resistance to a single agent. Toxicity is typically mild. Adverse side effects include gastrointestinal symptoms and allergic skin rashes. Patients with acquired immunodeficiency syndrome (AIDS) develop side effects more often than healthy individuals.
Mechanisms of Antibiotic Resistance
Principles
Successful bacterial resistance to antimicrobial action requires interruption or disturbance of one or more steps essential for effective antimicrobial action (see Figure 11-1). These disturbances or resistance mechanisms can occur as a result of various processes, but the end result is partial or complete loss of antibiotic effectiveness. Different aspects of antimicrobial resistance mechanisms discussed include biologic versus clinical antimicrobial resistance, environmentally mediated antimicrobial resistance, and microorganism-mediated antimicrobial resistance.
Environmentally Mediated Antimicrobial Resistance
Antimicrobial resistance is the result of nearly inseparable interactions involving the drug, the microorganism, and the environment in which they coexist. Characteristics of the antimicrobial agents, other than the mode and spectrum of activity, include important aspects of each drug’s pharmacologic attributes. However these factors are beyond the scope of this text. Microorganism characteristics are discussed in subsequent sections of this chapter (see Microorganism-Mediated Antimicrobial Resistance). The environmental impact on antimicrobial activity is considered here, and its importance cannot be overstated.
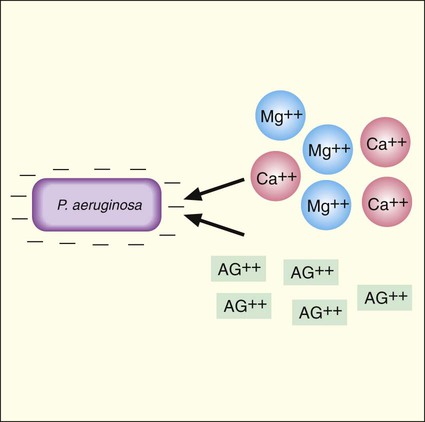
Aminoglycoside activity is also affected by the concentration of cations in the environment, such as calcium and magnesium (Ca++ and Mg++). This effect is most notable with P. aeruginosa. As shown in Figure 11-1, an important step in antimicrobial activity is the adsorption of the antibiotic to the bacterial cell surface. Aminoglycoside molecules have a net positive charge, and as is true for most gram-negative bacteria, the outer membrane of P. aeruginosa has a net negative charge. This electrostatic attraction facilitates attachment of the drug to the surface before internalization and subsequent inhibition of protein synthesis (Figure 11-7). However, calcium and magnesium cations compete with the aminoglycosides for negatively charged binding sites on the cell surface. If the positively charged calcium and magnesium ions outcompete aminoglycoside molecules for these sites, the amount of the drug taken up is decreased and antimicrobial activity is diminished. For this reason, aminoglycoside activity against P. aeruginosa tends to decrease as environmental cation concentrations increase.
The presence of certain metabolites or nutrients in the environment may also affect antimicrobial activity. For example, enterococci are able to use thymine and other exogenous folic acid metabolites to circumvent the activities of the sulfonamides and trimethoprim, which are folic acid pathway inhibitors (see Figure 11-6). In essence, if the environment supplies other metabolites for the microorganism, the activities of antibiotics that target pathways for producing those metabolites are greatly diminished, if not entirely lost. In the absence of the metabolites, full susceptibility to the antibiotics may be restored.
Microorganism-Mediated Antimicrobial Resistance
Intrinsic Resistance
Antimicrobial resistance resulting from the normal genetic, structural, or physiologic state of a microorganism is referred to as intrinsic resistance (Table 11-3). Such resistance is considered a natural and consistently inherited characteristic associated with the vast majority of strains in a particular bacterial group, genus, or species. Therefore, this resistance pattern may be predictable, leading to identification of the organism. Intrinsic resistance profiles are useful for determining which antimicrobial agents should be included in the battery of drugs tested against specific types of organisms. For example, referring to the information given in Table 11-3, aztreonam would not be included in antibiotic batteries tested against gram-positive cocci. Similarly, vancomycin would not be routinely tested against gram-negative bacilli. As is discussed in Chapter 7, intrinsic resistance profiles are also useful markers to aid the identification of certain bacteria or bacterial groups.
TABLE 11-3
Examples of Intrinsic Resistance to Antibacterial Agents
| Natural Resistance | Mechanism |
| Anaerobic bacteria versus aminoglycosides | Lack of oxidative metabolism to drive uptake of aminoglycosides |
| Gram-positive bacteria versus aztreonam (β-lactam) | Lack of penicillin-binding proteins (PBPs) that bind and are inhibited by this β-lactam antibiotic |
| Gram-negative bacteria versus vancomycin | Lack of uptake resulting from inability of vancomycin to penetrate outer membrane |
| Pseudomonas aeruginosa versus sulfonamides, trimethoprim, tetracycline, or chloramphenicol | Lack of uptake resulting from inability of antibiotics to achieve effective intracellular concentrations |
| Klebsiella spp. versus ampicillin (a β-lactam) targets | Production of enzymes (β-lactamases) that destroy ampicillin before the drug can reach the PBP |
| Aerobic bacteria versus metronidazole | Inability to anaerobically reduce drug to its active form |
| Enterococci versus aminoglycosides | Lack of sufficient oxidative metabolism to drive uptake of aminoglycosides |
| Enterococci versus all cephalosporin antibiotics | Lack of PBPs that effectively bind and are inhibited by these lactams |
| Lactobacilli and Leuconostoc sp. versus vancomycin | Lack of appropriate cell wall precursor target to allow vancomycin to bind and inhibit cell wall synthesis |
| Stenotrophomonas maltophilia versus imipenem (a beta-lactam) | Production of enzymes (β-lactamases) that destroy imipenem before the drug can reach the PBP targets |
Common Pathways for Antimicrobial Resistance
Whether resistance is intrinsic or acquired, bacteria share similar pathways or strategies to effect resistance to antimicrobial agents. Of the pathways listed in Figure 11-8, those that involve enzymatic destruction or alteration of the antibiotic, decreased intracellular uptake or accumulation of drug, and altered antibiotic target are the most common. One or more of these pathways may be expressed by a single cell successfully avoiding and protecting itself from the action of one or more antibiotics.

Resistance to Beta-Lactam Antibiotics
As discussed earlier, bacterial resistance to beta-lactams may be mediated by enzymatic destruction of the antibiotics (β-lactamase); altered antibiotic targets, resulting in low affinity or decreased binding of antibiotic to the target PBPs; or decreased intracellular uptake or increased cellular efflux of the drug (Table 11-4). All three pathways play an important role in clinically relevant antibacterial resistance, but bacterial destruction of β-lactams through the production of β-lactamases is by far the most common method of resistance. Extended spectrum β-lactamases are derived from β-lactamases and confer resistance to both penicillins and cephalosporins; carbapenemases are active against carbapenem drugs, such as imipenem. β-lactamases open the drug’s β-lactam ring, and the altered structure prevents subsequent effective binding to PBPs; consequently, cell wall synthesis is able to continue (Figure 11-9).
TABLE 11-4
Summary of Resistance Mechanisms for Beta-Lactams, Vancomycin, Aminoglycosides, and Fluoroquinolones
| Antimicrobial Class | Resistance Pathway | Specific Mechanism | Examples |
| β-lactams (e.g., penicillin, ampicillin, mezlocillin, piperacillin, cefazolin, cefotetan, ceftriaxone, cefotaxime, ceftazidime, aztreonam, imipenem) | Enzymatic destruction | β-lactamase enzymes destroy β-lactam ring, thus antibiotic cannot bind to penicillin-binding protein (PBP) and interfere with cell wall synthesis (see Figure 11-9) | Staphylococcal resistance to penicillin; resistance of Enterobacteriaceae and Pseudomonas aeruginosa to several penicillins, cephalosporins, and aztreonam |
| Altered target | Mutational changes in original PBPs or acquisition of different PBPs that do not bind β-lactams sufficiently to inhibit cell wall synthesis | Staphylococcal resistance to methicillin and other available β-lactams Penicillin and cephalosporin resistance in Streptococcus pneumoniae and viridans streptococci |
|
| Decreased uptake | Porin channels (through which β-lactams cross the outer membrane to reach PBPs of gram-negative bacteria) change in number or character so that β-lactam uptake is substantially diminished | P. aeruginosa resistance to imipenem | |
| Glycopeptides (e.g., vancomycin) | Altered target | Alteration in the molecular structure of cell wall precursor components decreases binding of vancomycin so that cell wall synthesis is able to continue | Enterococcal and Staphylococcus aureus resistance to vancomycin |
| Target overproduction | Excess peptidoglycan | Vancomycin-intermediate staphylococci | |
| Aminoglycosides (e.g., gentamicin, tobramycin, amikacin, streptomycin, kanamycin) | Enzymatic modification | Modifying enzymes alter various sites on the aminoglycoside molecule so that the ability of drug to bind the ribosome and halt protein synthesis is greatly diminished or lost | Gram-positive and gram-negative resistance to aminoglycosides |
| Decreased uptake | Porin channels (through which aminoglycosides cross the outer membrane to reach the ribosomes of gram-negative bacteria) change in number or character so that aminoglycoside uptake is substantially diminished | Aminoglycoside resistance in a variety of gram-negative bacteria | |
| Altered target | Mutational changes in ribosomal binding site diminish ability of aminoglycoside to bind sufficiently and halt protein synthesis | Enterococcal resistance to streptomycin (may also be mediated by enzymatic modifications) | |
| Quinolones (e.g., ciprofloxacin, ofloxacin, levofloxacin, norfloxacin, lomefloxacin) | Decreased uptake | Alterations in the outer membrane diminish uptake of drug and/or activation of an “efflux” pump that removes quinolones before an intracellular concentration sufficient to inhibit DNA metabolism can be achieved | Gram-negative and staphylococcal (efflux mechanism only) resistance to various quinolones |
| Altered target | Changes in the DNA gyrase subunits decrease ability of quinolones to bind this enzyme and interfere with DNA processes | Gram-negative and gram-positive resistance to various quinolones | |
| Macrolides (e.g., erythromycin, azithromycin, clarithromycin) | Efflux | Pumps drug out of cell before target binding | Various streptococci and staphylococci |
| Altered target | Enzymatic alteration of ribosomal target reduces drug binding | Various streptococci and staphylococci |
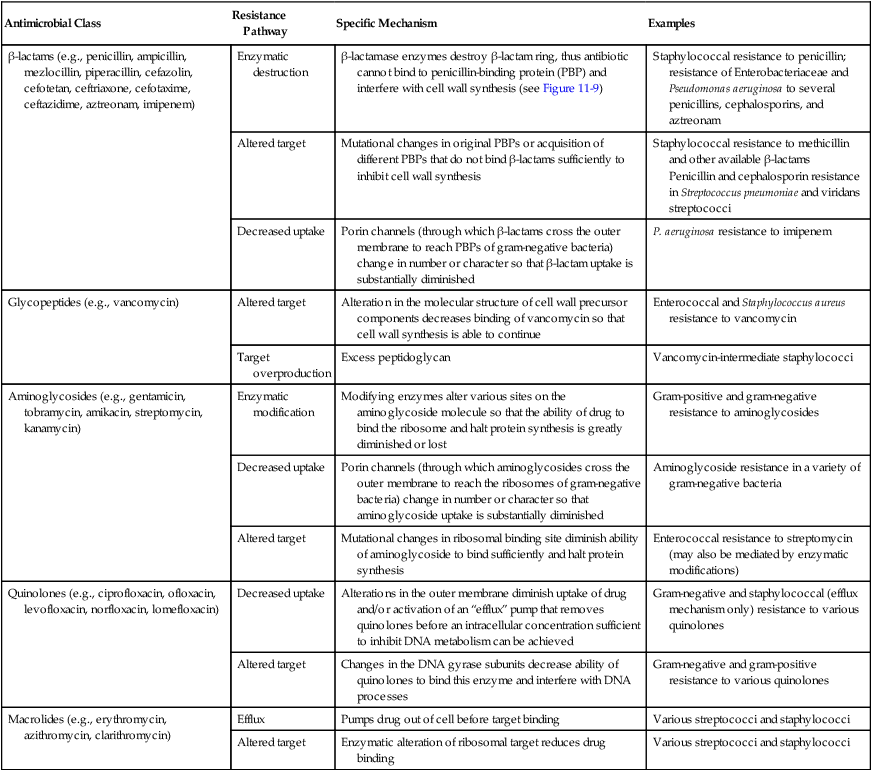
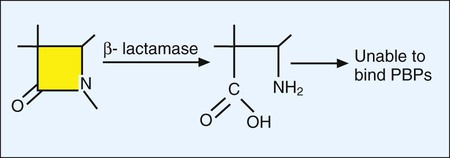
Although the basic mechanism for β-lactamase activity shown in Figure 11-9 is the same for all types of these enzymes, there are distinct differences. For example, β-lactamases produced by gram-positive bacteria, such as staphylococci, are excreted into the surrounding environment, where the hydrolysis of β-lactams takes place before the drug can bind to PBPs in the cell membrane (Figure 11-10). In contrast, β-lactamases produced by gram-negative bacteria remain intracellular, in the periplasmic space, where they are strategically positioned to hydrolyze beta-lactams as they traverse the outer membrane through water-filled, protein-lined porin channels (see Figure 11-10). β-lactamases also vary in their spectrum of substrates; that is, not all β-lactams are susceptible to hydrolysis by every β-lactamase. For example, staphylococcal β-lactamase can readily hydrolyze penicillin and penicillin derivatives (e.g., ampicillin, mezlocillin, and piperacillin); however, it cannot effectively hydrolyze many cephalosporins or imipenem.
Altered targets also play a key role in clinically relevant β-lactam resistance (see Table 11-4). Through this pathway the organism changes, or acquires from another organism, genes that encode altered cell wall–synthesizing enzymes (i.e., PBPs). These “new” PBPs continue their function even in the presence of a β-lactam antibiotic, usually because the beta-lactam lacks sufficient affinity for the altered PBP. This is the mechanism by which staphylococci are resistant to methicillin and all other β-lactams (e.g., cephalosporins and imipenem). Methicillin-resistant S. aureus produces an altered PBP called PBP2a. PBP2a is encoded by the gene mecA. Because of the decreased binding between β-lactam agents and PBP2a, cell wall synthesis proceeds. Therefore, strains exhibiting this mechanism of resistance must be challenged with a non–β-lactam agent, such as vancomycin, another cell wall–active agent. Changes in PBPs are also responsible for ampicillin resistance in Enterococcus faecium and in the widespread β-lactam resistance observed in S. pneumoniae and viridans streptococci.
Because gram-positive bacteria do not have outer membranes through which β-lactams must pass before reaching their PBP targets, decreased uptake is not a pathway for β-lactam resistance among these bacteria. However, diminished uptake can contribute significantly to β-lactam resistance seen in gram-negative bacteria (see Figure 11-10). Changes in the number or characteristics of the outer membrane porins through which β-lactams pass contribute to absolute resistance (e.g., P. aeruginosa resistance to imipenem). Additionally, porin changes combined with the presence of certain β-lactamases in the periplasmic space may result in clinically relevant levels of resistance.
Resistance to Glycopeptides
To date, acquired, high-level resistance to vancomycin has been commonly encountered among enterococci, rarely among staphylococci, and not at all among streptococci. The mechanism involves the production of altered cell wall precursors unable to bind vancomycin with sufficient avidity to allow inhibition of peptidoglycan-synthesizing enzymes. The altered targets are readily incorporated into the cell wall, allowing synthesis to progress (see Table 11-4). A second mechanism of resistance to glycopeptides, described only among staphylococci to date, results in a lower level of resistance; this mechanism is thought to be mediated by overproduction of the peptidoglycan layer, resulting in excessive binding of the glycopeptide molecule and diminished ability of the drug to exert its antibacterial effect.
Resistance to Aminoglycosides
Analogous to beta-lactam resistance, aminoglycoside resistance is accomplished by enzymatic, altered target, or decreased uptake pathways (see Table 11-4). Gram-positive and gram-negative bacteria produce several different aminoglycoside-modifying enzymes. Three general types of enzymes catalyze one of the following modifications of an aminoglycoside molecule (see Figure 11-4):
Resistance to Quinolones
Enzymatic degradation or alteration of quinolones has not been fully described as a key pathway for resistance. Resistance is most frequently mediated either by a decrease in uptake or in accumulation or by production of an altered target (see Table 11-4). Components of the gram-negative cellular envelope can limit quinolone access to the cell’s interior location where DNA processing occurs. Other bacteria, notably staphylococci, exhibit a mechanism by which the drug is “pumped” out of the cell, thus keeping the intracellular quinolone concentration sufficiently low to allow DNA processing to continue relatively unaffected. This “efflux” process, therefore, is a pathway of diminished accumulation of drug rather than of diminished uptake.
Emergence and Dissemination of Antimicrobial Resistance
The resistance pathways that have been discussed are not necessarily new mechanisms that have recently evolved among bacteria. By definition, antibiotics originate from microorganisms. Therefore, antibiotic resistance mechanisms have always been part of the evolution of bacteria as a means of survival among antibiotic-producing competitors. However, with the introduction of antibiotics into medical practice, clinically relevant bacteria have adopted resistance mechanisms as part of their survival strategy. As a result of the increased use of antimicrobial agents, a survival of the fittest strategy has been documented as bacteria adapt to the pressures of antimicrobial attack (Figure 11-11).
All bacterial resistance strategies are encoded on one or more genes. These resistance genes are readily shared between strains of the same species, between species of different genera, and even between more distantly related bacteria. When a resistance mechanism arises, either by mutation or gene transfer, in a particular bacterial strain or species, it is possible for this mechanism to be passed on to other organisms using commonly described paths of genetic communication (see Figure 2-10). Therefore, resistance may spread to a wide variety of clinically relevant bacteria, and any single organism may acquire multiple genes and become resistant to the full spectrum of available antimicrobial agents. For example, strains of enterococci and P. aeruginosa already exist for which there are few effective therapeutic choices. Also, a gene encoding a single, very potent resistance mechanism may mediate multiple resistances. One such example is the mecA gene, which encodes staphylococcal resistance to methicillin and to all other beta-lactams currently available for use against these organisms; this leaves vancomycin as the only available and effective cell wall–inhibiting agent.
In summary, antibiotic use, coupled with the formidable repertoire bacteria have for thwarting antimicrobial activity and their ability to genetically share these strategies, drives the ongoing process of resistance emergence and dissemination (see Figure 11-11). This has been manifested by the emergence of new genes of unknown origin (e.g., methicillin-resistant staphylococci and vancomycin-resistant enterococci), the movement of old genes into new bacterial hosts (e.g., penicillin-resistant N. gonorrhoeae [PPNG]), mutations in familiar resistance genes that result in greater potency (e.g., beta-lactamase–mediated resistance to cephalosporins in Escherichia coli), and the emergence of new pathogens for which the most evident virulence factor is intrinsic or natural resistance to many of the antimicrobial agents used in the hospital setting (e.g., Stenotrophomonas maltophilia).






