CHAPTER 15 PRIMARY DISORDERS OF SLEEP
Difficulty with the regulation of sleep and wake states is present in up to 25% of the general population on a chronic basis and in up to one half of all individuals on occasion. For some, the primary concern is difficulty falling asleep, whereas for others it may be maintaining sleep or awakening feeling unrefreshed, even after a full night’s rest. Other individuals report excess daytime sleepiness, having difficulty maintaining alertness at inopportune or embarrassing times, or interference of sleepiness at times with productivity or even safety. The evaluation and treatment of such patients are the domains of sleep disorders medicine, a field that combines elements of neurology, psychiatry, pulmonary medicine, and otolaryngology.
A nosology of sleep disorders, the International Classification of Sleep Disorders, now in its second edition (ICSD-2),1 developed by the American Academy of Sleep Medicine (Table 15-1), has existed for more than 20 years. Its codes are consistent with the existing codes of the International Classification of Disease, 10th edition. The ICSD-2 organizes sleep disorders in eight categories on the basis of their predominant manifesting symptom and/or etiological basis: the insomnias; the sleep-related breathing disorders; hypersomnia not caused by a sleep-related breathing disorder; the circadian rhythm disorders; the parasomnias; the sleep-related movement disorders; and two miscellaneous categories comprising normal variants, isolated symptoms, and other sleep disorders. Readers are referred to Chapters 16 and 37 for detailed descriptions of obstructive sleep apnea and restless legs syndrome (RLS).
TABLE 15-1 International Classification of Sleep Disorders, 2nd Edition
NREM, non–rapid eye movement; REM, rapid eye movement.
Reprinted from American Academy of Sleep Medicine: International Classification of Sleep Disorders: Diagnostic and Coding Manual, 2nd ed. Rochester, MN: American Academy of Sleep Medicine, 2005.
INSOMNIA
Epidemiology, Consequences, and Diagnosis of Insomnia
The point prevalence of insomnia that lasts more than a few weeks is approximately 10% to 15% of the general population.2 However, because of its association with medical and psychiatric illnesses, up to 50% of individuals seen in medical practices report at least mild insomnia.3 Results of studies in individuals older than 65 suggest a 5% incidence and a 5% to 15% yearly rate of remission of insomnia.4,5 Female gender, increasing age, psychiatric and medical illnesses, substance use, low income, unemployment, and being single are all risk factors for having insomnia, although some of these may be consequences of insomnia rather than vulnerability factors.6–8
There is increasing recognition of the adverse consequences of insomnia. Multiple studies have demonstrated that persistent insomnia is associated with a substantial increased risk of incident depression.9 Insomnia is also associated with globally worsened quality of life, even when psychiatric illness10 or medical comorbidity3 is accounted for. The decrements in physical functioning, general health perception, and vitality are as substantial as, or more so than, those observed with congestive heart failure.3 Furthermore, there are suggestions that insomnia is associated with an increased risk of work-related and motor vehicle accidents, as well as falls by elderly persons.11 Finally, health costs in individuals with insomnia are elevated, even when comorbid medical and psychiatric illnesses are accounted for.12
The concept of hyperarousal is being used to unify the understanding of the pathophysiology of primary insomnia.13–15 From a physiological perspective, individuals with insomnia have elevated evening cortisol levels,16 increased 24-hour whole body metabolic rate,17 increases in both waking and sleep-related global cerebral glucose metabolism (Fig. 15-1),18 and high-frequency electroencephalographic (EEG) activity during sleep.19 It is unclear which neural circuits are responsible for these disparate findings. Similarly, cognitive arousal is considered to be central to the generation and maintenance of insomnia. It is hypothesized that cognitive and physiological hyperarousal become paired with the sleep environment, which gradually worsens sleep and increases these arousal processes in that setting, creating a vicious cycle of insomnia.15,20 Maladaptive compensatory strategies, such as spending excess time in bed, daytime napping, and alcohol and caffeine intake can then exacerbate this process.

Figure 15-1 Areas in which metabolism did not decrease from sleep to wakefulness in insomniac patients.
(From Nofzinger EA, Buysse DJ, Germain A, et al: Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry 2004; 161:2126-2168.)
Identification of potential medical, sleep-related, and psychiatric causes of insomnia is essential for optimal treatment, because treatment of such causes may at times eliminate the insomnia complaint. Insomnia in elderly persons, in whom frequent nocturnal awakenings are the most common complaint, is particularly related to medical illness,21 and careful attention to patients’ medical problems may provide guides to the etiology of insomnia in this group. The most common medical disorders associated with insomnia are listed in Table 15-2. In addition, all psychiatric disorders can and frequently do cause insomnia, and assessments for depression and anxiety disorders are an essential feature of the insomnia evaluation. However, it should be made clear that approximately 40% of individuals with insomnia do not have a psychiatric disorder,22 and thus the assumption that insomnia is necessarily caused by psychiatric illness is ill founded.
TABLE 15-2 Medical Disorders or Conditions Commonly Associated with Insomnia
Polysomnography can also assist with the assessment of insomnia in some cases. This diagnostic procedure is not recommended for most individuals with insomnia23; however, when the clinician suspects sleep apnea or periodic limb movements of sleep (PLMSs), or when the patient reports frequent brief awakenings, polysomnography is indicated for further evaluation.
Treatment of Insomnia
In individuals with chronic primary insomnia, and in some individuals with secondary insomnia, first-line treatments are modification of sleep-related behaviors and attitudes, called cognitive-behavioral therapy. Cognitive-behavioral therapy has a number of components: (1) limitation of time in bed (sleep restriction and stimulus control), which produces mild sleep deprivation, thus allowing shorter sleep onset and reduction in the number and duration of awakenings, and reduces the duration of time awake in bed, limiting negative associations to the sleep environment; (2) relaxation techniques, which reduce physiological and cognitive arousal in the sleep setting by use of yoga, meditation, and/or biofeedback; (3) cognitive restructuring, which addresses catastrophic beliefs and attitudes regarding sleeplessness, replacing them with more rational expectations of sleep and effects of insomnia; and (4) sleep hygiene, which refers to a variety of habits that promote good sleep such as regular bedtimes and waking times, daily exercise, avoidance of napping, careful use of alcohol and caffeine, and reduction in behaviors that promote nocturnal emotional and physical arousal (e.g., work, emotional stimulation, nighttime exercise). Cognitive-behavioral therapy has been shown to produce consistent reduction in sleep onset latency and wake time during the night, as well as smaller increases in total sleep time.24,25 These gains have generally been maintained over periods of up to 24 months.
Pharmacological therapies for insomnia have evolved since the 1950s from barbiturates to long-acting benzodiazepines, then to shorter acting benzodiazepines, and, since the mid-1990s, to nonbenzodiazepine receptor agonists (BzRAs). In addition, there has been a trend away from these approved medications for insomnia and toward the use of sedating medications with original indications for other disorders (e.g., antidepressants, anticonvulsants, antipsychotics), to the point at which antidepressants constitute more than 50% of all prescription medications for insomnia.26 Recommendations as to the appropriate use of hypnotics in the treatment of insomnia are evolving, and this and other treatment issues in insomnia were reviewed in a state-of-the-science National Institute of Mental Health consensus statement.27
Benzodiazepines and BzRAs bind at an allosteric site on the γ-amino butyric acid A (GABAA) receptor complex, influencing GABA binding and chloride flux. The BzRAs demonstrate relatively selective binding for GABAA receptors that contain α1 subunits. The α1 subunits mediate the sedative, amnestic, and anticonvulsant properties of these agents but few of the muscle relaxant and anxiolytic aspects (Fig. 15-2).28 However, it is unclear whether the relative receptor selectivity of the BzRAs have clinical significance in terms of efficacy or short- or long-term tolerability.

Figure 15-2 γ-Amino butyric acid A (GABAA) receptor subtypes, localization and function in mouse brain.
(From Mohler H, Fritschy J, Rudolph U: A new benzodiazepine pharmacology. J Pharmacol Exp Ther 2002; 300:2-8.)
More important than the receptor-binding characteristics of these agents are the major differences between the half-lives of these agents, which, when combined with dosage, determine the duration of the medication’s effects. Half-lives of hypnotics in this class vary from 1 to more than 100 hours (Table 15-3). Because of the variability of sleep complaints, medication choices in this class are usually based on matching the patient’s sleep complaint with an appropriate half-life agent, so as to maximize the opportunity for sleep but minimize waking hangover effects.
TABLE 15-3 Benzodiazepine Receptor Agonists Commonly Used for the Treatment of Insomnia
| Agent (Brand Name) | Dosage Range | Half-Life |
|---|---|---|
| Flurazepam (Dalmane)* | 15-30 mg | 50-100 hours |
| Estazolam (Prosom)* | 1.0-2.0 mg | 10-20 hours |
| Temazepam (Restoril)* | 7.5-30 mg | 4-18 hours |
| Triazolam (Halcion)* | 0.125-0.25 mg | 2-3 hours |
| Eszopiclone (Lunesta) | 1-3 mg | 5.5-8 hours |
| Zolpidem (Ambien) | 5-10 mg | 2-3 hours |
| Zaleplon (Sonata) | 5-10 mg | 1-2 hours |
Benzodiazepines.
Meta-analyses have demonstrated the efficacy of benzodiazepines and BzRAs in reducing sleep onset latency, decreasing the amount of wakefulness after sleep onset, and in increasing total sleep time in patients with primary insomnia.29 However, when a meta-analysis of benzodiazepines alone was performed, the absolute size of this effect for sleep onset latency was not dramatic: a reduction of 4.2 minutes when assessed by polysomnography and of 14.3 minutes by self-report. On the other hand, total sleep time was increased by a mean of 61.8 minutes.30
The majority of these efficacy data come from short-duration studies. For instance, the median duration of the studies in the benzodiazepine and BzRA meta-analysis was 7 days; the common duration of insomnia complaints, in contrast, is often months to years. Studies addressing the longer term efficacy of these medications in continuous and intermittent use have been performed. Eszopiclone, the S-isomer of the commonly prescribed hypnotic zopiclone, has been shown to produce persistent benefits for sleep onset latency, wakefulness after sleep onset, total sleep time, and daytime functioning for 6 months of nightly use in comparison with placebo in patients with primary insomnia.31
PERIODIC LIMB MOVEMENT DISORDER
PLMSs are commonly recorded movements during sleep consisting of repetitive dorsiflexion of the foot and/or lower leg. Movements are generally subtle and may not be recognized by a bed partner, although in more severe forms, they are more obvious. PLMS may or may not be associated with arousals from sleep, and indices of the number of movements with and without arousal per hour of sleep are derived. The term periodic limb movement of sleep is derived from the strict periodicity of movements, which occur at 15- to 30-second intervals during sleep. Movements are roughly 2 seconds in duration (Fig. 15-3). When a sleep complaint occurs in the presence of PLMS, in the absence of other known causes of sleep disruption, a diagnosis of periodic limb movement disorder is given.
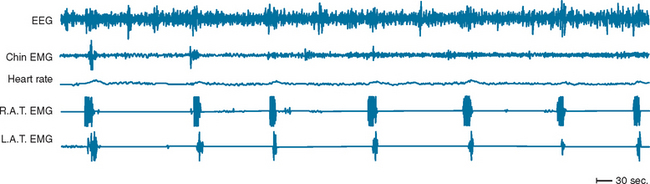
PLMSs are commonly recorded on overnight polysomnography, and population estimates of the prevalence of PLMSs exceeding five per hour range from 11% to 58%.32 PLMSs are more commonly recorded in elderly persons, in patients taking antidepressants, and in a number of medical conditions (end-stage renal disease, congestive heart failure, diabetes) and neurological or sleep disorders (obstructive sleep apnea, narcolepsy, Parkinson’s disease, multiple sclerosis). Although approximately 80% of individuals with RLS demonstrate PLMS, only a small proportion of those with PLMS describe symptoms of RLS. Controversy exists regarding the clinical importance of PLMS for sleep quality or daytime alertness; some studies show a lack of correlation between PLMS index and subjective or objective sleep quality or daytime sleepiness, and others show some mild associations.33
There is substantial evidence that PLMSs are associated with dopaminergic dysregulation at either spinal or higher central nervous system levels. Dopaminergic antagonists can produce PLMS,34 whereas dopaminergic agonists are extremely effective in reducing PLMS.35 Disorders characterized by dopaminergic deficiency (e.g., narcolepsy, rapid eye movement [REM] sleep behavior disorder [RBD]) are accompanied by high rates of PLMS. Functional imaging of the brain has demonstrated small but consistent reductions in dopaminergic function in PLMS. Finally, dopaminergic metabolites have been observed to be correlated with the number of PLMSs.36 The presence of PLMS in quadriplegic patients suggests that the motor programs for these movements exist in the spinal cord and are somehow disinhibited in patients with excessive movements during sleep.
Treatment of periodic limb movement disorder begins with an accurate diagnosis and proceeds to consideration of eliminating potential precipitating or exacerbating agents (e.g., antidepressants). PLMS can be dramatically reduced with the addition of dopaminergic agents, at least within the context of RLS. However, there is some suggestion that EEG arousals may persist even with elimination of the manifest motor activity. For this reason, coadministration of substitution of a benzodiazepine has also been advocated. Although studies of triazolam in patients with PLMS did not reveal a reduction in the periodic limb movement index, improvements in leg movements associated with arousal, sleep architecture, and daytime alertness were all demonstrated,37 even after 12 weeks of nightly use.38 Use of clonazepam in small numbers of patients was effective in reducing the number of PLMs, as well as improving scores on sleep continuity measures.39
EXCESS DAYTIME SLEEPINESS
Excess Daytime Sleepiness as a Result of Medical and Neurological Diseases
Multiple neurological diseases can cause sleepiness: either by disrupting the mechanisms involved in sleep homeostasis or by simply disrupting nighttime sleep. For example, cerebral traumatic injury or thalamic lesions (such as bilateral medial thalamic infarcts) can impair the central mechanisms of sleep-wake regulation, while pain from diabetic neuropathy of multiple sclerosis can cause sleep fragmentation and thus result in excessive sleepiness. Some specific examples are described as follows.
Stroke
Common Comorbid Conditions
One common cause of excessive sleepiness in the general population is sleep apnea. This condition is also quite common in patients with stroke.40 Symptoms of sleepiness and snoring may in fact be associated with higher risk of first-ever stroke.41,42 Prevalence after stroke may be even higher: Harbison and associates42 reported that up to 94% of patients had a respiratory disturbance index of 10 or above on polysomnography, performed in the 2 weeks after a stroke. Patients more likely to have more severe sleep apnea were older and more likely to have lacunar infarcts and greater prestroke disability. Sleep-disordered breathing improved over time, but about 72% of the patients had clinically important sleep apnea 6 weeks later.
As good-quality sleep may improve recovery from illness, treatment of sleep apnea can also hasten recovery from stroke. Patients with sleep apnea may have more residual symptoms of stroke after rehabilitation,43 whereas treatment of sleep apnea, when present in a patient with stroke, may hasten the rehabilitation process.44
Role of Specific Vascular Lesions
Sleepiness after stroke is common.45–47 Hemispheric stroke can result in insomnia, hypersomnia, or sleep disruption, but most EEG changes are transient.46,48,49 There can be alterations of sleep architecture, including REM sleep, especially within the first 3 days after the event.49 Increased slow-wave activity may be seen in the contralateral hemisphere.46 Consolidated sleep and high sleep efficiency are likely to herald a good clinical outcome.48,49
Rare alterations of sleep architecture include REM sleep abnormalities. For example, there are reports of dream loss with bilateral posterior cerebral artery infarcts50 and lesions of the pontine tegmentum can lead to absence of REM sleep,51 as well as to hypersomnia.52
Multiple Sclerosis
Impaired Sleep as a Result of Pain, Spasticity, or Nocturia
Spasticity may be associated with nocturnal pain and consequently sleep fragmentation. Muscle relaxants can effectively improve sleep. Because urinary symptoms are common in multiple sclerosis, nocturia can also fragment sleep. Treatment with desmopressin may be effective in reducing the nocturnal voids by 31% to 54% and, in one study, increased the initial sleep period or mean maximum period of uninterrupted sleep by approximately 2 hours.53
Associated Psychiatric Disorders
Many patients with multiple sclerosis have associated depressive or other psychiatric symptoms.54–56 These symptoms may vary in intensity, depending on the short-term risk of disability or wheelchair dependence.56 Because both depression and anxiety are associated with sleep disturbance, they can contribute to sleep impairments in patients with multiple sclerosis.
Immunological Factors
Immunological factors, which are involved in the pathogenesis of multiple sclerosis, may also have somnogenic effects. These include interleukin-1,57 which is known to be associated with sleepiness. Fatigue may be more prominent in patients who have markers of immune activation, including inductors of lymphocyte B cells, increase in helper T cells, interleukin-2 receptor cells, or other markers.54
Impaired Sleep-Wake Regulation as a Result of Plaques
Because demyelination can involve various pathways involved in the regulation of sleep and wakefulness, it would be logical to expect an independent effect of the focal dysfunction, depending on the location of the multiple sclerosis plaques. Indeed, case reports have suggested some such effects. For example, Oka and colleagues58 reported signs of narcolepsy associated with multiple sclerosis. Plazzi and Montagna59 reported RBD as a first symptom of multiple sclerosis. However, plaque location and burden are variable, and most sleep problems are multifactorial.
Medication Effect
Most muscle relaxants have sedating properties. Although their use at night improves sleep continuity, daytime use may be associated with undesirable sleepiness. This effect is more pronounced at the beginning of treatment, and some tolerance may develop over time. Pain management, when necessary, may lead to further sedation. Steroid treatment may be associated with decreased slow-wave sleep, increased sleep onset latency, and increased wakefulness after sleep onset.60 Certain sleep architecture abnormalities, resembling the ones seen in depression (shorter REM latency and increase in REM density), have also been reported.61 Interferon treatment may also be associated with increased somnolence.62,63
Treatment
In most cases, treatment should be targeted to the cause of sleep disruption. For example, treatment of the urinary frequency will probably improve sleep continuity as a result of fewer episodes of nocturia. Relief of spasticity and pain with gabapentin or baclofen may improve sleep as well.64 Most muscle relaxants can help additionally with sleep onset and continuity through their sedating properties.
When sleepiness continues despite optimal treatment of the underlying symptoms, the addition of modafinil can safely and effectively improve vigilance.65,66
Parkinson’s Disease
Patients with Parkinson’s disease frequently report sleepiness. This is reported on standardized instruments67,68 and also confirmed in standardized laboratory tests of sleepiness (i.e., a multiple sleep latency test), as well as a maintenance of wakefulness test. These impairments are correlated with poor sleep.69 Sleepiness should be distinguished from fatigue, which is also prevalent among patients with Parkinson’s disease.70,71 Longer disease duration, as well as anticholinergic medications, are associated with especially impaired sleep.72
Sleep Disruption from Parkinson’s Disease Itself
One cause of sleep fragmentation in Parkinson’s disease is the degeneration of dopaminergic neurons in the central nervous system. Because dopamine is involved in the regulation of the sleep-wake cycle, dopamine depletion, as well as dopaminergic stimulation in a dopamine-depleted state, may lead to sleepiness. In addition, alterations of levels of the hypothalamic peptide hypocretin, which have been reported to be extremely low or absent in narcolepsy, have been implicated in the pathogenesis of sleepiness in Parkinson’s disease, because hypocretin may have a role in the dopamine release mechanism.73,74 In addition, reports have implicated genetic polymorphism in D2 receptors in sleep attacks.75
Vivid Dreams
Vivid dreams are another potential cause of sleep disruption. This phenomenon may be more prevalent with the use of dopamine agonists and may be more likely to occur in patients who also have hallucinations. In a 6-year prospective study, patients with hallucinations had similar sleep patterns as did those without hallucinations, but vivid dreams were associated with significantly poorer sleep.76
RBD Sleep Behavior Disorder
RBD is common in patients with Parkinson’s disease. For example, Schenck and associates77 reported that 38% of the patients with Parkinson’s disease develop RBD. Possible causes in both disorders include loss of striatal dopamine transporters78 and resulting abnormal muscle tone, including loss of REM atonia.
Treatment
The first step of treatment is control of sleep-disrupting factors. In patients with refractory sleepiness, modafinil can be helpful to control residual sleepiness with minimal side effects.79 Counseling about driving may be appropriate as well, because episodes of irresistible sleepiness may occur, not preceded by obvious warning.
Other Neurological Diseases
Multisystem Atrophy
Multisystem atrophy may be associated with a high prevalence of apnea, mainly central.80,81 In addition, patients may have laryngeal stridor, which may lead to vocal cord paralysis and risk for sudden death.82–84 Depending on the clinical circumstances, the patients may require assisted ventilation or surgical procedures.
Dystonia
Sleep alterations with cervical dystonia are correlated with the severity of the disease, especially frequency of spasms.85,86 Spasms can persist in sleep even in the absence of EEG arousals but become progressively less severe with sleep depth.
Cerebellar Atrophy
Among patients with spinocerebellar ataxia, sleep complaints seem most common in spinocerebellar ataxia type 3.87 Contributing factors include higher frequencies of neuropathy and RLS with this form. Thus, treatment includes pain relief in the case of neuropathy and dopamine agonists or gabapentin if RLS is present.
Poliomyelitis and Postpolio Syndrome
Among patients with postpolio syndrome, the incidence of both obstructive and central apneas during sleep is higher than that in the general population. These disturbances are more prominent in patients who have had respiratory involvement during the initial illness.88 Thus, treatment is targeted at treatment of the sleep breathing disorder.
Cervical Myelopathy
Patients with cervical myelopathy have a higher prevalence of respiratory disturbances during sleep. In a study of 50 randomly selected tetraplegic patients, 55% of the men and 20% of the women had a respiratory disturbance index of 5 or higher.89 Mid- and low cervical lesions may also lead to delayed apneas, and these can sometimes be very severe.90 In an isolated case, anterior spinal artery syndrome led to continuous central apneas during sleep.91 Additional problems may include bradycardia, with or without hypotension.91 Thus, treatment should involve careful evaluation and treatment of sleep breathing disturbance (e.g., with continuous positive airway pressure).
Other causes of sleep impairment in these patients involve neurological deficits, pain, spasticity, and injury to the pathways involved in melatonin secretion. A study of patients with tetraplegia caused by cervical and upper thoracic injuries demonstrated near absence of melatonin in the patients with cervical lesions.92 To date, however, there are no reports of successful treatment with exogenous melatonin in these patients.
Dementia
Dementia can be the result of various conditions, including neurodegenerative, vascular, infectious, and other causes. The most common form is Alzheimer’s disease. Patients may have reduced sleep efficiency and increased number of arousals, and the severity of these findings tends to parallel that of the dementia itself. Abnormalities of sleep architecture, such as increased REM latency and decreased slow-wave sleep, have also been reported, but these findings are less consistent. Circadian rhythm abnormalities are also seen, discussed in more detail in the section “Circadian Rhythm Disorders.”
Epilepsy
Effects of Seizures on Sleep
Sleepiness is common among patients with epilepsy. As in other neurological conditions, sleepiness is multifactorial, secondary to the effects of sleep fragmentation from the disorder itself, as well as from effects of antiepileptic medications, most of which have sedative properties (Table 15-4). Nocturnal seizures can be associated with sleep fragmentation, and arousal or awakening may occur before or after the event. Frequently, temporal lobe seizures occur after awakening, and frontal lobe seizures occur during sleep. However, the causal relationship is still debated.
TABLE 15-4 Medications Associated with Sleepiness and Insomnia
| Medications Associated with Sleepiness | Medications Associated with Insomnia |
|---|---|
| Anticonvulsants | β Blockers |
| Dopamine agonists or precursors | Steroids |
| Interferons | SSRIs |
| SSRIs | Antimigraine medications (especially ones containing caffeine) |
| Tricyclic antidepressants | |
| Opiates | |
SSRI, selective serotonin reuptake inhibitor.
Effects of Sleep on Seizures
Diurnal and nocturnal variations in seizure rate have led to examinations of the relationship of seizures to sleep. In two prospective studies, researchers examined the distribution of seizures in relation to sleep stage and depth. Both Herman and colleagues in 200193 and Minecan and coworkers in 200294 reported that of all sleep stages, non-REM sleep, especially stage 2 sleep, is associated with the highest proportion of seizures.
Occurrence of seizures during sleep (versus wakefulness) may depend on epileptogenic region. A study of intracranial recordings in patients with TLE revealed a tendency toward arousal before a seizure in almost all of the patients.95 However, patients with frontal lobe epilepsy tended to have seizures during sleep, and these were usually not associated with abrupt arousal from sleep.93,96
Effects of Sleep on Interictal Discharges
Interictal discharges distinguish patients with epilepsy from healthy individuals. Interictal discharges are not evenly distributed through all sleep stages. Multiple studies have reported a higher rate of interictal discharges during stages 3 and 4 sleep than in stages 1 and 2 sleep in patients with TLE97,98 and those with generalized epilepsy.99,100 The effect of sleep stage on interictal discharges in patients with TLE was robust: most patients had a higher interictal discharge rate in deep non-REM sleep, and this rate was up to nine times higher than the interictal discharge rate during wakefulness. A caveat to this interpretation is that sleep preferentially occurs at specific circadian times; therefore, it is possible that these sleep-related effects are at least partially caused by an underlying circadian rhythm in interictal discharge propensity.
Seizures and Circadian Rhythm
Pavlova and associates101 analyzed data from 26 consecutive patients with confirmed TLE or other localization-related epilepsy. To test for any systematic day/night pattern in seizure frequency, they divided the 24-hour period into six 4-hour “bins” and compared the proportion of seizures across bins. In the TLE group, there was a clear peak in the time of occurrence of seizures: 50% occurred between the hours of 15:00 and 19:00 (see Fig. 15-1). In the patients with other localization-related epilepsies, there was a peak in seizure frequency between the hours of 19:00 and 23:00.
NARCOLEPSY
Clinically, REM dysregulation in narcolepsy is characterized by the inappropriate appearance of the REM phenomena muscle paralysis and dreams during wakefulness or at the sleep-wake transition. Cataplexy is a sudden onset of muscle atonia in the antigravity and facial muscles, resulting in falls, difficulty holding objects, or twitching of agonist and antagonist muscles in these areas. Cataplexy is usually stimulated by laughter, telling a joke, anger, surprise, or other emotional processes. Sleep paralysis is the appearance of REM atonia in wakefulness, leading to brief (seconds to minutes), usually frightening inability to move voluntary musculature in the presence of full alertness, either on awakening or at the transition from wakefulness to sleep. Concomitant paralysis of the accessory muscles of inspiration may result in the sensations of dyspnea. Hypnogogic (at sleep onset) or hypnopompic (at awakening) hallucinations is the appearance of the hallucinatory phenomena of dreams during wakefulness. Usually these are fragmentary and brief (hearing the telephone or one’s name being called) or seeing a shadow of a person, although in rare cases they may be more elaborate. Although these REM phenomena are occasionally reported in isolation by individuals without narcolepsy, they are common, and frequently observed as a group, in this disorder. Narcolepsy is currently classified as existing with or without cataplexy. The exact percentage of individuals with the excess daytime sleepiness of narcolepsy who also have cataplexy is unclear but is thought to be 50% to 80% of cases.
Major progress in the understanding of the pathophysiology of narcolepsy has been made since the mid-1990s, stimulated by findings derived from molecular biology. A mutation in the gene that codes for the receptor for the hypothalamic peptide hypocretin was determined to be responsible for the Doberman pinscher model of narcolepsy.102 At the same time, Chemelli and associates103 found that when the gene for the same peptide (which they called orexin) was knocked out in mice, the mice exhibited behavioral states consistent with narcolepsy. In humans, the dramatic reduction in hypothalamic neurons responsible for the production of hypocretin (orexin)104 and the absence of this ligand in the cerebrospinal fluid105 of narcoleptic patients have confirmed the importance of hypocretin (orexin) in human narcolepsy with cataplexy. The excess expression of the specific human leukocyte antigen genotype DQB1*0602 in individuals with narcolepsy (85% of narcoleptics versus 25% of the general population) is suggestive of an immunological etiology of narcolepsy. However, neither immunological abnormalities nor antigenic targets have been identified in human narcolepsy.
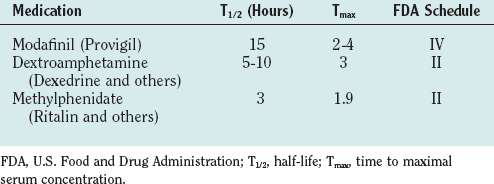
Treatment of narcolepsy is directed independently for the daytime sleepiness and REM dysregulation (Table 15-5). It is essential to stress the importance of adequate nocturnal sleep and the value of daytime napping, if feasible, as means of minimizing excess daytime sleepiness in narcolepsy. Stimulant medications, which have been available since the 1950s, have been the traditional mainstay of narcolepsy pharmacological treatment. These medications both release the catecholamines norepinephrine and dopamine and block their reuptake into their releasing neurons, enhancing their effects. They are effective in promoting wakefulness in narcolepsy, allowing a more normal level of professional and social functioning. In addition, controlled-release preparations of methylphenidate and amphetamines have been developed, allowing once- to twice-per-day dosing. However, there are persistent concerns regarding their potential for abuse and the not uncommon side effects of headache, anorexia, mood alterations, and blood pressure and pulse elevations. First-line treatment of excess daytime sleepiness has become modafinil, a long-acting agent that only partially acts on the dopaminergic system, and thus has substantially less risk of abuse, and that has fewer sympathomimetic side effects.
Treatment of the REM dysregulation–related symptoms (principally cataplexy) is achieved with REM suppressants. Tricyclic antidepressants, which once had a primary role in treatment, have been replaced by the better tolerated and safer selective serotonin reuptake inhibitors (Tables 15-6 and 15-7). Both of these classes of medications suppress cataplexy, sleep paralysis, and hynogogic hallucinations. Cataplexy, which does not respond to these agents, may be successfully treated with γ-hydroxybutyrate, a short-acting sedating medication that is given twice during the night and has demonstrated benefit in reducing daytime cataplectic attacks, as well as daytime sleepiness.
| Non-REM Parasomnias | REM-Related Parasomnias | |
|---|---|---|
| Stage of arousal | II, III, IV | REM |
| Time of night | First third | Any time |
| EEG with event | N.A. | Characteristic of REM |
| EMG with event | Low | High, variable |
| Relative unresponsiveness during event | Yes | Yes |
| Autonomic activity | Low (confusional arousal) | High |
| High (sleep terror) | ||
| Amnesia | Yes | No |
| Confusion after episode | Yes | No |
| Family history of parasomnias | Yes | No |
EEG, electroencephalography; EMG, electromyography; N.A., not applicable; REM, rapid eye movement.
TABLE 15-7 Pharmacological Treatment of Parasomnias
| Drug | Dosage |
|---|---|
| Non-REM Parasomnias | |
| Triazolam | 0.125-0.5 mg |
| Zolpidem | 5-10 mg |
| Lorazepam | 1-2 mg |
| Clonazepam | 0.5-2.0 mg |
| REM-Related Parasomnias | |
| Clonazepam | 0.5-2.0 mg |
| Lorazepam | 1.0-2.0 mg |
| Melatonin | 3-15 mg |
| Pramipexole | 0.5-1.0 mg |
REM, rapid eye movement.
IDIOPATHIC HYPERSOMNIA
A number of less common causes of excess daytime sleepiness are recognized. Principal among these is idiopathic hypersomnolence; patients experience the excess daytime sleepiness of narcolepsy but do not have any of the REM-related symptoms. Individuals report normal nocturnal sleep (in contrast to narcolepsy) but severe difficulty arousing from sleep in the morning or from daytime naps. These naps are longer than the ones in patients with narcolepsy and may take 2 to 3 hours. Even after these long naps, patients are only partially refreshed. Diagnosis is made by polysomnography and results of the multiple sleep latency test. Overnight polysomnography demonstrates high sleep efficiency, and the multiple sleep latency test reveals pathologically shortened sleep latency (similar to narcolepsy), but without the appearance of REM periods during daytime naps. Because medical and neurological disorders (described previously) can lead to excess daytime sleepiness, this is generally a diagnosis of exclusion. As suggested by its name, the cause of idiopathic hypersomnolence is unknown.
KLEINE-LEVIN SYNDROME
The Kleine-Levin syndrome is characterized by periodic, sudden-onset episodes of hypersomnia, compulsive hyperphagia, and hypersexuality, lasting from a few days to a few weeks, with complete remission in between. Various other behavioral disturbances may occur during the episodes. The cause and pathogenesis of Kleine-Levin syndrome remain unknown. It is more common in men, but female patients have been described as well, and the ratio is probably 4:1.106–108 When seen in young women, it can have a catamenial pattern.108 Diagnosis is made on the presence of the classic triad after other causes of excessive sleepiness are ruled out. Treatment can include stimulants or modafinil for hypersomnolence and possibly lithium salts.109,110
IDIOPATHIC RECURRENT STUPOR
Idiopathic recurring stupor is a syndrome of spontaneous stupor or coma that is not associated with known metabolic, toxic, or structural abnormalities. Electroencephalograms can be characterized by fast (14- to 16-Hz), nonreactive background activity.111 Plasma and cerebrospinal fluid may show a marked increase in a benzodiazepine-like endogenous substance, endozepine-4.111,112 Flumazenil, a benzodiazepine receptor antagonist, may promptly resolve the syndrome.
CIRCADIAN RHYTHM DISORDERS
Circadian Rhythm Effects on Normal and Abnormal Neurophysiological Functions
Normal neurophysiological functions are affected by the circadian system independently of sleep or wakefulness state and time awake. Subjective alertness, cognitive performance, and short-term memory are lowest close to the time of the temperature minimum, or the “biological night” (e.g., see Johnson et al,74 and Dijk et al113). Mood in healthy subjects is also modulated by a nonadditive interaction between the sleep-wake cycle and the circadian phase, and this modulation may be implicated in mood disorders.114
Disorders of the Circadian Rhythm
Delayed Sleep Phase Syndrome
Delayed sleep phase syndrome (DSPS) is a disorder of the phase relationships between the desired sleep times and the circadian system manifesting as a tendency to fall asleep much later than desired and awakening later than the desired time. As a result, these patients frequently come to medical attention with complaints of insomnia. DSPS is an especially frequent cause of insomnia in the young adult.115
Diagnosis
The International Classification of Sleep Disorders1 has established the following “minimal criteria” for diagnosis: (1) The patient is unable to initiate sleep at the desired time and difficulty awakening; (2) timing of the habitual sleep episode is delayed (late); (3) symptoms are present for 1 month or more; (4) when constraints permit (e.g., when not working or attending classes), the patient opts for delayed timing of the major sleep episode, which is believed to be of good quality and quantity, and can awaken from this sleep episode without difficulty and remains on this delayed sleep-wake schedule without difficulty; and (5) subjective sleep data (e.g., sleep-wake diary) for 2 weeks or more verify the presence of the delayed, habitual sleep-wake schedule.
Treatment
The most powerful factors that entrain the circadian rhythm are (1) light, which provides information about the time of the “day,” and (2) melatonin, which provides information about the time of “night.” On the basis of these, several major approaches have been proposed:
Advanced Sleep Phase Syndrome
In advanced sleep phase syndrome, as in DSPS, the “biological night” of the patient is believed to be “locked” in an adverse time in relation to the desired bedtime but occurs hours earlier rather than later. This disturbance is more frequent among older individuals. Occasional familial forms exist as well.125–127 Like that of DSPS, diagnosis is based on clinical history and can be confirmed by sleep diary or objective measures, such as wrist actigraphy. Treatment options are similar to those for DSPS. Phototherapy, as evening bright light, at 2000 to 2500 lux in the hours between 8:00 and 11:00 p.m. for 2 to 3 hours, can be used.123 However, the effectiveness of bright light has been questioned in one study.128
Non–24-Hour Rhythm Disturbance
Light is the primary stimulus that synchronizes the endogenous circadian rhythm with the environmental conditions. It is perceived by the retinal cells, and the signal is transmitted to the circadian pacemaker, the suprachiasmatic nucleus, via the retinohypothalamic tract. Because the circadian period tends to be close to but slightly longer than 24 hours, daily exposure to light ensures that sleep occurs during the night time for most people. Impairment or absence of light perception can lead to disruption or lack of synchronization between the circadian sleep promoting mechanisms and the scheduled time to sleep. This can occur in blind people. Klein and colleagues129 described such a cause of insomnia in a blind person, confirming that sleep was most likely to occur close to the subject’s temperature minimum.
Jet Lag
A similar desynchronization between the environmental night and the “biological night” occurs during travel across time zones in a short period of time; this desynchronization is commonly known as jet lag. Like non–24-hour rhythm disturbance, it can manifest as insomnia, difficulty with concentration, or sleepiness and could be logically expected to modify disorders that have a circadian pattern. Typically, adjustment to eastbound travel is found to be more difficult. Treatment can include phase advance before travel,130,131 use of bright light, or use of melatonin.132,133
PARASOMNIAS
The term parasomnia is derived from the Latin para, meaning “next to,” and somnus, referring to sleep. In the International Classification of Sleep Disorders, 2nd edition, parasomnias are defined as “undesirable physical or experiential events that accompany sleep.”1
Non-REM Parasomnias
The understanding of non-REM parasomnias is based on the concept that arousal from sleep is not an all-or-none phenomenon but rather a continuum of alertness, judgment, and control over behavior. Behaviors or affective expression can occur during full or partial sleep states, which are at least partially divorced from full awareness, both during the event and on awakening. Most commonly, such behaviors are dissociated motor activities (walking, eating, sexual behavior) or emotional responses (fear, anger, sexual excitement).134 They are distinct from waking behavior in that complex mentation is usually not present, feedback from the environment is usually given less salience, and sound judgment is usually not present. It is unclear to what extent these behaviors or emotional states are related to waking motivation, psychological state, or psychopathology. It is clear that these behaviors run in families.135 Phenotypically, they share many features: They are commonly brief, are more frequently expressed in children, are associated with amnesia, and occur in the first 1 to 2 hours of sleep, usually arising during slow-wave sleep. Non-REM parasomnias are best conceptualized along a continuum of emotional/motoric/autonomic arousal, in which confusional arousals have the least arousal and sleep terrors the most.
Confusional arousals are usually brief, simple, motor behaviors, which usually occur without substantial affective expression. Mental confusion with automatic behavior, indistinct speech, and relative unresponsiveness to the environment are hallmarks of a confusional arousal.136 Sitting up in bed with simple vocalization or picking at bedclothes are common examples. If interrupted by family members, responses may be absent, incomplete, or inappropriate.
Sleepwalking involves more elaborate behavior than simple confusional arousals, but it forms a continuum with the latter. Simple motivations without substantial emotional involvement, such as attempts to use the bathroom, go to the kitchen, or, in some cases, leave the home, are usually pursued. Although the walker’s eyes are open, behavior may be clumsy.137,138 Dreaming is usually not present, and individuals (if awakened) report only simple mentation. As in confusional arousals, sleepwalkers usually return to sleep, but if aroused by family members or as a result of their inappropriate behavior, sleep inertia may be present. In rare cases, individuals may become agitated if sleepwalking episodes are interrupted.
Sleep terrors have many of the properties of other non-REM parasomnias but are characterized by more intense autonomic, motor, and affective expression (and experience). In children, sleep terrors are classically heralded by a piercing scream, with extreme fear, crying, and inconsolability.139 In adults, agitation is common, frequently with the belief that there is an imminent threat, with the requirement of escape or defense.140 For this reason, sleep terror sufferers may cause injury to themselves, to others, or to property in their highly agitated state. As in sleepwalking, dreaming is usually not reported, but simple thoughts are present (“The room is on fire” or “I am being attacked”), which can be difficult to dispel, even after the sufferer has awakened. They may incorporate an individual into the threatening scenario if they are interfered with, potentially harming that individual. For this reason, it is recommended that individuals experiencing a sleep terror be gently redirected in an attempt to raise their level of consciousness.
Non-REM parasomnia variants have also been identified in adults: excessive sleep inertia (or “sleep drunkenness”),141 abnormal sleep-related sexual behavior (“sexsomnia”),142 and sleep-related violence.143
Amnesia for non-REM parasomnias is often so dense that without a bed partner’s or parent’s report, or evidence from the episode, these episodes might go unnoticed. Epidemiological information is therefore unreliable. In view of this caveat, approximately 10% to 20% of children and 2% to 5% of adults report a history of confusional arousals.136 Sleepwalking occurs in 10% to 20% of children and 1% to 4% of adults.136,144 Sleep terrors are less common than sleepwalking; approximately 5% of children and 1% to 2% of adults report a history of such events.136 In approximately 80% of adults with sleepwalking, this parasomnia is a continuation of a childhood behavior, although many such persons do not come to medical attention until their 20s or 30s. There is a wide range of sleepwalking frequency; most sleepwalkers present with only occasional episodes, although those who frequently sleepwalk are the ones who usually come to medical attention.
The expression of all non-REM parasomnias appears to depend on a genetic predisposition combined with a precipitating event, which may be endogenous (e.g., respiratory obstructive event, pain, leg movement of sleep) or exogenous (e.g., forced awakening or environmental disruption).144,145 In predisposed individuals, sleep deprivation, medications, sleep disorders, stress, and circadian misalignment may all aggravate or expose this underlying parasomnia. It is unclear why such partial arousals are more common in children. Nevertheless, genetic factors in non-REM parasomnias are evidenced by both epidemiological studies and studies of twins.145,146 Risk of sleepwalking is approximately doubled if one parent has a sleepwalking history and tripled when both parents have such a history. There do not appear to be gender or racial differences in the prevalence rates of these parasomnias.
Even in individuals with frequent episodes, parasomnia episodes are often not observed in the sleep laboratory.147 Sleep studies, however, are often performed in such patients (particularly in an adult with new-onset sleepwalking) to determine whether there are potential precipitating events occurring during sleep, such as a sleep-related breathing disorder, PLMSs, nocturnal seizures, or RBD. When they are observed, the electroencephalogram may show delta waves (characteristic of slow-wave sleep), theta or alpha activity, or alternation between sleep and waking activity.148
There is an unclear relationship between psychiatric disorders and non-REM parasomnias.134 Although childhood sleepwalking does not appear to be associated with psychiatric disorders, a variety of psychiatric disorders may increase the risk of persistent sleepwalking into adulthood.136,149 However, it is not believed that sleepwalking represents latent psychopathology.150 Nonetheless, psychiatric medications may raise the risk of sleepwalking, because of their sleep-disruptive or sleep-enhancing properties.151 Similarly, stress, sleep deprivation,152 and chaotic sleep schedules may increase the risk of sleepwalking, and each of these precipitants may be more common in the psychiatric patient.
Treatment of Non-REM Parasomnias
The decision to treat non-REM parasomnias is based on the frequency of the event, the risk of associated injury to self or others, and the distress the behavior is causing the patient or family members.136 Fortunately, for the majority of adult sufferers, parasomnias occur infrequently, but unfortunately, their appearance is unpredictable. Therefore, the decision to treat must be carefully considered, particularly when the sleepwalker engages in high-risk behaviors.
When treatment of sleepwalking or sleep terrors in an adult is warranted, a three-step approach is used: modification of predisposing and precipitating factors, enhancing safety of the sleeping environment, and, when these are not successful, pharmacotherapy. Sleep disorders (e.g., sleep apnea, PLMSs), symptoms of medical disorders (pain, nocturia, dyspnea), or medications that are thought to be contributing to sleep instability should be modified to the extent possible. As described previously, the safety of the environment should be maximized. The majority of data on the treatment of non-REM parasomnias exist for clonazepam (0.5 to 1.0 mg one hour before bed), which has been used successfully for sleepwalking and sleep terrors for extended periods without the development of tolerance in most patients.153 However, if the parasomnia occurs within the first half of the sleep period, short-acting benzodiazepine receptor agonists such as triazolam (0.125 to 0.25 mg) or zolpidem (5 to 10 mg) are recommended, to minimize daytime carryover effects. It is unclear whether these medications work by suppressing arousals during sleep or decreasing slow-wave sleep, and no controlled trials testing their efficacy have been performed. However, because of favorable clinical experience, they are first-line agents in the treatment of these disorders.
REM Sleep Behavior Disorder
RBD is characterized by pathological appearance of the normal features of REM sleep. In RBD, the usual atonia of REM sleep is absent; this allows the sleeper to enact dreams, which, when agitated or violent, can result in injury to the sleeper or bed partner.154 During such episodes, the sleeper’s eyes are closed, and the sleeper is unresponsive to the environment until awakened, at which point he or she achieves rapid and full alertness and reports a dream that usually corresponds to the exhibited behavior. It is this agitation and/or injury that bring the patient to medical attention, usually at the behest of the bed partner. Episodes of full-blown RBD are intermittent, but sleeptalking, shouting, vivid dreams, or fragmentary motor activity may commonly occur between such events.
RBD is a chronic disorder, usually observed in men older than 50 and in individuals with certain neurological disorders. In particular, RBD is often present in individuals with α-synucleinopathies (Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy).155 RBD may also be a heralding symptom of neurological illness: In one study, two-thirds of patients with RBD monitored for 10 years developed Parkinson’s disease.156 RBD may also be precipitated by treatment with serotonergic antidepressants.
An animal model of RBD, in which lesions around the locus ceruleus produced “REM sleep without atonia” was developed well before the discovery of RBD and implicates these brainstem areas in the control of motor activity in REM sleep.157 In patients with RBD, dopamine transporter abnormalities in the nigrostriatal system have been demonstrated.158 Similarly, a reduction in neurons around the locus ceruleus has been seen.159 However, more widespread central nervous system dysfunction is suggested by data showing slowing of the EEG pattern during wakefulness as well as subtle neuropsychological dysfunction in patients with idiopathic RBD.160
The diagnosis of RBD is made by polysomnography, which demonstrates elevated muscle tone or excessive phasic muscle activity in the submental and anterior tibialis electromyogram during REM sleep.1 At times, body movements are manifest during REM on sleep study. Excess PLMSs may also be observed during both REM and non-REM sleep. Otherwise, polysomnography findings are generally normal.
First-line treatment of RBD consists of benzodiazepine receptor agonists. The most commonly used agent is clonazepam (0.5 to 1.0 mg), which has been shown to substantially decrease the number and extent of pathological dream-enacting behaviors.153 In general, the medication is well tolerated for this indication; however, because of the age of most of the patients with RBD and the long half-life of clonazepam, excess daytime sleepiness and/or cognitive impairments may occur. In this case, shorter acting benzodiazepines (e.g., lorazepam, 1 to 2 mg) may be used. Other medications, particularly melatonin (3 to 15 mg one hour before bed)161 and pramipexole (0.5 to 1.0 mg one hour before bed), have also been used with some success.162 These alternatives are appropriate for patients for whom a benzodiazepine is associated with cognitive or motor side effects or is contraindicated because of substance abuse. Certainly, removal of potentially offending medications, such as antidepressants, should be attempted if clinically possible. In addition, as with the non-REM parasomnias, safety of the sleeping environment for both the patient and the bed partner is essential.
Krystal AD, Walsh JK, Laska E, et al. Sustained efficacy of eszopiclone over 6 months of nightly treatment: results of a randomized, double-blind, placebo-controlled study in adults with chronic insomnia. Sleep. 2003;26:793-799.
Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991-997.
Riemann D, Voderholzer U. Primary insomnia: a risk factor to develop depression? J Affect Disord. 2003;76:255-259.
Ripley B, Overeem S, Fujiki N, et al. CSF. hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology. 2001;57:2253-2258.
Schenck CH, Bundlie SR, Mahowold MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder. Neurology. 1996;46:388-393.
1 American Academy of Sleep Medicine. International Classification of Sleep Disorders: Diagnostic and Coding Manual, 2nd ed. Rochester, MN: American Academy of Sleep Medicine, 2005.
2 Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6:97-111.
3 Katz DA, McHorney CA. The relationship between insomnia and health-related quality of life in patients with chronic illness. J Fam Pract. 2002;51:229-235.
4 Foley DJ, Monjan A, Simonsick EM, et al. Incidence and remission of insomnia among elderly adults: an epidemiologic study of 6800 persons over three years. Sleep. 1999;22:S366-S372.
5 Dodge R, Cline MG, Quan SF. The natural history of insomnia and its relationship to respiratory symptoms. Arch Intern Med. 1995;155:1797-1800.
6 Klink ME, Quan SF, Kaltenborn WT, et al. Risk factors associated with complaints of insomnia in a general adult population. Influence of previous complaints of insomnia. Arch Intern Med. 1992;152:1634-1637.
7 Foley DJ, Monjan AA, Brown SL, et al. Sleep complaints among elderly persons: an epidemiologic study of three communities. Sleep. 1995;18:425-432.
8 Ohayon MM, Roth T. What are the contributing factors for insomnia in the general population? J Psychosom Res. 2001;51:745-755.
9 Riemann D, Voderholzer U. Primary insomnia: a risk factor to develop depression? J Affect Disord. 2003;76:255-259.
10 Léger D, Scheuermaier K, Philip P, et al. SF-36: evaluation of quality of life in severe and mild insomniacs compared with good sleepers. Psychosom Med. 2001;63:49-55.
11 Avidan AY, Fries BE, James ML, et al. Insomnia and hypnotic use, recorded in the minimum data set, as predictors of falls and hip fractures in Michigan nursing homes. J Am Geriatr Soc. 2005;53:955-962.
12 Simon GE, VonKorff M. Prevalence, burden, and treatment of insomnia in primary care. Am J Psychiatry. 1997;154:1417-1423.
13 Bonnet MH, Arand DL. Hyperarousal and insomnia. Sleep Med Rev. 1997;1:97-108.
14 Drake C, Richardson G, Roehrs T, et al. Vulnerability to stress-related sleep disturbance and hyperarousal. Sleep. 2004;27:285-291.
15 Tang NK, Harvey AG. Effects of cognitive arousal and physiological arousal on sleep perception. Sleep. 2004;27:69-78.
16 Vgontzas AN, Bixler EO, Lin HM, et al. Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: clinical implications. J Clin Endocrinol Metab. 2001;86:3787-3794.
17 Bonnet MH, Arand DL. Situational insomnia: consistency, predictors, and outcomes. Sleep. 2003;26:1029-1036.
18 Nofzinger EA, Buysse DJ, Germain A, et al. Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry. 2004;161:2126-2128.
19 Krystal AD, Edinger JD, Wohlgemuth WK, et al. NREM sleep EEG frequency spectral correlates of sleep complaints in primary insomnia subtypes. Sleep. 2002;25:630-640.
20 Perlis ML, Giles DE, Mendelson WB, et al. Psychophysiological insomnia: the behavioural model and a neurocognitive perspective. J Sleep Res. 1997;6:179-188.
21 Vitiello MV, Moe KE, Prinz PN. Sleep complaints cosegregate with illness in older adults: clinical research informed by and informing epidemiological studies of sleep. J Psychosom Res. 2002;53:555-559.
22 Ford DE, Kamerow DB. Epidemiologic study of sleep disturbances and psychiatric disorders. An opportunity for prevention? JAMA. 1989;262:1479-1484.
23 Sateia MJ, Doghramji K, Hauri PJ, et al. Evaluation of chronic insomnia. An American Academy of Sleep Medicine review. Sleep. 2000;23:243-308.
24 Morin CM, Culbert JP, Schwartz SM. Nonpharmacological interventions for insomnia: a meta-analysis of treatment efficacy. Am J Psychiatry. 1994;151:1172-1180.
25 Edinger JD, Glenn DM, Bastian LA, et al. Sleep in the laboratory and sleep at home II: comparisons of middle-aged insomnia sufferers and normal sleepers. Sleep. 2001;24:761-770.
26 Walsh JK. Pharmacologic management of insomnia [Review]. J Clin Psychiatry. 2004;65(Suppl 16):41-45.
27 National Institutes of Health. State-of-the-science conference statement: manifestations and management on chronic insomnia in adults. Available at: http://consensus.nih.gov/ta/026/InsomniaDraftStatement061505.pdf. (accessed March 9, 2006).
28 Mohler H, Fritschy J, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2-8.
29 Nowell PD, Mazumdar S, Buysse DJ, et al. Benzodiazepines and zolpidem for chronic insomnia: a meta-analysis of treatment efficacy. JAMA. 1997;278:2170-2177.
30 Holbrook AM, Crowther R, Lotter A, et al. Meta-analysis of benzodiazepine use in the treatment of insomnia. CMAJ. 2000;162:225-233.
31 Krystal AD, Walsh JK, Laska E, et al. Sustained efficacy of eszopiclone over 6 months of nightly treatment: results of a randomized, double-blind, placebo-controlled study in adults with chronic insomnia. Sleep. 2003;26:793-799.
32 Morrish E, King MA, Pilsworth SN, et al. Periodic limb movement in a community population detected by a new actigraphy technique. Sleep Med. 2002;3:489-495.
33 Carrier J, Frenette S, Montplaisir J, et al. Effects of periodic leg movements during sleep in middle-aged subjects without sleep complaints. Mov Disord. 2005;20:1127-1132.
34 Cohrs S, Rodenbeck A, Guan Z, et al. Sleep-promoting properties of quetiapine in healthy subjects. Psychopharmacology (Berl). 2004;174:421-429.
35 Montplaisir J, Nicolas A, Denesle R, et al. Restless legs syndrome improved by pramipexole: a double-blind randomized trial. Neurology. 1999;52:938-943.
36 Cohrs S, Guan Z, Pohlmann K, et al. Nocturnal urinary dopamine excretion is reduced in otherwise healthy subjects with periodic leg movements in sleep. Neurosci Lett. 2004;360:161-164.
37 Doghramji K, Browman CP, Gaddy JR, et al. Triazolam diminishes daytime sleepiness and sleep fragmentation in patients with periodic leg movements in sleep. J Clin Psychopharmacol. 1991;11:284-290.
38 Bonnet MH, Arand DL. Chronic use of triazolam in patients with periodic leg movements, fragmented sleep and daytime sleepiness. Aging (Milano). 1991;3:313-324.
39 Peled R, Lavie P. Double-blind evaluation of clonazepam on periodic leg movements in sleep. J Neurol Neurosurg Psychiatry. 1987;50:1679-1681.
40 Parish JM, Somers VK. Obstructive sleep apnea and cardiovascular disease. Mayo Clin Proc. 2004;79:1036-1046.
41 Davies DP, Rodgers H, Walshaw D, et al. Snoring, daytime sleepiness and stroke: a case-control study of first-ever stroke. J Sleep Res. 2003;12:313-318.
42 Harbison J, Ford GA, James OF, et al. Sleep-disordered breathing following acute stroke. Q J Med. 2002;95:741-747.
43 Cherkassky T, Oksenberg A, Froom P, et al. Sleep-related breathing disorders and rehabilitation outcome of stroke patients: a prospective study. Am J Phys Med Rehabil. 2003;82:452-455.
44 Kaneko Y, Hajek VE, Zivanovic V, et al. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep. 2003;26:293-297.
45 De Groot MH, Phillips SJ, Eskes GA. Fatigue associated with stroke and other neurologic conditions: implications for stroke rehabilitation. Arch Phys Med Rehabil. 2003;84:1714-1720.
46 Muller C, Achermann P, Bischof M, et al. Visual and spectral analysis of sleep EEG in acute hemispheric stroke. Eur Neurol. 2002;48:164-171.
47 Bliwise DL, Rye DB, Dihenia B, et al. Greater daytime sleepiness in subcortical stroke relative to Parkinson’s disease and Alzheimer’s disease. J Geriatr Psychiatry Neurol. 2002;15:61-67.
48 Vock J, Achermann P, Bischof M, et al. Evolution of sleep and sleep EEG after hemispheric stroke. J Sleep Res. 2002;11:331-338.
49 Bassetti CL, Aldrich MS. Sleep electroencephalogram changes in acute hemispheric stroke. Sleep Med. 2001;2:185-194.
50 Bischof M, Bassetti CL. Total dream loss: a distinct neuropsychological dysfunction after bilateral PCA stroke. Ann Neurol. 2004;56:583-586.
51 Gironell A, de la Calzada MD, Sagales T, et al. Absence of REM sleep and altered non-REM sleep caused by a haematoma in the pontine tegmentum. J Neurol Neurosurg Psychiatry. 1995;59:195-196.
52 Arpa J, Rodriguez-Albarino A, Izal E, et al. Hypersomnia after tegmental pontine hematoma: case report. Neurologia. 1995;10:140-144.
53 Cvetkovic RS, Plosker GL. Desmopressin: in adults with nocturia. Drugs. 2005;65:99-107.
54 Irarte J, Subira M, Castro PL. Modalities of fatigue in multiple sclerosis: correlation with clinical and biologic factors. Mult Scler. 2000;6:124-130.
55 Janssens AC, van Doorn PA, de Boer JB, et al. Impact of recently diagnosed multiple sclerosis on quality of life, anxiety, depression and distress of patients and partners. Acta Neurol Scand. 2003;108:389-395.
56 Janssens AC, van Doorn PA, de Boer JB, et al. Perception of prognostic risk in patients with multiple sclerosis: the relationship with anxiety, depression, and disease-related distress. J Clin Epidemiol. 2004;57:180-186.
57 Rothwell NJ, Luheshi GN. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 2000;23:618-625.
58 Oka Y, Kanbayashi T, Mezaki T, et al. Low CSF hypocretin-1/orexin-A associated with hypersomnia secondary to hypothalamic lesion in a case of multiple sclerosis. J Neurol. 2004;251:885-886.
59 Plazzi G, Montagna P. Remitting REM sleep behavior disorder as the initial sign of multiple sclerosis. Sleep Med. 2002;3:437-439.
60 Vgontzas AN, Chrousos GP. Sleep, the hypothalamic-pituitary-adrenal axis, and cytokines: multiple interactions and disturbances in sleep disorders. Endocrinol Metab Clin North Am. 2002;31:15-36.
61 Antonijevic IA, Steiger A. Depression-like changes of the sleep-EEG during high dose corticosteroid treatment in patients with multiple sclerosis. Psychoneuroendocrinology. 2003;28:780-795.
62 Kimura M, Majde JA, Toth LA, et al. Somnogenic effects of rabbit and recombinant human interferons in rabbits. Am J Physiol. 1994;267:R53-R61.
63 Munschauer FE3rd, Kinkel RP. Managing side effects of interferon-beta in patients with relapsing-remitting multiple sclerosis. Clin Ther. 1997;19:883-893.
64 Mueller ME, Gruenthal M, Olson WL, et al. Gabapentin for relief of upper motor neuron symptoms in multiple sclerosis. Arch Phys Med Rehabil. 1997;78:521-524.
65 Zifko UA, Rupp M, Schwarz S, et al. Modafinil in treatment of fatigue in multiple sclerosis. Results of an open-label study. J Neurol. 2002;249:983-987.
66 Rammohan KW, Rosenberg JH, Lynn DJ, et al. Efficacy and safety of modafinil (Provigil) for the treatment of fatigue in multiple sclerosis: a two centre phase 2 study. J Neurol Neurosurg Psychiatry. 2002;72:150.
67 Johns M. Daytime sleepiness, snoring, and obstructive sleep apnea. The Epworth sleepiness scale. Chest. 1993;103:30-36.
68 Tan EK, Lum SY, Fook-Chong SMC, et al. Evaluation of somnolence in Parkinson’s disease: comparison with age- and sex-matched controls. Neurology. 2002;58:465-468.
69 Stevens S, Cormella CL, Stepanski EJ. Daytime sleepiness and alertness in patients with Parkinson disease. Sleep. 2004;27:967-972.
70 Alves G, Wentzel-Larsen T, Larsen JP. Is fatigue an independent and persistent symptom in patients with Parkinson disease? Neurology. 2004;63:1908-1911.
71 Serrano C, Garcia-Borreguero D. Fluctuations in cognition and alertness in Parkinson’s disease and dementia. Neurology. 2004;63:S31-S34.
72 Nausieda PA, Weiner WJ, Kaplan LR, et al. Sleep disruption in the course of chronic levodopa therapy: an early feature of the levodopa psychosis. Clin Neuropharmacol. 1982;5:183-194.
73 Ripley B, Overeem S, Fujiki N, et al. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology. 2001;57:2253-2258.
74 Johnson MP, Duffy JF, Dijk DJ, et al. Short-term memory, alertness and performance: a reappraisal of their relationship to body temperature. J Sleep Res. 1992;1:24-29.
75 Rissling I, Geller F, Brandmann O, et al. Dopamine receptor gene polymorphisms in Parkinson’s disease patients reporting “sleep attacks.”. Mov Disord. 2004;19:1279-1284.
76 Goetz CG, Wuu J, Curgian LM, et al. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology. 2005;64:81-86.
77 Schenck CH, Bundlie SR, Mahowold MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder. Neurology. 1996;46:388-393.
78 Eisensehr I, Linke R, Tatsch K, et al. Increased muscle activity during rapid eye movement sleep correlates with decrease of striatal presynaptic dopamine transporters. IPT and IBZM SPECT imaging in subclinical and clinically manifest idiopathic REM sleep behavior disorder, Parkinson’s disease, and controls. Sleep. 2003;26:507-512.
79 Hogl B, Saletu M, Brandauer E, et al. Modafinil for the treatment of daytime sleepiness in Parkinson’s disease: a double-blind, randomized, crossover, placebo-controlled polygraphic trial. Sleep. 2002;25:905-909.
80 Guilleminault C, Lehrman K, Forno L, et al. Sleep apnoea syndrome: states of sleep and autonomic dysfunction. J Neurol Neurosurg Psychiatry. 1977;40:718-725.
81 Castaigne P, Laplane D, Autret A, et al. [Shy-Drager syndrome with disturbances of the respiratory rhythm and consciousness. A propos of an anatomo-clinical case]. Rev Neurol. 1977;113:455-466.
82 Munschauer FE, Loh L, Bannister R, et al. Abnormal respiration and sudden death during sleep in multiple system atrophy with autonomic failure. Neurology. 1990;40:677-679.
83 Isozaki E, Hayashi M, Hayashida T, et al. [Vocal cord abductor in multiple system atrophy—paradoxical movement of vocal cords during sleep]. Rinsho Shinkeigaku. 1996;36:529-533.
84 Sadaoka T, Kakitsuba N, Fujiwara Y, et al. Sleep-related breathing disorders in patients with multiple system atrophy and vocal fold palsy. Sleep. 1996;19:479-484.
85 Sforza E, Montagna P, Defazio G, Lugaresi E. Sleep and cranial dystonia. Electroencephalogr Clin Neurophysiol. 1991;79:166-169.
86 Silvestri R, De Domenico P, Di Rosa AE, et al. The effect of nocturnal physiological sleep on various movement disorders. Mov Disord. 1990;5:8-14.
87 Schols L, Haan J, Riess O, et al. Sleep disturbance in spinocerebellar ataxias: is the SCA3 mutation a cause of restless legs syndrome? Neurology. 1998;51:1603-1607.
88 Steljes DG, Kryger MH, Kirk BW, et al. Sleep in postpolio syndrome. Chest. 1990;98:133-140.
89 Stockhammer E, Tobon A, Michel F, et al. Characteristics of sleep apnea syndrome in tetraplegic patients. Spinal Cord. 2002;40:286-294.
90 Lu K, Lee TC, Liang CL, et al. Delayed apnea in patients with mid- to lower cervical spinal cord injury. Spine. 2000;25:1332-1338.
91 Manconi M, Mondini S, Fabiani A, et al. Anterior spinal artery syndrome complicated by the Ondine curse. Arch Neurol. 2003;60:1787-1790.
92 Kneisley LW, Moskowitz MA, Lynch HG. Cervical spinal cord lesions disrupt the rhythm in human melatonin excretion. J Neural Transm Suppl. 1978;13:311-323.
93 Herman ST, Walczak TS, Bazil CW. Distribution of partial seizures during the sleep-wake cycle: differences by seizure onset site. Neurology. 2001;56:1453-1459.
94 Minecan D, Natarajan A, Marzec M, et al. Relationship of epileptic seizures to sleep stage and sleep depth. Sleep. 2002;25:899-904.
95 Malow BA, Kushwala R, Lin X, et al. Relationship of interictal epileptiform discharges to sleep depth in partial epilepsy. Electroencephalogr Clin Neurophysiol. 1997;102:20-26.
96 Crespel A, Baldy-Moulinier M, Coubes P. The relationship between sleep and epilepsy in frontal and temporal lobe epilepsies: practical and physiopathologic considerations. Epilepsia. 1998;39:150-157.
97 Malow BA, Lin X, Kushwaha R, et al. Interictal spiking increases with sleep depth in temporal lobe epilepsy. Epilepsia. 1998;39:1309-1316.
98 Sammaritano M, Gigli GL, Gotman J. Interictal spiking during wakefulness and sleep and the localization of foci in temporal lobe epilepsy. Neurology. 1991;41:290-297.
99 Ross JJ, Johnson LC, Walter RD. Spike and wave discharges during stages of sleep. Arch Neurol. 1966;14:399-407.
100 Sato S, Dreifus FE, Penry JK. The effect of sleep on spike-wave discharges in absence seizures. Neurology. 1973;23:1335-1345.
101 Pavlova MK, Shea SA, Bromfield EB. Day/night patterns of focal seizures. Epilepsy Behav. 2004;5:44-49.
102 Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365-376.
103 Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437-451.
104 Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991-997.
105 Nishino S, Ripley B, Overeem S, et al. Low cerebrospinal fluid hypocretin (orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol. 2001;50:381-388.
106 Mayer G, Leonhard E, Krieg J, et al. Endocrinological and polysomnographic findings in Kleine-Levin syndrome: no evidence for hypothalamic and circadian dysfunction. Sleep. 1998;21:278-284.
107 Billiard M, Guilleminault C, Dement WC. A menstruation-linked periodic hypersomnia. Kleine-Levin syndrome or new clinical entity? Neurology. 1975;25:436-443.
108 Kesler A, Gadoth N, Vainstein G, et al. Kleine-Levin syndrome (KLS) in young females. Sleep. 2000;23:1-5.
109 Muratori F, Bertini N, Masi G. Efficacy of lithium treatment in Kleine-Levin syndrome. Eur Psychiatry. 2002;17:232-233.
110 Visscher F, Smit LM, Smith F, et al. The Kleine-Levin syndrome. Tijdschr Kindergeneeskd. 1989;57:218-221.
111 Tinuper P, Montagna P, Plazzi G, et al. Idiopathic recurring stupor. Neurology. 1994;44:621-625.
112 Rothstein JD, Guidotti A, Tinuper P, et al. Endogenous benzodiazepine receptor ligands in idiopathic recurring stupor. Lancet. 1992;340:1002-1004.
113 Dijk DJ, Duffy JF, Czeisler CA. Circadian and sleep/wake dependent aspects of subjective alertness and cognitive performance. J Sleep Res. 1992;1:112-117.
114 Boivin D. Influence of sleep-wake and circadian rhythm disturbances in psychiatric disorders. J Psychiatry Neurosci. 2000;25:446-458.
115 Okawa M, Uchiyama M, Ozaki S, et al. Circadian rhythm sleep disorders in adolescents: clinical trials of combined treatments based on chronobiology. Psychiatry Clin Neurosci. 1998;52:483-490.
116 Czeisler CA, Richardson GS, Coleman RM, et al. Chronotherapy: resetting the circadian clocks of patients with delayed sleep phase insomnia. Sleep. 1981;4:1-21.
117 Dijk DJ, Beersma DG, Daan S, et al. Bright morning light advances the human circadian systems without affecting the NREM sleep homeostasis. Am J Physiol. 1989;256:R100-R111.
118 Czeisler CA, Kronauer RE, Allan JS, et al. Bright light induction of strong (type 0) resetting of the human circadian pacemaker. Science. 1989;244:1328-1333.
119 Minors DS, Waterhouse JM, Wirz-Justice A. A human phase-response curve to light. Neurosci Lett. 1991;133:36-40.
120 Morris M, Lack L, Dawson D. Sleep-onset insomniacs have delayed temperature rhythms. Sleep. 1990;13:1-14.
121 Rosenthal NE, Joseph-Vanderpool JR, Levendosky AA, et al. Phase-shifting effects of bright morning light as treatment for delayed sleep phase syndrome. Sleep. 1990;13:354-361.
122 Chesson ALJr, Littner M, Davila D, et al. Practice parameters for the use of light therapy in the treatment of sleep disorders. Sleep. 1999;22:641-660.
123 Dahlitz M, Alvarez B, Vignau J, et al. Delayed sleep phase syndrome response to melatonin. Lancet. 1991;337:1121-1124.
124 Dagan Y, Yovel I, Hallis D, et al. Evaluating the role of melatonin in the long-term treatment of delayed sleep phase syndrome (DSPS). Chronobiol Int. 1998;15:181-190.
125 Reid KJ, Chang AM, Dubocovich ML, et al. Familial advanced sleep phase syndrome. Arch Neurol. 2001;58:1089-1094.
126 Zucconi M. Familial advanced sleep phase syndrome. Sleep Med. 2002;3:177-178.
127 Satoh K, Mishima K, Inoue Y, et al. Two pedigrees of familial advanced sleep phase syndrome in Japan. Sleep. 2003;26:416-417.
128 Palmer CR, Kripke DF, Savage HCJr, et al. Efficacy of enhanced evening light for advanced sleep phase syndrome. Behav Sleep Med. 2003;1:213-226.
129 Klein T, Martens H, Dijk DJ, et al. Circadian sleep regulation in the absence of light perception: chronic non-24-hour circadian rhythm sleep disorder in a blind man with a regular 24-hour sleep-wake schedule. Sleep. 1993;16:333-343.
130 Eastman CI, Gazda CJ, Burgess HJ, et al. Advancing circadian rhythms before eastward flight: a strategy to prevent or reduce jet lag. Sleep. 2005;28:33-44.
131 Burgess HJ, Crowley SJ, Gazda CJ, et al. Preflight adjustment to eastward travel: 3 days of advancing sleep with and without morning bright light. J Biol Rhythms. 2003;18:318-328.
132 Oxenkrug GF, Requintina PJ. Melatonin and jet lag syndrome: experimental model and clinical implications. CNS Spectr. 2003;8:139-148.
133 Herxheimer A, Waterhouse J. The prevention and treatment of jet lag. BMJ. 2003;326:296-297.
134 Schenck CH, Mahowald MW. Parasomnias. Managing bizarre sleep-related behavior disorders. Postgrad Med. 2000;107:145-156.
135 Mahowald MW. Parasomnias. Med Clin North Am. 2004;88:669-678.
136 Ohayon MM, Guilleminault C, Priest RG. Night terrors, sleepwalking, and confusional arousals in the general population: their frequency and relationship to other sleep and mental disorders. J Clin Psychiatry. 1999;60:268-276.
137 Kavey NB, Whyte J, Resor SRJr, et al. Somnambulism in adults. Neurology. 1990;40:749-752.
138 Crisp AH. The sleepwalking/night terrors syndrome in adults. Postgrad Med J. 1996;72:599-604.
139 Mehlenbeck R, Spirito A, Owens J, et al. The clinical presentation of childhood partial arousal parasomnias. Sleep Med. 2000;1:307-312.
140 Schenck CH, Boyd JL, Mahowald MW. A parasomnia overlap disorder involving sleepwalking, sleep terrors, and REM sleep behavior disorder in 33 polysomnographically confirmed cases. Sleep. 1997;20:972-981.
141 Roth B, Nevsimalova S, Rechtschaffen A. Hypersomnia with “sleep drunkenness.”. Arch Gen Psychiatry. 1972;26:456-462.
142 Shapiro CM, Trajanovic NN, Fedoroff JP. Sexsomnia—a new parasomnia? Can J Psychiatry. 2003;48:311-317.
143 Cartwright R. Sleepwalking violence: a sleep disorder, a legal dilemma, and a psychological challenge [Review]. Am J Psychiatry. 2004;161:1149-1158.
144 Laberge L, Tremblay RE, Vitaro F, et al. Development of parasomnias from childhood to early adolescence. Pediatrics. 2000;106:67-74.
145 Hublin C, Kaprio J, Partinen M, et al. Parasomnias: cooccurrence and genetics. Psychiatr Genet. 2001;11:65-70.
146 Hublin C, Kaprio J, Partinen M, et al. Prevalence and genetics of sleepwalking: a population-based twin study. Neurology. 1997;48:177-181.
147 Broughton RJ. Sleep disorders: disorders of arousal? Enuresis, somnambulism, and nightmares occur in confusional states of arousal, not in “dreaming sleep.”. Science. 1968;159:1070-1078.
148 Gaudreau H, Joncas S, Zadra A, et al. Dynamics of slow-wave activity during the NREM sleep of sleepwalkers and control subjects. Sleep. 2000;23:755-760.
149 Gau SF, Soong WT. Psychiatric comorbidity of adolescents with sleep terrors or sleepwalking: a case-control study. Aust N Z J Psychiatry. 1999;33:734-739.
150 Hartman D, Crisp AH, Sedgwick P, et al. Is there a dissociative process in sleepwalking and night terrors? Postgrad Med J. 2001;77:244-249.
151 Landry P, Warnes H, Nielsen T, et al. Somnambulistic-like behaviour in patients attending a lithium clinic. Int Clin Psychopharmacol. 1999;14:173-175.
152 Joncas S, Zadra A, Paquet J, et al. The value of sleep deprivation as a diagnostic tool in adult sleepwalkers. Neurology. 2002;58:936-940.
153 Schenck CH, Mahowald MW. REM sleep parasomnias. Neurol Clin. 1996;14:697-720.
154 Schenck CH, Mahowald MW. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25:120-138.
155 Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61:40-45.
156 Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46:388-393.
157 Hendricks JC, Morrison AR, Farnbach GL, et al. A disorder of rapid eye movement sleep in a cat. J Am Vet Med Assoc. 1981;178:55-57.
158 Eisensehr I, Linke R, Noachtar S, et al. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson’s disease and controls. Brain. 2000;123:1155-1160.
159 Turner RS, D’Amato CJ, Chervin RD, et al. The pathology of REM sleep behavior disorder with comorbid Lewy body dementia. Neurology. 2000;55:1730-1732.
160 Gagnon JF, Fantini ML, Bedard MA, et al. Association between waking EEG slowing and REM sleep behavior disorder in PD without dementia. Neurology. 2004;62:401-406.
161 Boeve BF, Silber MH, Ferman TJ. Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: results in 14 patients. Sleep Med. 2003;4:281-284.
162 Fantini ML, Gagnon JF, Filipini D, et al. The effects of pramipexole in REM sleep behavior disorder. Neurology. 2003;61:1418-1420.






