Chapter 14 Primary Pulmonary Hypertension
Pulmonary arterial pressure was measured in 1852,1 but a century elapsed before cardiac catheterization provided the means for studying the physiology of the lesser circulation. Elevated pulmonary arterial pressure—pulmonary hypertension—results from disorders of the pulmonary vascular bed, the pulmonary parenchyma, the respiratory system, and the pulmonary venous bed and from chronic thromboembolic disease (Box 14-1).2 This chapter focuses on the pulmonary vascular bed, specifically on primary (idiopathic) pulmonary hypertension, a disorder that originates in the terminal muscular pulmonary arteries and arterioles.3,4
The earliest description of idiopathic pulmonary hypertension was a report in 1891 of the cardiac pathology in a patient with pulmonary artery sclerosis and right ventricular hypertrophy of unknown cause.5 Primary pulmonary hypertension, a term coined by Dresdale in 1951, referred to an idiopathic disorder residing in the terminal pulmonary arteries and arterioles. The prevalence rate of primary pulmonary hypertension is estimated at one or two cases per million in the general population.3 Shortly after Dresdale’s report, Wood6 characterized the clinical manifestations of primary pulmonary hypertension. Two decades later, the World Health Organization (WHO) proposed a classification based on three histopathologic patterns: plexogenic pulmonary arteriopathy, microthrombotic pulmonary arteriopathy, and pulmonary venoocclusive disease.7 In 1975, WHO defined primary pulmonary hypertension as a disorder with no identifiable cause in which resting mean pulmonary artery pressure in adults at sea level is above 25 mm Hg and above 30 mm Hg with exercise. Pulmonary vascular resistance does not exceed three Wood units, and pulmonary capillary wedge pressure is normal.3,4,7 In 1981, the National Heart, Lung, and Blood Institute established a Registry to collect information on primary pulmonary hypertension in accordance with a uniform database.8 Enlightening histopathologic studies abound.8–13
The pulmonary vascular bed consists of an elaborately branched system of arteries, arterioles, capillaries, venules, and veins that accommodate the cardiac output at low pressures. Approximately 17 gradations of arterial vascular channels separate the main pulmonary artery from the pulmonary arterioles. In the fetus, the pulmonary arteriole is structurally analogous to the systemic arteriole and is therefore equipped to meet the full force of systemic pressure the moment the neonatal lungs expand and pulmonary blood flow commences. The thick-walled neonatal pulmonary arterioles rapidly involute, and the pulmonary vascular bed remodels, establishing the low-resistance lesser circulation. Remodeling continues for 1 or 2 months as the lungs adapt to extrauterine life and then proceeds slowly throughout childhood as the pulmonary vascular bed matures.14
The major airways and the major pulmonary arterial branches are formed by 16 weeks of gestation. An understanding of the density per unit area of small peripheral pulmonary arteries relative to alveolar density is necessary to understand normal and abnormal fetal and postnatal pulmonary arterial development.15 The number of peripheral acinar units increases in proportion to the number of intraacinar arteries until lung growth is completed at 8 to 10 years of age. Two thirds of the normal number of intraacinar arteries are formed by 18 months, and most of the remainder are formed by 5 years of age.15 Also important is the normal age-related distal extension of medial smooth muscle.15 At birth, the vascular channels beyond the terminal bronchioles are devoid of medial smooth muscle, but within 2 to 3 years, medial smooth muscle extends to the junction of respiratory bronchioles and alveolar ducts and, by the mid-teens, extends among alveoli into precapillary vessels.
The histologic findings in primary pulmonary hypertension (see previous discussion) include luminal hyperplasia, medial hypertrophy, adventitial proliferation and fibrosis, occlusion of small arteries, in situ thrombosis, and infiltration of inflammatory and progenitor cells.4 In 1958, Heath and Edwards16 published elegant descriptions of the histology of pulmonary vascular disease together with a comprehensive classification.17 The earliest abnormality was muscular thickening of small pulmonary arteries from medial hyperplasia, followed by intimal thickening (hyperplasia) that ranged from minimal to virtual luminal occlusion. The end stage was the angioproliferative plexiform lesion that corresponds to one of the three major histopathologic types of primary pulmonary hypertension in the WHO classification.7 Originally, plexiform lesions were thought to be unique to primary pulmonary hypertension,4 but now the belief is that plexogenic pulmonary arteriopathy is a nonspecific response of the pulmonary vasculature to hemodynamic injury.3 The progression of lesion severity from medial hypertrophy to plexiform arteriopathy and the functional relevance of plexiform lesions remain uncertain.4
Plexogenic pulmonary arteriopathy resides in small muscular arteries 100 to 200 μm in diameter and is a complex process that involves intimal cell proliferation, migration of medial smooth muscle cells, recruitment of inflammatory cells, and deposition of extracellular matrix proteins.8,12,18 Intimal proliferation is disproportionate to medial hypertrophy and culminates in complete luminal obliteration. Plexiform lesions are found in 70% to 85% of patients with primary pulmonary hypertension in whom lung tissue is available.3,12
Microthrombotic pulmonary arteriopathy occurs in 20% to 50% of cases of primary pulmonary hypertension12 and is represented by recanalized in situ microthrombi that consist of fibrous webs with eccentric intimal fibrosis together with medial hypertrophy but without plexiform lesions.8,12,18
Pulmonary venoocclusive disease is found in less than 7% of cases of primary pulmonary hypertension12 and is characterized by fibrosis and intimal proliferation of intrapulmonary venules, culminating in complete obliteration of venous channels that tend to recanalize.8,12,18,19 Studies with use of light and electron microscopy have confirmed and extended these observations of pulmonary vascular pathophysiology (Box 14-2).20 The clinical manifestations of primary pulmonary hypertension do not distinguish among the histopathologic types of pulmonary vascular disease. Early vasoreactivity ultimately becomes fixed,11 but once the disorder is established, it rarely regresses.12,16,17,21,22
Advances in pulmonary vascular biology continue to shed light on the pathogenesis of primary pulmonary hypertension.23–25 Endothelial abnormalities have been identified (vasoconstrictor endothlin-1 homeostasis),26–28 together with defects in voltage-gated potassium channels,4 abnormalities in metalloproteinase and elastase activity,4,29 transforming growth factor beta,30,31 and bone morphogenetic protein.32 Mediators of inflammation can cause pulmonary vasoconstriction, but the role of inflammation in chronic pulmonary hypertension is less clear. Genetic mechanisms are generally accepted to play a pathogenetic role in familial and sporadic cases of primary pulmonary hypertension (see section The History).4,32–36 Persistent fetal circulation or persistent pulmonary hypertension of the newborn, a disorder of unknown etiology that was described in 1969,37 is characterized by muscularization of small pulmonary arteries in the fetal lung, high neonatal pulmonary vascular resistance that fails to regress, and persistent right-to-left shunts through the ductus arteriosus and foramen ovale.37–40 A relationship between persistent fetal circulation and primary pulmonary hypertension is tenuous.41Portal hypertension can be associated with pulmonary hypertension presumably because the pulmonary vascular bed is exposed to vasoreactive substances normally metabolized by the liver.42,43 An association between human immunodeficiency infection and hypertensive pulmonary arteriopathy has been reported.44Alveolar hypoventilation and sleep apnea caused by chronic upper airway obstruction of hypertrophied tonsils and adenoids in children result in an increase in pulmonary vascular resistance (see Box 14-1).45–48 High-altitude pulmonary hypertension clinically and histologically resembles primary pulmonary hypertension, which is more common at high altitudes and which is aggravated by the low partial pressure of oxygen (see section The History).49–51
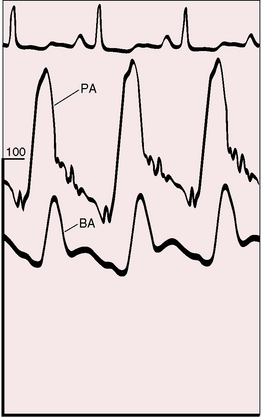
The physiologic consequences of primary pulmonary hypertension are direct reflections of the increased resistance to blood flow through the lesser circulation. The pressure in the pulmonary arteries can exceed systemic pressure (Figure 14-1). Pulmonary vasoreactivity can be present initially but diminishes and is ultimately lost. Resting pulmonary blood flow and cardiac output fall and either fail to increase during exercise or fall still further.52 Asymptomatic gene carriers for primary pulmonary hypertension have been identified with abnormal responses to exercise.53
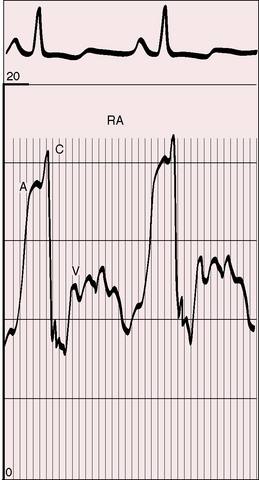
Forceful right atrial contraction generates a large A wave and an increase in right ventricular end-diastolic segment length. Right ventricular end-diastolic pressure rises in response, right atrial mean pressure rises, and a right-to-left shunt is established through a stretched patent foramen ovale. Tricuspid regurgitation increases mean right atrial pressure still further.54 Systemic venous pressure can be sufficiently elevated to cause venous stasis retinopathy.55,56
The atrial natriuretic peptide system is activated in primary pulmonary hypertension in response to increased right atrial pressure.57 The polypeptide hormone is secreted by atrial myocytes; is involved in the homeostatic control of body water, sodium, and potassium; and correlates significantly with right ventricular preload and afterload.57Brain natriuretic peptide levels increase in proportion to right ventricular dysfunction.58
A mild albeit significant reduction in total lung capacity occurs in females with primary pulmonary hypertension, and a reduction in forced vital capacity occurs in both males and females.8 Hypoxemia and hypocapnia are almost invariable because of a ventilation-perfusion mismatch, not because of a right-to-left shunt across a patent foramen ovale.8 The diffusing capacity for carbon monoxide is significantly less than predicted.8
The ventricular septum encroaches on the left ventricular cavity, sometimes appreciably.59 End-systolic and early diastolic deformations of the left ventricular cavity result in impaired filling in early diastole and in redistribution of filling in late diastole.59–61 Early diastolic filling of the right ventricle is also encroached upon because high pulmonary vascular resistance prolongs the duration of right ventricular systole and prolongs isovolumetric relaxation.61
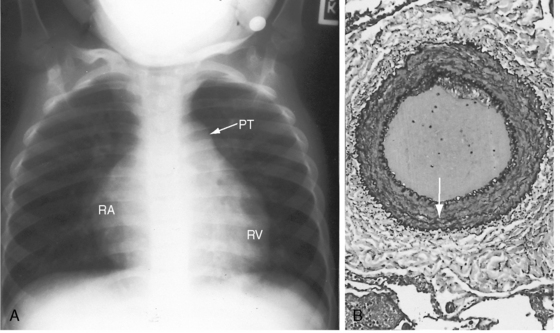
The time course of pulmonary hypertension can be established from the histology of the pulmonary trunk compared with the histology of the aortic root.17,62,63 When pulmonary hypertension begins at birth, the histology of the pulmonary trunk and the histology of the aortic root are virtually identical (Figures 14-2C and 14-2D). When pulmonary hypertension begins after birth, the histology of the pulmonary trunk and aortic root differ significantly (Figures 14-2A and 14-2B). Primary pulmonary hypertension is rarely present at birth,17,62,63 but when that is the case, it serves as a model of isolated pure pulmonary hypertension with which pulmonary hypertension with congenital heart disease can be compared.

History
The frequency of primary pulmonary hypertension in young females was cited as early as 1927,64 with a female:male ratio subsequently reported as high as 3:1.9 In the National Institutes of Health (NIH) registry, however, the gender incidence rate in children was equal; and in adults, the female:male ratio was 1.7:1, with a relatively constant incidence rate decade by decade.8 Oral contraceptives are a consideration in light of evidence that birth control pills can aggravate if not initiate pulmonary vascular disease.65
Variations in gender incidence may in part reflect differences among the three histopathologic subgroups. Plexogenic pulmonary arteriopathy, the most frequent of the three histopathologic subgroups of primary pulmonary hypertension,8,18 has a female:male ratio of 3:1. In the less common microthrombotic pulmonary hypertension, the female:male ratio is approximately equal,18 and in the venoocclusive subgroup, which accounts for less than 7% of cases, a slight male predominance is seen.12
Familial primary pulmonary hypertension is well recognized (see Figure 14-12A) and occasionally recurs in more than one generation.66–71 In the NIH registry, 6% of patients with primary pulmonary hypertension had a first-degree relative with primary pulmonary hypertension.3 Inheritance is autosomal dominant with incomplete penetrance.53 The genetic basis has been assigned to mutations in the bone morphogenetic protein receptor.72–74
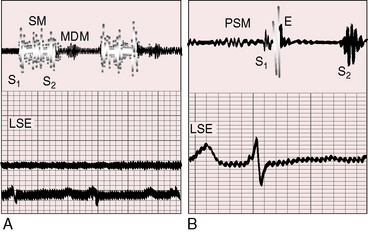
Figure 14-12 A, Phonocardiogram from a 20-year-old man with primary pulmonary hypertension. His younger sister also had primary pulmonary hypertension. The tracing at the lower left sternal edge (LSE) shows a holosystolic murmur of tricuspid regurgitation and a short middiastolic murmur (MDM). B, Phonocardiogram from the 29-year-old woman referred to in Figure 14-5. A short presystolic murmur (PSM) and a prominent pulmonary ejection sound (E) are shown at the lower left sternal edge. (S2 = second heart sound.)
Mean patient age in three large studies of primary pulmonary hypertension was 21 to 30 years, with a range of 2 to 56 years.75 In the NIH registry, only 8% of patients reached the seventh decade,8 although primary pulmonary hypertension of long duration has been reported. Regression has been suspected, albeit rarely.21,22 Short-term survival is related to the severity of endothelial dysfunction.27 Median survival after diagnosis is approximately 2.8 years,3,76 with right ventricular failure the most common cause of death.76–78 Longevity and morbidity are significantly influenced by lifestyle. Abrupt, strenuous, or isometric exercise should be avoided, and isotonic exercise should cease at the very onset of dyspnea, light-headedness, dizziness, or chest pain. Exposure to high altitude is an avoidable risk, and certain constraints are appropriate for air travel. A commercial jetliner flying at 33,000 to 36,000 ft has a cabin atmosphere equivalent to approximately 6000 to 8000 ft, which is comparable with breathing 15% oxygen at sea level.17,79 Airline passengers experience a significant decrease in arterial PO2, but only a mild decrease in arterial oxygen saturation.79 Low humidity causes dehydration and an undesirable fall in systemic blood pressure. Just as important are the risks incurred by non–flight-related stress and travel fatigue. Rushing at the last minute, carrying heavy baggage, and transferring from one terminal to another at large airports are recognized risks.
The most common symptoms associated with primary pulmonary hypertension are effort dyspnea, fatigue, light-headedness, and chest pain. An increase in ventilatory response to exercise—hyperventilatory dyspnea—is attributed chiefly to a ventilation/perfusion mismatch.12,52
Muscle fatigue is attributed to a reduction in the rate of aerobic regeneration of adenosine triphosphate (ATP).52 Hypothyroidism is surprisingly prevalent in primary pulmonary hypertension80 and should be considered in patients with inappropriate fatigue. Stress-related or exercise-related light-headedness, giddiness, dizziness, or faintness reflects an inability to achieve sufficient cardiac output and systemic blood pressure to maintain cerebral blood flow. The chest pain of myocardial ischemic originates in the hypertrophied hypoperfused right ventricle.81 Histologic evidence of right ventricular infarction occurs in patients with primary pulmonary hypertension and normal coronary arteries. Myocardial ischemia has also been attributed to compression of the left main coronary artery by a dilated hypertensive pulmonary trunk.82,83 Syncope, which is usually provoked by effort or excitement, is ominous and heralds sudden death.8,12,84 Sudden death can also follow relatively innocuous stress, such as bone marrow aspiration and cardiac catheterization, especially in children.77,85 Surgery, anesthesia, and even sedatives are poorly tolerated. Symptoms may first appear during pregnancy, which warrants special emphasis.86–90 Fixed pulmonary vascular resistance blunts or precludes adaptive responses to the hemodynamic fluctuations of labor, delivery, and the puerperium. Maternal mortality rate is as high as 50%.88 A hypercoagulable state in the third trimester reinforces the propensity for in situ thromboses in small terminal pulmonary arteries (see previous discussion).
Hemoptysis is a feature of Eisenmenger’s syndrome (see Chapter 17) but not primary pulmonary hypertension. Raynaud’s phenomenon is occasionally associated with primary pulmonary hypertension and calls attention to autoimmunity (see previous discussion).24 Hoarseness, Ortner’s syndrome, results from compression of the left recurrent laryngeal nerve by a dilated hypertensive pulmonary trunk.12,69 Patients are sometimes unpleasantly aware of visible neck pulsations caused by giant jugular A waves (Figure 14-3; see previous discussion).
Arterial pulse and jugular venous pulse
The systemic arterial pulse is small with a narrow pulse pressure (see Figure 14-1) because left ventricular stroke volume is reduced.6 The small arterial pulse is in striking contrast to the large if not giant jugular venous A wave (Figures 14-3 and 14-4A), which Paul Wood aptly described: “This presystolic venous pulse is abrupt and collapsing in quality; is little influenced by change in posture and may be more noticeable on inspiration. Thus it is best seen when the patient sits bolt upright or stands up, when the V wave usually disappears altogether.”6 An increase in V wave awaits the advent of tricuspid regurgitation,54 which attenuates the X descent and exaggerates the Y trough. Giant V waves reach the mandible when tricuspid regurgitation is accompanied by a ruptured tricuspid papillary muscle.91
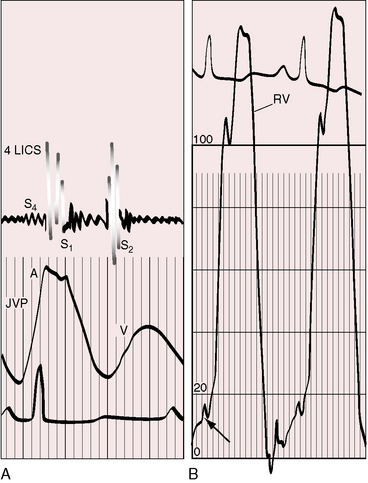
Figure 14-4 Recordings from the 18-year-old man referred to in Figure 14-1. A, Fourth heart sound (S4) was present in the fourth left intercostal space (4 LICS). The jugular venous pulse (JVP) exhibits a correspondingly prominent A wave. B, Increased force of right atrial contraction resulted in presystolic distention of the right ventricle (arrow, lower left). (RV = right ventricle; S1 = first heart sound; S2 = second heart sound.)
Precordial movement and palpation
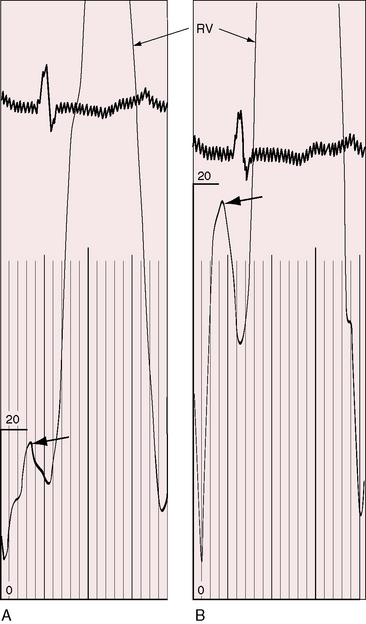
The right ventricular impulse varies in prominence with the severity and duration of pulmonary hypertension and with the size and function of the right ventricle. An enlarged right ventricle displaces the left ventricle from the apex. An increased force of right atrial contraction causes presystolic distention, which is palpated at the lower left sternal edge and subxyphoid area (Figures 14-4 and 14-5). Palpation in the second left intercostal space detects the systolic impulse of a dilated hypertensive pulmonary trunk together with a loud pulmonary ejection sound and, as Graham Steell wrote in 1888, “… the closure of the semilunar valve being generally perceptible to the hand placed over the pulmonary area as a sharp thud.”92
Auscultation
The eight auscultatory signs are reflections of pure pulmonary hypertension:93,94 the pulmonary ejection sound, the pulmonary midsystolic murmur, the tricuspid holosystolic murmur, the second heart sound, the Graham Steell murmur, third and fourth heart sounds, and a mid-diastolic–presystolic murmur (Figure 14-6).94 The pulmonary ejection sound is identified by its high-pitched sharp clicking quality, by its maximal intensity in the second left intercostal space, and occasionally but distinctively by its selective decrease during inspiration (Figures 14-6, 14-7, and 14-8A). The timing of the ejection sound coincides with the interval between the onset of right ventricular systole and the opening of the pulmonary valve (isovolumetric contraction). The slower the rate of right ventricular contraction and the higher the pulmonary arterial diastolic pressure, the later the ejection sound (see Figures 14-7 and 14-8A).93 A loud ejection sound radiates to the lower left sternal edge and apex, especially when the right ventricle occupies the apex. A pulmonary midsystolic murmur results from ejection into the dilated hypertensive pulmonary trunk (see Figures 14-6 and 14-7). The murmur is confined to the second left intercostal space, is introduced by the pulmonary ejection sound, and is symmetric, short, impure, and grade 1/6 or 2/6. The tricuspid regurgitation murmur is maximal at the lower left sternal edge (see Figures 14-6, 14-7 and 14-8B); but when the right ventricle occupies the apex, the murmur is well heard at the apex, and when the right atrium is enlarged, the murmur is heard to the right of the sternum. The tricuspid murmur is holosystolic and high-pitched because regurgitant flow is holosystolic and high velocity.94 Intensity can be sufficient to generate a thrill or barely sufficient to achieve audibility. An increase in intensity during active inspiration, Rivero-Carvallo sign, is diagnostically important (see Figure 14-6).93–95 The murmur is sometimes audible only during deep inspiration. Amplification during inspiration depends on a right ventricle that is functionally capable of converting an inspiratory increase in venous return into an increase in stroke volume and regurgitant flow.93 This capacity is lost with right ventricular failure, so Carvallo’s sign disappears.94
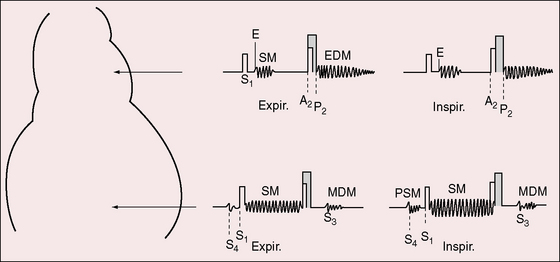
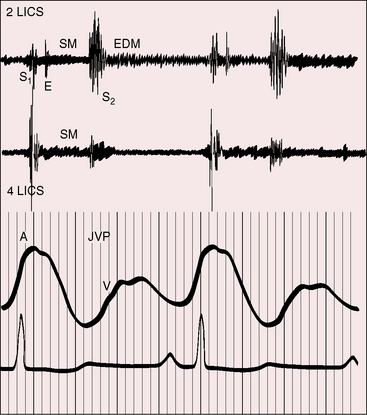
Figure 14-7 Phonocardiograms with jugular venous pulse (JVP) from the 18-year-old man with primary pulmonary hypertension referred to in Figure 14-1. In the second left intercostal space (2 LICS), a pulmonary ejection sound (E) begins at a long interval after the first heart sound and introduces a soft, short, midsystolic murmur (SM). The single second heart sound (S2) is prolonged and loud because the pulmonary component was of great amplitude. A soft early diastolic murmur (EDM) issues from the loud second sound. A high-frequency holosystolic murmur of tricuspid regurgitation appears in the fourth left intercostal space (4 LICS). The A wave in the jugular venous pulse is prominent.
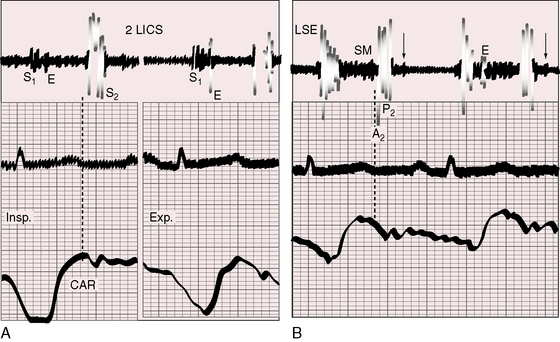
The pulmonary component of the second heart sound is altered in its timing, intensity, and precordial location.93,94 The degree of splitting is the net effect of two variables: 1, the decreased capacitance and increased resistance in the pulmonary vascular bed that serves to narrow the split; and 2, prolongation of right ventricular systole that serves to increase the split.96–98 When right ventricular function is normal, inspiratory splitting is normal or close (see Figure 14-6).98 A functionally depressed right ventricle cannot increase its stroke volume with inspiration, so the split becomes fixed.96,98 A loud pulmonary component in the second left interspace obscures a closely preceding aortic component (see Figures 14-7 and 14-8), but auscultation at the right base, lower left sternal edge, or apex permits analysis of the transmitted but attenuated pulmonary component and allows detection of splitting (Figures 14-8B and 14-9).93 Of all the auscultatory signs of pulmonary hypertension, the loud pulmonary component of the second sound is the most consistent (Figures 14-6 through 14-10). Graham Steell wrote that “accentuation of the pulmonary second sound is always present, the closure of the semilunar valves being generally perceptible to the hand placed over the pulmonary area as a sharp thud”92 (see section Precordial Palpation). Detection of the loud pulmonary component transmitted to the apex is a useful sign of elevated pressure in the pulmonary artery.93
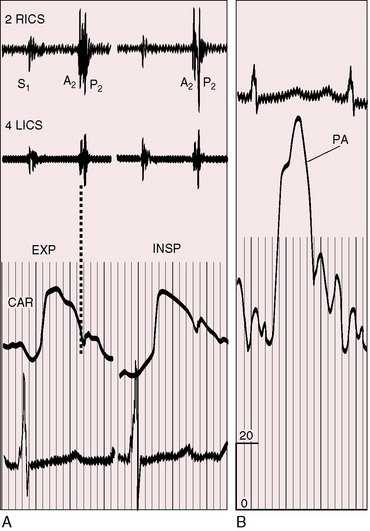
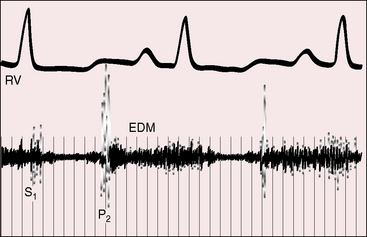
Figure 14-10 Intracardiac phonocardiogram from the right ventricular (RV) outflow tract of the 18-year-old man with primary pulmonary hypertension referred to in Figure 14-1. A high-frequency early diastolic Graham Steell murmur (EDM) begins with the loud pulmonary component of the second sound (P2). The murmur lasts throughout diastole. The configuration varies from beat to beat.
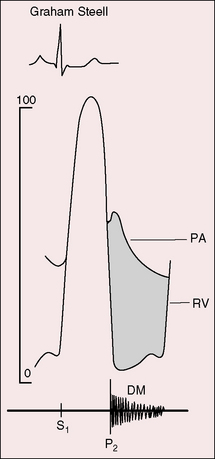
“There is occasionally heard over the pulmonary area … and below this region for the distance of an inch or two along the left border of the sternum, and rarely over the lowest part of the bone itself, a soft blowing diastolic murmur immediately following or more exactly running off the accentuated second sound, while the usual indications of aortic regurgitation afforded by the pulse, etc., are absent. When the second sound is reduplicated, the murmur proceeds from its latter part. That such a murmur as I have described does exist, there can, I think, be no doubt.”92 Graham Steell made this auscultatory observation with a light boxwood monaural stethoscope.99 The Graham Steell murmur is typically located in the second and third left intercostal spaces adjacent to the sternum (see Figures 14-6 and 14-7), but when it is loud, it is heard at the lower left sternal edge or even to the right of the sternum. Elevated diastolic pressure is exerted on the incompetent pulmonary valve at the inscription of the dichotic notch when the right ventricular and pulmonary arterial pressure pulses diverge, so the murmur begins with or immediately after the accentuated pulmonary component of the second sound (Figures 14-10 and 14-11). The marked difference between the diastolic pressure in the pulmonary artery and right ventricle exists from the beginning to the end of diastole, so the accompanying murmur is prolonged and high frequency (see Figures 14-10 and 14-11). The configuration is decrescendo (see Figures 14-7 and 14-11), but vibrations can be almost equal throughout diastole, or the murmur is sometimes crescendo-decrescendo (see Figure 14-10). Intensity can be sufficient to generate a thrill or soft and variable to the point of inaudibility. Steell was adept at detecting these murmurs despite his monaural boxwood stethoscope because of the “absolute quiet which prevailed during his lengthy round.”100
Fourth heart sounds accompany presystolic distention of the right ventricle (see Figures 14-4 and 14-6) and are best detected with the bell of the stethoscope applied lightly at the lower left sternal edge, but they are heard at the apex when the apex is formed by the right ventricle. Fourth heart sounds are distinguished from split first heart sounds or pulmonary ejection sounds by their quality, precordial location, and response to respiration.93,94 The low-frequency fourth sound can be more readily palpated than heard, a feature recognized by Potain who stated that “if one applies the ear to the chest, it affects the tactile sensation more than the auditory sense.”101 Right-sided fourth sounds become louder and occur earlier during inspiration (see Figure 14-6) because the greater force of right atrial contraction is translated into earlier and more vigorous presystolic filling of the right ventricle.
Third heart sounds occur during the rapid filling phase of the cardiac cycle and are signs of right ventricular failure (see Figure 14-6).94 With the advent of tricuspid regurgitation, third sounds intensify because high right atrial V waves are followed by accelerated atrioventricular flow. Third heart sounds are low-frequency events best detected with the bell of the stethoscope applied over the body of the right ventricle, but they are audible at the apex when the apex is occupied by the right ventricle.
In 1931, MacCallum reported a mid-diastolic rumbling murmur in a young woman with pulmonary hypertension.102 The occasional occurrence of mid-diastolic/presystolic murmurs (Figures 14-6 and 14-12) represents prolonged vibrations of third or fourth heart sounds (see Figure 14-6).93,94 Mid-diastolic murmurs increase during inspiration and are more apt to occur when the tricuspid valve is incompetent and the rate of atrioventricular flow is rapid (see Figure 14-12A). Right-sided Austin Flint mid-diastolic/presystolic murmurs have been described with pulmonary hypertensive pulmonary regurgitation.103
In a 1957 article on solitary pulmonary hypertension, Bedford, Evans, and Short104 wrote that “a sound that resembles a mitral opening snap was heard in early diastole.” McKusick recorded an “early diastolic snap” in a patient with necropsy-confirmed primary pulmonary hypertension and assigned the sound to the tricuspid valve. On one occasion, a soft early diastolic sound was recorded but not heard (see Figure 14-8B).93
Electrocardiogram
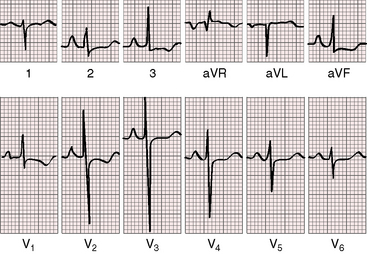
The electrocardiogram plays a useful role in the clinical assessment of primary pulmonary hypertension (Figure 14-13).12,105 Abnormal P waves display pure right atrial configurations (see Figure 14-13). The PR interval is normal or slightly prolonged. Atrial fibrillation is exceptional.12 The QRS axis varies from normal to right axis deviation, and the QRS duration is normal or slightly increased (see Figure 14-13). The degree of right ventricular hypertrophy reflects the severity and duration of right ventricular (pulmonary) hypertension. At one end of the spectrum, the electrocardiogram shows little more than a rightward QRS axis. At the other end of the spectrum, there is marked right axis deviation, right precordial leads that exhibit tall monophasic R waves with ST segment depressions and asymmetric T wave inversions, and left precordial that exhibit deep S waves (see Figure 14-13).
X-ray
Enlargement of the pulmonary trunk and its proximal branches is characteristic in primary pulmonary hypertension and varies from moderate to marked but is seldom aneurismal (Figures 14-14 through 14-16).12 The lucent pruned appearance of the peripheral lung fields stands out in sharp contrast to the prominence of the pulmonary trunk and its central branches (see Figures 14-15 and 14-16). Pulmonary venous congestion is reserved for the occasional patient with venoocclusive pulmonary hypertension.12 The ascending aorta is inconspicuous, especially when compared with the dilated pulmonary trunk (see Figures 14-14 and 14-15). Enlargement of the right ventricle reflects the degree and chronicity of right ventricular failure (see Figure 14-15). The size of the right atrium varies from a slight convexity at the right lower cardiac border (see Figure 14-14) to striking enlargement provoked by right ventricular failure with tricuspid regurgitation (Figures 14-15, 14-16, and 14-17).
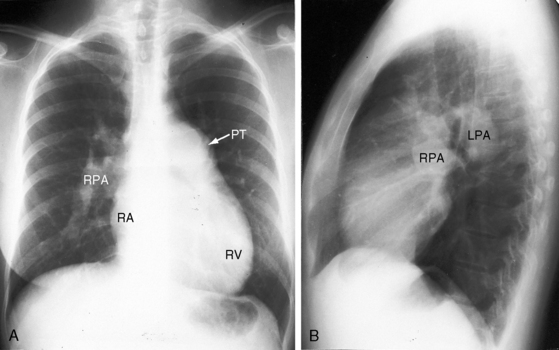
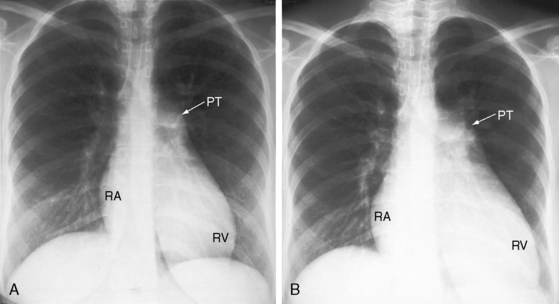
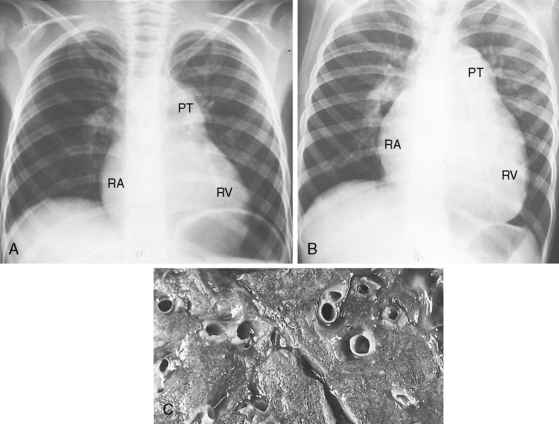
Figure 14-16 A, X-ray at age 6 years from the patient with primary pulmonary hypertension referred to in Figure 14-2. Pulmonary vascularity is reduced. The pulmonary trunk (PT) is dilated, the right atrial silhouette (RA) is increased, and a dilated convex right ventricle (RV) occupies the apex and extends below the left hemidiaphragm. B, X-ray at age 10 years after the onset of right ventricular failure. There is a striking increase in size of the pulmonary trunk, right atrium, and right ventricle. C, The patient died at age 11 years. Cross section of the lung at necropsy shows thick-walled intrapulmonary arteries that rise well above the surface. See also Figure 14-2.
Echocardiogram
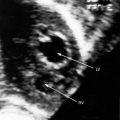
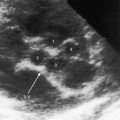
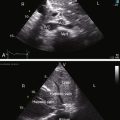
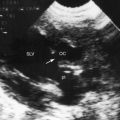
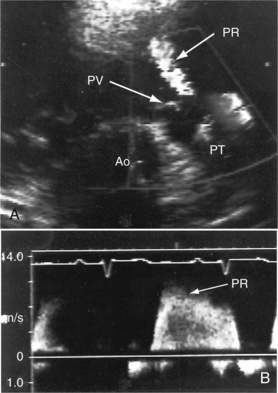
Transthoracic and transesophageal echocardiography with Doppler interrogation and color flow imaging exclude congenital or acquired heart disease as causes of the pulmonary hypertension and establish the physiologic and morphologic consequences of elevated pressure in the lesser circulation.54,106–110 Intravascular ultrasound scan is another method of assessing the pulmonary circulation in pulmonary hypertension.111 Echocardiography defines the size of the right atrium and right ventricle, characterizes right ventricular free wall motion, provides an estimate of right ventricular ejection fraction, establishes the position and motion of the ventricular septum, and determines the effect of ventricular septal position on the diastolic size and shape of the left ventricle (Figure 14-18).59 The ventricular septum flattens toward the left ventricular cavity at end systole and in early diastole,60 resulting in deformation of the left ventricular cavity, underfilling of the left ventricle in early diastole, and redistribution of filling into late diastole (Figures 14-18 and 14-19).60,61 Color flow imaging establishes the presence and degree of pulmonary and tricuspid regurgitation (Figures 14-19 through 14-21). Continuous-wave Doppler scan across the right ventricular outflow tract and tricuspid valve permits estimates of the pulmonary arterial diastolic pressure and the right ventricular systolic pressure (see Figures 14-19 through 14-21).
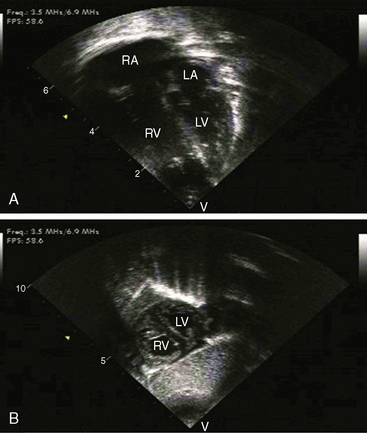
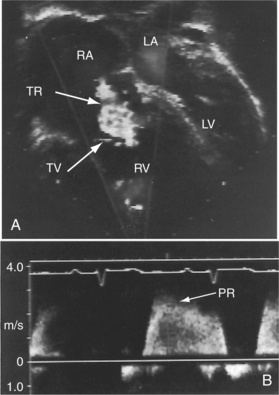
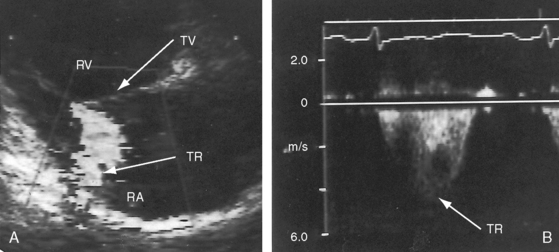
Figure 14-19 A, Black and white print of a color flow image from the 11-year-old girl referred to in Figures 14-2, 14-16, and 14-18. A high-velocity regurgitant jet (TR) originates at the tricuspid valve (TV) and enters an enlarged right atrium (RA). A dilated right ventricle (RV) encroaches on the left ventricular cavity (LV). B, Continuous-wave Doppler scan across the pulmonary valve records a peak velocity of pulmonary regurgitation (PR) that indicates a diastolic pressure of 45 mm Hg. Peak velocity across the tricuspid valve (not shown) indicated a right ventricular systolic pressure of 100 mm Hg. (LA = left atrium.)
1 Beutner A. ZF rat. Medicine. 1852;2:97.
2 Gaine S. Pulmonary hypertension. JAMA. 2000;284:3160-3168.
3 Helmersen D.S., Ford G.T., Viner S.M., Auger W.R. POEMS syndrome: a clue to understanding primary pulmonary hypertension? A review of current insights into the pathogenesis of primary pulmonary hypertension. Can J Cardiol. 2000;16:975-981.
4 Archer S., Rich S. Primary pulmonary hypertension: a vascular biology and translational research “Work in progress.”. Circulation. 2000;102:2781-2791.
5 Romberg E. Ueber sklerose der lungen arterie. Dtsch Arch Klin Med. 1891;48:197-206.
6 Wood P. Pulmonary hypertension. Br Med Bull. 1952;8:348-353.
7 Hatano S., Strasser T. Primary pulmonary hypertension: report on a WHO meeting, Geneva, 15–17 October 1973. World Health Organization; 1975.
8 Rich S., Dantzker D.R., Ayres S.M., et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216-223.
9 Bjornsson J., Edwards W.D. Primary pulmonary hypertension: a histopathologic study of 80 cases. Mayo Clin Proc. 1985;60:16-25.
10 Heath D., Smith P., Gosney J., et al. The pathology of the early and late stages of primary pulmonary hypertension. Br Heart J. 1987;58:204-213.
11 Palevsky H.I., Schloo B.L., Pietra G.G., et al. Primary pulmonary hypertension. Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation. 1989;80:1207-1221.
12 Rich S. Primary pulmonary hypertension. Prog Cardiovasc Dis. 1988;31:205-238.
13 Yamaki S., Wagenvoort C.A. Plexogenic pulmonary arteriopathy: significance of medial thickness with respect to advanced pulmonary vascular lesions. Am J Pathol. 1981;105:70-75.
14 Haworth S.G. Pulmonary vascular remodeling in neonatal pulmonary hypertension. State of the art. Chest. 1988;93:133S-138S.
15 Hislop A., Reid L. Pulmonary arterial development during childhood: branching pattern and structure. Thorax. 1973;28:129-135.
16 Heath D., Edwards J.E. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18:533-547.
17 Wagenvoort C.A. The pathology of the pulmonary vasculature. Springfield, Illinois: Charles C Thomas; 1964.
18 Pietra G.G., Edwards W.D., Kay J.M., et al. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation. 1989;80:1198-1206.
19 Resten A., Maitre S., Humbert M., et al. Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. AJR Am J Roentgenol. 2004;183:65-70.
20 Ye C., Rabinovitch M. New developments in the pulmonary circulation in children. Curr Opin Cardiol. 1992;7:124-133.
21 Bourdillon P.D., Oakley C.M. Regression of primary pulmonary hypertension. Br Heart J. 1976;38:264-270.
22 Fujii A., Rabinovitch M., Matthews E.C. A case of spontaneous resolution of idiopathic pulmonary hypertension. Br Heart J. 1981;46:574-577.
23 Rich S., Brundage B.H. Pulmonary hypertension: a cellular basis for understanding the pathophysiology and treatment. J Am Coll Cardiol. 1989;14:545-550.
24 Rich S., Kieras K., Hart K., Groves B.M., Stobo J.D., Brundage B.H. Antinuclear antibodies in primary pulmonary hypertension. J Am Coll Cardiol. 1986;8:1307-1311.
25 Snyder S.H. Nitric oxide: first in a new class of neurotransmitters. Science. 1992;257:494-496.
26 Chen Y.F., Oparil S. Endothelin and pulmonary hypertension. J Cardiovasc Pharmacol. 2000;35:S49-S53.
27 Lopes A.A., Maeda N.Y., Goncalves R.C., Bydlowski S.P. Endothelial cell dysfunction correlates differentially with survival in primary and secondary pulmonary hypertension. Am Heart J. 2000;139:618-623.
28 Rich S. Clinical insights into the pathogenesis of primary pulmonary hypertension. Chest. 1998;114:237S-241S.
29 Lepetit H., Eddahibi S., Fadel E., et al. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur Respir J. 2005;25:834-842.
30 Yeager M.E., Halley G.R., Golpon H.A., Voelkel N.F., Tuder R.M. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88:E2-E11.
31 Morrell N.W., Yang X., Upton P.D., et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104:790-795.
32 Machado R.D., Pauciulo M.W., Thomson J.R., et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92-102.
33 Geraci M.W., Moore M., Gesell T., et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555-562.
34 Thomson J.R., Trembath R.C. Primary pulmonary hypertension: the pressure rises for a gene. J Clin Pathol. 2000;53:899-903.
35 Loscalzo J. Genetic clues to the cause of primary pulmonary hypertension. N Engl J Med. 2001;345:367-371.
36 Grunig E., Mereles D., Arnold K., et al. Primary pulmonary hypertension is predominantly a hereditary disease. Chest. 2002;121:81S-82S.
37 Gersony W.M., Duc G.V., Sinclair J.C. PFC syndrome (persistence of the fetal circulation). Circulation. 1969;40:87.
38 Gersony W.M. Neonatal pulmonary hypertension: pathophysiology, classification, and etiology. Clin Perinatol. 1984;11:517-524.
39 Hageman J.R., Adams M.A., Gardner T.H. Persistent pulmonary hypertension of the newborn. Trends in incidence, diagnosis, and management. Am J Dis Child. 1984;138:592-595.
40 Murphy J.D., Rabinovitch M., Goldstein J.D., Reid L.M. The structural basis of persistent pulmonary hypertension of the newborn infant. J Pediatr. 1981;98:962-967.
41 Widlitz A., Barst R.J. Pulmonary arterial hypertension in children. Eur Respir J. 2003;21:155-176.
42 Robalino B.D., Moodie D.S. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol. 1991;17:492-498.
43 Ruttner J.R., Bartschi J.P., Niedermann R., Schneider J. Plexogenic pulmonary arteriopathy and liver cirrhosis. Thorax. 1980;35:133-136.
44 Speich R., Jenni R., Opravil M., Pfab M., Russi E.W. Primary pulmonary hypertension in HIV infection. Chest. 1991;100:1268-1271.
45 Ainger L.E. Large tonsils and adenoids in small children with cor pulmonale. Br Heart J. 1968;30:356-362.
46 Bland J.W.Jr, Edwards F.K. Pulmonary hypertension and congestive heart failure in children with chronic upper airway obstruction. New concepts of etiologic factors. Am J Cardiol. 1969;23:830-837.
47 Brouillette R.T., Fernbach S.K., Hunt C.E. Obstructive sleep apnea in infants and children. J Pediatr. 1982;100:31-40.
48 Mauer K.W., Staats B.A., Olsen K.D. Upper airway obstruction and disordered nocturnal breathing in children. Mayo Clin Proc. 1983;58:349-353.
49 Khoury G.H., Hawes C.R. Primary pulmonary hypertension in children living at high altitude. J Pediatr. 1963;62:177-185.
50 O’Neill D., Morton R., Kennedy J.A. Progressive primary pulmonary hypertension in a patient born at high altitude. Br Heart J. 1981;45:725-728.
51 Sime F., Banchero N., Penaloza D., Gamboa R., Cruz J., Marticorena E. Pulmonary hypertension in children born and living at high altitudes. Am J Cardiol. 1963;11:143-149.
52 Sun X.G., Hansen J.E., Oudiz R.J., Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation. 2001;104:429-435.
53 Grunig E., Janssen B., Mereles D., et al. Abnormal pulmonary artery pressure response in asymptomatic carriers of primary pulmonary hypertension gene. Circulation. 2000;102:1145-1150.
54 Hinderliter A.L., Willis P.W.T., Long W.A., et alGroup PPHS. Frequency and severity of tricuspid regurgitation determined by Doppler echocardiography in primary pulmonary hypertension. Am J Cardiol. 2003;91:1033-1037.
55 Bhan A., Rennie I.G., Higenbottam T.W. Central retinal vein occlusion associated with primary pulmonary hypertension. Retina. 2001;21:83-85.
56 Saran B.R., Brucker A.J., Bandello F., Verougstraete C. Familial primary pulmonary hypertension and associated ocular findings. Retina. 2001;21:34-39.
57 Wiedemann R., Ghofrani H.A., Weissmann N., et al. Atrial natriuretic peptide in severe primary and nonprimary pulmonary hypertension: response to iloprost inhalation. J Am Coll Cardiol. 2001;38:1130-1136.
58 Nagaya N., Nishikimi T., Uematsu M., et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102:865-870.
59 Marcus J.T., Vonk Noordegraaf A., Roeleveld R.J., et al. Impaired left ventricular filling due to right ventricular pressure overload in primary pulmonary hypertension: noninvasive monitoring using MRI. Chest. 2001;119:1761-1765.
60 Louie E.K., Rich S., Brundage B.H. Doppler echocardiographic assessment of impaired left ventricular filling in patients with right ventricular pressure overload due to primary pulmonary hypertension. J Am Coll Cardiol. 1986;8:1298-1306.
61 Stojnic B.B., Brecker S.J., Xiao H.B., Helmy S.M., Mbaissouroum M., Gibson D.G. Left ventricular filling characteristics in pulmonary hypertension: a new mode of ventricular interaction. Br Heart J. 1992;68:16-20.
62 Heath D., Edwards J.E. Configuration of elastic tissue of pulmonary trunk in idiopathic pulmonary hypertension. Circulation. 1960;21:59-62.
63 Roberts W.C. The histologic structure of the pulmonary trunk in patients with “primary” pulmonary hypertension. Am Heart J. 1963;65:230-236.
64 Clarke R.C., Coombs C.F., Hadfield G., Todd A.T. On certain abnormalities, congenital and acquired, of the pulmonary artery. Q J Med. 1927;21:51-68.
65 Kleiger R.E., Boxer M., Ingham R.E., Harrison D.C. Pulmonary hypertension in patients using oral contraceptives. A report of six cases. Chest. 1976;69:143-147.
66 Hood W.B.Jr, Spencer H., Lass R.W., Daley R. Primary pulmonary hypertension: familial occurrence. Br Heart J. 1968;30:336-343.
67 Husson G.S. Primary pulmonary hypertension in siblings. Am J Dis Child. 1956;92:506.
68 Parry W.R., Verel D. Familial primary pulmonary hypertension. Br Heart J. 1966;28:193-198.
69 Rogge J.D., Mishkin M.E., Genovese P.D. The familial occurrence of primary pulmonary hypertension. Ann Intern Med. 1966;65:672-684.
70 Thompson P., Mcrae C. Familial pulmonary hypertension. Evidence of autosomal dominant inheritance. Br Heart J. 1970;32:758-760.
71 Eddahibi S., Morrell N., D’Ortho M.P., Naeije R., Adnot S. Pathobiology of pulmonary arterial hypertension. Eur Respir J. 2002;20:1559-1572.
72 Deng Z., Morse J.H., Slager S.L., et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737-744.
73 Morse J.H., Jones A.C., Barst R.J., Hodge S.E., Wilhelmsen K.C., Nygaard T.G. Familial primary pulmonary hypertension locus mapped to chromosome 2q31-q32. Chest. 1998;114:57S-58S.
74 Newman J.H., Wheeler L., Lane K.B., et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319-324.
75 Wagenvoort C.A. Lung biopsy specimens in the evaluation of pulmonary vascular disease. Chest. 1980;77:614-625.
76 D’alonzo G.E., Barst R.J., Ayres S.M., et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343-349.
77 Rhodes J., Barst R.J., Garofano R.P., Thoele D.G., Gersony W.M. Hemodynamic correlates of exercise function in patients with primary pulmonary hypertension. J Am Coll Cardiol. 1991;18:1738-1744.
78 Rozkovec A., Montanes P., Oakley C.M. Factors that influence the outcome of primary pulmonary hypertension. Br Heart J. 1986;55:449-458.
79 Harinck E., Hutter P.A., Hoorntje T.M., et al. Air travel and adults with cyanotic congenital heart disease. Circulation. 1996;93:272-276.
80 Curnock A.L., Dweik R.A., Higgins B.H., Saadi H.F., Arroliga A.C. High prevalence of hypothyroidism in patients with primary pulmonary hypertension. Am J Med Sci. 1999;318:289-292.
81 Gomez A., Bialostozky D., Zajarias A., et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol. 2001;38:1137-1142.
82 Patrat J.F., Jondeau G., Dubourg O., et al. Left main coronary artery compression during primary pulmonary hypertension. Chest. 1997;112:842-843.
83 Kawut S.M., Silvestry F.E., Ferrari V.A., et al. Extrinsic compression of the left main coronary artery by the pulmonary artery in patients with long-standing pulmonary hypertension. Am J Cardiol. 1999;83:984-986. A910
84 Mikhail G.W., Gibbs J.S., Yacoub M.H. Pulmonary and systemic arterial pressure changes during syncope in primary pulmonary hypertension. Circulation. 2001;104:1326-1327.
85 Taylor C.J., Derrick G., McEwan A., Haworth S.G., Sury M.R.J. Risk of cardiac catheterization under anaesthesia in children with pulmonary hypertension. Br J Anaesth. 2007;98:657-661.
86 Dawkins K.D., Burke C.M., Billingham M.E., Jamieson S.W. Primary pulmonary hypertension and pregnancy. Chest. 1986;89:383-388.
87 Mccaffrey R.M., Dunn L.J. Primary pulmonary hypertension in pregnancy. Obstet Gynecol Surv. 1964;19:567-591.
88 Perloff J.K. Pregnancy in congenital heart disease: the mother and the fetus. In Perloff J.K., Child J.S., Aboulhosn J., editors: Congenital heart disease in adults, 3rd ed, Philadelphia: W.B. Saunders, 2009.
89 Roberts N.V., Keast P.J. Pulmonary hypertension and pregnancy—a lethal combination. Anaesth Intensive Care. 1990;18:366-374.
90 Takeuchi T., Nishii O., Okamura T., Yaginuma T. Primary pulmonary hypertension in pregnancy. Int J Gynaecol Obstet. 1988;26:145-150.
91 Kunhali K., Cherian G., Bakthaviziam A., Abraham M.T., Krishnaswami S. Rupture of a papillary muscle of the tricuspid valve in primary pulmonary hypertension. Am Heart J. 1980;99:225-229.
92 Steell G. The murmur of high pressure in the pulmonary artery. Medical Chronicle. 1888;9:182.
93 Perloff J.K. Auscultatory and phonocardiographic manifestations of pulmonary hypertension. Prog Cardiovasc Dis. 1967;9:303-340.
94 Perloff J.K. The physical examination of the heart and circulation, 4th ed. Shelton, Connecticut: People’s Medical Publishing House; 2009.
95 Rivero-Carvallo J.M. New diagnostic sign of tricuspid insufficiency. Arch Inst Cardiol Mex. 1946;16:531-540.
96 Perloff J.K., Harvey W.P. Mechanisms of fixed splitting of the second heart sound. Circulation. 1958;18:998-1009.
97 Shapiro S., Clark T.J., Goodwin J.F. Delayed closure of the pulmonary valve in obliterative pulmonary hypertension. Lancet. 1965;2:1207-1211.
98 Shaver J.A., Nadolny R.A., O’Toole J.D., et al. Sound pressure correlates of the second heart sound. An intracardiac sound study. Circulation. 1974;49:316-325.
99 Silverman B.D. Graham Steell. Clin Cardiol. 1995;18:54-55.
100 Major R.H. Classic descriptions of disease: with biographical sketches of the authors, 3rd ed. Springfield, Illinois: Charles C Thomas; 1948.
101 Potain P.C. Concerning the cardiac rhythm called gallop rhythm. Bull Mem Soc Med Hop Paris. 1876;12:137.
102 MacCallum W.G. Obliterative pulmonary arteriolosclerosis. Bull Johns Hopkins Hosp. 1931;49:37.
103 Green E.W., Agruss N.S., Adolph R.J. Right-sided Austin Flint murmur. Documentation by intracardiac phonocardiography, echocardiography and postmortem findings. Am J Cardiol. 1973;32:370-374.
104 Bedford D.E., Evans W., Short D.S. Solitary pulmonary hypertension. Br Heart J. 1957;19:93-116.
105 Bossone E., Paciocco G., Iarussi D., et al. The prognostic role of the ECG in primary pulmonary hypertension. Chest. 2002;121:513-518.
106 Bossone E., Duong-Wagner T.H., Paciocco G., et al. Echocardiographic features of primary pulmonary hypertension. J Am Soc Echocardiogr. 1999;12:655-662.
107 Child J.S. Transthoracic and transesophageal imaging: anatomic and hemodynamic assessment. In: Perloff J.K., Child J.S., editors. Congenital heart disease in adults. Philadelphia: W.B. Saunders; 1998:91.
108 Amaki M., Nakatani S., Kanzaki H., et al. Usefulness of three-dimensional echocardiography in assessing right ventricular function in patients with primary pulmonary hypertension. Hypertens Res Clin Exp. 2009;32:419-422.
109 Ghio S., Raineri C., Scelsi L., et al. Usefulness and limits of transthoracic echocardiography in the evaluation of patients with primary and chronic thromboembolic pulmonary hypertension. J Am Soc Echocardiogr. 2002;15:1374-1380.
110 Raymond R.J., Hinderliter A.L., Willis P.W., et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol. 2002;39:1214-1219.
111 Ivy D.D., Neish S.R., Knudson O.A., et al. Intravascular ultrasonic characteristics and vasoreactivity of the pulmonary vasculature in children with pulmonary hypertension. Am J Cardiol. 1998;81:740-748.