Chapter 30 Primary Myelofibrosis
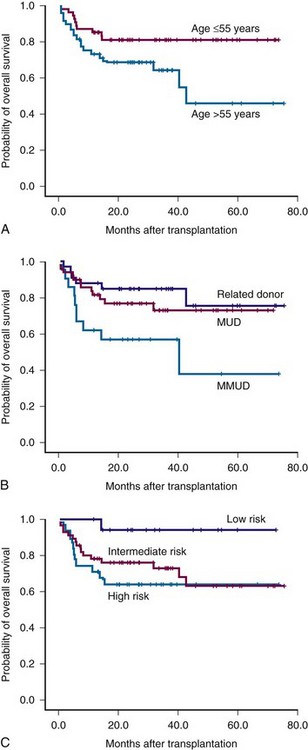
Figure 30-1 Survival of patients with myelofibrosis after reduced-intensity allogeneic stem cell transplantation according to age (A), donor (B), and Lille risk profile (C).
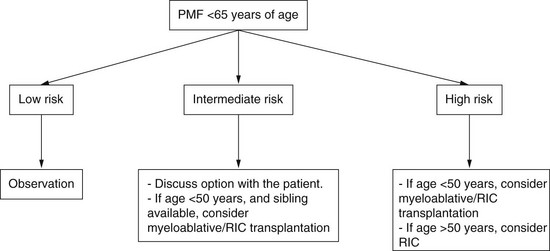
Algorithm for Selection of Appropriate Patients for Stem Cell Transplantation Stratified According to Risk Status
Case Vignettes
How Should PMF Patients with Thrombocytopenia Be Treated?
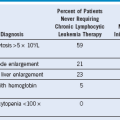
Table 30-2 Summary of Symptoms and Physical Findings of Patients With Primary Myelofibrosis Detected at Diagnosis
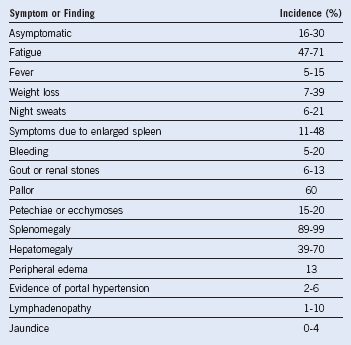
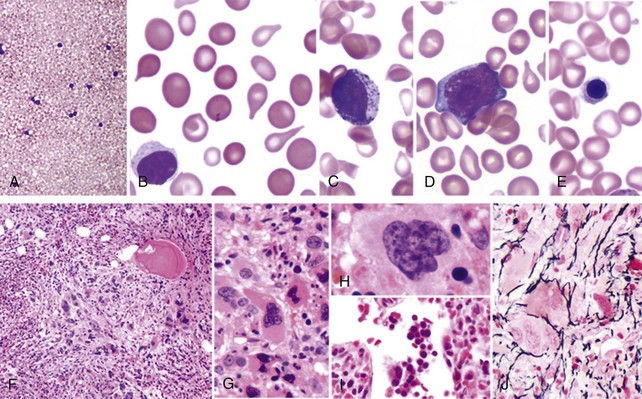
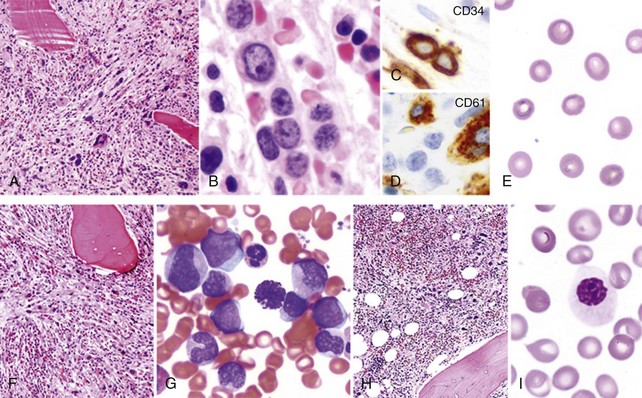
Table 30-3 Proposed Revised World Health Organization Criteria for Primary Myelofibrosis*
| MAJOR CRITERIA |
|
1. Presence of megakaryocyte proliferation and atypia,† usually accompanied by either reticulin or collagen fibrosis, or, in the absence of significant reticulin fibrosis, the megakaryocyte changes must be accompanied by an increased bone marrow cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (i.e., prefibrotic cellular-phase disease) 2. Not meeting WHO criteria for PV,‡ CML,§ MDS,‖ or other myeloid neoplasm 3. Demonstration of JAK2617V >F or other clonal marker (e.g., MPL515W >L/K) or in the absence of a clonal marker, no evidence of bone marrow fibrosis caused by underlying inflammatory or other neoplastic diseases¶ |
| MINOR CRITERIA |
CML, Chronic myeloid leukemia; MDS, myelodysplastic syndrome; PV, polycythemia vera; WHO, World Health Organization.
*Diagnosis requires meeting all three major criteria and two minor criteria.
†Small to large megakaryocytes with an aberrant nuclear-to-cytoplasmic ratio and hyperchromatic, bulbous, or irregularly folded nuclei and dense clustering.
‡Requires the failure of iron replacement therapy to increase hemoglobin level to the polycythemia vera range in the presence of decreased serum ferritin. Exclusion of polycythemia vera is based on hemoglobin and hematocrit levels. Red blood cell mass measurement is not required.
§Requires the absence of BCR-ABL.
‖Requires the absence of dyserythropoiesis and dysgranulopoiesis.
¶Secondary to infection, autoimmune disorder or other chronic inflammatory condition, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies. It should be noted that patients with conditions associated with reactive myelofibrosis are not immune to primary myelofibrosis, and the diagnosis should be considered in such cases if other criteria are met.
**Degree of abnormality could be borderline or marked.
Data from Tefferi A, Thiele J, Orazi A, et al: Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: Recommendations from an ad hoc international expert panel, Blood 110:1092, 2007.
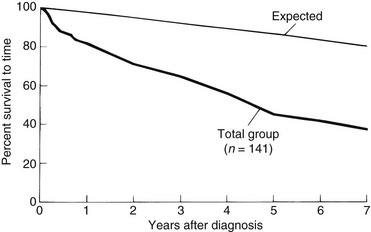
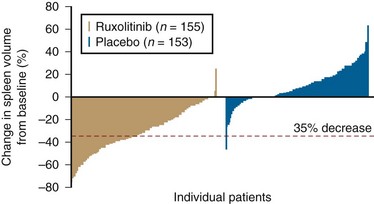
Figure 30-4 OVERALL SURVIVAL FROM THE TIME OF DIAGNOSIS OF 141 PATIENTS WITH PRIMARY MYELOFIBROSIS.
(Data from Silverstein MN: Agnogenic myeloid metaplasia, Acton, MA, 1975, Publishing Sciences Group, p 197.)
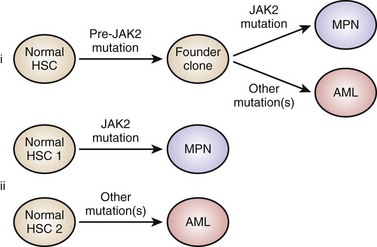
Figure 30-5 MODELS TO EXPLAIN PROGRESSION FROM A JANUS KINASE 2 (JAK2) MUTANT MYELOPROLIFERATIVE NEOPLASM TO A JAK2 WILD-TYPE LEUKEMIA.
(From Beer PA, Delhommeau F, LeCouédic JP, et al: Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 115:2891, 2010.)