[level-membership-for-pediatrics-category]
Chapter 118 Primary Defects of Antibody Production
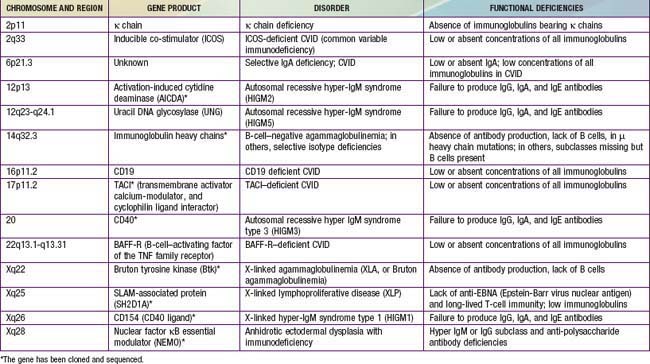
Of all of the primary immunodeficiency diseases, those affecting antibody production are most frequent. Selective absence of serum and secretory IgA is the most common defect, with rates ranging from 1/333 to 1/18,000 persons among different races. By contrast, agammaglobulinemia is estimated to occur with a frequency of only 1/10,000 to 1/50,000 persons. Patients with antibody deficiency are usually recognized because they have recurrent infections with encapsulated bacteria predominantly in the upper and lower respiratory tracts; some individuals with selective IgA deficiency or infants with transient hypogammaglobulinemia may have few or no infections (see Table 116-4). The defective gene products for many primary antibody deficiency disorders have been identified (Table 118-1) and localized (Fig. 118-1). Sometimes the defect is not in the B cell itself but in T cells, which are required for complete B-cell function.
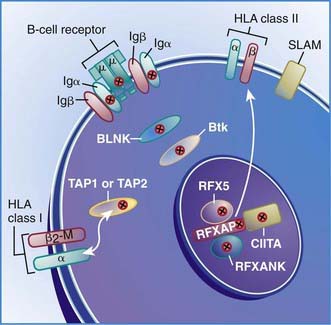
X-Linked Agammaglobulinemia (XLA)
Genetics and Pathogenesis
The abnormal gene in XLA maps to q22 on the long arm of the X chromosome and encodes the B-cell protein tyrosine kinase Btk (Bruton tyrosine kinase). Btk is a member of the Tec family of cytoplasmic protein tyrosine kinases and is expressed at high levels in all B-lineage cells, including pre–B cells. It appears to be necessary for pre–B-cell expansion and maturation into surface Ig-expressing B cells, but probably has a role at all stages of B-cell development; it has also been found in cells of the myeloid series. More than 500 different mutations in the human Btk gene are recognized; they encompass most parts of the coding portions of the gene. There is not a clear correlation between the location of the mutation and the clinical phenotype (Fig. 118-2). Carriers are detected by mutation analysis, and prenatal diagnosis of affected male fetuses is possible if the mutation is known in the family.
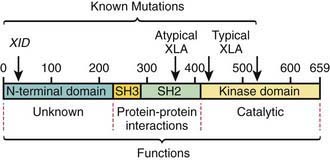
Six autosomal recessive defects have also been shown to result in agammaglobulinemia with an absence of circulating B cells (see Fig. 118-2), including mutations in the genes encoding: (1) the µ heavy chain gene; (2) the Igα and Igβ signaling molecules; (3) B-cell linker adaptor protein (BLNK); (4) the surrogate light chain, λ5/14.1; and (5) leucine-rich repeat-containing 8 (LRRC8).
Diagnosis
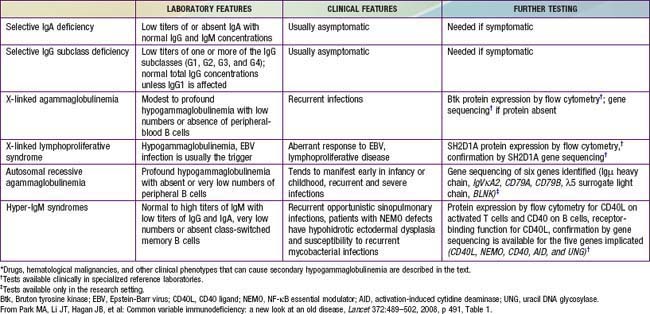
The diagnosis of XLA should be suspected if lymphoid hypoplasia is found on physical examination (minimal or no tonsillar tissue and no palpable lymph nodes), and serum concentrations of IgG, IgA, IgM, and IgE are far below the 95% confidence limits for appropriate age- and race-matched controls (Chapter 708), usually with total immunoglobulins <100 mg/dL. Levels of natural antibodies to type A and B red blood cell polysaccharide antigens (isohemagglutinins) and antibodies to antigens given during routine immunizations are abnormally low in this disorder, whereas they are normal in transient hypogammaglobulinemia of infancy. Flow cytometry is an important test to demonstrate the absence of circulating B cells, which will distinguish this disorder from common variable immunodeficiency, the hyper-IgM syndrome and transient hypogammaglobulinemia of infancy.
Common Variable Immunodeficiency
Common variable immunodeficiency (CVID) is a syndrome characterized by hypogammaglobulinemia with phenotypically normal B cells. It has also been called “acquired hypogammaglobulinemia” because of a generally later age of onset of infections. CVID patients may appear similar clinically to those with XLA in the types of infections experienced and bacterial etiologic agents involved, except that echovirus meningoencephalitis is rare in patients with CVID (Table 118-2). In contrast to XLA, the sex distribution in CVID is almost equal, the age of onset is later (although it may be present in infancy), and infections may be less severe.
Table 118-2 DIFFERENTIAL DIAGNOSIS FOR PATIENTS WITH SUSPECTED CVID (HYPOGAMMAGLOBULINEMIA WITH RECURRENT INFECTIONS)*
Hyper-IgM Syndrome
X-Linked Hyper-IgM Caused by Mutations in the CD40 Ligand: Hyper-IgM Type 1 (HIGM1)
X-Linked Hyper-IgM Caused by Mutations in the Gene Encoding Nuclear Factor κB (NF-κB) Essential Modulator (NEMO, OR IKKγ); XHM-ED
Autosomal Recessive Hyper-IgM Caused by Mutations in the Gene for Activation-Induced Cytidine Deaminase (AICDA): Hyper-IgM Type 2 (HIGM2)
Autosomal Recessive Hyper-IgM Caused by Mutations in the Gene for Uracil DNA Glycosylase (UNG); Hyper-IgM Type 5 (HIGM5)
Another cause of the hyper-IgM syndrome is a deficiency of uracil DNA glycosylase.
X-Linked Lymphoproliferative Disease
Genetics and Pathogenesis
The defective gene in XLP was localized to Xq25, cloned, and the gene product was initially named SAP (for SLAM-associated protein), but is now known officially as SH2D1A. SLAM (signaling lymphocyte activation molecule) is an adhesion molecule that is upregulated on both T and B cells with infection and other stimulation. SH2D1A is highly expressed in thymocytes and peripheral blood T and NK cells, with a prevalent expression on Th1 cells. Its presence on B lymphocytes is unclear. Thus, although antibody deficiency is frequently present, this is really a T- and NK-cell defect. SH2D1A competes with SHP-2 for binding to SLAM and, as such, is a regulatory molecule (see Fig. 119-1). In XLP patients, the absence of SH2D1A can lead to an uncontrolled cytotoxic T-cell immune response to EBV. The SH2D1A protein associates permissively with 2B4 on NK cells; thus, selective impairment of 2B4-mediated NK-cell activation also contributes to the immunopathology of XLP.
118.1 Treatment of B-Cell Defects
Fasth A, Nystrom J. Quality of life and health-care resource utilization among children with primary immunodeficiency receiving home treatment with subcutaneous human immunoglobulin. J Clin Immunol. 2008;28:370-378.
The Medical Letter: Subcutaneous immune globulin (SGIC). Med Lett. 2007;49:31-32.
Moore ML, Quinn JM. Subcutaneous immunoglobulin replacement therapy for primary antibody deficiency: advancements into the 21st century. Ann Allerg Asthma Immunol. 2008;101:114-121.
Aghamohammadi A, Mohammadi J, Parvaneh N, et al. Progression of selective IgA deficiency to common variable immunodeficiency. Int Arch Allergy Immunol. 2008;147:87-92.
Bonilla FA, Geha RS. Common variable immunodeficiency. Pediatr Res. 2009;65:13R-19R.
Durandy A, Taubenheim N, Peron S, et al. Pathophysiology of B-cell intrinsic immunoglobulin class switch recombination deficiencies. Adv Immunol. 2007;94:275-306.
Fried AJ, Bonilla FA. Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Rev. 2009;22:396-414.
Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776-794.
Lougaris V, Badolato R, Ferrari S, et al. Hyper immunoglobulin M syndrome due to CD40 deficiency: clinical, molecular, and immunological features. Immunol Rev. 2005;203:48-66.
Oksenhendler E, Gérard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 46, 2008. 1547–1154
Park MA, Li JT, Hagan JB, et al. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008;372:489-502.
Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308-316.
Salzer U, Bacchelli C, Buckridge S, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967-1976.
Urschel S, Kayikci L, Wintergerst U, et al. Common variable immunodeficiency disorders in children: delayed diagnosis despite typical clinical presentation. J Pediatr. 2009;154:888-894.
van Zelm MC, Reisli I, van der BM, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901-1912.
Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85:193-202.
Wood P, Stanworth S, Burton J, et al. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol. 2007;149:410-423.
Yong PF, Salzer U, Grimbacher B. The role of costimulation in antibody deficiencies: ICOS and common variable immunodeficiency. Immunol Rev. 2009;229:101-113.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 118 Primary Defects of Antibody Production
Of all of the primary immunodeficiency diseases, those affecting antibody production are most frequent. Selective absence of serum and secretory IgA is the most common defect, with rates ranging from 1/333 to 1/18,000 persons among different races. By contrast, agammaglobulinemia is estimated to occur with a frequency of only 1/10,000 to 1/50,000 persons. Patients with antibody deficiency are usually recognized because they have recurrent infections with encapsulated bacteria predominantly in the upper and lower respiratory tracts; some individuals with selective IgA deficiency or infants with transient hypogammaglobulinemia may have few or no infections (see Table 116-4). The defective gene products for many primary antibody deficiency disorders have been identified (Table 118-1) and localized (Fig. 118-1). Sometimes the defect is not in the B cell itself but in T cells, which are required for complete B-cell function.
X-Linked Agammaglobulinemia (XLA)
Genetics and Pathogenesis
The abnormal gene in XLA maps to q22 on the long arm of the X chromosome and encodes the B-cell protein tyrosine kinase Btk (Bruton tyrosine kinase). Btk is a member of the Tec family of cytoplasmic protein tyrosine kinases and is expressed at high levels in all B-lineage cells, including pre–B cells. It appears to be necessary for pre–B-cell expansion and maturation into surface Ig-expressing B cells, but probably has a role at all stages of B-cell development; it has also been found in cells of the myeloid series. More than 500 different mutations in the human Btk gene are recognized; they encompass most parts of the coding portions of the gene. There is not a clear correlation between the location of the mutation and the clinical phenotype (Fig. 118-2). Carriers are detected by mutation analysis, and prenatal diagnosis of affected male fetuses is possible if the mutation is known in the family.
Six autosomal recessive defects have also been shown to result in agammaglobulinemia with an absence of circulating B cells (see Fig. 118-2), including mutations in the genes encoding: (1) the µ heavy chain gene; (2) the Igα and Igβ signaling molecules; (3) B-cell linker adaptor protein (BLNK); (4) the surrogate light chain, λ5/14.1; and (5) leucine-rich repeat-containing 8 (LRRC8).
Diagnosis
The diagnosis of XLA should be suspected if lymphoid hypoplasia is found on physical examination (minimal or no tonsillar tissue and no palpable lymph nodes), and serum concentrations of IgG, IgA, IgM, and IgE are far below the 95% confidence limits for appropriate age- and race-matched controls (Chapter 708), usually with total immunoglobulins <100 mg/dL. Levels of natural antibodies to type A and B red blood cell polysaccharide antigens (isohemagglutinins) and antibodies to antigens given during routine immunizations are abnormally low in this disorder, whereas they are normal in transient hypogammaglobulinemia of infancy. Flow cytometry is an important test to demonstrate the absence of circulating B cells, which will distinguish this disorder from common variable immunodeficiency, the hyper-IgM syndrome and transient hypogammaglobulinemia of infancy.
Common Variable Immunodeficiency
Common variable immunodeficiency (CVID) is a syndrome characterized by hypogammaglobulinemia with phenotypically normal B cells. It has also been called “acquired hypogammaglobulinemia” because of a generally later age of onset of infections. CVID patients may appear similar clinically to those with XLA in the types of infections experienced and bacterial etiologic agents involved, except that echovirus meningoencephalitis is rare in patients with CVID (Table 118-2). In contrast to XLA, the sex distribution in CVID is almost equal, the age of onset is later (although it may be present in infancy), and infections may be less severe.
Table 118-2 DIFFERENTIAL DIAGNOSIS FOR PATIENTS WITH SUSPECTED CVID (HYPOGAMMAGLOBULINEMIA WITH RECURRENT INFECTIONS)*
