Chapter 106 Primary Bony Spinal Lesions
General Discussion
Primary tumors of the spine are extraordinarily uncommon. The incidence of primary spinal neoplasms has been estimated to be between 2.5 and 8.5 per 100,000 per year,1 the equivalent of an estimated 7500 new cases per year in the United States.2 Overall, primary spinal tumors are more common in men than women. Osteoid osteoma, osteoblastoma, osteochondroma, plasmacytoma, chordoma, and chondrosarcoma all occur more commonly in men at a nearly 2:1 ratio compared with women.2 In a review of 6221 bone tumors at the Mayo Clinic, Dahlin and Coventry3 found that less than 10% of all primary tumors involved the spine. In a recent, even more extensive review at the Mayo Clinic, Unni et al. (unpublished data, personal communication, 2000) reviewed 8091 skeletal bone tumors in patients who underwent surgery. Of these 8091 skeletal tumors distributed throughout the skeleton, 2334 were benign and 5757 were malignant. A further analysis of this group revealed that 510 tumors involving the spine were malignant and only 145 were benign. A more detailed grouping of the benign tumors is presented in Table 106-1. Not all patients with benign or malignant tumors undergo surgery. Therefore, these figures likely underestimate the true incidence of the disease.
| Number of patients with | ||
|---|---|---|
| Tumor Type | Tumors Involving the Spine | Benign Skeletal Tumors |
| Giant cell tumor | 32 | 574 |
| Osteoid osteoma | 30 | 332 |
| Osteoblastoma | 29 | 87 |
| Hemangioma | 28 | 109 |
| Osteochondroma | 19 | 748 |
| Chondroma | 5 | 290 |
| Chondroblastoma | 1 | 119 |
| Chondromyxoid fibroma | 1 | 45 |
| Neurilemmoma | 0 | 14 |
| Fibrous histiocytoma | 0 | 9 |
| Lipoma | 0 | 7 |
| Total | 145 | 2334 |
Only those patients undergoing surgery and who, therefore, have tissue available for pathologic review are included in this table.
From Unni KK et al: Unpublished data, personal communication, 2000.
The presenting symptoms of night pain, pain at rest, or progressive neurologic deficit should prompt the clinician to entertain the diagnosis of benign or malignant disease of the spine. The primary complaint of most patients with primary tumors of the spine is pain. In a recent review, more than 84% of the patients complained of pain, either localized to the back (60.2%) or radicular (24%). There was no apparent difference between the pain symptoms in patients with benign disease involving the spine and those with malignant disease involving the spine.4 Fifty-five percent of the patients with malignant spine tumors and 35% of the patients with benign tumors demonstrated objective evidence of neurologic deficits. It is postulated that with the advent and increased availability of MRI, the number of patients presenting with neurologic deficits will decrease secondary to earlier diagnosis.
The data presented in Table 106-1, contrasted with those in Table 106-2, demonstrate the relative frequency or infrequency of these primary tumors involving the spine. In spinal tumors the distinction between tumors considered benign or malignant can be somewhat misleading. Chordoma is listed among the malignant tumors involving the spine, although tumor growth in some patients with chordoma is extremely slow. Conversely, giant-cell tumor is considered a benign tumor, although this particular lesion, in some cases, can be aggressive in nature. Early recurrence following surgery for giant-cell tumors is common, and there is even the potential for metastasis. Furthermore, progression to a high-grade sarcoma can occur in up to 10% of patients following postoperative radiation therapy.5
Table 106-2 Skeletal Distribution of Malignant Tumors
| Number of patients with | ||
|---|---|---|
| Tumor Type | Tumors Involving the Spine | Malignant Skeletal Tumors |
| Myeloma | 232 | 803 |
| Lymphoma | 82 | 694 |
| Chondrosarcoma | 54 | 892 |
| Chordoma | 51 | 356 |
| Osteosarcoma | 37 | 1649 |
| Ewing tumor | 16 | 514 |
| Hemangioendothelioma | 13 | 80 |
| Fibrosarcoma | 9 | 255 |
| Secondary chondrosarcoma | 7 | 121 |
| Mesenchymal chondrosarcoma | 4 | 25 |
| Malignant giant-cell tumor | 2 | 35 |
| Malignant fibrous histiocytoma | 2 | 83 |
| Dedifferentiated chondrosarcoma | 1 | 120 |
| Hemangiopericytoma | 1 | 13 |
| Periosteal osteosarcoma | 0 | 69 |
| Adamantinoma | 0 | 36 |
| Desmoid fibroma | 0 | 12 |
| Total | 511 | 5757 |
Only those patients undergoing surgery and who, therefore, have tissue available for pathologic review are included in this table.
From Unni KK et al: Unpublished data, personal communication, 2000.
Management
Staging
Once a primary spinal tumor is suspected, thorough imaging of the lesion is required, including plain radiographs, CT, and MRI to narrow the differential diagnosis. In addition, a complete systemic workup should also be performed. A CT of the chest, abdomen, and pelvis, or a positron emission tomography scan can help to identify any additional distant pathology. Ultimately, however, a tissue diagnosis is typically required to aid in preoperative planning. The decision to perform a biopsy must be a part of a comprehensive management strategy to reduce the risk of local recurrence. It has been suggested that resecting the tissue along the biopsy tract during the definitive resection could prevent recurrence caused by the contamination of surrounding tissues during the biopsy.6
After a histopathologic diagnosis is made, staging can be completed to aid in presurgical planning. The Enneking classification system assists in determining the goals of surgery and is a guide to adjuvent therapy. Both benign and malignant tumors can be staged and are classified as either high- or low-grade, depending on the local extent of disease and the presence of metastasis. Management strategies range from no surgical intervention to palliative surgery based on the stage.7 The Weinstein, Boriani, Biagini (WBB) classification8 was developed to guide a surgeon in determining the most appropriate surgical approach. In the WBB system, the vertebra is divided into 12 radiating zones (numbered 1 through 12, clockwise starting dorsally at the spinous process) and into five layers (A–E, from paravertebral to dural involvement). In addition, the longitudinal extent of the tumor is noted. Depending on the area of involvement, a surgical approach is recommended. For tumors involving only the vertebral body (zones 4–9), an anterior approach is recommended, whereas a dorsal approach is ideal for tumors involving the pedicles, facets, spinous process, and lamina (zones 3–10).
Surgery
Spine tumor resection can be en bloc or intralesional.1 En bloc resection, or spondylectomy, is the resection of the entire tumor in one piece. En bloc resections are further subdivided into marginal or wide. Marginal resection dissects through the pseudocapsule of the tumor, and wide resection provides a cuff of normal tissue (>2 mm of healthy bone, reactive periosteum, or pleura) with a margin of healthy surrounding tissue. For primary tumors, the long-term local tumor control, survival, and cure are dependent on en bloc tumor resection. For this reason, there has been resurgent interest in treating primary spinal tumors with en bloc resection. Intralesional resection is the incision into the tumor, and debulking from within. Although intralesional resection of spine tumors results in good neurologic outcomes, local recurrence rates remain high. Wide resection occurs when the excision is inclusive of the pseudocapsule. As applied to the spine, marginal en bloc resection or intralesional resection with an adjuvant (e.g., phenol, liquid nitrogen, methyl methacrylate) may be curative for aneurysmal bone cysts, giant-cell tumors, osteoid osteomas, and osteoblastomas. Evidence is mounting that wide en bloc resections for primary tumors such as chondrosarcoma, chordoma, osteogenic sarcoma, and Ewing sarcoma may effect a longer disease-free interval and a potential cure.
However, en bloc resections often require extensive procedures associated with high rates of morbidity and mortality.9 Bandiera et al.9 recently published the largest study to date examining the complication rates in en bloc resections of spinal tumors. They reviewed 134 consecutive attempted en bloc resections from 1990 to 2007 at a single institution. Major complications occurred in 43 cases, including three deaths. Major complications were also more common in patients who had undergone a previous failed resection at a prior institution. Of those previously treated, 72% suffered a major complication, whereas 20% with a new presentation suffered a major complication. Furthermore, with an average follow-up period of 37 months, the local recurrence rate was higher in patients treated previously elsewhere (40% vs. 16%). Thus, surgical management of primary spinal tumors is associated with high morbidity and recurrence rates, both of which can be reduced if treatment from biopsy to resection occurs at the same institution by a dedicated multidisciplinary team.
When the biopsy is performed prior to the definitive procedure, if possible, the biopsy path should be well marked and the soft tissue along the biopsy path should be resected along with the tumor at the time of surgery. Also, one must always be cautious to avoid contamination of surrounding tissues with tumor cells. In addition, resecting the dura as a margin may increase the risk of intradural seeding.10 When a fusion is planned, bone graft should be obtained through a separate surgical setup.5
Radiation
Primary tumors may benefit from neoadjuvant or postoperative adjuvant radiation or chemotherapy. In general, the more benign tumors (e.g., osteoid osteoma, osteoblastoma, osteochondroma) have a poor response rate to these therapeutic modalities, and gross resection of the tumor will effect a cure. A significant concern in patients receiving radian therapy for the more benign tumors is the development of postradiation sarcomas. In a review of 59 patients who underwent operation for spinal sarcoma at Memorial Sloan-Kettering Cancer Center, 7 patients had postradiation sarcomas at a median interval of 14 years from the time of radiation.11,12 Other tumors, such as osteogenic sarcoma and Ewing sarcoma, may benefit from neoadjuvant chemotherapy followed by resection. Chondrosarcoma and chordoma are extremely radiation therapy resistant and still chemotherapy resistant, but positive surgical margins are irradiated.
A great challenge in radiation therapy to the spine is that the dose the spinal cord can tolerate is significantly lower than the dose required to achieve local tumor control. Experience with extremity sarcomas has demonstrated good local control with 60 Gy for postoperative radiation therapy in patients with close surgical margins. For patients with gross residual disease after resection, 70 Gy is typically delivered in 200-cGy fractions. The spinal cord is thought to tolerate no more than 50 Gy when delivered in 200-cGy fractions.11 Several strategies have evolved in an attempt to deliver tumoricidal doses of radiation while avoiding radiation-induced myelopathy. These advances include intraoperative radiation therapy, brachytherapy, proton beam therapy, high-dose conformal photon therapy, and stereotactic radiation. Each of these techniques provides a higher tumoral dose of radiation with reduced damage to surrounding tissues and potentially smaller fields than do conventional external beam techniques.
Intraoperative radiation therapy (IORT) involves the delivery of a custom-designed electron beam or high-dose brachytherapy that precisely demarcates the tumor volume. Lead shields and gold foil are used to shield the spinal cord.13 Unfortunately, this technique is somewhat labor intensive, and dosimetry considerations are difficult to predict around the spinal cord. An alternative radiation approach is brachytherapy, or the direct application of radioisotopes within the resection cavity. Earlier attempts with the use of iodine-125 were disappointing due to difficulties with dosing near the spinal cord. However, intraoperative therapy with yttrium-90 has recently been approved by the U.S. Food and Drug Administration and has been demonstrated to be effective in the treatment of sarcoma. Yttrium-90 is a pure β-emitter with limited penetrance. DeLaney et al.14 treated five patients with yttrium-90 plaques placed intraoperatively, including two chondrosarcomas and one osteosarcoma. With a median follow-up of 24 months, no local recurrences were observed in the chondrosarcoma or osteosarcoma patients and no treatment-related myelopathy or neuropathy was detected.14
Proton beam therapy15,16 has an inherent geometric advantage over therapy with photons and electrons because of the finite range of penetration in tissues (Bragg-peak effect). Proton beams can be designed so that a uniform dose is administered to the target volume (i.e., tumor) and a minimal dose is delivered to the critical surrounding tissues (e.g., spinal cord, bowel, esophagus). Proton beam treatment plans are often supplemented with additional photon beam therapy to improve tumoral coverage. Studies have shown outstanding results for the control of chordoma and chondrosarcoma with proton beam therapy. Hug17 reported on 33 patients with skull base chordomas and 25 patients with skull base chondrosarcomas treated to a mean dose of 70.7 cobalt gray equivalents (CGEs) with a mean follow-up period of 33 months. Local control was achieved in 76% of the chordoma patients and 82% of the chondrosarcoma patients.17 A major drawback for proton beam therapy is the limited availability of treatment centers in the United States to accommodate the demands for treating primary tumors, particularly chordomas and chondrosarcomas. Currently, proton beam centers are located at the Loma Linda University Medical Center, Massachusetts General Hospital, Indiana University, University of Florida, M.D. Anderson Cancer Center in Houston, and INTEGRIS Cancer Campus in Oklahoma.
High-dose conformal photon therapy (3D-CRT) has made it possible to deliver cytotoxic doses to tumor volume, doses similar to proton beam radiation, without the side effect of radiation-induced myelopathy.18–22 Similar to proton beam therapy, 3D-CRT is a method of irradiating a tumor volume with an array of photon beams that are individually shaped to conform to a 3D rendering of the target. Treatment planning considers dose inhomogeneities caused by the differing electron densities of various tissues and calculates the resulting dose distribution using sophisticated algorithms. Intensity-modulated radiation therapy (IMRT) represents an advanced form of 3D-CRT in which multileaf collimators are used to dynamically change the field shape during treatment, thus permitting the delivery of an inhomogeneous dose that conforms more tightly to the target region. Because of the precise dosimetry demands of IMRT, accurate delivery requires reproducible patient setup and positioning. Recent data suggest that IMRT may improve the clinical outcome of inoperable tumors and those tumors requiring a boost after surgical resection. Yamada et al.23 reported on 14 patients with primary spinal malignancies and 21 with metastatic lesions treated with IMRT at Memorial Sloan-Kettering Cancer Center. Patients had unresectable disease near the spinal cord and either previously received radiation therapy or were prescribed doses beyond spinal cord tolerance. With a mean follow-up period of 11 months, local control was achieved in 81% of primary malignancies and 75% of metastatic lesions. No radiation-induced myelopathy was observed, and more than 90% reported palliation from pain, weakness, or paresthesia.23
Chemotherapy
Chemotherapy has not been found to be beneficial in the majority of spinal tumors. However, recent evidence has shown promise in the treatment of osteogenic sarcoma. A recent prospective randomized trial demonstrated 3-year event-free survival rates of 71% and 78% for patients with osteogenic sarcoma treated with either cisplatin, doxorubicin, and methotrexate versus muramyl tetrapeptide following resection, respectively.24 Chordoma, also previously thought to be chemoresistant, has also been shown to be sensitive to a new class of tyrosine kinase inhibitors. Imatinib mesylate (Gleevac) is one such tyrosine kynase inhibitor that has shown promise in early clinical trials. In initial studies, imaging revealed extensive tumor necrosis in six out of six patients treated with imatinib mesylate.2 Sunitinib (Sutent) is another tyrosine kinase inhibitor recently developed that is currently being tested on chordoma patients in clinical trials.11 As new therapeutic strategies continue to be developed, chemotherapy holds great promise for the treatment of primary spinal tumors in the future.
Hemangioma
Hemangioma is one of the common benign lesions involving the spinal axis. It is often discovered incidentally during evaluation of patients with back or neck pain. The relatively low incidence, noted in Table 106-1, confirms that most patients with hemangioma are not treated surgically. Several studies have demonstrated that this entity may affect as much as 10% to 12% of the population.25–33 Less than 5% of patients with hemangiomas develop symptoms.34 Spine surgeons become involved with the treatment of hemangioma when the lesion causes spinal cord or nerve root compression. In general, decompressive surgery should be reserved for this specific group, because such surgery is usually not required for the management of pain that is not associated with neurologic involvement. In a 1993 review of spinal hemangioma from the Mayo Clinic, it was demonstrated that, in fact, it is rare for incidental hemangiomas associated with pain alone to progress to spinal cord compression.28 Only 2 of 59 patients with previously diagnosed asymptomatic or painful lesions later developed spinal cord compression. Symptomatic hemangiomas are usually observed during adulthood and found to occur in the thoracic region.29 Patients with asymptomatic lesions do not require further evaluation unless pain or neurologic deficits develop. Patients with painful lesions should be followed closely, with a combination of radiographic studies and periodic neurologic evaluations.
Imaging
Plain radiography, tomography, and CT often clearly demonstrate the typical coarsened trabeculae within the involved vertebrae, with a characteristic honeycombed appearance. Gadolinium enhancement and MRI evidence of a soft tissue component are often observed (Fig. 106-1).
Histology
Most of the trabeculae are atrophic because of the abnormal blood vessels, although some become thickened and sclerotic. Microscopically, there are two main types of trabeculae. These are characterized by cavernous or capillary vessels. In some cases, adipose tissue may be found within the lesion.27 Spinal cord compression may arise from the expansile nature of the vertebral body, an associated soft tissue component of the tumor that rests within the spinal canal, a compression fracture of the weakened vertebral body, or, rarely, an epidural hemorrhage.
Management
The management scheme should depend on the size, extent, and location of the lesion; the patient’s general age and health; and the patient’s clinical course and neurologic findings. Surgical decompression is recommended if there is progressive neurologic decline. It is important for the spine surgeon to be familiar with the variety of available surgical approaches so that the most appropriate technique can be used to remove the tumor. Many patients for whom laminectomy and postoperative radiation therapy would have been recommended can now be treated using a lateral or ventral surgical approach to the lesion. Laminectomy followed by radiation therapy of lesions involving the vertebral body yielded a 93% rate of neurologic recovery, without recurrent symptoms, in a 52-month follow-up period.31 Laminectomy without radiation therapy for subtotal tumor resections resulted in tumor control rates of 70% to 80%.29–31 It appears that postoperative irradiation reduces the risk of tumor recurrence in patients after subtotal tumor removal. Nevertheless, because of the potential morbidity and relative lack of efficacy associated with radiation therapy, total lesion removal often should be attempted.
Vertebroplasty and kyphoplasty have also been advocated for the treatment of symptomatic hemangiomas. Studies have recently reported excellent pain relief with no evidence of posttreatment instability and preservation of vertebral body height. Such a less invasive approach may be ideal for patients who are not good surgical candidates.35
Eosinophilic Granuloma (Langerhans Cell Histiocytosis)
Eosinophilic granulomas are vertebral lesions found in 10% to 15% of cases of Langerhans cell histiocytosis. Most commonly, eosinophilic granulomas are identified in children younger than age 10, and spinal lesions have been reported in 6.5% to 25% of cases involving bone.36 Eosinophilic granulomas involving the adult lumbar spine are very rare, and to date only 13 cases have been reported.37 These self-limiting, benign lesions cause bony destruction38 secondary to the local proliferation of histiocytes. Occasionally, multiple levels are involved and can rarely result in pain, but are more often identified incidentally.5
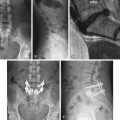
Radiographically, eosinophilic granulomas are identified as destructive bony lesions with well-demarcated borders and no evidence of a soft tissue mass. The adjacent disc spaces are well preserved (Fig. 106-2). These findings differentiate eosinophilic granulomas from other lesions in the differential diagnosis (e.g., infection, benign tumor, or malignancy).5
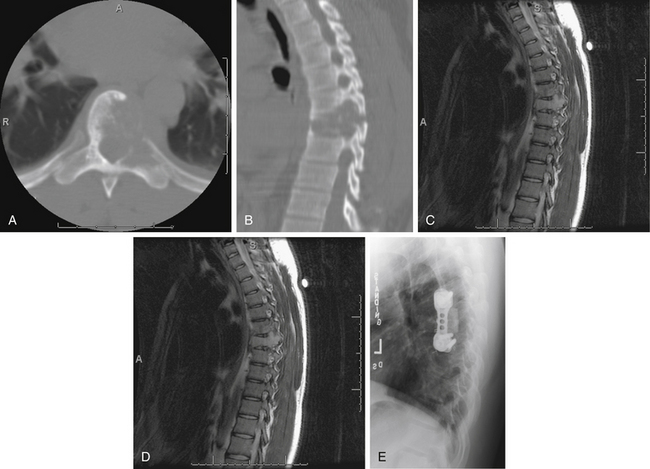
Management
Prior to any invasive treatment, a biopsy is indicated. In many cases, symptoms will resolve over time, and a conservative approach should always be considered first. The vertebral body height can be restored spontaneously if the areas of endochondral ossification were preserved and the child is young. Therefore, treatment is generally conservative with activity limitations and bracing.36,38 A recent report described the use of percutaneous vertebroplasty to treat eosinophilic granuloma involving the cervical spine of a child, though this approach was taken only after conservative measures failed and a more aggressive approach was declined by the family.39 In cases where vertebral body collapse results in loss of neurologic function, decompression and biopsy are warranted.5 If the bony destruction leads to instability that persists despite a course of conservative management, arthrodesis is required.38
Aneurysmal Bone Cysts
Aneurysmal bone cysts (ABCs) are benign, proliferative non-neoplastic lesions that may occur in any part of the skeleton. Although this is not a tumor per se, its classical appearance and presentation should be familiar to clinicians dealing with spine lesions. ABCs make up 1% to 6% of primary spinal neoplasms, with approximately 40% to 45% involving the lumbar spine, 30% involving the thoracic spine, and 25% to 30% involving the cervical spine.40 Lesions are often not confined to a single vertebra; instead they bridge two or more levels in approximately 40% of cases.41,42 Although primary ABCs are of unknown cause, a secondary form of ABC has been described that arises within eosinophilic granulomas, simple bone cysts, osteosarcomas, chondroblastomas, or giant-cell tumors.42
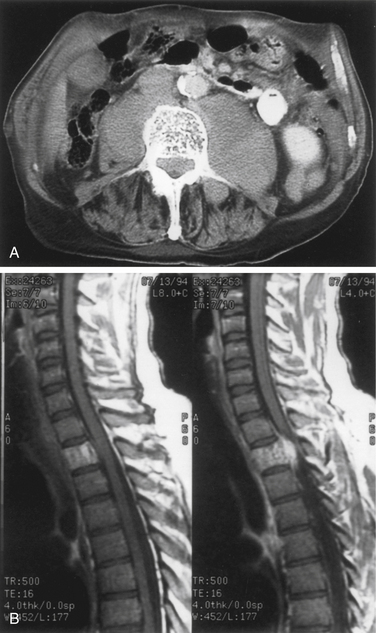
ABCs of the spine typically present in young patients in their second decade, with a slight predominance in women.43 In one series, 60% of lesions arose in the neural arch and 40% arose in the body. Pain that occurs especially at night and that is localized to the site of the mass is the most common presenting complaint.44 The presence or absence of a neurologic deficit depends on the site of the tumor and on the degree of compression of adjacent neural elements. Symptoms and signs may vary from cord compression with myelopathic findings to radicular features of single-root involvement. The clinical course is commonly progressive over several months because of the slow growth of these lesions, although rapid growth can also occur. Imaging the anatomic delineation of ABCs is often best achieved with plain radiography and CT, which accurately define the degree of bone destruction and full extent of the lesion (Figs. 106-3A and B). MRI can be helpful for defining a spinal cord compressive component, and it readily demonstrates the full epidural extent of the mass. The rather classical appearance of the involved vertebra is that of a multiloculated, expansile, highly vascular mass with eggshell-like cortical bone and blood product fluid levels. Collapse of involved bodies and involvement of adjacent ribs may also be observed.
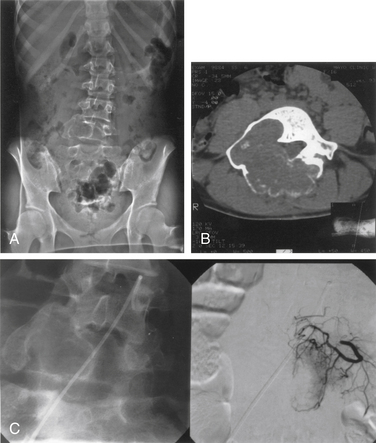
Selective spinal angiography has both diagnostic and, potentially, therapeutic value.44 In addition to defining the relationship of the arterial supply of the lesion to the arterial supply of the cord, angiography also defines the involvement of the vertebral arteries with cervical lesions. The anatomic location of the artery of Adamkiewicz in lower thoracic or upper lumbar lesions can be defined clearly with spinal angiography. Finally, preoperative embolization is a useful adjunct that may decrease the intraoperative blood loss (Fig. 106-3C).
Management
Rarely, spontaneous disappearance of ABCs has been reported to occur, and when discovered incidentally, conservative management could be considered. Diagnosis is generally established based on diagnostic studies (CT and MRI) without the need for a biopsy. When a biopsy is required, an open biopsy is preferred to decrease the risk of hemorrhage and improve the diagnostic value of the sample. Once a diagnosis has been established based on imaging or biopsy, several therapeutic options for ABCs have been described in the literature. The treatment options for ABCs include percutaneous injection of a fibrosing agent, arterial embolization, radiation therapy, curettage with or without bone grafting, or resection. Percutaneous injection of fibrosing agents has been shown to successfully treat ABCs. Injection of zein alcohol (Ethibloc) with histoacryl glue or of methylprednisolone with calcitonin has been shown to lead to successful destruction of the lesion with low recurrence rates.40 However, injection therapies must be performed cautiously because of the presence of abnormal vascular channels and the risk of migration of the material into the vasculature. Embolic stroke resulting in death has been reported.40
More recently, some investigators have suggested that selective arterial embolization is the treatment of choice in cases where neither spinal instability nor neurologic deficits are identified. In such cases, it is sometimes preferable to repeat the embolization at least two times in an effort to avoid open surgery.5 Arterial embolization preoperatively can also significantly reduce the risk of bleeding during surgery.40
Because of the destructive nature of ABCs and the risk of progressive instability coupled with the frequent presence of neurologic deficits secondary to compression of neural elements, complete surgical resection is often considered the treatment of choice. The approach (ventral, dorsal, or dorsolateral) depends on the exact location and extent of the lesion. An eggshell-thin cyst of subperiosteal new bone that is continuous with adjacent cortex is observed at surgery. This delineates the extent of the lesion, and its removal often results in intense bleeding. The core of the tumor consists of soft, fleshy, vascular tissue, as well as a cystic trabeculation of the interior of the mass containing unclotted blood. The mass may invade adjacent soft tissue or surround the thecal sac. Although there may be some concern about using a cell saver when removing tumors from any location in the body, there appears to be no contraindication to this procedure when removing ABCs. Despite the inherent technical difficulties of excision that are related to location and extent of the lesion, the prognosis is excellent with complete excision and recurrence rates are rare in most series.40 Subtotal surgical excision, conversely, is followed by a high incidence of recurrence, which is usually rapid (within 1 year, and often within 4 months).43 Likewise, curettage and bone grafting is associated with a 20% or greater recurrence rate.40 Therefore, en bloc resection should be the goal of surgical intervention. As with all resections of spinal neoplasms, postoperative spinal instability should always be considered and instrumented fusion performed when identified.
Giant-Cell Tumor
Giant-cell tumors of the spine are locally aggressive benign primary bone tumors that constitute 4.2% of bone tumors in Dahlin and Coventry’s series.3 Approximately 6.5% of all giant-cell tumors occur in the spine,45 with half occurring in the sacrum, followed by the thoracic and cervical spine in frequency.46 The mean age of involvement is approximately 30 years, with a range of 13 to 62 years.46,47 In most reviews, the incidence in women appears to outnumber that in men by a ratio of 2:1.45 Most patients present with pain localized to the site of the lesion and, occasionally, with a neurologic deficit, depending on the location. Malignant transformation occurs in approximately 10% of cases. Some giant-cell tumors are biologically aggressive lesions. In patients with these tumors, local recurrence is common after incomplete resection.
Imaging
The diagnosis of giant-cell tumors may be made from plain radiographs and CTs (Fig. 106-4). The lesion is characterized as a destructive, expansile mass within the sacrum or other vertebrae. MRI reveals a heterogeneous, cystic, compartmentalized mass that may contain blood degradation products.
Management
In a review of 24 patients with giant-cell tumor of the spine, pain was the presenting symptom in all cases, and half of the patients also had a neurologic deficit.45 All patients were treated with surgical curettage or en bloc resection, depending on the location and extent of the tumor. Ten patients had recurrences, and seven of these were treated with radiation. The most acceptable treatment approach seems to be an attempted wide resection. Preoperative tumor embolization may be beneficial and advisable according to some authors.5
Radiation therapy may be considered with subtotal surgical excision. Giant-cell tumors can recur relatively early after even the most radical surgical excision. In such cases, it may be reasonable to consider radiation therapy if it is believed that further excisional attempts will also be unsuccessful. The 10-year success rate of radiation alone was 69% compared with 83% for postoperative radiation therapy.5 Unfortunately, progression to a high-grade sarcoma occurs in 5% to 15% of cases following radiation treatment.5 Therefore, radiation therapy should be reserved for patients with recurrent or residual disease after attempted resection.48 In the spine, wide resection may cause destabilization and may necessitate instrumentation of the spine and fusion.23 However, total excision is curative if it is achieved.48
Osteoid Osteoma
Osteoid osteoma is a relatively common benign neoplasm that occurs in the spine and accounts for 21% of surgically managed benign lesions.49,50 Typically, it has a central nidus of interlacing osteoid and woven bone within a loose vascular stroma, surrounded by an osteosclerotic rim. These lesions are sharply demarcated from surrounding bone (Fig. 106-5). The nidus is rarely greater than 1.5 cm in diameter, and lesions larger than this are categorized as osteoblastomas. The distinction between the two lesions is often unclear, and they may represent a continuum.
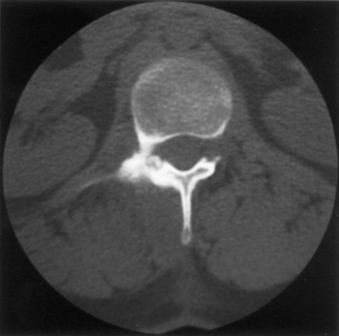
Most patients are young, with half of all cases occurring in the second decade, with a marked male predominance. Typically, the neural arch elements are affected, most commonly in the lumbar spine. In a series of 33, the patients invariably presented with pain, which increased with exercise and was classically relieved with aspirin.51 Associated features included the presence of radicular symptoms referable to the underlying root and antalgic scoliosis. A neurologic deficit was present in only two patients (both with cervical lesions). Osteoid osteoma is the most common cause of painful scoliosis in adolescents. This deformity can be corrected with resection of the osteoid osteoma alone if surgery is within 15 months of onset of symptoms or before the development of a structural curve.51 Fifteen months appears to be the critical cut-off point after which spontaneous correction does not occur after surgery.
Management
Because treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) is so effective for pain relief, conservative therapy is an option. Long-term NSAID therapy is particularly attractive when surgical morbidity would be high due to the complexity of a given case. However, chronic NSAID treatment is associated with significant side effects, and in a small number of cases the lesion may ultimately be found to be an osteoblastoma. Therefore, the definitive treatment for osteoid osteomas is complete surgical excision.50,52 In more than 95% of cases, resection results in almost immediate pain relief. When a complete resection is not achieved and the lesion recurs, reoperation is recommended. Radiation is not recommended, either alone or following surgery.5 In patients who are not good surgical candidates, radiofrequency ablation has recently emerged as an additional therapeutic option. Initially avoided due to concerns of causing thermal injury to neural structures, radiofrequency ablation has since been shown to provide complete pain relief in a number of patients. The reported pain relief rates range between 77% and 100%, complication rates between 5% and 24%, and recurrence rates between 5% and 12%.35
Osteoblastoma
Osteoblastomas are uncommon lesions, constituting approximately 0.36% of all primary bone tumors that are treated with surgery. Thirty-three percent of these lesions occur in the spine. Osteoblastomas represent a histologic continuum of osteoid osteoma, the difference being the size of the lesions. Osteoblastomas are lesions that are greater than 1.5 cm in diameter. Patients are usually in their second or third decade at presentation,52 and there is a male:female predominance of 2:1.7.52 Osteoblastomas are distributed throughout the spine, and in the series by Boriani et al.,53 16 of 30 lesions occurred in the lumbar spine, 8 in the thoracic spine, and 6 in the cervical spine. Two thirds of these lesions are confined to the dorsal elements. As observed with other benign spinal neoplasms, pain is the most common presenting complaint, and it may be associated with scoliosis and neurologic deficit. Unlike osteoid osteomas, osteoblastomas can progressively enlarge. The radiologic workup of these lesions should include plain radiography and CT. These scans may show a well-defined, lobulated, lytic, expansile mass that usually involves the neural arch structures. Fifty percent of lesions are radiolucent with a sclerotic rim. As with osteoid osteomas, MRI is often less informative than CT for defining these lesions.54 Unlike osteoid osteomas, bone scans often are not necessary for diagnosis but may be helpful with smaller lesions. Osteoblastomas should be treated surgically with total resection. Approximately 10% of lesions recur after surgery.
Management
Both osteoid osteomas and osteoblastomas may cause scoliosis and neurologic deficit, the latter being more common with osteoblastoma. Surgical resection is the first line of treatment, and when total is associated with cure.55,56 With larger, aggressive lesions, radiation therapy and embolization may be considered as adjuncts to surgical resection, but the potential risks of radiation therapy must be considered, including the risk of sarcomatous transformation.
Osteochondromas
Osteochondromas constitute 9.2% of all primary bone tumors that are treated surgically, but they rarely occur in the spine (2.5% of all osteochondromas). They consist of cartilage-covered cortical bone with underlying medullary bone, both types of bone being contiguous with their counterparts in the parent bone. The cartilaginous cap undergoes ossification to form the osteochondroma.
Histologically, the cartilaginous cap and underlying bone are identical to normal bone. The tumors can be solitary or multiple, and most present in the third decade. There is a male:female predominance of 2:1. The lesions affect the transverse or spinous processes, and half of all lesions occur in the cervical spine. Lesions are best diagnosed by plain radiography and CT. Patients usually have localized pain, although neurologic deficit can also occur.57 Rarely, osteochondromas may also be a manifestation of hereditary multiple osteochondromas. Only 1% of solitary lesions undergo malignant transformation. Surgical excision is the treatment of choice.58–60
Other Benign Primary Spine Tumors
The other benign tumors that affect the spine are uncommon, as evidenced from Table 106-1. However, the indications for surgical intervention for these rare lesions and the goals of treatment are similar to those for the more common spine lesions already discussed.
Plasma Cell Tumors
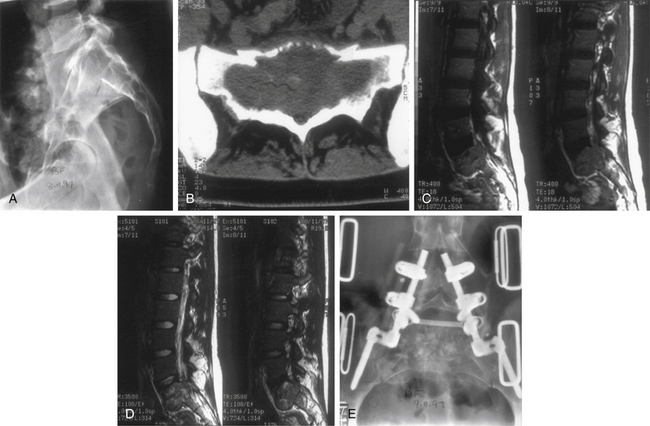
Plasma cell tumors of the spine (multiple myeloma and solitary plasmacytoma) are the most common type of malignant primary tumors involving the spine (see Table 106-2). Multiple myeloma is the most common plasma cell tumor, characterized by multiple bony lesions, infiltration of the bone marrow by plasma cells, and a marked reduction of normal immunoglobulins.26,61,62 Conversely, solitary plasmacytoma constitutes only 3% of plasma cell tumors. Up to 50% of plasmacytomas occur in the spine and most commonly occur in the thoracic spine, accounting for 24 of 33 cases in a recent series, though they have been reported throughout the spine.63 Fifty percent of patients diagnosed with plasmacytoma ultimately develop multiple myeloma, most commonly within 2 years64 (Fig. 106-6). Patients with this condition are characterized by one or, at most, two bony lesions. A bone scan can be used to identify additional lesions.64 The incidence of multiple myeloma is the same in both males and females, whereas with solitary plasmacytoma, a twofold to threefold higher incidence is encountered in males. A small monoclonal spike that disappears with treatment may reflect local lesions only.
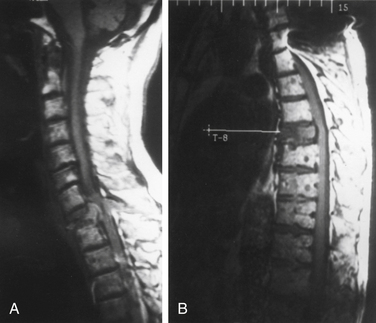
Plasma cell tumors of the spine usually present with pain and, in advanced cases, with myelopathy. The duration of symptoms before diagnosis may vary; however, in general, symptoms worsen considerably in the 6 to 12 months before presentation. The diagnosis of solitary plasmacytoma is established histologically by either a needle biopsy or an open procedure. In plasmacytoma, the bone marrow is negative for plasma cell infiltrates, and serum protein electrophoresis results are normal. Multiple myeloma is usually diagnosed definitively by a bone marrow biopsy, the presence of multiple bony lesions on bone survey, and an abnormal monoclonal immunoglobulin spike on serum or urine electrophoresis.64 Most patients with multiple myeloma present with Bence Jones proteinuria, reflecting the spillover of monoclonal immunoglobulin fragments into the urine.
Management
The treatment of choice for solitary plasmacytoma in the absence of instability or rapid paralysis is radiation therapy. The dose of radiation to the spine for plasmacytoma varies from 35 to 50 Gy, although some oncologists favor larger doses. Local control rates of up to 96% and survival rates of up to 11 years have been reported.64 It is recommended that patients with a diagnosis of solitary plasmacytoma be followed closely for the development of indices characteristic of multiple myeloma. Chemotherapy is generally withheld until progression to multiple myeloma is documented.62 When the diagnosis of multiple myeloma is established, chemotherapy is indicated, although prognosis at that time is poor, with a median survival rate thereafter of 2 years and a 5-year survival rate of 18%.47,62 Whether chemotherapy should be instituted after the diagnosis of solitary plasmacytoma of bone remains unclear.62 Vertebroplasty can help relieve pain and improve the quality of life in patients without neurologic deficit.65
Because plasmacytomas are highly radiosensitive, surgical decompression and fusion are generally reserved for patients with progressive neurologic deficit and deformity. Studies have demonstrated that one can predict the risk of pathologic collapse by determining the extent of the involvement of the vertebral body.66 It has been shown that patients with osteolytic lesions involving greater than 50% of the vertebral body are likely to require instrumentation. It has been recommended that surgical intervention be considered when the likelihood of future instability exceeds 50%, local kyphosis is greater than 20%, or there is translational deformity.67
Chordoma
Chordoma constitutes approximately 5% of malignant tumors involving the spine (see Table 106-2). The tumor originates from primitive remnants of the notochord. Fifty percent of all chordomas are sacrococcygeal, 40% are sphenoccipital, and the remaining 10% involve the mobile intermediate regions of the spine.26,47,68,69 These tumors, considered to be of low-grade malignancy, are extremely difficult to resect because of their proximity to the spinal cord and cauda equina. In addition, in 5% to 10% of cases, they tend to metastasize within 1 to 10 years of the diagnosis. Fifty percent of all chordomas occur in the fifth to seventh decades of life, with a mean age of onset of approximately 50 years.26,68,69 Men are twice as likely to be afflicted with this tumor as women.70
Chordomas arise from the vertebral body, with ensuing ventral and dorsal extension. Pain is the presenting symptom in 75% of cases and a radicular component in 10% of patients. More than two thirds of patients present with weakness or other neurologic deficits. Mean duration of symptoms at presentation is 14 months, with a range of 4 to 24 months.71 In chordomas arising from the sacrum, 40% complain of rectal dysfunction, including constipation, tenesmus, or bleeding hemorrhoids. A palpable tumor on rectal examination can be identified in most patients.1 These tumors can also affect two adjacent vertebral bodies, while sparing the intervertebral disc.
Histology
Histologically, chordomas consist of two cell types: a small, compact stellate cell that is considered to be the precursor of the more prevalent and larger physaliferous cell containing mucinous vacuoles.1,68 These tumors have a characteristic lobular appearance on MRI. They can often be diagnosed by radiologic studies, including plain radiography. On plain radiographs, calcification can be observed in 40% of cases, and it is twice as likely to be observed by CT. Chordomas are generally avascular and are not associated with excessive intraoperative blood loss.
Grossly, chordomas appear lobulated and are often gray or partially translucent. The consistency varies from firm and focally ossified to a soft, myxoid, or semifluid material. There is generally a pseudocapsule separating the tumor from adjacent soft tissue, but the tumor is diffusely invasive within adjacent bone without clear margins.1
Management
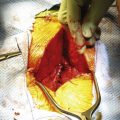
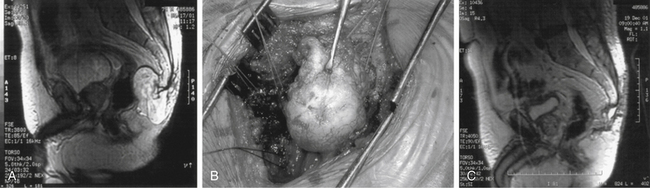
Percutaneous CT-guided biopsy is generally recommended to establish a definitive working diagnosis. The optimal treatment of chordomas is wide en bloc resection.69,70,72 The location of these lesions (close to neural structures) renders cure extremely unlikely. Some authors recommend a staged approach where first a posterior approach is performed to mobilize the posterior elements, free the dura from the tumor pseudocapsule, and place posterior instramentation, followed by an anterior approach to perform an en bloc vertebrectomy.1 With involvement of the sacrococcygeal spine, resection can be accomplished by ventral, dorsal (Fig. 106-7), or combined approaches.1,69,73–75 These approaches are geared for the widest possible resection of tumor, with sparing of as many nerves as possible.69 Generally, sparing the S3 nerve root on either side may be sufficient to preserve bladder and fecal continence. Loss of sexual function also needs to be discussed with the patient preoperatively. Local recurrence in patients undergoing en bloc resection was 28%. Local recurrence in those where the tumor capsule was violated was 64%.76
Chordomas are not generally radiosensitive. However, because of the high risk of recurrence, postoperative radiation therapy is often recommended.69,77,78 A retrospective study involving 21 patients with spinal chordomas favored a combination of surgery and irradiation over surgery alone.78 There is no evidence that doses higher than 40 to 55 Gy are more likely to be associated with better results.77 Neither is there evidence in support of treatment with more than a single fraction per day. The 40-year experience at the M.D. Anderson Cancer Center from 1954 to 1994 yielded 27 patients.72 The median Kaplan-Meier survival estimate for the entire group was 7.4 years. The disease-free interval for patients undergoing radical resection was 2.3 years compared with 8 months for those undergoing subtotal excision (P < .0001). The addition of radiation to the group undergoing subtotal resection increased survival from 8 months to 2 years (P < .02). The often-quoted 5-year survival rate has ranged, depending on the source, from 50% to 77%, with a 10-year survival rate of 50%.1,79 Approximately 30% of spinal chordomas develop metastases.70
Proton therapy has also been used as an adjuvant treatment; however; there is currently insufficient evidence to demonstrate that proton therapy is superior to conventional radiation therapy. In a review of the available literature, Brada et al. found the 5-year local progression-free survival rate of skull base chordomas to range from 54% to 64%.80
Chordomas are well known to be resistant to chemotherapy. In cases where all surgical and radiation therapy options have been exhausted, reports have been published showing responses to adriamycin-cisplatin or ifosfamide-adriamycin-platinum combinations, particularly in high-grade spindle cell sarcomas. More recently, evidence has emerged demonstrating that chordomas may be susceptible to agents targeting tyrosine kinase and angiogenesis pathways. Agents such as imatinib, erlotinib, and gefitinib have all shown promise in small series.1
Primary Spinal Ewing Sarcoma
Ewing sarcoma constitutes 6% of all primary malignant bone tumors26,81; however, primary spinal involvement occurs in only 3.5% of all patients with Ewing sarcomas.81 This tumor afflicts males more commonly than females (2:1 ratio), and 88% of cases present in the first 2 decades of life.26,81 In terms of the level of involvement, frequency decreases in a caudal-to-rostral progression, with more than 50% of all cases occurring in the sacrum.26,81,82 The most common presenting feature is pain, with or without radicular involvement, depending on the level. In general, two of three patients present with a neurologic deficit (Fig. 106-8). The duration of symptoms can range from 1 to 30 months; most symptoms are 1 year or less in duration. A rectal mass may be encountered on examination, considering that there is sacral involvement in one of four patients with primary spinal Ewing sarcoma. In half of the cases, a lytic process is observed on plain radiographs, whereas other cases show blastic or mixed features.81 A paravertebral soft tissue mass may occur independently or concurrently with bony involvement. This may be best appreciated on CT or MRI (see Figs. 106-8C and D). Confirmation of diagnosis has been accomplished more commonly by laminectomy, particularly in the presence of neurologic deficit. With the advent of CT-guided biopsies, needle aspiration has been undertaken in some patients.
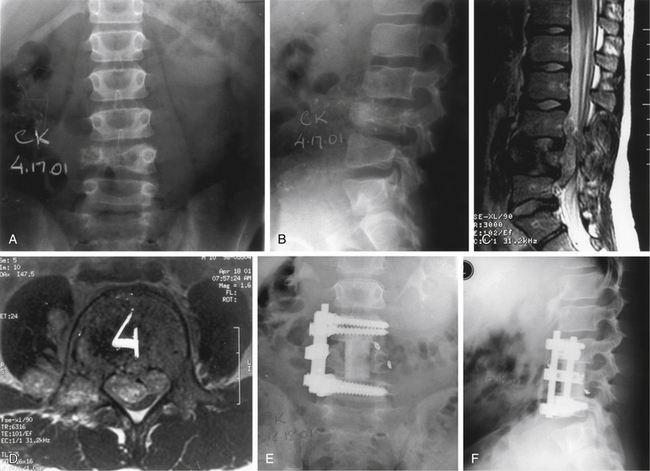
Histology
Histologically, Ewing sarcoma consists of infiltrating sheets of small, round to oval cells with a scant amount of cytoplasm that tests positive for glycogen.26 These tumors are fragile and vascular. Intraoperative bleeding can be extensive, particularly with spinal involvement. Grossly, these tumors are gelatinous in consistency and gray-white in color. The borders of these lesions are poorly outlined, with extension into the bony trabeculae as well as into the paravertebral soft tissues.
Management
Optimal treatment after diagnosis is provided by a combination of radiation therapy and chemotherapy.82,83 Local radiation therapy to the spine is usually given at a dose of 50 to 55 Gy, with inclusion of an adequate margin as deemed appropriate by CT or MRI. Higher doses, when delivered to the spine, can be associated with postradiation myelopathy.84 In a recent review of the data collected in the Cooperative Ewing’s Sarcoma Study and the European Intergroup Cooperative Ewing’s Sarcoma Study, local recurrence occurs in 22% of patients receiving radiation alone and 18.7% of those receiving surgery and radiation.85 These recurrence rates were similar to those found in patients with Ewing sarcoma not involving the spine. Further analysis confirmed that surgical debulking prior to radiation did not improve outcomes and that surgery should be performed only if a wide resection is possible.85
The accepted chemotherapy protocol developed by the Intergroup Ewing’s Sarcoma Study (IESS) consists of cyclophosphamide, vincristine, dactinomycin, and doxorubicin. This regimen, when administered in addition to local radiation therapy, has proven superior to a three-drug regimen with local irradiation or to a four-drug regimen with bilateral pulmonary radiation therapy.82 The IESS protocol was tested in 342 patients and involved mostly appendicular, pelvic, and rib tumors. As expected, the most favorable results were encountered with distal appendicular disease, and the worst were encountered with pelvic involvement. Younger patients (<10 years of age) were observed to have an associated 71% 5-year survival, compared with 46% in patients older than 15 years of age. A more intense chemotherapeutic and radiation therapy protocol (the IESS-II protocol) was developed for the treatment of pelvic and sacral Ewing sarcoma.83 With this regimen consisting of four-agent chemotherapy before and after high-dose local irradiation, a survival pattern was achieved that is comparable to that achieved for disease in nonpelvic sites. A large randomized trial was recently published that revealed that cyclophosphamide resulted in similar survival rates compared with ifosfamide when used in conjunction with vincristine, dactinomycin, and doxorubicin.86 Furthermore, the addition of etoposide with ifosfamide improved survival in high-risk patients.86
The role of surgery in the treatment of spinal Ewing sarcoma remains controversial and is best performed after initial treatment with chemotherapy and radiation. Neoadjuvant therapy can reduce the size of the tumor and allow for adjacent bone to heal prior to surgical intervention. Indications for operation include high-grade epidural compression with symptomatic neurologic progression, stabilization, poor response to neoadjuvant chemotherapy or radiation therapy, and radiographic residual tumor postneoadjuvant treatment. When resection is attempted, maximal resection should be attempted. Evidence has shown that en bloc resection may improve local control but does not significantly improve survival.87 When surgical excision is attempted, preoperative embolization is recommended26,84 and routinely requires bony fusions with spinal implants to restore stability and prevent progressive deformity and neurologic deficit. Decompressive laminectomy alone may be associated with significant complications, including instability and progressive angulation.81 When an adequate surgical margin is not achievable, postoperative radiation should be added to the surgical bed.
In an attempt to identify certain prognostic factors associated with Ewing sarcoma, a retrospective study was conducted with 46 patients, 43 with osseous involvement and 3 with extraosseous involvement. An attempted resection was performed in 12 patients but not in the remaining 34.88 This analysis demonstrated that survival is improved in patients with local disease, tumors less than 500 mL in volume, peripheral involvement only, and who undergo gross total resection, compared with patients who have metastatic involvement, tumor volume greater than 500 mL, central involvement, and who do not undergo resection. This analysis suggested that aggressive surgery with radiation therapy might be an important prognostic factor. The best prognosis is provided by local irradiation and chemotherapy. A retrospective review of 36 patients who had primary involvement of the spine with Ewing sarcoma yielded a 5-year survival of 33%, with a mean survival of 2.9 years. Nine patients treated with the IESS regimen remained disease free at follow-up.88
In summary, all patients with Ewing sarcoma should initially be treated with chemotherapy. Surgical resection should be considered in all cases where the surgeon feels it is possible to achieve a complete resection. If the margins of the resection are less than 1 cm, as is often the case in spinal tumors, radiation therapy is added. Alternatively, chemotherapy and radiation treatment can be utilized alone when an adequate resection is not possible.87
Osteosarcoma
Osteosarcoma is the most common sarcoma of the spine, accounting for 3% to 15% of all primary tumors of the spine.89 Among all sarcomas, however, spinal involvement is uncommon, present in only 37 of 1649 osteosarcomas (2.2%) (see Table 106-2). Osteosarcomas occur most commonly in the fourth decade of life,89 have a slight predilection for males,3 and are the most common primary malignant bone tumor in the pediatric population. In addition, osteosarcoma has one of the lowest survival rates among pediatric cancers.87 In adults, prognosis with this tumor is especially poor. Shives et al.90 reviewed 27 cases of spinal osteosarcoma and reported a median survival of 10 months from diagnosis.
Osteosarcomas can occur throughout the spine, though they are more common in the thoracic spine and sacrum and typically arise in the posterior elements.89 Osteosarcomas usually arise de novo or may occur secondarily in previously irradiated bone, usually several years later (Fig. 106-9). Postirradiation osteosarcomas occurred in 16 of 600 cases in Dahlin and Coventry’s series.3 Paget disease is also a known risk factor for the development of osteosarcoma. Though as few as 1% of Paget disease patients go on to develop osteosarcoma, these patients make up as much as 50% of the patients in some registries.89
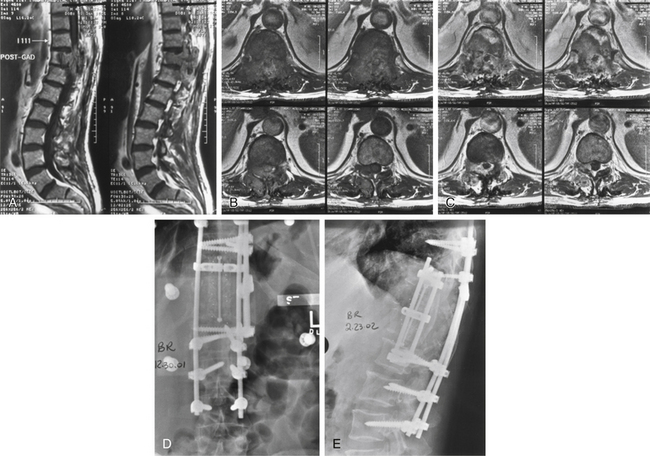
Diagnosis is often delayed due to the nonspecific nature of the symptoms. Pain is the most common presenting feature and may be axial or radicular. Weakness is present in more than half of patients at the time of diagnosis, whereas loss of bowel and bladder function is generally seen only in advanced cases.
Pathologically, osteosarcomas are firm and calcified and consist mostly of sarcomatous connective tissue that forms osteoid tissue or bone. Histologically there are several different subtypes, but all osteosarcomas produce an osteoid matrix. Conventional osteosarcomas can be broken into three subtypes based on the matrix produced: osteoblastic (55%), fibroblastic (23%), and chondroblastic (22%) types.3 Telangiectatic osteosarcomas are a separate classification of osteosarcoma that more closely resembles an ABC. In addition to the production of an osteoid matrix, there are also numerous blood-filled sinusoids.89 Small-cell osteosarcoma is another subtype characterized by the presence of small cells with hyperchromatic nuclei and is positive for CD99. Sometimes confused for a Ewing sarcoma, small-cell osteosarcomas can be distinguished by the presence of an osteoid matrix. Similarily, epitheliod osteosarcoma is another subtype that is sometimes mistaken for a carcinoma, and again, the presence of an osteoid matrix leads to a correct diagnosis. Other reported variants include giant-cell osteosarcoma or osteoblastoma-like osteosarcoma; however, these are exceedingly rare.89
Management
Initial treatment with radiation as the primary treatment modality produced poor results. More recently, improvement in surgical techniques allowing more complete resections with improvement in adjuvant therapies has appeared to improve survival rates from the 10 months originally reported.89 Multimodality therapy to treat spinal osteogenic sarcoma was originally described by Sundaresan et al.91 in 1988. Eleven patients underwent neoadjuvant chemotherapy followed by aggressive resection and postoperative irradiation. In this limited series, there were five long-term survivors. A more recent series by Ozaki et al.50 reported on 22 patients who received a neoadjuvant chemotherapy regimen according to the Cooperative Osteosarcoma Study Group (COSS) followed by resection. Four different chemotherapy protocols were used. All patients received preoperative high-dose methotrexate and doxorubicin in combination with a variety of other agents, including cisplatin, ifosfamide, bleomycin, actinomycin D, and alfa-interferon. COSS 96 used both preoperative and postoperative chemotherapy. Twelve patients underwent tumor resection, including two wide, three marginal, and seven intralesional excisions. Eight patients received radiation therapy. The median survival was 23 months. Patients with metastases (p = .004), large tumors (>10 cm) (p = .010), and sacral tumors (p = .048) had a worsened survival rate compared with those with no metastases, small tumors, and nonsacral tumors. There was a significant difference between the 17 patients who underwent intralesional excision or no surgery and the 5 patients who underwent marginal or wide excisions (p = .033). Postoperative irradiation extended overall survival in patients who underwent intralesional or no resection (p = .059).50 In this series, patients received conventional external photon beam irradiation (median tumoral dose 45 Gy). No patient underwent either proton beam or IMRT irradiation.
Based on the available information, the Spine Oncology Study Group recommends that all patients with osteosarcoma of the spine are treated with neoadjuvant chemotherapy. Surgical resection should be attempted when it is felt a complete resection can be achieved and is associated with an improvement in survival and local control.87 Postoperative radiation should also be considered to further improve survival.
Chondrosarcoma
Chondrosarcomas are primary malignant tumors arising from cartilaginous elements. They are rare tumors, constituting 892 of the 5751 malignant skeletal tumors (see Table 106-2). In this series, 54 tumors (6%) occurred in the spine. Like osteosarcomas, chondrosarcomas show a predilection for males; however, unlike osteosarcomas, they occur in middle-aged and older patients. Chondrosarcoma may arise de novo as a primary lesion or may occur as a secondary tumor from a preexisting solitary osteochondroma (1%) or from hereditary multiple exostosis (20%).92
Imaging
Plain radiographs and CT scans demonstrate osteolytic lesions with a calcified matrix. The amount of calcification correlates with the degree of differentiation. MRI is the study of choice for demonstrating the adjacent soft tissue and epidural extent of tumor spread. MRI also demonstrates the heterogeneity of the lesion (Fig. 106-10). Of note, lesions that are more malignant tend to have larger amounts of soft tissue components, more irregular calcification, and more extensive bone destruction.
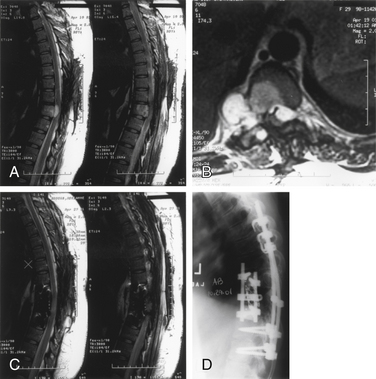
Management
In a series of predominantly low-grade chondrosarcomas, Boriani et al.93 showed improved local control with marginal or wide resections compared with intralesional resection. In this series, 17 of 18 patients undergoing intralesional resection had a local recurrence within 36 months. Two patients who had en bloc resections but contaminated margins (i.e., intralesional resections) experienced recurrence at 12 and 32 months, respectively. At a median follow-up of 81 months, only one patient undergoing marginal or wide resection had recurrence at 48 months. The patients in this series had predominantly posterior-element tumors and no epidural extension.
Avoidance of Complications
One of the disadvantages of using metal implants is their interference with CT and MRI. Some of the new alloys produce fewer imaging artifacts, and these alternatives, although more expensive, should be considered in some cases. Titanium implants have major advantages over stainless steel in MRI tumor recurrence. Titanium implants should be standard for spine tumor reconstruction. Other materials such as carbon fiber have been adopted to lessen artifact and improve imaging. They readily show tumor recurrence, as well as osteointegration of bone graft, on imaging.94
Management of Complications
A deep wound infection often complicates stabilization efforts. Clearing an infection in a previously irradiated wound may be almost impossible. Spinal instability makes it necessary to leave the instrumentation in place while the infection is brought under control. This allows time for the bony fusion to mature. In some cases, the implant can be left in place, and the construct salvaged.
Bilsky M.H., Gerszten P., Laufer I., et al. Radiation for primary spine tumors. Neurosurg Clin North Am. 2008;19:119-123.
Boriani S., Weinstein J.N., Biagini R. Primary bone tumors of the spine. Terminology and surgical staging. Spine (Phila Pa 1976). 1997;22:1036-1044.
Fenoy A.J., Greenlee J.D., Menezes A.H., et al. Primary bone tumors of the spine in children. J Neurosurg. 2006;105:252-260.
McMaster M.L., Goldstein A.M., Bromley C.M., et al. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12:1-11.
Ozaki T., Flege S., Liljenqvist U., et al. Osteosarcoma of the spine: experience of the Cooperative Osteosarcoma Study Group. Cancer. 2002;94:1069-1077.
Sundaresan N., Rosen G., Boriani S. Primary malignant tumors of the spine. Orthop Clin North Am. 2009;40:21-36.
Weinstein J.N. Surgical approach to spine tumors. Orthopedics. 1989;12:897-905.
1. Sundaresan N., Rosen G., Boriani S. Primary malignant tumors of the spine. Orthop Clin North Am. 2009;40:21-36.
2. Casali P.G., Messina A., Stacchiotti S., et al. Imatinib mesylate in chordoma. Cancer. 2004;101:2086-2097.
3. Dahlin D.C., Coventry M.B. Osteogenic sarcoma. A study of six hundred cases. J Bone Joint Surg [Am]. 1967;49:101-110.
4. Weinstein J.N. Surgical approach to spine tumors. Orthopedics. 1989;12:897-905.
5. Gasbarrini A., Cappuccio M., Donthineni R., et al. Management of benign tumors of the mobile spine. Orthop Clin North Am. 2009;40:9-19.
6. Yamazaki T., McLoughlin G., Patel S., et al. Feasibility and safety of en bloc resection for primary spine tumors. A systematic review by the Spine Oncology Study Group. Spine (Phila Pa 1976). 2009;34:S31-S38.
7. Enneking W.F. A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res. 1986;204:9-24.
8. Boriani S., Weinstein J.N., Biagini R. Primary bone tumors of the spine. Terminology and surgical staging. Spine (Phila Pa 1976). 1997;22:1036-1044.
9. Bandiera S., Boriani S., Donthineni R., Amendola, et al. Complications of en bloc resections in the spine. Orthop Clin North Am. 2009;40:125-131.
10. Bilsky M.H., Boland P.J., Panageas K.S., et al. Intralesional resection of primary and metastatic sarcoma involving the spine: outcome analysis of 59 patients. Neurosurgery. 2001;49:1277-1286. discussion 86–87
11. Bilsky M.H., Gerszten P., Laufer I., Yamada Y. Radiation for primary spine tumors. Neurosurg Clin North Am. 2008;19:119-123.
12. Brady M.S., Gaynor J.J., Brennan M.F. Radiation-associated sarcoma of bone and soft tissue. Arch Surg. 1992;127:1379-1385.
13. Seichi A., Kondoh T., Hozumi T., Karasawa K. Intraoperative radiation therapy for metastatic spinal tumors. Spine (Phila Pa 1976). 1999;24:470-473. discussion 474–475
14. DeLaney T.F., Chen G.T., Mauceri T.C., et al. Intraoperative dural irradiation by customized 192iridium and 90yttrium brachytherapy plaques. Int J Radiat Oncol Biol Phys. 2003;57:239-245.
15. Levin W.P., Kooy H., Loeffler J.S., DeLaney T.F. Proton beam therapy. Br J Cancer. 2005;93:849-854.
16. DeLaney T.F., Trofimov A.V., Engelsman M., Suit H.D. Advanced-technology radiation therapy in the management of bone and soft tissue sarcomas. Cancer Control. 2005;12:27-35.
17. Hug E.B. Review of skull base chordomas: prognostic factors and long-term results of proton-beam radiotherapy. Neurosurg Focus. 2001;10:E11.
18. Bilsky M.H., Yenice K., Lovelock M., Yamada J. Stereotactic intensity-modulation radiation therapy for vertebral body and paraspinal tumors. Neurosurg Focus. 2001;11:E7.
19. De Salles A.A., Pedroso A.G., Medin P., et al. Spinal lesions treated with Novalis shaped beam intensity-modulated radiosurgery and stereotactic radiotherapy. J Neurosurg. 2004;101(Suppl 3):435-440.
20. Guckenberger M., Meyer J., Wilbert J., et al. Precision required for dose-escalated treatment of spinal metastases and implications for image-guided radiation therapy (IGRT). Radiother Oncol. 2007;84:56-63.
21. Wright J.L., Lovelock D.M., Bilsky M.H., et al. Clinical outcomes after reirradiation of paraspinal tumors. Am J Clin Oncol. 2006;29:495-502.
22. Yamada Y., Lovelock D.M., Yenice K.M., et al. Multifractionated image-guided and stereotactic intensity-modulated radiotherapy of paraspinal tumors: a preliminary report. Int J Radiat Oncol Biol Phys. 2005;62:53-61.
23. Yamada Y., Lovelock D.M., Bilsky M.H. A review of image-guided intensity-modulated radiotherapy for spinal tumors. Neurosurgery. 2007;61:226-235. discussion 235
24. Meyers P.A., Schwartz C.L., Krailo M., et al. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol. 2005;23:2004-2011.
25. Blankstein A., Spiegelmann R., Shacked I., et al. Hemangioma of the thoracic spine involving multiple adjacent levels: case report. Paraplegia. 1988;26:186-191.
26. Sundaresan N., Schmidek H.H., Schiller A.L., Rosenthal D.I. Tumors of the spine: diagnosis and clinical management. Philadelphia: Saunders; 1990.
27. Faria S.L., Schlupp W.R., Chiminazzo H.Jr. Radiotherapy in the treatment of vertebral hemangiomas. Int J Radiat Oncol Biol Phys. 1985;11:387-390.
28. Fox M.W., Onofrio B.M. The natural history and management of symptomatic and asymptomatic vertebral hemangiomas. J Neurosurg. 1993;78:36-45.
29. Krueger E.G., Sobel G.L., Weinstein C. Vertebral hemangioma with compression of spinal cord. J Neurosurg. 1961;18:331-338.
30. Lang E.F.Jr., Peserico L. Neurologic and surgical aspects of vertebral hemangiomas. Surg Clin North Am. 1960;40:817-823.
31. Nguyen J.P., Djindjian M., Pavlovitch J.M., et al. [Vertebral hemangioma with neurologic signs. Therapeutic results. Survey of the French Society of Neurosurgery]. Neurochirurgie. 1989;35:299-303. 305–308
32. Nguyen J.P., Djindjian M., Badiane S. [Vertebral hemangioma with neurologic signs. Clinical presentation. Results of a survey by the French Society of Neurosurgery]. Neurochirurgie. 1989;35:270-274. 305–308
33. Laredo J.D., Reizine D., Bard M., Merland J.J. Vertebral hemangiomas: radiologic evaluation. Radiology. 1986;161:183-189.
34. Chi J.H., Bydon A., Hsieh P., et al. Epidemiology and demographics for primary vertebral tumors. Neurosurg Clin North Am. 2008;19:1-4.
35. Gottfried O.N., Dailey A.T., Schmidt M.H. Adjunct and minimally invasive techniques for the diagnosis and treatment of vertebral tumors. Neurosurg Clin North Am. 2008;19:125-138.
36. Fenoy A.J., Greenlee J.D., Menezes A.H., et al. Primary bone tumors of the spine in children. J Neurosurg. 2006;105:252-260.
37. Nakamura H., Nagayama R. Eosinophilic granuloma presenting with local osteolysis in an adult lumbar spine. J Clin Neurosci. 2008;15:1398-1400.
38. Greenlee J.D., Fenoy A.J., Donovan K.A., Menezes A.H. Eosinophilic granuloma in the pediatric spine. Pediatr Neurosurg. 2007;43:285-292.
39. Tan H.Q., Li M.H., Wu C.G., Gu Y.F., et al. Percutaneous vertebroplasty for eosinophilic granuloma of the cervical spine in a child. Pediatr Radiol. 2007;37:1053-1057.
40. Burch S., Hu S., Berven S. Aneurysmal bone cysts of the spine. Neurosurg Clin North Am. 2008;19:41-47.
41. Karparov M., Kitov D. Aneurysmal bone cyst of the spine. Acta Neurochir (Wien). 1977;39:101-113.
42. Vandertop W.P., Pruijs J.E., Snoeck I.N., van den Hout J.H. Aneurysmal bone cyst of the thoracic spine: radical excision with use of the cavitron. A case report. J Bone Joint Surg [Am]. 1994;76:608-611.
43. Hay M.C., Paterson D., Taylor T.K. Aneurysmal bone cysts of the spine. J Bone Joint Surg [Br]. 1978;60:406-411.
44. Mohan V., Arora M.M., Gupta R.P., Izzat F. Aneurysmal bone cysts of the dorsal spine. Arch Orthop Trauma Surg. 1989;108:390-393.
45. Sanjay B.K., Sim F.H., Unni K.K., et al. Giant-cell tumours of the spine. J Bone Joint Surg [Br]. 1993;75:148-154.
46. Hart R.A., Boriani S., Biagini R., et al. A system for surgical staging and management of spine tumors. A clinical outcome study of giant cell tumors of the spine. Spine (Phila Pa 1976). 1997;22:1773-1782. discussion 1783
47. Weinstein J.N., McLain R.F. Primary tumors of the spine. Spine (Phila Pa 1976). 1987;12:843-851.
48. Shikata J., Yamamuro T., Shimizu K., Kotoura Y. Surgical treatment of giant-cell tumors of the spine. Clin Orthop Relat Res. 1992;278:29-36.
49. Kan P., Schmidt M.H. Osteoid osteoma and osteoblastoma of the spine. Neurosurg Clin North Am. 2008;19:65-70.
50. Ozaki T., Flege S., Liljenqvist U., et al. Osteosarcoma of the spine: experience of the Cooperative Osteosarcoma Study Group. Cancer. 2002;94:1069-1077.
51. Pettine K.A., Klassen R.A. Osteoid-osteoma and osteoblastoma of the spine. J Bone Joint Surg [Am]. 1986;68:354-361.
52. Ozaki T., Liljenqvist U., Hillmann A., et al. Osteoid osteoma and osteoblastoma of the spine: experiences with 22 patients. Clin Orthop Relat Res. 2002;397:394-402.
53. Boriani S., Capanna R., Donati D., et al. Osteoblastoma of the spine. Clin Orthop Relat Res. 1992;278:37-45.
54. Shaikh M.I., Saifuddin A., Pringle J., et al. Spinal osteoblastoma: CT and MR imaging with pathological correlation. Skeletal Radiol. 1999;28:33-40.
55. Bruneau M., Cornelius J.F., George B. Osteoid osteomas and osteoblastomas of the occipitocervical junction. Spine (Phila Pa 1976). 2005;30:E567-E571.
56. Denaro V., Denaro L., Papalia R., et al. Surgical management of cervical spine osteoblastomas. Clin Orthop Relat Res. 2007;455:190-195.
57. Song K.J., Lee K.B. Solitary osteochondroma of the thoracic spine causing myelopathy. Eur J Pediatr Surg. 2007;17:210-213.
58. Gille O., Pointillart V., Vital J.M. Course of spinal solitary osteochondromas. Spine (Phila Pa 1976). 2005;30:E13-E19.
59. Srikantha U., Bhagavatula I.D., Satyanarayana S., et al. Spinal osteochondroma: spectrum of a rare disease. J Neurosurg Spine. 2008;8:561-566.
60. Yagi M., Ninomiya K., Kihara M., Horiuchi Y. Symptomatic osteochondroma of the spine in elderly patients. Report of 3 cases. J Neurosurg Spine. 2009;11:64-70.
61. Loftus C.M., Michelsen C.B., Rapoport F., Antunes J.L. Management of plasmacytomas of the spine. Neurosurgery. 1983;13:30-36.
62. Poor M.M., Hitchon P.W., Riggs C.E.Jr. Solitary spinal plasmacytomas: management and outcome. J Spinal Disord. 1988;1:295-300.
63. Kelley S.P., Ashford R.U., Rao A.S., Dickson R.A. Primary bone tumours of the spine: a 42-year survey from the Leeds Regional Bone Tumour Registry. Eur Spine J. 2007;16:405-409.
64. Singh H., Meyer S.A., Jenkins A.L.3rd. Treatment of primary vertebral tumors. Mt Sinai J Med. 2009;76:499-504.
65. McDonald R.J., Trout A.T., Gray L.A., et al. Vertebroplasty in multiple myeloma: outcomes in a large patient series. AJNR Am J Neuroradiol. 2008;29:642-648.
66. Taneichi H., Kaneda K., Takeda N., et al. Risk factors and probability of vertebral body collapse in metastases of the thoracic and lumbar spine. Spine (Phila Pa 1976). 1997;22:239-245.
67. Mendoza S., Urrutia J., Fuentes D. Surgical treatment of solitary plasmocytoma of the spine: case series. Iowa Orthop J. 2004;24:86-94.
68. Bjornsson J., Wold L.E., Ebersold M.J., Laws E.R. Chordoma of the mobile spine. A clinicopathologic analysis of 40 patients. Cancer. 1993;71:735-740.
69. Samson I.R., Springfield D.S., Suit H.D., Mankin H.J. Operative treatment of sacrococcygeal chordoma. A review of twenty-one cases. J Bone Joint Surg [Am]. 1993;75:1476-1484.
70. Bergh P., Kindblom L.G., Gunterberg B., et al. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer. 2000;88:2122-2134.
71. Chandawarkar R.Y. Sacrococcygeal chordoma: review of 50 consecutive patients. World J Surg. 1996;20:717-719.
72. York J.E., Kaczaraj A., Abi-Said D., et al. Sacral chordoma: 40-year experience at a major cancer center. Neurosurgery. 1999;44:74-79. discussion 79–80
73. Jackson R.J., Gokaslan Z.L. Spinal-pelvic fixation in patients with lumbosacral neoplasms. J Neurosurg. 2000;92:61-70.
74. Localio S.A., Eng K., Ranson J.H. Abdominosacral approach for retrorectal tumors. Ann Surg. 1980;191:555-560.
75. Mac C.C., Waugh J.M., Mayo C.W., Coventry M.B. The surgical treatment of presacral tumors: a combined problem. Proc Staff Meet Mayo Clin. 1952;27:73-84.
76. Kaiser T.E., Pritchard D.J., Unni K.K. Clinicopathologic study of sacrococcygeal chordoma. Cancer. 1984;53:2574-2578.
77. Cummings B.J., Hodson D.I., Bush R.S. Chordoma: the results of megavoltage radiation therapy. Int J Radiat Oncol Biol Phys. 1983;9:633-642.
78. Keisch M.E., Garcia D.M., Shibuya R.B. Retrospective long-term follow-up analysis in 21 patients with chordomas of various sites treated at a single institution. J Neurosurg. 1991;75:374-377.
79. McMaster M.L., Goldstein A.M., Bromley C.M., et al. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12:1-11.
80. Brada M., Pijls-Johannesma M., De Ruysscher D. Proton therapy in clinical practice: current clinical evidence. J Clin Oncol. 2007;25:965-970.
81. Grubb M.R., Currier B.L., Pritchard D.J., Ebersold M.J. Primary Ewing’s sarcoma of the spine. Spine (Phila Pa 1976). 1994;19:309-313.
82. Nesbit M.E.Jr., Gehan E.A., Burgert E.O.Jr., et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol. 1990;8:1664-1674.
83. Evans R.G., Nesbit M.E., Gehan E.A., et al. Multimodal therapy for the management of localized Ewing’s sarcoma of pelvic and sacral bones: a report from the second intergroup study. J Clin Oncol. 1991;9:1173-1180.
84. Sharafuddin M.J., Haddad F.S., Hitchon P.W., et al. Treatment options in primary Ewing’s sarcoma of the spine: report of seven cases and review of the literature. Neurosurgery. 1992;30:610-618. discussion 618–619
85. Schuck A., Ahrens S., von Schorlemer I., et al. Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2005;63:1562-1567.
86. Paulussen M., Craft A.W., Lewis I., et al. Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment—cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol. 2008;26:4385-4393.
87. Sciubba D., Okuno S., Dekutoski M., Gokaslan Z., Ewing and osteogenic sarcoma. evidence for multidisciplinary management. Spine. 2009;34:S58-S68.
88. Sailer S.L., Harmon D.C., Mankin H.J., et al. Ewing’s sarcoma: surgical resection as a prognostic factor. Int J Radiat Oncol Biol Phys. 1988;15:43-52.
89. Wang V.Y., Potts M., Chou D. Sarcoma and the spinal column. Neurosurg Clin North Am. 2008;19:71-80.
90. Shives T.C., Dahlin D.C., Sim F.H., et al. Osteosarcoma of the spine. J Bone Joint Surg [Am]. 1986;68:660-668.
91. Sundaresan N., Rosen G., Huvos A.G., Krol G. Combined treatment of osteosarcoma of the spine. Neurosurgery. 1988;23:714-719.
92. Shives T.C., McLeod R.A., Unni K.K., Schray M.F. Chondrosarcoma of the spine. J Bone Joint Surg [Am]. 1989;71:1158-1165.
93. Boriani S., De Iure F., Bandiera S., et al. Chondrosarcoma of the mobile spine: report on 22 cases. Spine (Phila Pa 1976). 2000;25:804-812.
94. Boriani S., Biagini R., Bandiera S., et al. Reconstruction of the anterior column of the thoracic and lumbar spine with a carbon fiber stackable cage system. Orthopedics. 2002;25:37-42.