Chapter 119 Primary Defects of Cellular Immunity
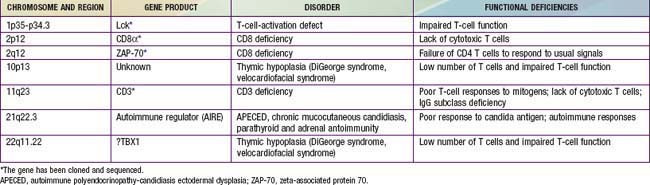
In general, patients with defects in T-cell function have infections or other clinical problems that are more severe than in patients with antibody deficiency disorders (see Table 116-4). The defective gene products for some primary T-cell diseases are identified (Table 119-1). These individuals rarely survive beyond infancy or childhood. Transplantation of thymic tissue, or of major histocompatibility complex (MHC)-compatible sibling or haploidentical (half-matched) parental hematopoietic stem cell, is the treatment of choice for patients with primary T-cell defects (Chapter 129).
Thymic Hypoplasia (DIgeorge Syndrome)
Thymic hypoplasia results from dysmorphogenesis of the 3rd and 4th pharyngeal pouches during early embryogenesis, leading to hypoplasia or aplasia of the thymus and parathyroid glands. Other structures forming at the same age are also frequently affected, resulting in anomalies of the great vessels (right-sided aortic arch), esophageal atresia, bifid uvula, congenital heart disease (conotruncal, atrial, and ventricular septal defects), a short philtrum of the upper lip, hypertelorism, an antimongoloid slant to the eyes, mandibular hypoplasia, and low-set, often notched ears (Chapters 76 and 102). The diagnosis is often first suggested by hypocalcemic seizures during the neonatal period.
Genetics and Pathogenesis
DiGeorge syndrome occurs in both males and females. Microdeletions of specific DNA sequences from chromosome 22q11.2, the DiGeorge chromosomal region (DGCR), are found in a majority of cases. Several candidate genes have been identified in this region. A T-box transcription family member, TBX1, has been implicated as an etiology for most of the major signs of DGS. There appears to be an excess of 22q11.2 deletions of maternal origin. Polymerase chain reaction (PCR)-based genotyping using microsatellite DNA markers located within the commonly deleted region permits rapid detection of such microdeletions. Conotruncal heart defects and 22q deletions are observed in DiGeorge syndrome, velocardiofacial syndrome (VCFS), and conotruncal anomaly face syndrome (CTAFS). The CATCH 22 syndrome (cardiac, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia) includes the broad clinical spectrum of conditions with 22q11.2 deletions. Other deletions associated with DiGeorge and velocardiofacial syndromes have been identified on chromosome 10p13 (Chapter 76).
Defective Expression of the T-Cell Receptor–CD3 Complex (Ti-CD3)
The first type of this disorder was found in two brothers in a Spanish family. The proband presented with severe infections and died at 31 mo of age with autoimmune hemolytic anemia and viral pneumonia. His lymphocytes had responded poorly to mitogens and to anti-CD3 in vitro and could not be stimulated to develop cytotoxic T cells. His antibody responses to protein antigens had been normal, indicating normal T-helper-cell function. His 12 yr old brother was healthy but had almost no CD3-bearing T cells and had IgG2 deficiency similar to his sibling. The defect in this family was due to mutations in the gene encoding the CD3γ chain (Fig. 119-1).
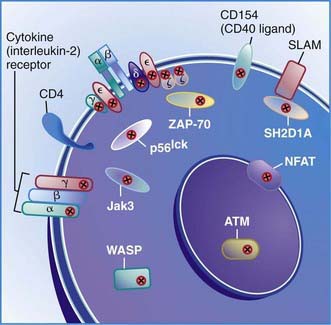
The second type of this disorder was diagnosed in a 4 yr old French boy who had recurrent Haemophilus influenzae pneumonia and otitis media in early life but is now healthy. He had a partial defect in expression of Ti-CD3, and thus the percentage of CD3 cells was about half-normal, but the level of expression is markedly decreased. The defect was shown to be due to two independent CD3ε gene mutations, leading to defective CD3ε chain synthesis. There was a splice site mutation on one allele that did not totally abrogate the normal intron 7 splicing, thus there was partial expression of CD3 on the T cells. His T cells did not proliferate normally in response to anti-CD3 or anti-CD2, but did respond normally to stimulation with anti-CD28 or antigens, such as tetanus toxoid. Thus, this mutation did not result in failure of T-cell development, whereas mutations in the portions of the gene that encode the extracellular component of CD3ε result in a profound deficiency of circulating mature CD3 T cells (Chapter 120).
CD8 Lymphocytopenia Due to Mutations in the Gene Encoding Zeta-Associated Protein 70 (ZAP-70)
Patients with this T-cell activation defect present during infancy with severe, recurrent, and often fatal infections. The majority of cases are reported among Mennonites. These patients have normal or elevated numbers of blood B cells and low to elevated serum immunoglobulin concentrations. Their blood lymphocytes exhibit normal expression of the T-cell surface antigens CD3 and CD4, but CD8 cells are almost totally absent. These cells fail to respond to mitogens or to allogeneic cells in vitro or to generate cytotoxic T lymphocytes. NK cell activity is normal. The thymus of one patient exhibited normal architecture with normal numbers of CD4:CD8 double-positive thymocytes but an absence of CD8 single-positive thymocytes. This condition is due to mutations in the gene encoding zeta-associated protein 70 (ZAP-70), a non-src family protein tyrosine kinase important in T-cell signaling that is localized to chromosome 2q12 (see Fig. 119-1). The normal number of CD4:CD8 double-positive T cells results because the thymocytes can use the other member of the same tyrosine kinase family, Syk, to facilitate positive selection. Syk is present at fourfold higher levels in thymocytes than in peripheral T cells, possibly accounting for the lack of normal responses by the CD4 blood T cells.
Autoimmune Polyendocrinopathy-Candidiasis Ectodermal Dysplasia (Apeced)
Most pediatric patients are identified by the presence of mucocutaneous candidiasis and hypoparathyroidsim and later develop insidious signs of Addison disease (Chapter 565).
Al-Tamemi S, Mazer B, Mitchell D, et al. Complete DiGeorge anomaly in the absence of neonatal hypocalcemia and velofacial and cardiac defects. Pediatrics. 2005;116:e457-e460.
Davis CM, Kancherla VS, Reddy A, et al. Development of specific T-cell responses to Candida and tetanus antigens in partial DiGeorge syndrome. J Allergy Clin Immunol. 2008;122:1194-1199.
Driscoll DA, Sullivan KE. DiGeorge syndrome: a chromosome 22q11.2 deletion syndrome. In: Ochs HD, Smith, CIE, Puck JM. Primary immunodeficiency diseases: a molecular and genetic approach. New York: Oxford University Press; 2007:485-495.
Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776-794.
Gennery AR, Slatter MA, Rice J, et al. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol. 2008;153:75-80.
Haerynick F, Holland SM, Rosenzweig SD, et al. Disseminated Mycobacterium avium infection in a patient with a novel mutation in the interleukin-12 receptor β1 chain. J Pediatr. 2008;153:721-722.
Jean-Philippe P, Freedman A, Chang MW, et al. Severe varicella caused by varicella-vaccine strain in a child with significant T-cell dysfunction. Pediatrics. 2007;120:e1345-e1349.
Lankisch TO, Jaeckel E, Strassburg CP. The autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy or autoimmune polyglandular syndrome type 1. Semin Liver Dis. 2009;29:307-314.
McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome-velocardiofacial syndrome). Medicine. 2011;90(1):1-18.
Perez EE, Bokszczanin A, McDonald-McGinn D, et al. Safety of live viral vaccines in patients with chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Pediatrics. 2003;112:e325-e327.
Rieux-Laucat F, Hivroz C, Lim A, et al. Inherited and somatic CD3ζ mutations in a patient with T-cell deficiency. N Engl J Med. 2006;18:1913-1921.
Roifman CM. Studies of patients’ thymi aid in the discovery and characterization of immunodeficiency in humans. Immunol Rev. 2005;203:143-155.
Rope AF, Cragun DL, Saal HM, et al. DiGeorge anomaly in the absence of chromosome 22q11.2 deletion. J Pediatr. 2009;155:560-565.
Shikama N, Nusspaumer G, Hollander GA. Clearing the AIRE: on the pathophysiological basis of the autoimmune polyendocrinopathy syndrome type-1. Endocrinol Metab Clin North Am. 2009;38:273-288. vii





