[level-membership-for-allergy-and-immunology-category]
Chapter 16 Primary Immunodeficiencies
• Primary immunodeficiency diseases result from intrinsic defects in cells and mediators of the innate and adaptive immune system.
• Defects in B cell function result in recurrent pyogenic infections. Defective antibody responses are due to failure of B cell function, as occurs in X-linked agammaglobulinemia, or failure of proper T cell signals to B cells, as occurs in hyper-IgM (HIgM) syndrome and common variable immunodeficiency (CVID).
• Defects in T cell function due to ineffective antigen presentation or immune recognition result in susceptibility to opportunistic infections. Other abnormalities of T cells may also lead to immune dysregulation with autoimmunity or overactive immune responses.
• Hereditary complement component defects cause a number of clinical syndromes; the most common affects C1 inhibitor, which results in hereditary angioedema (HAE). Deficiencies of the terminal complement components (C5, C6, C7, and C8) and the alternative pathway proteins (factor H, factor I, and properdin) lead to increased susceptibility to infections with N. gonorrheae and N. meningitidis.
• Phagocyte defects, due to reduced numbers or impaired function, can result in overwhelming bacterial and fungal infections. Failure to kill bacteria and persistence of bacterial products in phagocytes leads to abscesses or granulomas, depending on the pathogen.
• Leukocyte adhesion deficiency (LAD) is associated with a persistent leukocytosis because phagocytic cells cannot migrate into the tissues.
• Antibody deficiencies reflect impaired function of B lymphocytes as a result of intrinsic B cell abnormalities or of defects in T lymphocytes that affect activation and terminal maturation of B lymphocytes
• Combined immunodeficiencies are characterized by impaired development and/or function of T lymphocytes, and functional B cell abnormalities
• Phagocytic cell disorders include defects in development and/or function of myeloid cells (granulocytes, macrophages)
• Complement deficiencies are represented by genetically-determined defects of functional or regulatory components of the complement system
• Disorders of immune regulation include diseases characterized by abnormalities in the mechanisms that control autoimmunity, apoptosis, or extinction of immune responses
• Immunodeficiency syndromes represent a heterogeneous group of PIDs in which defects of one or more components of the immune system are associated with extra-immune manifestations.
• patients with antibody deficiencies are highly susceptible to recurrent pyogenic infections sustained by encapsulated bacteria (Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus);
• combined immunodeficiencies are characterized by broad susceptibility to infections, that includes not only bacteria, but also viruses and opportunistic pathogens (i.e. ubiquitous germs that do not pose significant harm to immunocompetent individuals);
• patients with disorders of neutrophils are prone to bacterial and fungal infections;
• defects of macrophages result in increased susceptibility to mycobacterial disease;
• defects of Toll-like receptors (TLRs), that act as microbial sensors, cause selective susceptibility to specific types of pathogens;
• defects of complement may lead to increased risk of pyogenic infections, but also of autoimmunity, consistent with the role played by complement in removal of immune complexes.
B lymphocyte deficiencies
Congenital agammaglobulinemia results from defects of early B cell development
B lymphocytes develop in the bone marrow from the hematopoietic stem cell (HSC), through various stages of maturation (see Fig. 9.w1) during which time they rearrange their immunogloulin genes to generate the pre-B cell receptor (see Fig. 9.w2)
during which time they rearrange their immunogloulin genes to generate the pre-B cell receptor (see Fig. 9.w2) . Defects in the expression and/or signaling through the pre-BCR cause congenital agammaglobulinemia with lack of circulating B lymphocytes.
. Defects in the expression and/or signaling through the pre-BCR cause congenital agammaglobulinemia with lack of circulating B lymphocytes.
X-linked agammaglobulinemia (XLA) is the prototype of these disorders, and was described by Dr Bruton in 1952. Affected males suffer from recurrent pyogenic infections. They lack serum IgA, IgM, IgD and IgE, and IgG levels are extremely low, usually <100 mg/dL. Circulating B lymphocytes are absent or markedly reduced (<1% of peripheral lymphocytes). Tonsils are absent and lymph nodes are unusually small. XLA is caused by mutations of the Bruton tyrosine kinase (BTK) gene, that encodes an enzyme involved in signaling through the pre-BCR and the BCR (Fig. 16.1). BTK mutations cause an incomplete, but severe, block at the pre-B cell stage in the bone marrow (Fig. 16.2). The BTK protein is also expressed by other cells (including monocytes and megakaryocytes), but its defect does not affect development of these cell types.
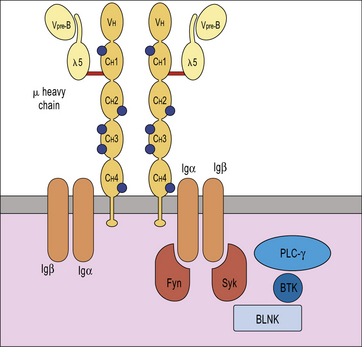
More rarely, congenital agammaglobulinemia is inherited as an autosomal recessive trait, due to mutations of other genes that encode for components of the pre-BCR or of the adaptor molecule BLNK (see Fig. 16.1). In all of these cases, there is a severe block in B-cell development at the pre-B cell stage in the bone marrow. The clinical phenotype is virtually identical to that of XLA.
Defects in terminal differentiation of B cells produces selective antibody deficiencies
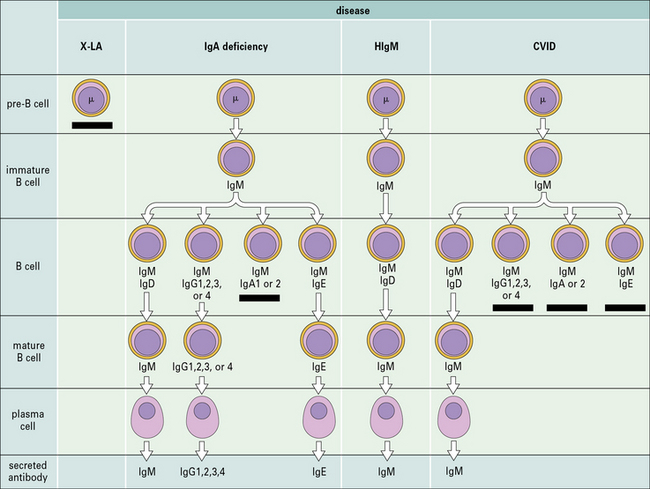
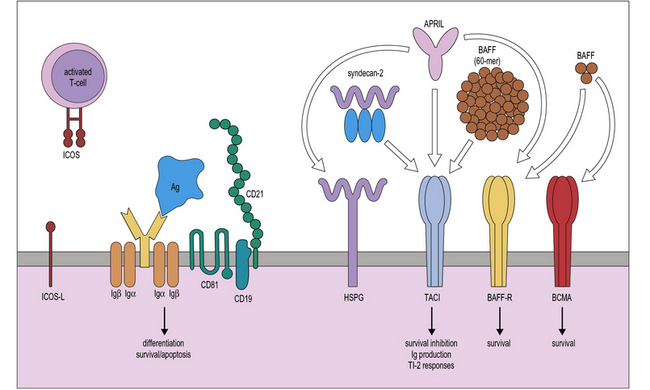
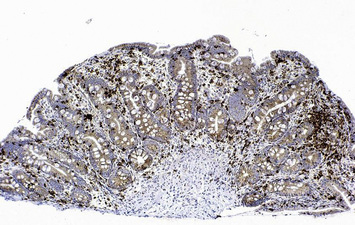
Terminal maturation of B lymphocytes is marked by their differentiation into antibody-secreting plasma cells. Generation of plasma cells is markedly reduced in patients with CVID (see Fig. 16.2), who typically develop progressive hypogammaglobulinemia in the second and third decades of life. CVID is the most common primary immunodeficiency (1:10 000 affected individuals in the general population), characterized by extensive clinical and immunologic heterogeneity. Some patients have a reduced number of circulating B cells, and especially of CD27+ memory B lymphocytes; others show impaired function of T lymphocytes. CVID is usually sporadic, and the underlying molecular defect remains unknown in most cases. However, in some families CVID is inherited as an autosomal dominant or an autosomal recessive trait. A minority of CVID patients carry mutations in genes that play a key role in T-B cell interaction and B cell signaling (Fig. 16.3).
CVID is characterized by reduced levels of specific antibody isotypes
Individuals with CVID have impaired antibody production in response to immunization or to natural infections and there is a virtual absence of plasma cells in lymphoid tissues and in the bone marrow. They suffer from recurrent infections of the respiratory tract (sinusitis, otitis, bronchitis, and pneumonia) sustained by common bacteria (non typeable H. influenzae, S. pneumoniae, etc.); lack of mucosal antibodies results in increased risk of gastrointestinal infection due to Giardia lamblia (Fig. 16.4). They are also highly prone to autoimmmune manifestations (cytopenias, inflammatory bowel disease), granulomatous lesions, lymphoid hyperplasia, and tumors (especially lymphomas). Treatment is based on immunoglobulin replacement therapy and antibiotics. Immunosuppressive and anti-inflammatory drugs may be needed in patients with autoimmune or inflammatory complications.
Defects of class switch recombination (CSR)
Class switch recombination (CSR) is the mechanism by which the μ chain of immunoglobulins is replaced by other heavy chains, resulting in the production of IgG, IgA, and IgE. The process occurs in germinal centres and is accompanied by affinity maturation as described in Chapter 9.
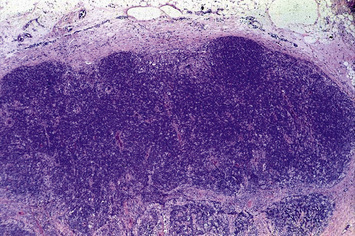
Deficiency of CD40L (X-linked) or more rarely of CD40 (autosomal recessive) results in failure of CSR, with very low or undetectable levels of IgG, IgA, and IgE and normal to increased levels of serum IgM (see Fig. 16.2). In the past, this condition was also known as ‘hyper-IgM syndrome’. In the lymph nodes, primary follicles are present, but germinal centers are absent (Fig. 16.5). Binding of CD40L to CD40 is also important to promote interaction between activated T cells and dendritic cells or monocytes/macrophages. This promotes T cell priming, production of IFNγ and activation of macrophages, that are important in the immune defense against intracellular pathogens. Consistent with this, the clinical phenotype of CD40L and of CD40 deficiency is characterized not only by recurrent bacterial infections, but also by increased risk of early-onset opportunistic infections (Pneumocystis jiroveci pneumonia, cytomegalovirus infection, protracted and watery diarrhoea due to Cryptosporidium). Neutropenia and severe liver disease are frequent. Therefore, CD40L and CD40 deficiency are not pure antibody deficiency, but rather represent examples of combined immunodeficiency. Treatment of these disorders is based on administration of immunoglobulins and antibiotics, but often requires hematopoietic stem cell transplantation (HSCT).
T lymphocyte deficiencies
Severe combined immunodeficiency (SCID) can be caused by many different genetic defects
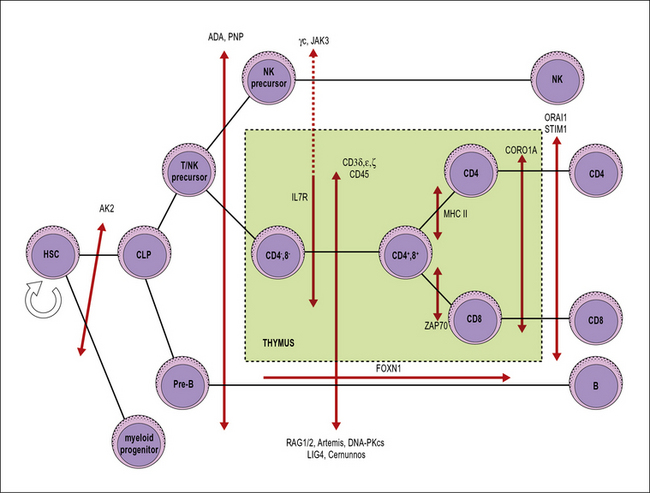
SCID includes a heterogeneous group of genetic disorders that affect various stages of T lymphocyte development or function (Fig. 16.6). The main pathophysiology mechanisms of SCID (and the associated diseases) are:
• impaired survival of thymocytes and T lymphocytes (reticular dysgenesis, adenosine deaminase deficiency, purine nucleoside phosphorylase deficiency);
• defective cytokine-mediated expansion of lymphoid progenitors (X-linked SCID, JAK3 deficiency, interleukin-7 receptor deficiency);
• defective expression of the pre-T cell receptor (deficiency of RAG1, RAG2, and of other components of the V(D)J recombination machinery);
• defective signaling through the pre-T cell receptor (deficiency of CD3 chains, CD45 deficiency);
• impaired positive selection of CD4+ or of CD8+ lymphocytes (HLA class II deficiency and ZAP-70 deficiency, respectively);
• defective egress of T lymphocytes from the thymus (coronin 1A deficiency);
• impairment of calcium flux and of T lymphocyte activation (Stim1, Orai1 deficiencies).
Accordingly, patients with X-SCID have a T−B+NK− phenotype.
ADA is a ubiquitously expressed enzyme involved in purine metabolism. Also purine nucleoside phosphorylase (PNP) is involved in the same metabolic pathway (Fig. 16.7). Lack of ADA results in accumulation of adenosine, deoxyadenosine, and their phosphorylated derivatives. Among them, dATP is particularly toxic; it inhibits the enzyme ribonucleotide reductase, that is required for DNA synthesis and hence for cell replication.
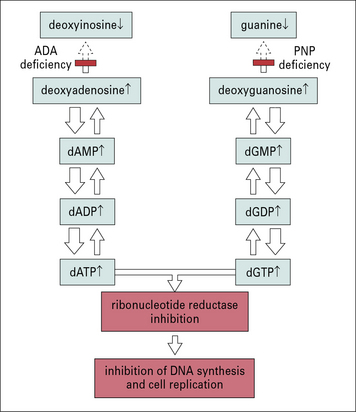
Fig. 16.7 Possible role of adenosine deaminase and purine nucleoside phosphorylase deficiency in SCID
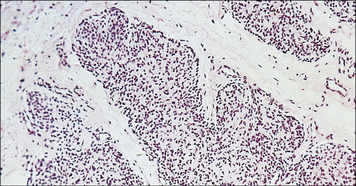
The thymus of SCID infants is very small and typically devoid of lymphoid elements (Fig. 16.8); lymph nodes are often absent or – when present – contain mostly stromal cells. Although B cells are normally present in some forms of SCID, antibody responses are profoundly impaired and immunoglobulin levels are usually reduced.
Clinically, SCID is apparent in the first months of life. Interstitial pneumonia (due to Pneumocystis jiroveci or to viral infections: cytomegalovirus, syncytial respiratory virus, adenovirus, parainfluenzae virus type 3), protracted diarrhea leading to failure to grow, and persistent candidiasis (Fig. 16.9) are common clinical findings; however, other infections (meningitis, sepsis) are also possible. Use of live vaccines in SCID infants often leads to severe consequences and should be strictly avoided; in particular, administration of rotavirus vaccine may cause intractable diarrhea, and immunization with BCG may lead to disseminated infection.

Fig. 16.9 Candida albicans in the mouth of a patient with SCID
C. albicans grows luxuriantly in the mouth and on the skin of patients with SCID.
TH cell deficiency results from HLA class II deficiency
Because the development of CD4+ helper T cells (TH) depends on positive selection by HLA class II molecules in the thymus (see Chapter 2), HLA class II molecule-deficient infants have a deficiency of CD4+ T cells. This lack of TH cells leads to a deficiency in antibodies as well. The HLA class II deficiency results from defects in transcription factors that bind to the 5′ untranslated promoter region of the class II HLA genes.
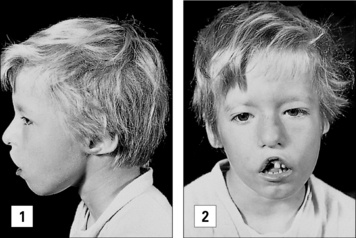
The DiGeorge anomaly arises from a defect in thymus embryogenesis
A congenital defect in the organs derived from the third and fourth pharyngeal pouches results in the DiGeorge anomaly. The T cell deficiency is variable, depending on how badly the thymus is affected; only in <1% of the patients, the T cell deficiency is so severe to cause SCID. Infants with DiGeorge anomaly have distinctive facial features (Fig. 16.10). They also have congenital malformations of the heart or aortic arch and neonatal tetany due to hypocalcemia resulting from the hypoplasia or aplasia of the parathyroid glands.
Disorders of immune regulation
Defective function of regulatory T (Treg) cells causes severe autoimmunity
Regulatory T cells (CD4+ CD25+) suppress immune responses to self-antigens in the periphery.
Mutations of the FOXP3 gene cause Immune dysregulation-polyendocrinopathy-enteropathy-X-linked (IPEX) syndrome, a severe, X-linked form of autoimmunity. Males with IPEX syndrome present in the first months of life with intractable diarrhoea, insulin-dependent diabetes and skin rash. Severe infections may follow because of breakage of the cutaneous and mucosal barriers. There is a lack of functional CD4+ CD25+ FOXP3+ lymphocytes. Activated, self-reactive T lymphocytes infiltrate target organs (Fig. 16.11) and there are high levels of autoantibodies against insulin, other pancreatic antigens and enterocytes. In typical cases, the disease evolves rapidly. Treatment with immunosuppressive drugs is required to control autoimmune manifestations, however stem cell therapy is the only curative approach.
Impaired apoptosis of self-reactive lymphocytes causes autoimmune lymphoproliferative syndrome (ALPS)
Congenital defects of lymphocyte cytotoxicity result in persistent inflammation and severe tissue damage
The cytotoxic activity of T cells and NK cells depends on the expression of cytolytic proteins that are assembled into granules and transported through microtubules to the lytic synapse that is formed upon contact with target cells. Familial hemophagocytic lymphohistiocytosis (FHL) includes a group of disorders characterized by impairment of the mechanisms of transport, docking or release of the lytic granules. Deficiency of perforin (see Fig. 10.12) is the most common form of FHL. In these diseases, persistence of the pathogen (most often, a virus) causes expansion of CD8+ T cells that, while unable to mount a cytotoxic response, secrete increased amounts of TH1 cytokines, and IFNγ. Excessive amounts of IFNγ trigger macrophage activation, causing phagocytosis of blood elements and tissue damage. The disease is usually fatal, and treatment is based on immunosuppressive drugs (to reduce immune activation) and HSCT.
Affected males appear normal until they encounter EBV, when they develop either:
Immunodeficiency syndromes
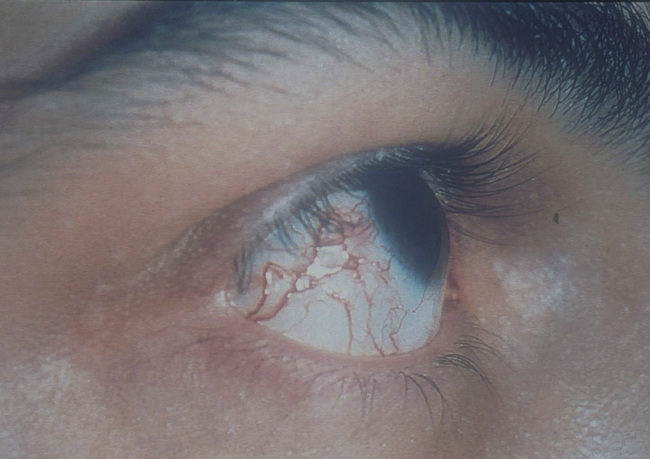
Chromosomal breaks occur in TCR and immunoglobulin genes in hereditary ataxia telangiectasia
Hereditary ataxia telangiectasia (AT) is inherited as an autosomal recessive trait. Affected infants develop a wobbly gait (ataxia) at about 18 months and ultimately are wheel-chaired. Dilated capillaries (telangiectasia) appear in the eyes and on the skin by 6 years of age (Fig. 16.12). AT is accompanied by a variable T cell deficiency. About 70% of patients with AT are also IgA deficient and some also have IgG2 and IgG4 deficiency.
Q. In what other ways are IgA, IgG2, and IgG4 related (as opposed to IgM, IgG1, and IgG3)?
A. The IgA1, IgA2, IgG2, and IgG4 heavy chain genes all lie further downstream of the recombined VDJ gene than IgM, IgG1, and IgG3 (see Fig. 9.16). This may account for the selective deficiency in making the class switch to IgA, IgG2, and IgG4. Class switching involves the production and resolution of double-stranded DNA breaks.
T cell defects and abnormal immunoglobulin levels occur in Wiskott–Aldrich syndrome
The Wiskott–Aldrich syndrome (WAS) is an X-linked immunodeficiency disease. Affected males with WAS:
Genetic defects of phagocytes
Two groups of genetic defects affect phagocyte function without altering their development:
Chronic granulomatous disease results from a defect in the oxygen reduction pathway
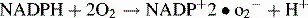 . They are therefore incapable of forming superoxide anions (O2–) and hydrogen peroxide in their phagocytes following the ingestion of microorganisms.
. They are therefore incapable of forming superoxide anions (O2–) and hydrogen peroxide in their phagocytes following the ingestion of microorganisms.
Enzyme defects in CGD
When phagocytosis occurs, the p47phox and p67phox cytosolic proteins become phosphorylated, move to the membrane of the phagosome, and bind to flavocytochrome. The complex formed acts as an enzyme, NADPH oxidase, catalyzing the NADPH oxidation reaction, production of oxygen radical, and activation of lytic enzymes (cathepsin G, elastase), thereby permitting intracellular killing of bacteria and fungi (Fig. 16.w1).
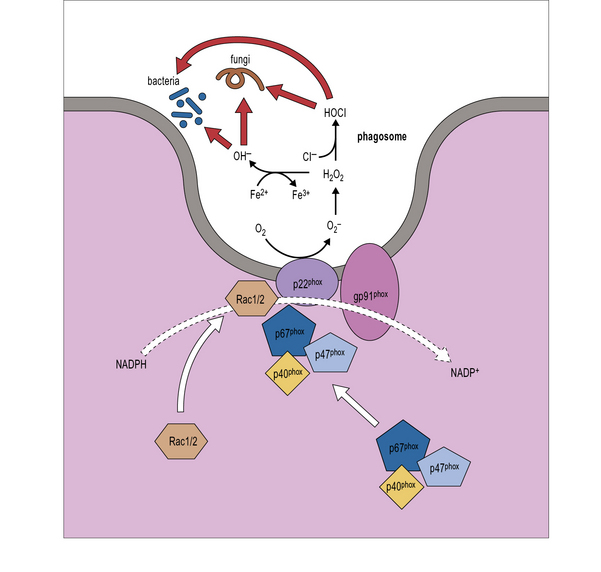
Fig. 16.w1 NADPH oxidase
(Adapted from Rosenzweig SD, Uzel G and Holland SM. Phagocyte disorders. In: Steihm ER, Ochs HD, Winkelstein JA Eds., Immunological disorders in infants and children, 5th Edition, Elsevier, 2004, pp.618–651.)
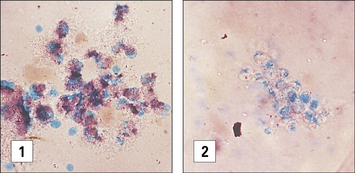
Diagnosis of CGD is most commonly performed by flow cytometry, evaluating dihydrorhodamine-123 (DHR-123) oxidation upon in vitro activation. In the past, diagnosis of CGD was made by demonstrating inability of phagocytes to reduce nitroblue tetrazolium (NBT) dye after a phagocytic stimulus. NBT is a pale, clear, yellow dye taken up by phagocytes when they are ingesting a particle. When NBT accepts H+ and is reduced as a result of NADPH oxidation it forms a deep purple precipitate inside the phagocytes; precipitation does not occur in the phagocytes of patients with CGD (Fig. 16.w2).
LAD is due to defects of leukocytes trafficking
In LAD1, there is a genetic defect of the β chain, encoded by a gene on chromosome 21.
Two other integrin proteins share the same β chain as CR3 – namely lymphocyte functional antigen (LFA-1) and p150,95 (see Chapter 6); these proteins are also defective in LAD1.
When leukocytes in the circulation enter an area of inflammation their speed of movement is greatly retarded by the interaction of selectins with their ligands (see Fig. 6.6). E-selectin interacts with Sialyl Lewisx (SLeX), a fucosylated molecule that is expressed on the surface of neutrophils and monocytes. In LAD2 a genetic defect of intracellular fucose transporter prevents fucosylation of membrane glycoproteins, including SLeX. Consequently, the leukocytes of patients with LAD2 cannot roll on the endothelium and fail to extravasate and reach inflamed tissues. Since fucose metabolism is important also in the central nervous system, patients with LAD2 also show mental retardation and dysmorphisms in addition to infections.
Immunodeficiencies with selective susceptibility to infections
Defects of TLR-signaling cause susceptibility to pyogenic infections
Toll-like receptors (TLR) are a series of molecules that are expressed at the cell surface or at the membrane of endosomes, and mediate recognition of pathogen-associated molecular patterns, such as lipopolysaccharide, glycolipids, single- or double-stranded RNA (see Fig. 6.20). The classical pathway of TLR activation involves the adaptor molecules MyD88 and the intracellular kinases IRAK-4 and IRAK-1. Activation of this pathway upon binding of TLRs to their ligands, results in the induction of NFκB and production of inflammatory cytokines (IL-1, IL-6, TNFα, IL-12). Mutations of IRAK4 and MyD88 cause severe and invasive pyogenic infections early in life, often without significant inflammatory response. Infections tend to become less frequent later in life, when the adaptive immune system has matured.
Genetic deficiencies of complement proteins
The proteins of the complement system and their interactions with the immune system are discussed in Chapter 4. Genetic deficiencies of almost all the complement proteins have been found in humans (Fig. 16.13) and these deficiencies reveal much about the normal function of the complement system.
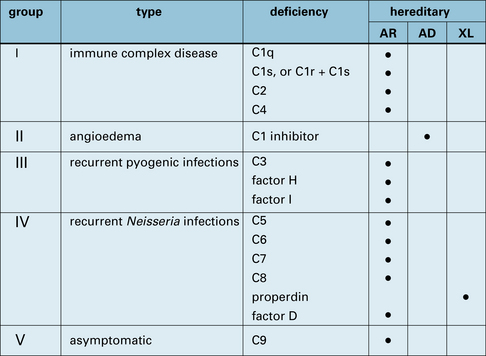
Immune complex clearance, inflammation, phagocytosis, and bacteriolysis can be affected by complement deficiencies
Q. Why should these deficiencies result in immune complex disease?
A. The classical complement pathway is required for the dissolution of immune complexes by covalent binding of C4b and C3b to components of the complex. It is also required for the transport of complexes on erythrocytes in humans (see Fig. 25.6).
Hereditary angioneurotic edema (HAE) results from C1 inhibitor deficiency
The C1 inhibitor (C1INH) is responsible for dissociation of activated C1, by binding to C1r2C1s2. Deficiency of C1INH results in HAE (Fig. 16.14), that is inherited as an autosomal dominant trait. Patients have recurrent episodes of swelling of various parts of the body (angioedema):
• when the edema involves the intestine, excruciating abdominal pains and cramps result, with severe vomiting;
• when the edema involves the upper airway, the patients may choke to death from respiratory obstruction – angioedema of the upper airway therefore presents a medical emergency, which requires rapid action to restore normal breathing.
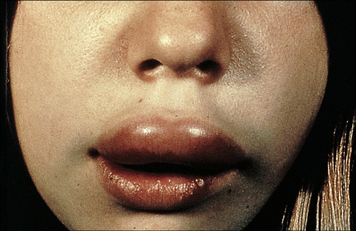
Fig. 16.14 Hereditary angioneurotic edema
This clinical photograph shows the transient localized swelling that occurs in this condition.
• a peptide derived from the activation of C2, called C2 kinin; and
• bradykinin derived from the activation of the contact system (Fig. 16.15).
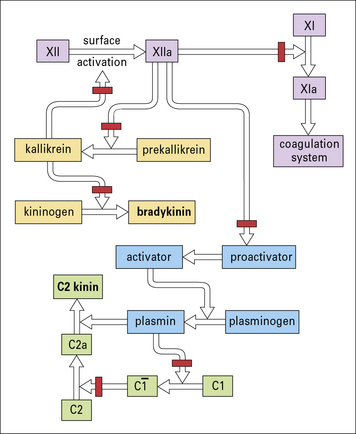
The effect of these peptides is on the postcapillary venule, where they cause endothelial cells to retract, forming gaps that allow leakage of plasma (see Chapter 6).
There are two genetically determined forms of HAE:
• in type I, the C1INH gene is defective and no transcripts are formed; and
• in type II, there are point mutations in the C1INH gene resulting in the synthesis of defective molecules.
Critical thinking: Hyper-IgM immunodeficiency (see p. 438 for explanations)
1. What clinical and laboratory tests lead to the suspicion that this child has hyper-IgM (HIgM) immunodeficiency and how do you conclude that it is not due to a mutation in CD40 ligand?
2. What is the most likely diagnosis in this case?
3. How do you explain that this child had no response to tetanus immunization and yet has a high titer for her age of antibody to blood group substance B?
4. What treatment would you recommend to the parents for this child and what would you say is her prognosis?
Conley M.E., Dobbs A.K., Farmer D.M., et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227.
Fischer A. Human primary immunodeficiency diseases. Immunity. 2007;27:835–845.
Fischer A., Le Deist F., Hacein-Bey-Abina S., et al. Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Immunol Rev. 2005;203:98–109.
Frank M.M. Complement disorders and hereditary angioedema. J Allergy Clin Immunol. 2010;125:S262–S271.
Holland S.M. Chronic granulomatous disease. Clin Rev Allergy Immunol. 2010;38:3–10.
Klein C. Congenital neutropenia. Hematology Am Soc Hematol Educ Program. 2009:344–350.
Notarangelo L.D. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125:S182–S194.
Pachlopnik Schmid J., Côte M., Ménager M.M., et al. Inherited defects in lymphocyte cytotoxic activity. Immunol Rev. 2010;235:10–23.
Sancho-Shimizu V., Zhang S.Y., Abel L., et al. Genetic susceptibility to herpes simplex virus 1 encephalitis in mice and humans. Curr Opin Allergy Clin Immunol. 2007;7:495–505.
Thrasher A.J., Burns S.O. WASP: a key immunological multitasker. Nat Rev Immunol. 2010;10:182–192.
Zhang S.Y., Boisson-Dupuis S., Chapgier A., et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40.
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Chapter 16 Primary Immunodeficiencies
• Primary immunodeficiency diseases result from intrinsic defects in cells and mediators of the innate and adaptive immune system.
• Defects in B cell function result in recurrent pyogenic infections. Defective antibody responses are due to failure of B cell function, as occurs in X-linked agammaglobulinemia, or failure of proper T cell signals to B cells, as occurs in hyper-IgM (HIgM) syndrome and common variable immunodeficiency (CVID).
• Defects in T cell function due to ineffective antigen presentation or immune recognition result in susceptibility to opportunistic infections. Other abnormalities of T cells may also lead to immune dysregulation with autoimmunity or overactive immune responses.
• Hereditary complement component defects cause a number of clinical syndromes; the most common affects C1 inhibitor, which results in hereditary angioedema (HAE). Deficiencies of the terminal complement components (C5, C6, C7, and C8) and the alternative pathway proteins (factor H, factor I, and properdin) lead to increased susceptibility to infections with N. gonorrheae and N. meningitidis.
• Phagocyte defects, due to reduced numbers or impaired function, can result in overwhelming bacterial and fungal infections. Failure to kill bacteria and persistence of bacterial products in phagocytes leads to abscesses or granulomas, depending on the pathogen.
• Leukocyte adhesion deficiency (LAD) is associated with a persistent leukocytosis because phagocytic cells cannot migrate into the tissues.
• Antibody deficiencies reflect impaired function of B lymphocytes as a result of intrinsic B cell abnormalities or of defects in T lymphocytes that affect activation and terminal maturation of B lymphocytes
• Combined immunodeficiencies are characterized by impaired development and/or function of T lymphocytes, and functional B cell abnormalities
• Phagocytic cell disorders include defects in development and/or function of myeloid cells (granulocytes, macrophages)
• Complement deficiencies are represented by genetically-determined defects of functional or regulatory components of the complement system
• Disorders of immune regulation include diseases characterized by abnormalities in the mechanisms that control autoimmunity, apoptosis, or extinction of immune responses
• Immunodeficiency syndromes represent a heterogeneous group of PIDs in which defects of one or more components of the immune system are associated with extra-immune manifestations.
• patients with antibody deficiencies are highly susceptible to recurrent pyogenic infections sustained by encapsulated bacteria (Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus);
• combined immunodeficiencies are characterized by broad susceptibility to infections, that includes not only bacteria, but also viruses and opportunistic pathogens (i.e. ubiquitous germs that do not pose significant harm to immunocompetent individuals);
• patients with disorders of neutrophils are prone to bacterial and fungal infections;
• defects of macrophages result in increased susceptibility to mycobacterial disease;
• defects of Toll-like receptors (TLRs), that act as microbial sensors, cause selective susceptibility to specific types of pathogens;
• defects of complement may lead to increased risk of pyogenic infections, but also of autoimmunity, consistent with the role played by complement in removal of immune complexes.
B lymphocyte deficiencies
Congenital agammaglobulinemia results from defects of early B cell development
B lymphocytes develop in the bone marrow from the hematopoietic stem cell (HSC), through various stages of maturation (see Fig. 9.w1) during which time they rearrange their immunogloulin genes to generate the pre-B cell receptor (see Fig. 9.w2). Defects in the expression and/or signaling through the pre-BCR cause congenital agammaglobulinemia with lack of circulating B lymphocytes.
X-linked agammaglobulinemia (XLA) is the prototype of these disorders, and was described by Dr Bruton in 1952. Affected males suffer from recurrent pyogenic infections. They lack serum IgA, IgM, IgD and IgE, and IgG levels are extremely low, usually <100 mg/dL. Circulating B lymphocytes are absent or markedly reduced (<1% of peripheral lymphocytes). Tonsils are absent and lymph nodes are unusually small. XLA is caused by mutations of the Bruton tyrosine kinase (BTK) gene, that encodes an enzyme involved in signaling through the pre-BCR and the BCR (Fig. 16.1). BTK mutations cause an incomplete, but severe, block at the pre-B cell stage in the bone marrow (Fig. 16.2). The BTK protein is also expressed by other cells (including monocytes and megakaryocytes), but its defect does not affect development of these cell types.
More rarely, congenital agammaglobulinemia is inherited as an autosomal recessive trait, due to mutations of other genes that encode for components of the pre-BCR or of the adaptor molecule BLNK (see Fig. 16.1). In all of these cases, there is a severe block in B-cell development at the pre-B cell stage in the bone marrow. The clinical phenotype is virtually identical to that of XLA.
Defects in terminal differentiation of B cells produces selective antibody deficiencies
Terminal maturation of B lymphocytes is marked by their differentiation into antibody-secreting plasma cells. Generation of plasma cells is markedly reduced in patients with CVID (see Fig. 16.2), who typically develop progressive hypogammaglobulinemia in the second and third decades of life. CVID is the most common primary immunodeficiency (1:10 000 affected individuals in the general population), characterized by extensive clinical and immunologic heterogeneity. Some patients have a reduced number of circulating B cells, and especially of CD27+ memory B lymphocytes; others show impaired function of T lymphocytes. CVID is usually sporadic, and the underlying molecular defect remains unknown in most cases. However, in some families CVID is inherited as an autosomal dominant or an autosomal recessive trait. A minority of CVID patients carry mutations in genes that play a key role in T-B cell interaction and B cell signaling (Fig. 16.3).
CVID is characterized by reduced levels of specific antibody isotypes
Individuals with CVID have impaired antibody production in response to immunization or to natural infections and there is a virtual absence of plasma cells in lymphoid tissues and in the bone marrow. They suffer from recurrent infections of the respiratory tract (sinusitis, otitis, bronchitis, and pneumonia) sustained by common bacteria (non typeable H. influenzae, S. pneumoniae, etc.); lack of mucosal antibodies results in increased risk of gastrointestinal infection due to Giardia lamblia (Fig. 16.4). They are also highly prone to autoimmmune manifestations (cytopenias, inflammatory bowel disease), granulomatous lesions, lymphoid hyperplasia, and tumors (especially lymphomas). Treatment is based on immunoglobulin replacement therapy and antibiotics. Immunosuppressive and anti-inflammatory drugs may be needed in patients with autoimmune or inflammatory complications.
Defects of class switch recombination (CSR)
Class switch recombination (CSR) is the mechanism by which the μ chain of immunoglobulins is replaced by other heavy chains, resulting in the production of IgG, IgA, and IgE. The process occurs in germinal centres and is accompanied by affinity maturation as described in Chapter 9.
Deficiency of CD40L (X-linked) or more rarely of CD40 (autosomal recessive) results in failure of CSR, with very low or undetectable levels of IgG, IgA, and IgE and normal to increased levels of serum IgM (see Fig. 16.2). In the past, this condition was also known as ‘hyper-IgM syndrome’. In the lymph nodes, primary follicles are present, but germinal centers are absent (Fig. 16.5). Binding of CD40L to CD40 is also important to promote interaction between activated T cells and dendritic cells or monocytes/macrophages. This promotes T cell priming, production of IFNγ and activation of macrophages, that are important in the immune defense against intracellular pathogens. Consistent with this, the clinical phenotype of CD40L and of CD40 deficiency is characterized not only by recurrent bacterial infections, but also by increased risk of early-onset opportunistic infections (Pneumocystis jiroveci pneumonia, cytomegalovirus infection, protracted and watery diarrhoea due to Cryptosporidium). Neutropenia and severe liver disease are frequent. Therefore, CD40L and CD40 deficiency are not pure antibody deficiency, but rather represent examples of combined immunodeficiency. Treatment of these disorders is based on administration of immunoglobulins and antibiotics, but often requires hematopoietic stem cell transplantation (HSCT).
T lymphocyte deficiencies
Severe combined immunodeficiency (SCID) can be caused by many different genetic defects
SCID includes a heterogeneous group of genetic disorders that affect various stages of T lymphocyte development or function (Fig. 16.6). The main pathophysiology mechanisms of SCID (and the associated diseases) are:
• impaired survival of thymocytes and T lymphocytes (reticular dysgenesis, adenosine deaminase deficiency, purine nucleoside phosphorylase deficiency);
• defective cytokine-mediated expansion of lymphoid progenitors (X-linked SCID, JAK3 deficiency, interleukin-7 receptor deficiency);
• defective expression of the pre-T cell receptor (deficiency of RAG1, RAG2, and of other components of the V(D)J recombination machinery);
• defective signaling through the pre-T cell receptor (deficiency of CD3 chains, CD45 deficiency);
• impaired positive selection of CD4+ or of CD8+ lymphocytes (HLA class II deficiency and ZAP-70 deficiency, respectively);
• defective egress of T lymphocytes from the thymus (coronin 1A deficiency);
• impairment of calcium flux and of T lymphocyte activation (Stim1, Orai1 deficiencies).
