CHAPTER 124 Primitive Neuroectodermal Tumors
History
Through the years, many different names have been applied to the tumors that are now known as medulloblastomas or primitive neuroectodermal tumors (PNETs). First described by Bailey and Cushing in 1925,1 they were initially called spongioblastoma cerebelli, subsequently termed spongioblastomas, and eventually named medulloblastoma cerebelli, as they were thought to arise in the cerebellum from medulloblasts. Medulloblasts were thought to be pluripotential cells that eventually gave rise to these tumors in the cerebellum. Present terminology refers to medulloblastomas specifically as PNETs located in the posterior fossa, but it should be noted that although we will limit our discussion to tumors within the brain, PNETs may also occur outside the brain throughout the body, particularly as peripheral neuroblastomas and Ewing sarcomas.
Incidence
PNETs are found in both children and adults, although they are more common in children. Posterior fossa PNETs (medulloblastomas) are the most common malignant solid tumor and the most common malignant brain tumor diagnosed in children. Medulloblastomas constitute about 20% of childhood brain tumors and about 30% of all posterior fossa tumors. Medulloblastomas account for 1% of all adult brain tumors. The incidence of childhood brain tumors has risen during the past 10 years, and most recent evaluations suggest that between 250 and 350 children will be diagnosed with medulloblastoma in the United States each year. The age at diagnosis ranges between 5 and 77 years, and 80% are found in children younger than 15 years.2 Medulloblastomas, although more commonly seen in childhood, can occur in adults, in which case they are usually located in the cerebellar hemispheres and are histologically characterized as desmoplastic.3,4
Most studies indicate that there is a slight male predominance; it is 1.4 to 4.8 times more common in males than in females.5,6 The incidence is 42% higher among whites than blacks.7 A protective effect of maternal folate, iron, and multivitamin supplementation has been suggested to correlate with a decreased incidence of medulloblastomas seen in some studies.8,9 The effect of environmental factors on tumor formation has been debated for several years. N-nitroso compounds are found in beer. Mothers who report drinking one or more beers per week have been associated with a slightly increased incidence of medulloblastoma. Children born of mothers living on a farm during pregnancy also have a mildly increased disease incidence. This association was not significant in mothers who handled animals but was statistically significant with children who resided on a farm for more than 1 year. There were no significant associations with maternal or early childhood medication exposures.10
Several syndromes are associated with a familial increased incidence of medulloblastoma. Although the incidence of medulloblastoma has not been shown to be increased in patients with phakomatosis, such as neurofibromatosis or tuberous sclerosis, it has been shown to be increased in patients with Gorlin’s syndrome (nevoid basal cell carcinoma), Li-Fraumeni syndrome, and Turcot’s syndrome. Gorlin’s syndrome is characterized by the occurrence of multiple nevoid basal cell carcinomas, multiple skeletal anomalies (including jaw cysts), cutaneous anomalies (including pits on the hands and soles of the feet), calcifications of the dura, hydrocephalus, and developmental delay. In Gorlin’s syndrome, about 5% of children develop medulloblastoma when they are younger than 5 years.11,12 The medulloblastoma subtype in Gorlin’s syndrome is often desmoplastic and is thought to be due to inactivation of the tumor suppressor gene on chromosome 9q.13 Gorlin’s syndrome is an autosomal dominant disorder in which the gene mutated is the human homologue of the Drosophila melanogaster patched gene (PATCHED). PATCHED codes for a transmembrane receptor for the secreted ligand sonic hedgehog homologue (SHH). The SHH signaling pathway is vital during the development and formation of the central nervous system (CNS). PATCHED has been localized to chromosome 9q, and medulloblastomas have been shown to have a loss of heterozygosity on chromosome 9q.14
In Turcot’s syndrome, or multiple familial polyposis, the inheritance of medulloblastoma is variable and is believed to be either autosomal recessive or dominant. The frequency with which medulloblastomas develop is uncertain.6,15 In Turcot’s syndrome, the patient has a primary brain tumor but also develops multiple colorectal adenomas, colorectal adenocarcinoma, or both. These patients tend to have mutations in the adenomatous polyposis coli (APC) gene on chromosome 5q21.14
Supratentorial PNETs may also be seen in conjunction with retinoblastoma. Often termed pinealoblastomas because of their location, these tumors can be associated with bilateral retinoblastomas and are caused by germline mutations in the Rb gene. Suprasellar or parasellar PNET tumors may also be seen in these patients.14
Pathology
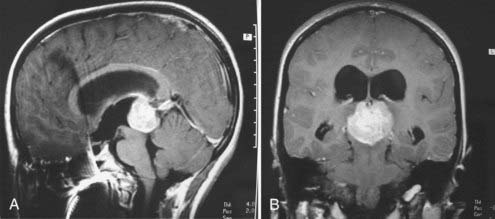
Historically, the terminology used to describe medulloblastomas or PNETs has been confusing. Initially termed spongioblastoma cerebelli by Bailey and Cushing,1 they had earlier been termed spongioblastoma multiforme. In 1983, Rorke16 named them primitive neuroectodermal tumors, recognizing that histologically these tumors were identical to those seen supratentorially and elsewhere. Histologically similar tumors found in the pineal gland are termed pinealoblastomas (Fig. 124-1), whereas those in the supratentorial space are termed either PNETs or neuroblastomas (Fig. 124-2), and those in the eye are termed retinoblastomas. Medulloblastoma is the term now applied to PNETs arising in the posterior fossa. Subcategories have been described based on differentiation, and thus medulloblastomas have been categorized as having or not having glial, ependymal, or neuronal differentiation.
About 50% of medulloblastomas demonstrate neuronal or glial differentiation.17 Homer Wright pseudorosettes, in which nuclei surround a central clear area made up of cell processes rather than a vessel, are present in more than 40% of medulloblastomas (Fig. 124-3). Astrocytic differentiation is seen in more than 50% of tumors and is identified by cell processes that stain positive for glial fibrillary acidic protein (GFAP).18,19
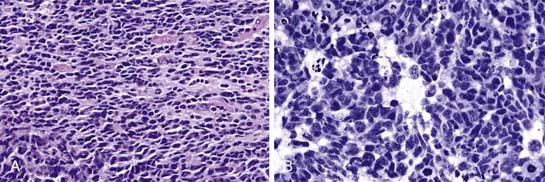
Grossly, medulloblastomas appear as soft, unencapsulated but relatively well-circumscribed, friable, purplish tumors, usually located in proximity to the fourth ventricle in the region of the vermis. They may invade the cerebellum, brainstem, or cerebellar peduncle. Unlike ependymomas, lateral extension into the cerebellopontine angle is a rare occurrence.20 The four histologic subtypes of medulloblastomas, according to the World Health Organization (WHO) classification of CNS tumors, include desmoplastic (or nodular) medulloblastomas, medulloblastomas with extensive nodularity, anaplastic medulloblastomas, and large cell medulloblastomas.21 The desmoplastic subtype, which represents 29% of all medulloblastomas, tends to be located within the cerebellar hemisphere and is usually firmer and often more well demarcated. After hematoxylin and eosin staining, they appear as small, round, blue-cell tumors with hyperchromatic nuclei and minimal cytoplasm.22 This variant is characterized by nodular reticulin-free zones surrounded by densely packed proliferative cells with hyperchromatic and pleomorphic nuclei. The nodules represent regions of neuronal maturation with a reduced nuclear-to-cytoplasmic ratio. Medulloblastoma with extensive nodularity occurs in infants. These tumors differ from the desmoplastic subtype in that the reticulin-free zones become enlarged and rich in a neuropil-like tissue. Anaplastic medulloblastomas display widespread cellular atypia, including marked nuclear pleomorphism, high mitotic activity, atypical cell shape, and cell-to-cell wrapping. Apoptosis is a prominent feature in this subtype. Large cell medulloblastoma represents 2% to 4% of all medulloblastomas. Characteristics of this subtype include large round cells with prominent nucleoli and varying amounts of cytoplasm. Apoptotic features and regions of anaplasia are common.21
The cell of origin in medulloblastomas remains controversial. The fetal external granule cell layer has been hypothesized as the area of origin. Scattered foci of this fetal external granule cell layer persist in the medullary velum, often considered a point of origin for medulloblastomas.23,24 Other candidate cells have included undifferentiated subependymal cells and neurons within the internal granular layer. It has been suggested that exposure to toxins or viruses during the perinatal period may induce some of these pluripotential cells to become medulloblastomas. Posterior fossa tumors similar in histopathology to medulloblastomas have been induced in hamsters by intracerebral injection of JC virus and in rats through the large T-antigen of SV40 during the perinatal period.24
A variety of chromosomal abnormalities have been reported in medulloblastomas, but no specific genetic abnormality has been consistently associated with its pathogenesis. The most consistent chromosomal abnormality reported involves chromosome 17. Abnormalities may include loss or gain of parts of chromosome 17.25,27 The most frequent aberration involves the coincidental loss of part of 17p and gain of 17q through formation of an isochromosome of the long arm of 17q.27 This can be seen in up to 66% of medulloblastomas, but it is thought to be related to tumor progression rather than tumor origin.25–27 The breakpoint is usually the proximal p-arm at the 17p18318880-19046234 region.29,30 It is now generally accepted that isochromosome 17q represents an unbalanced translocation between the two copies of chromosome 17.31 The breakpoint occurs within the EPN2 gene. This gene is expressed in the cerebellum and interacts with proteins related to protein transport. The tumor suppressor gene p53 is located in the 17p region and is included within the region of loss; however, it is only mutated in 5% to 10% of medulloblastomas.32 The p53 gene has therefore been postulated to contribute to the pathogenesis of medulloblastomas but is unlikely to be the cause.33,34 Other frequent losses are seen on chromosomes 8p, 10q, 16q, and 20p. Frequent gains are seen on chromosomes 2p, 4p, 7, and 19.27
Medulloblastoma tumor cells have been shown to overexpress various factors. Among these, platelet-derived growth factor-α receptor expression has been associated with neuronal differentiation.35 Various growth factors have been associated with medulloblastomas, but none has been found that is specific to medulloblastomas. The overamplification of MYC, occurring in 16% of medulloblastomas on chromosome 8, has been associated with unfavorable patient outcomes. Favorable outcomes for medulloblastoma have been associated with high levels of TrkC transcripts and low levels of MYC, MYCN, and erbB-2 transcription.27,36,37 Cytogenetic analysis has shown that some cells may be diploid, some tetraploid, and others aneuploid.38,39
A separate subtype of medulloblastoma was recognized by Rorke and associates40 in 1995. Termed atypical teratoid-rhabdoid tumors of the cerebellum, these tumors are often mistaken for medulloblastomas. They show loss of heterozygosity on chromosome 22q. The tumor suppressor gene altered in these tumors is on chromosome 22 and was identified as hSNF5/INI1. The hSNF5 gene has been identified as altered in sporadic medulloblastomas and PNETs.14,28,41,42
Clinical Evaluation
Signs and Symptoms
Children with medulloblastoma generally present with symptoms of increased intracranial pressure (ICP). Symptoms are generally present for several weeks to months, are typical for all posterior fossa tumors, and are not specific for medulloblastomas. Among the common symptoms are headaches, nausea, and vomiting. These symptoms are also related to hydrocephalus, which often occurs simultaneously with these tumors. Morning headaches and vomiting are often seen, and many children have undergone extensive gastrointestinal evaluation before a diagnosis of a brain tumor is made. Other symptoms include an unsteady gait, ataxia, and diminished coordination. Rarely, children may present with head tilt or torticollis. This can be related to cranial nerve palsy, particularly sixth nerve palsy or secondary to pain because of dural traction.43,44 In very young children, the diagnosis can be difficult, and such children may present only with macrocephaly, loss of milestones, irritability, and vomiting.
Radiographic Evaluation
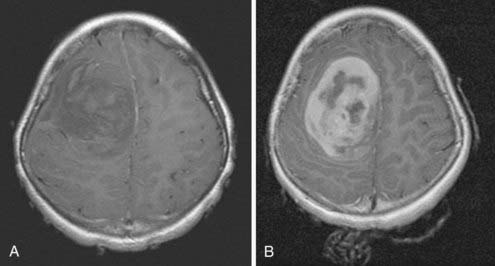
Children with PNETs or medulloblastomas are usually diagnosed by means of computed tomography (CT) or magnetic resonance imaging (MRI). On CT scans, medulloblastomas are typically hyperdense, showing homogeneous contrast enhancement, and may be partially cystic.45 They usually have smaller areas of calcification and small cysts; rarely, they may be extensively calcified.46,47 On MRI scans, the tumors are usually isointense or hypointense on T1-weighted images, hyperintense on T2-weighted images, and intensely enhancing after gadolinium injection (Fig. 124-4).48,49 About 10% to 15% of medulloblastomas are not contrast enhancing on an MRI scan, which makes postoperative assessment of residual disease more difficult. Unfortunately, there are no clear-cut radiologic features that absolutely differentiate a medulloblastoma from other posterior fossa masses; pathologic analysis is the only way to be certain.
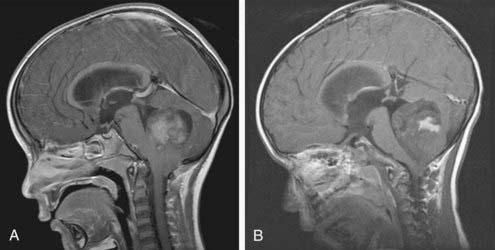
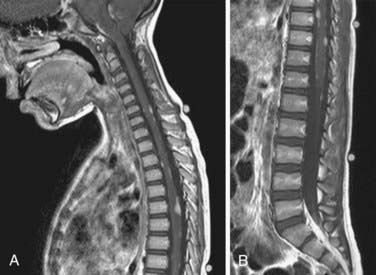
Because medulloblastomas can spread throughout the CNS, many physicians obtain an MRI scan of the spine as soon as the diagnosis of medulloblastoma is entertained (Fig. 124-5). Postsurgical blood and protein may confound spinal imaging for weeks after surgery, and thus preoperative spinal MRI may give the best assessment of spinal metastasis. Because most children have concomitant hydrocephalus, performing a lumbar puncture is dangerous and should be avoided until most of the mass has been removed.
Treatment
Management of Hydrocephalus
Hydrocephalus is a common presenting feature, particularly of posterior fossa PNETs or medulloblastomas. Treatment of hydrocephalus usually involves placement of an external ventricular drainage device (EVD) or ventriculostomy at the time of surgical excision of the lesion. Preoperatively, hydrocephalus may also be treated with dexamethasone to help reduce swelling associated with the tumor. The use of EVDs permits easy access to the CSF and allows intermittent fluid drainage at a desired height or pressure. Attention must be paid to the height or pressure at which the ventriculostomy or EVD drainage is maintained when the tumor is still in place. In less than 40% of cases, an EVD will be necessary over the long term after removal of the tumor, and preoperative shunting before removal of the tumor is discouraged to avoid upward herniation.50,51 Upward herniation occurs when there is an imbalance in pressure across the tentorium such that there is increased pressure in the posterior fossa relative to the supratentorial space. Upward herniation is a rare event, occurring in less than 3% of cases.52
The risk for propagation of medulloblastoma cells through a shunt appears to be minimal. Berger and associates51 found that extraneural metastases occurred in only 8 of 415 children with brain tumors, all of whom had medulloblastomas. However, 5 of the 8 children did not even have a shunt. Jamjoom and colleagues,53 in a review of 160 cases of medulloblastomas with systemic metastasis, reported that only 19% of patients had shunts and that propagation through the shunt could be ascribed to only 7% of those cases. The use of millipore filters in shunts to decrease such dissemination through the shunt has largely been abandoned because of the high rate of occlusion of such filters and because of the lack of any evidence suggesting efficacy in preventing metastasis.
In the postoperative period, EVD pressure is gradually increased to allow normal CSF pathways to resume absorption of CSF. Drainage is generally stopped for a time, and pressures are measured before the ventriculostomy catheter is removed. Symptoms of increased ICP, CSF leakage from the wound, and development of a pseudomeningocele can be indicators of failure to wean the patient from the EVD and the need for a shunt. The need for postoperative shunts has been correlated with younger patients, large ventricles at the outset, long-standing ventriculomegaly, and large tumors.54
Tumor Removal
Standard pediatric neurosurgical and anesthetic techniques are used in the removal of PNETs. Because these can be bloody tumors, adequate venous access as well as arterial pressure monitoring is important, and transfusion may be necessary. Such operations may be lengthy, particularly in the case of large tumors with brainstem involvement, and monitoring of urine output through a Foley catheter is useful. Doppler monitoring is rarely necessary because the head is positioned only slightly higher than the heart. Air embolism in this position is rare but must be considered if there is a sudden decrease of end-tidal PCO2 or a drop in O2 saturation. Electromyographic monitoring of the lateral rectus and facial muscles may provide assistance to the surgeon when working near the abducens and facial nuclei along the brainstem,55 but improvement in outcomes with use of such monitoring has not been generally established.
A craniotomy is appropriate for tumors in the supratentorial space, although most will be located in the posterior fossa. For those in the posterior fossa, the patient is usually in the prone position with the head slightly flexed.56,57 This position is also described as the angulated Concorde position. The sitting position has largely been abandoned because of the risk for air embolism, difficulty in positioning, and surgeon fatigue. Many surgeons will perform a suboccipital craniectomy, but with the development of improved high-speed drills, more neurosurgeons now opt for a suboccipital craniotomy and replacement of the bone flap.
The tumor is usually purplish and can be distinguished from normal cerebellum. Use of an ultrasonic aspirator greatly enhances the speed of tumor removal and assists with reducing blood loss. Blood loss can be significant, particularly in very young children, and can act as a limiting force in completing tumor resection. Debulking of the tumor can allow the surgeon to bring in the edges of the tumor to better identify the borders of the tumor. Gross total resection of the tumor is the goal of surgery, but it must be balanced against possible neurological deficit. The dura is then closed primarily or, if necessary, by employing a dural graft. The bone may then be replaced, and the wound closed in layers. Multi-institutional data indicate that in 80% of cases, greater than 90% of the tumor is resected. In only a small number is there less than 50% removal, and there is some indication that surgeons most familiar with these tumors achieve the greatest amount of resection.58,59
Complications
The tumor may invade the floor of the fourth ventricle or brainstem in as many as 15% to 40% of medulloblastomas.52,58,60 Infiltration of the brainstem may limit total resection of the tumor. Use of the operating microscope allows such tumors to be shaved down to the point that there is a thin film remaining. Such small amounts have been shown to be successfully treated with irradiation and chemotherapy.58 Dissection beyond the floor of the fourth ventricle into the brainstem can result in significant morbidity, especially cranial nerve deficits.61
Surgical mortality has decreased steadily from as high as 30% in the 1930s to as low as 2% in present studies.58,59 Albright and associates59 have shown that surgeons familiar with pediatric brain tumors had the lowest complication rate and achieved the greatest amount of resection, removing more than 90% of the tumor and leaving less than 1.5 cm3 of residual tumor. As is true in most aspects of neurosurgical practice, patients with the most severe preoperative deficits have the greatest intraoperative risk. Postoperative morbidity is seen in upward of 50% of patients. This includes either transient or permanent deficits, which can include transient nystagmus, ataxia, cranial nerve deficits, hemiparesis, and nausea and vomiting.56 Cochrane, in a single-institution assessment, indicated that preoperative deficits worsened after craniotomy in 41% of children with medulloblastomas and that only 14% of new postoperative deficits resolved completely.61
An interesting complication, which more recently has become increasingly appreciated as a postoperative complication of resection of medulloblastomas, is posterior fossa syndrome, which has also been termed cerebellar mutism. Initially described by Wisoff and Epstein,62 it is characterized by mutism, abulia, a high-pitched cry, oral motor apraxia, drooling, and ataxia. These children are often without deficit for the first 48 to 72 hours postoperatively but then develop symptoms later in the postoperative period. Various aspects of resection have been linked to the development of posterior fossa syndrome. Edema of the dentorubrothalamic tract, splitting of the inferior vermis, and extent of resection of the vermis have all been associated with the development of cerebellar mutism.63,64 The exact incidence of the syndrome has been reported to be between 15% and 25% of medulloblastoma cases.65 Preoperative brainstem invasion is the only feature that correlates with risk for cerebellar mutism syndrome. One year after diagnosis, nonmotor speech and language deficits, neurocognitive deficits, and ataxia persisted in a certain population of patients.65 There is no specific treatment for posterior fossa syndrome, and although corticosteroids are commonly used, there is no study that suggests that their use improves outcome. Symptoms usually begin to abate about 2 to 6 weeks after onset and generally are largely gone within 3 months; however, some persist.
Outcomes and Adjuvant Therapy
The outcome for patients with medulloblastoma is dependent on the stage of the tumor. The Chang system is the most commonly employed staging system. Tumors are graded based on size (T stage) and metastases (M stage) (Table 124-1).66 Recent studies using improved radiation techniques and adjuvant chemotherapy indicate that tumor size, or T stage, is much less important than the finding of metastatic dissemination in the CNS.58,67,68 Multiple studies have now confirmed the importance of age at the time of diagnosis, evidence of metastatic disease, site of tumor, histopathology, and time of diagnosis as being consistently important for outcome.7 There is no difference in survival time according to race or sex.
| Tumor (T) Stage |
| T1: Tumor < 3 cm in diameter, involving 1 posterior fossa structure |
| T2: Tumor < 3 cm in diameter, invading 2 or more posterior fossa structures |
| T3a: Tumor > 3 cm in diameter, invading 2 or more posterior fossa structures |
| T3b:Tumor invading the floor of the fourth ventricle |
| T4: Tumor extending out of the fourth ventricle, upward into the third ventricle, caudally into the cisterna magna, or associated with severe hydrocephalus |
| METASTASES (M) STAGE |
| M0: No evidence of tumor dissemination |
| M1: Positive lumbar cerebrospinal fluid cytology |
| M2: Intracranial tumor dissemination |
| M3: Intraspinal tumor dissemination |
| M4: Systemic dissemination |
From Chang CH, Housepian EM, Herbert C Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastoma. Radiology. 1969;93:1351-1359.
Determination of residual disease is best done by MRI 24 to 48 hours after surgery, before any enhancement attributable to postoperative inflammation or gliosis can cloud the imaging of residual neoplasm. Cytologic examination of CSF is done by lumbar puncture about 2 weeks after surgery, after sufficient time has passed to avoid contamination by operative debris. Similarly, intraoperative samples may be taken at the beginning of the procedure. A CSF cytology determination that is positive for tumor cells, either preoperatively or postoperatively, predicts a poor outcome, similar to that observed in stage M2/3 patients. A negative cytology test, however, does not preclude more advanced disease.69
Spinal MRI should be done after surgery if it was not performed before. Postoperative imaging can occasionally be difficult because of the presence of postoperative debris and blood.70 Myelography has largely been replaced by MRI for determination of drop metastasis and is not as sensitive for detection of “sugar-coating” of the leptomeninges with tumor.71 Most tumors are stage M0 at diagnosis; however, 15% to 20% of patients are found to have a CSF cytologic determination positive for tumor cells, and 30% have supratentorial or spinal metastases.58 Less than 1% have a diagnosis of systemic cancer, and thus a bone scan is no longer recommended as part of the staging process.
The degree of surgical excision has been shown to correlate with outcome, with a residual tumor volume of less than 1.5 cm3 associated with improved outcome.56,58,59 Earlier studies confirmed that even patients undergoing gross total resection will have recurrence without adjuvant therapy.2 The tumors tend to recur locally but can disseminate throughout the CSF pathways. Standard treatment has usually included radiation therapy. Cushing was the first to use craniospinal irradiation to treat these tumors,1 recognizing their tendency for CSF propagation. Failure to treat the entire neuraxis with radiation results in a near uniform failure. Paterson and Farr72 reported a doubling in survival time when tumors were treated with radiation. Survival rates improved from 12% to 50% in the mid-1970s to the present rates of 50% to 70%, with 5400 to 5800 cGy administered to the posterior fossa and 3500 cGy to the neuraxis.73,74
Attempts have been made to decrease the morbidity associated with craniospinal irradiation. Hyperfractionated irradiation and dose reduction of craniospinal irradiation have been used to try to decrease the global effects of irradiation. The Children’s Cancer Study Group and the Pediatric Oncology Group recently evaluated reduced-dose craniospinal irradiation. Low-stage or good-stage medulloblastoma patients were randomized to receive either the standard dose (3600 cGy) or a reduced dose (2340 cGy) of craniospinal irradiation.75 The study was terminated prematurely, after only 16 months, because there was clear evidence of an increase in relapses in the reduced-dose group. More recently, attempts at reducing the dose of irradiation have included the use of chemotherapy. The International Society of Pediatric Oncology compared craniospinal doses of 3600 cGy with 2400 cGy plus preirradiation chemotherapy for average-risk medulloblastoma patients and found that the reduced-dose patients had a 69% survival rate compared with 60% for those receiving the standard dose.76 Halberg and coworkers77 showed that procarbazine and hydroxyurea combined with 2400 to 3600 cGy delivered to the entire brain and 2400 to 2600 cGy to the neuraxis produced a 5-year disease-free survival rate of 77%. A more recent study from the Children’s Cancer Study group has shown acceptable survival times with reduced-dose irradiation.78 In this study, 65 children 3 to 10 years old were treated with postoperative reduced-dose craniospinal irradiation (23.4 Gy) and 55.8 Gy local irradiation. Adjuvant chemotherapy with vincristine was administered during irradiation, and lomustine (CCNU), vincristine, and cisplatin were administered subsequently. The progression-free survival rates were 86% at 3 years and 79% at 5 years. Stereotactic radiosurgery has been suggested for use as an adjuvant to conventional irradiation in patients with defined residual tumor.79
The first study to indicate clearly the usefulness of chemotherapy in treating medulloblastoma was done in 1990.68 In that study, patients with high-risk medulloblastomas, who had metastatic disease and large tumors, had a survival rate of 46% when they received chemotherapy (CCNU, vincristine, and prednisone) and irradiation, but there were no survivors among similar patients who received radiation therapy only.68 A second study, in 1991, demonstrated a 5-year survival rate of 74% in patients treated with chemotherapy (nitrogen mustard, vincristine, procarbazine, and prednisone) and irradiation compared with 56% in those treated with radiation therapy alone.80
Various chemotherapy regimens have been tried in association with radiation therapy. A Children’s Cancer Study Group report showed a 46% 4-year survival rate in those treated with eight drugs in 1 day (8-in-1) compared with a 63% survival rate in those receiving CCNU and vincristine.81 In conjunction with high-dose methotrexate, 8-in-1 therapy offered a disease-free survival interval of 5 years in 74% of patients with low-stage medulloblastomas and in 57% of patients with high-stage medulloblastomas.82 Perhaps the best results reported to date come from Packer and colleagues.83,84 Packer’s protocol using vincristine weekly during radiation therapy, then vincristine, cisplatin, and CCNU for eight cycles afterward, substantially improved survival. In this study, started as a single-institution trial, 108 children with high-stage (therefore high-risk) medulloblastomas were treated with this protocol, and the 5-year disease-free survival rate was 73%, better than the best previously available treatment for low-stage medulloblastoma patients. The protocol was then expanded to two additional centers, and the progression-free survival rate in these high-stage medulloblastoma patients was 85%. This was true even in children younger than 5 years who received only 2400 cGy of spinal irradiation.85 The results for children with metastatic disease were encouraging, with a 5-year survival rate of 67%. Those with only localized disease at diagnosis had a 90% 5-year survival rate. These results contrast with the results from the 1970s, with 5-year survival rates of 12% to 50% in patients who had received surgery and radiation therapy alone.73
Although radiation therapy has been the mainstay of treatment in medulloblastoma patients, its use comes with a significant price. The most notable effects are seen in patients who are younger than 3 years. In these patients, standard-dose irradiation produces significant cognitive deficits. New protocols have been designed to determine whether radiation therapy may be delayed for several years by using chemotherapy first. In a Pediatric Oncology Group study, children younger than 3 years with malignant brain tumors were treated with cyclophosphamide plus vincristine, followed by cisplatin plus etoposide.86 Progression-free survival for all tumors was 40%, and children with medulloblastomas had even higher response rates.
Survival from medulloblastoma correlates strongly with five factors: age at diagnosis, whether there is evidence of dissemination, tumor location, tumor histopathology, and diagnosis after 1985. Children 0 to 3 years old have the worst survival times. Children diagnosed with supratentorial medulloblastomas have a poorer median survival interval (2.5 years) than children with infratentorial medulloblastomas (7.2 years).7,87 Also, children diagnosed after 1985 have a longer median survival time.7 Duration of symptoms, severity of hydrocephalus, size of tumor, and even invasion of the brainstem have not been shown to correlate with survival.58,68 The extent of tumor resection does appear to influence survival. Analysis of postoperative scans in good-risk children (older than 3 years without evidence of metastasis) has confirmed that there is a significant difference in survival time between patients undergoing total and those those undergoing near total resection (<1.5 cm3).58 To date, no study has shown a difference in survival time between patients undergoing total and those undergoing near-total resection, which is probably attributable to the responsiveness of these tumors to radiation and chemotherapy.
Using the Surveillance, Epidemiology, and End Results (SEER) registry database, Lai88 published the largest survival study to date of adult patients with medulloblastomas. This study reviewed 454 cases from 1973 to 2003. The 5- and 10-year relative survival rates were 64.9% and 52.1%, respectively. This 5-year survival rate was lower than the 72% previously reported in the French multicenter study of 253 patients.89 Based on the results of this study, patients diagnosed after age 40 years have the worst prognosis, with a median survival time of 7.7 years. Factors associated with better survival in adult patients include diagnosis after 1985, being younger than 20 years at diagnosis, undergoing gross total resection, and receiving cranial irradiation. Large cell histopathology is associated with a worse prognosis.88
A variety of biologic indicators have been examined in patients with respect to survivability. Table 124-2 lists these various factors. None has had sufficient correlation to be widely used in staging these tumors or designing treatment strategies. A significant push is on, however, to determine whether such tumor biology studies may one day influence therapy. The Children’s Oncology Group, which now represents the combined efforts of the Pediatric Oncology Group and the Children’s Cancer Study Group, has made collection and banking of tumor data a priority in all future protocols.
TABLE 124-2 Tumor Factors and Their Correlation with Patient Survival
| FACTOR | CORRELATION | STUDY |
|---|---|---|
| GFAP | GFAP positive, 5-year survival rate of 82% GFAP negative, 5-year survival rate of 30% |
Goldberg-Stern et al, 1991106 |
| DNA ploidy | Aneuploid tumors recur more frequently than diploid or tetraploid; patients with hyperdiploid tumors survived longer than those with diploid tumors | Zerbini et al, 1993107; Tait et al, 1990108 |
| Chromosome 17p heterozygosity | Loss of heterozygosity corresponds with shorter survival | Cogen et al, 199030; Batra et al, 1995109 |
| Chromosome 22 | Loss of chromosome 22 corresponds with poor prognosis | Nicholson et al, 1999110 |
| c-erbB-2 oncogene | Worse survival with > 50% c-erbB-2–positive tumor cells | Gilbertson et al, 1995111 |
| TrkC neurotrophin receptor | Worse survival with low TrkC levels | Segal et al, 1994112 |
| BUDR labeling index | No correlation seen | Ito et al, 1992113 |
| HER2 and HER4 genes | Coexpression correlates with improved survival | Gilbertson et al, 1997114 |
| Mitotic percentage index | Increased mitotic percentage index correlates negatively with survival | Gilbertson et al, 1997115 |
| p53 gene | p53 expression correlates with poor outcome | Adesina et al, 2000116 |
| Apoptosis | Higher apoptosis index correlates with improved outcome | Haslam et al, 1998117 |
| PSA-NCAM in cerebrospinal fluid | PSA-NCAM concentration was higher in patients who relapsed | Figarella-Branger et al, 1996118 |
| MYC and MYCN genes | Favorable outcome associated with low expression levels | Eberhart et al, 200436 |
BUDR, bromodeoxyuridine; GFAP, glial fibrillary acidic protein; PSA-NCAM, polysialic-neural cell adhesion molecule.
Recurrence of medulloblastoma after initial treatment is usually incurable, and only a small number of patients have responded to additional therapy. The practice of routinely performing imaging studies in patients for several years after treatment has recently been called into question.90 In the study by Torres and associates,90 794 scans were obtained from 86 children treated for medulloblastoma between 1980 and 1991. Twenty-three children had recurrent tumor, but only 4 tumors were detected on surveillance scanning, and 19 were associated with some kind of symptoms. Patients with asymptomatic recurrences survived longer than those with symptomatic recurrences, but all who had recurrence died. There are additional reports indicating that detection of tumors with surveillance scans could be valuable in providing a therapeutic window for adjuvant therapy.91,92 Most patients who have a recurrence experience it within the first 2 years after presentation. It has been suggested that if there is no recurrence after 8 years, the patient may be considered cured.93
Seventy percent of recurrences occur in the posterior fossa and can be detected by the observation of focal nodular enhancement on CT or MRI.94 Unlike other CNS tumors, medulloblastomas can metastasize systemically. Eighty percent of these systemic metastases are to bone or bone marrow, 30% to lymph nodes, 15% to lungs or pleura, and 14% to the liver.94,95 Tarbell and associates95 have shown that chemotherapy may decrease the incidence of bone metastases. In this report, 12% of patients who received only craniospinal irradiation had bony metastases, whereas none of those who received craniospinal irradiation and chemotherapy had bony metastases. Similarly, even in high-risk patients, none of the children who received chemotherapy had metastases outside the CNS.96
Treatment of patients with relapsed medulloblastoma has included a variety of chemotherapeutic agents.97,98 High-dose chemotherapy with stem cell rescue and even bone marrow transplantation have been tried with moderate success.98 An event-free survival rate of 30% has been achieved in relapsed patients using stem cell rescue after chemotherapy.
Larger issues regarding the quality of life after treatment are now being addressed.99 Neurocognitive and endocrine sequelae have been well documented.100,101 It has been shown that children younger than 7 years have a substantially greater loss of intellectual function and drop in IQ scores than do older children.102 Farwell and associates101 showed that IQ was less than 90 in 89% of children treated with craniospinal irradiation. This was not true in the case of other tumors, such as cerebellar astrocytomas, for which cranial spinal irradiation is not given. Thus, even after surviving their cancer, 80% to 90% of children will reveal serious neurocognitive sequelae on psychometric testing.103,104 Similarly, dysfunction is seen in pituitary function, and most children will require endocrine replacement therapy of some type.
The risk for a second cancer after surviving medulloblastoma is also increased. A recent population-based study found a 5.4-fold increase for secondary cancers in these survivors.105 These tumors occurred 6 to 7 years after completion of initial therapy, and 50% occurred within the field of the radiation therapy. As survival rates increase, the ultimate long-term burden of secondary tumors and long-term complications in these children remains to be determined.
Berger MS, Baumeister B, Geyer JR, et al. The risks of metastases from shunting in children with primary central nervous system tumors. J Neurosurg. 1991;74:872-877.
Chapman CA, Waber DP, Bernstein JH, et al. Neurobehavioral and neurologic outcome in long-term survivors of posterior fossa brain tumors: role of age and perioperative factors. J Child Neurol. 1995;10:209-212.
Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
Eberhart CG, Kratz J, Wang Y, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol. 2004;63:441-449.
Gilbertson RJ, Perry RH, Kelly PJ, et al. Prognostic significance of HER2 and HER4 coexpression in childhood medulloblastoma. Cancer Res. 1997;57:3272-3280.
Haie-Meder C, Song PY. Medulloblastoma: differences in adults and children—regarding Frost et al., IJROBP 32:951-957; 1995 and Prados et al., IJROBP 32:1145-1152; 1995. Int J Radiat Oncol Biol Phys. 1995;32:1255-1257.
Hornbek NB. Radiation therapy in treatment of medulloblastoma in childhood. In: Zeltzer PM, Pochedly C, editors. Medulloblastomas in Children. New Concepts in Tumor Biology, Diagnosis and Treatment. New York: Praeger; 1986:164-181.
Johnson DL, McCabe MA, Nicholson HS, et al. Quality of long-term survival in young children with medulloblastoma. J Neurosurg. 1994;80:1004-1010.
Jung HL, Wang KC, Kim SK, et al. Loss of heterozygosity analysis of chromosome 17p13.1-13.3 and its correlation with clinical outcome in medulloblastomas. J Neurooncol. 2004;67:41-46.
Lai R. Survival of patients with adult medulloblastoma: a population-based study. Cancer. 2008;112:1568-1574.
Laurent JP, Chang CH, Cohen ME. A classification system for primitive neuroectodermal tumors (medulloblastoma) of the posterior fossa. Cancer. 1985;56:1807-1809.
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
McNeil DE, Cote TR, Clegg L, et al. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncol. 2002;39:190-194.
Pollack IF, Polinko P, Albright AL, et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery. 1995;37:885-893.
Ray A, Ho M, Ma J, et al. A clinicobiological model predicting survival in medulloblastoma. Clin Cancer Res. 2004;10:7613-7620.
Reddy AT, Janss AJ, Phillips PC, et al. Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer. 2000;88:2189-2193.
Robertson PL, Muraszko KM, Holmes EJ, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105:444-451.
Steinbok P, Hentschel S, Cochrane DD, et al. Value of postoperative surveillance imaging in the management of children with some common brain tumors. J Neurosurg. 1996;84:726-732.
Stevenson L, Echlin F. Nature and origin of some tumors of the cerebellum. Medulloblastoma. Arch Neurol. 1934;31:93-109.
Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888-901.
1 Bailey P, Cushing H. Medulloblastoma cerebelli, a common type of midcerebellar glioma of childhood. Arch Neurol Psychiatry. 1925;14:192-224.
2 Roberts RO, Lynch CF, Jones MP, et al. Medulloblastoma: a population-based study of 532 cases. J Neuropathol Exp Neurol. 1991;50:134-144.
3 Maleci A, Cervoni L, Delfini R. Medulloblastoma in children and in adults: a comparative study. Acta Neurochir (Wien). 1992;119:62-67.
4 Haie-Meder C, Song PY. Medulloblastoma: differences in adults and children—regarding Frost et al., IJROBP 32:951-957; 1995 and Prados et al., IJROBP 32:1145-1152; 1995. Int J Radiat Oncol Biol Phys. 1995;32:1255-1257.
5 Chatty EM, Earle KM. Medulloblastoma. A report of 201 cases with emphasis on the relationship of histologic variants to survival. Cancer. 1971;28:977-983.
6 Finlay JL. Natural history and epidemiology of medulloblastoma. In: Zelter PM, Pochedly C, editors. Medulloblastomas in Children: New Concepts in Tumor Biology, Diagnosis and Treatment. New York: Praeger; 1986:22-36.
7 McNeil DE, Cote TR, Clegg L, et al. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncol. 2002;39:190-194.
8 Young JLJr, Miller RW. Incidence of malignant tumors in U.S. children. J Pediatr. 1975;86:254-258.
9 Thorne RN, Pearson AD, Nicoll JA, et al. Decline in incidence of medulloblastoma in children. Cancer. 1994;74:3240-3244.
10 Bunin GR, Buckley JD, Boesel CP, et al. Risk factors for astrocytic glioma and primitive neuroectodermal tumor of the brain in young children: a report from the Children’s Cancer Group. Cancer Epidemiol Biomarkers Prev. 1994;3:197-204.
11 Evans DG, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959-961.
12 Lacombe D, Chateil JF, Fontan D, et al. Medulloblastoma in the nevoid basal-cell carcinoma syndrome: case reports and review of the literature. Genet Couns. 1990;1:273-277.
13 Schofield D, West DC, Anthony DC, et al. Correlation of loss of heterozygosity at chromosome 9q with histological subtype in medulloblastomas. Am J Pathol. 1995;146:472-480.
14 Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888-901.
15 Anseline PF. Turcot’s syndrome. Aust N Z J Surg. 1992;62:587-590.
16 Rorke LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1983;42:1-15.
17 Packer RJ, Sutton LN, Rorke LB, et al. Prognostic importance of cellular differentiation in medulloblastoma of childhood. J Neurosurg. 1984;61:296-301.
18 Mannoji H, Takeshita I, Fukui M, et al. Glial fibrillary acidic protein in medulloblastoma. Acta Neuropathol. 1981;55:63-69.
19 Taomoto K, Tomita T, Raimondi AJ, et al. Medulloblastomas in childhood: histological factors influencing patients’ outcome. Childs Nerv Syst. 1987;3:354-360.
20 Patterson A. Meningiomas and other nonglial neoplasms. In: Osborne AG, editor. Diagnostic Neuroradiology. St Louis: Mosby; 1994:613-620.
21 Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
22 Friedman HS, Oakes WJ, Bigner SH, et al. Medulloblastoma: tumor biological and clinical perspectives. J Neurooncol. 1991;11:1-15.
23 Kershman J. The medulloblast and the medulloblastoma: a study of human embryos. Arch Neurol Psychiatry. 1938;40:937-967.
24 Nagashima K, Yasui K, Kimura J, et al. Induction of brain tumors by newly isolated JC virus (Tokyo-1 strain). Am J Pathol. 1984;116:455-463.
25 Bigner SH, Mark J, Friedman HS, et al. Structural chromosomal abnormalities in human medulloblastoma. Cancer Genet Cytogenet. 1988;30:91-101.
26 Bigner SH, Mark J, Burger PC, et al. Specific chromosomal abnormalities in malignant human gliomas. Cancer Res. 1988;48:405-411.
27 Rossi MR, Conroy J, McQuaid D, et al. Array CGH analysis of pediatric medulloblastomas. Genes Chromosomes Cancer. 2006;45:290-303.
28 Biegel JA, Zhou JY, Rorke LB, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74-79.
29 Biegel JA, Rorke LB, Packer RJ, et al. Isochromosome 17q in primitive neuroectodermal tumors of the central nervous system. Genes Chromosomes Cancer. 1989;1:139-147.
30 Cogen PH, Daneshvar L, Metzger AK, et al. Deletion mapping of the medulloblastoma locus on chromosome 17p. Genomics. 1990;8:279-285.
31 Aldosari N, Bigner SH, Burger PC, et al. MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children’s Oncology Group. Arch Pathol Lab Med. 2002;126:540-544.
32 Ellison D. Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol. 2002;28:257-282.
33 Raffel C, Thomas GA, Tishler DM, et al. Absence of p53 mutations in childhood central nervous system primitive neuroectodermal tumors. Neurosurgery. 1993;33:302-306.
34 Badaiali M, Iolascon A, Loda M, et al. p53 Gene mutations in medulloblastoma. Immunohistochemistry, gel shift analysis, and sequencing. Diagn Mol Pathol. 1993;2:23-28.
35 Tohyama T, Lee VM, Rorke LB, et al. Monoclonal antibodies to a rat nestin fusion protein recognize a 220-kDa polypeptide in subsets of fetal and adult human central nervous system neurons and in primitive neuroectodermal tumor cells. Am J Pathol. 1993;143:258-268.
36 Eberhart CG, Kratz J, Wang Y, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol. 2004;63:441-449.
37 Tong CY, Hui AB, Yin XL, et al. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J Neurosurg. 2004;100:187-193.
38 Yasue M, Tomita T, Engelhard H, et al. Prognostic importance of DNA ploidy in medulloblastoma of childhood. J Neurosurg. 1989;70:385-391.
39 Giangaspero F, Chieco P, Ceccarelli C, et al. “Desmoplastic” versus “classic” medulloblastoma: comparison of DNA content, histopathology and differentiation. Virchows Arch A Pathol Anat Histopathol. 1991;418:207-214.
40 Rorke LB, Packer R, Biegel J. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol. 1995;24:21-28.
41 Muchardt C, Sardet C, Bourachot B, et al. A human protein with homology to Saccharomyces cerevisiae SNF5 interacts with the potential helicase hbrm. Nucleic Acids Res. 1995;23:1127-1132.
42 Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203-206.
43 Squires RHJr. Intracranial tumors. Vomiting as a presenting sign. A gastroenterologist’s perspective. Clin Pediatr (Phila). 1989;28:351-354.
44 Kayama T, Yoshimoto T, Shimizu H, et al. Neonatal medulloblastoma. J Neurooncol. 1993;15:157-163.
45 Nelson M, Diebler C, Forbes WS. Paediatric medulloblastoma: atypical CT features at presentation in the SIOP II trial. Neuroradiology. 1991;33:140-142.
46 Zee CS, Segall HD, Miller C, et al. Less common CT features of medulloblastoma. Radiology. 1982;144:97-102.
47 Prasad A, Madan VS, Buxi TB, et al. Medulloblastoma with extensive calcification. Neuroradiology. 1991;33:447-448.
48 Meyers SP, Kemp SS, Tarr RW. MR imaging features of medulloblastomas. AJR Am J Roentgenol. 1992;158:859-865.
49 Mueller DP, Moore SA, Sato Y, et al. MRI spectrum of medulloblastoma. Clin Imaging. 1992;16:250-255.
50 Goel A. Whither preoperative shunts for posterior fossa tumours? Br J Neurosurg. 1993;7:395-399.
51 Berger MS, Baumeister B, Geyer JR, et al. The risks of metastases from shunting in children with primary central nervous system tumors. J Neurosurg. 1991;74:872-877.
52 Tomita T. Medulloblastomas. In: Youmans JR, editor. Neurological Surgery. 4th ed. Philadelphia: Saunders; 1996:2570-2592.
53 Jamjoom ZA, Jamjoom AB, Sulaiman AH, et al. Systemic metastasis of medulloblastoma through ventriculoperitoneal shunt: report of a case and critical analysis of the literature. Surg Neurol. 1993;40:403-410.
54 Lee M, Wisoff JH, Abbott R, et al. Management of hydrocephalus in children with medulloblastoma: prognostic factors in shunting. Pediatr Neurosurg. 1994;20:240-247.
55 Grabb PA, Albright AL, Sclabassi RJ, et al. Continuous intraoperative electromyographic monitoring of cranial nerves during resection of fourth ventricular tumors in children. J Neurosurg. 1997;86:1-4.
56 Albright AL, Wisoff JH, Zeltzer PM, et al. Current neurosurgical treatment of medulloblastomas in children. A report from the Children’s Cancer Study Group. Pediatr Neurosci. 1989;15:276-282.
57 Albright AL. Medulloblastomas. In: Albright AL, Pollack IF, Adelson PD, editors. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:591-608.
58 Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: a report from the Children’s Cancer Group. Neurosurgery. 1996;38:265-271.
59 Albright AL, Sposto R, Holmes E, et al. Correlation of neurosurgical subspecialization with outcomes in children with malignant brain tumors. Neurosurgery. 2000;47:879-887.
60 Park TS, Hoffman HJ, Hendrick EB, et al. Medulloblastoma: clinical presentation and management. Experience at the Hospital for Sick Children, Toronto, 1950-1980. J Neurosurg. 1983;58:543-552.
61 Cochrane DD, Gustavsson B, Poskitt KP, et al. The surgical and natural morbidity of aggressive resection for posterior fossa tumors in childhood. Pediatr Neurosurg. 1994;20:19-29.
62 Wisoff JH, Epstein FJ. Pseudobulbar palsy after posterior fossa operation in children. Neurosurgery. 1984;15:707-709.
63 Pollack IF, Polinko P, Albright AL, et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery. 1995;37:885-893.
64 Dailey AT, McKhann GM2nd, Berger MS. The pathophysiology of oral pharyngeal apraxia and mutism following posterior fossa tumor resection in children. J Neurosurg. 1995;83:467-475.
65 Robertson PL, Muraszko KM, Holmes EJ, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105:444-451.
66 Chang CH, Housepian EM, Herbert CJr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastoma. Radiology. 1969;93:1351-1359.
67 Laurent JP, Chang CH, Cohen ME. A classification system for primitive neuroectodermal tumors (medulloblastoma) of the posterior fossa. Cancer. 1985;56:1807-1809.
68 Evans AE, Jenkin RD, Sposto R, et al. The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg. 1990;72:572-582.
69 Mirabell R, Bieri S, Huguenin P, et al. Prognostic value of cerebrospinal fluid cytology in pediatric medulloblastoma. Swiss Pediatric Oncology Group. Ann Oncol. 1999;10:239-241.
70 Wiener MD, Boyko OB, Friedman HS, et al. False-positive spinal MR findings for subarachnoid spread of primary CNS tumor in postoperative pediatric patients. AJNR Am J Neuroradiol. 1990;11:1100-1103.
71 O’Reilly G, Hayward RD, Harness WF. Myelography in the assessment of children with medulloblastoma. Br J Neurosurg. 1993;7:183-188.
72 Paterson E, Farr RF. Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta Radiol. 1953;39:323-336.
73 Jenkin RD. Medulloblastoma in childhood: radiation therapy. Can Med Assoc J. 1969;100:51-53.
74 Hornbek NB. Radiation therapy in treatment of medulloblastoma in childhood. In: Zeltzer PM, Pochedly C, editors. Medulloblastomas in Children. New Concepts in Tumor Biology, Diagnosis and Treatment. New York: Praeger; 1986:164-181.
75 Kun LE, Constine LS. Medulloblastoma: caution regarding new treatment approaches. Int J Radiat Oncol Biol Phys. 1991;20:897-899.
76 Bailey CC, Gnekow A, Wellek S, et al. Prospective randomized trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GOP): SIOP II. Med Pediatr Oncol. 1995;25:166-178.
77 Halberg FE, Wara WM, Fippin LF, et al. Low-dose craniospinal radiation therapy for medulloblastoma. Int J Radiat Oncol Biol Phys. 1991;20:651-654.
78 Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children’s Cancer Group Study. J Clin Oncol. 1999;17:2127-2136.
79 Woo C, Stea B, Lulu B, et al. The use of stereotactic radiosurgical boost in the treatment of medulloblastoma. Int J Radiat Oncol Biol Phys. 1997;37:761-764.
80 Krischer JP, Ragab AH, Kun L, et al. Nitrogen mustard, vincristine, procarbazine, and prednisone as adjuvant chemotherapy in the treatment of medulloblastoma. A Pediatric Oncology Group study. J Neurosurg. 1991;74:905-909.
81 Pendergrass TW, Milstein JM, Geyer JR, et al. Eight drugs in one day chemotherapy for brain tumors: experience in 107 children and rationale for preradiation chemotherapy. J Clin Oncol. 1987;5:1221-1231.
82 Gentet JC, Bouffet E, Doz F, et al. Preirradiation chemotherapy including “eight drugs in 1 day” regimen and high-dose methotrexate in childhood medulloblastoma: results of the M7 French Cooperative Study. J Neurosurg. 1995;82:608-614.
83 Packer RJ, Siegel KR, Sutton LN, et al. Efficacy of adjuvant chemotherapy for patients with poor-risk medulloblastoma: a preliminary report. Ann Neurol. 1988;24:503-508.
84 Packer RJ, Sutton LN, Goldwein JW, et al. Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg. 1991;74:433-440.
85 Packer RJ, Sutton LN, Elterman R, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81:690-698.
86 Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
87 Reddy AT, Janss AJ, Phillips PC, et al. Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer. 2000;88:2189-2193.
88 Lai R. Survival of patients with adult medulloblastoma: a population-based study. Cancer. 2008;112:1568-1574.
89 Padovani L, Sunyach MP, Perol D, et al. Common strategy for adult and pediatric medulloblastoma: a multicenter series of 253 adults. Int J Radiat Oncol Biol Phys. 2007;68:433-440.
90 Torres CF, Rebsamen S, Silber JH, et al. Surveillance scanning of children with medulloblastoma. N Engl J Med. 1994;330:892-895.
91 Mendel E, Levy ML, Raffel C, et al. Surveillance imaging in children with primitive neuroectodermal tumors. Neurosurgery. 1996;38:692-695.
92 Steinbok P, Hentschel S, Cochrane DD, et al. Value of postoperative surveillance imaging in the management of children with some common brain tumors. J Neurosurg. 1996;84:726-732.
93 Belza MG, Donaldson SS, Steinberg GK, et al. Medulloblastoma: freedom from relapse longer than 8 years—a therapeutic cure? J Neurosurg. 1991;75:575-582.
94 Meyers SP, Wildenhain S, Chess MA, et al. Postoperative evaluation for intracranial recurrence of medulloblastoma: MR findings with gadopentetate dimeglumine. AJNR Am J Neuroradiol. 1994;15:1425-1434.
95 Tarbell NJ, Loeffler JS, Silver B, et al. The change in patterns of relapse in medulloblastoma. Cancer. 1991;68:1600-1604.
96 Nicholson HS, Zeltzer PM, Boyett JM, et al. Patterns of relapse in high-risk posterior fossa PNET/medulloblastoma (MB) patients with radiotherapy (XRT) and chemotherapy (CT). Med Pediatr Oncol. 1995;24:238.
97 Friedman HS, Mahaley MSJr, Schold SCJr, et al. Efficacy of vincristine and cyclophosphamide in the therapy of recurrent medulloblastoma. Neurosurgery. 1986;18:335-340.
98 Dunkel IJ, Boyett JM, Yates A, et al. High-dose carboplatin, thiotepa, and etoposide with autologous stem-cell rescue for patients with recurrent medulloblastoma. Children’s Cancer Group. J Clin Oncol. 1998;16:222-228.
99 Chapman CA, Waber DP, Bernstein JH, et al. Neurobehavioral and neurologic outcome in long-term survivors of posterior fossa brain tumors: role of age and perioperative factors. J Child Neurol. 1995;10:209-212.
100 Ogilvy-Stuart AL, Clayton PE, Shalet SM. Cranial irradiation and early puberty. J Clin Endocrinol Metab. 1994;78:1282-1286.
101 Farwell JR, Dohrmann GJ, Flannery JT. Medulloblastoma in childhood: an epidemiological study. J Neurosurg. 1984;61:657-664.
102 Radcliffe J, Bunin GR, Sutton LN, et al. Cognitive deficits in long-term survivors of childhood medulloblastoma and other noncortical tumors: age-dependent effects of whole brain radiation. Int J Dev Neurosci. 1994;12:327-334.
103 Johnson DL, McCabe MA, Nicholson HS, et al. Quality of long-term survival in young children with medulloblastoma. J Neurosurg. 1994;80:1004-1010.
104 Kimmings E, Kleinlugtebeld AT, Casey A, et al. Medulloblastoma: factors influencing the educational potential of surviving children. Br J Neurosurg. 1995;9:611-617.
105 Goldstein AM, Yuen J, Tucker MA. Second cancers after medulloblastoma: population-based results from the United States and Sweden. Cancer Causes Control. 1997;8:865-871.
106 Goldberg-Stern H, Gadoth N, Stern S, et al. The prognostic significance of glial fibrillary acidic protein staining in medulloblastoma. Cancer. 1991;68:568-573.
107 Zerbini C, Gelber RD, Weinberg D. Prognostic factors in medulloblastoma, including DNA ploidy. J Clin Oncol. 1993;11:616-622.
108 Tait DM, Thornton-Jones H, Blood HJ, et al. Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer. 1990;26:464-469.
109 Batra SK, McLendon RE, Koo JS, et al. Prognostic implications of chromosome 17p deletions in human medulloblastomas. J Neurooncol. 1995;24:39-45.
110 Nicholson JC, Ross FM, Kohler JA, et al. Comparative genomic hybridization and histological variation in primitive neuroectodermal tumours. Br J Cancer. 1999;80:1322-1331.
111 Gilbertson RJ, Peason AD, Perry RH, et al. Prognostic significance of the c-erbB-2 oncogene product in childhood medulloblastoma. Br J Cancer. 1995;71:473-477.
112 Segal RA, Goumnerova LC, Kwon YK, et al. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc Natl Acad Sci U S A. 1994;91:12867-12871.
113 Ito S, Hoshino T, Prados MD, et al. Cell kinetics of medulloblastomas. Cancer. 1992;70:671-678.
114 Gilbertson RJ, Perry RH, Kelly PJ, et al. Prognostic significance of HER2 and HER4 coexpression in childhood medulloblastoma. Cancer Res. 1997;57:3272-3280.
115 Gilbertson RJ, Jaros E, Perry RH, et al. Mitotic percentage index: a new prognostic factor for childhood medulloblastoma. Eur J Cancer. 1997;33:609-615.
116 Adesina AM, Dunn ST, Moore WE, et al. Expression of p27kip1 and p53 in medulloblastoma: relationship with cell proliferation and survival. Pathol Res Pract. 2000;196:243-250.
117 Haslam RH, Lamborn KR, Becker LE, et al. Tumor cell apoptosis present at diagnosis may predict treatment outcome for patients with medulloblastoma. J Pediatr Hematol Oncol. 1998;20:520-527.
118 Figarella-Branger D, Dubois C, Chauvin P, et al. Correlation between polysialic-neural cell adhesion molecule levels in CSF and medulloblastoma outcomes. J Clin Oncol. 1996;14:2066-2072.






