CHAPTER 89 Primary Biliary Cirrhosis
EPIDEMIOLOGY
PBC occurs worldwide and predominantly in women, with a female-to-male ratio of 9 : 1. The diagnosis of PBC usually is made between the ages of 30 and 60 years, with a range of 21 to 93 years. The disease has been documented in even younger patients—two teenagers 15 and 16 years of age, respectively.1 Until the early 1970s, PBC was considered a rare condition that manifested with persistent jaundice and almost inevitably progressed to end-stage liver disease. A better understanding of its pathogenesis, along with subsequent clinical and epidemiologic studies, has modified current concepts regarding this condition. PBC seems to be more common than was formerly believed because of increasing awareness of the disease and because asymptomatic patients are identified through the widespread use of screening tests such as determination of serum cholesterol levels and liver biochemical test levels in otherwise healthy persons.
The reported prevalence of PBC varies among countries, with a range of 19 cases per 1 million population in Israel to 402 cases per 1 million population in Olmsted County, Minnesota.2 Whether the difference in prevalence is real or a result of different methodologies used to detect the disease is unknown. Inconsistency in case definition and case finding methods, as well as imprecision in defining the study area, the populations evaluated, and the dates of diagnosis, particularly in earlier reports, makes comparisons among studies difficult. Estimates of the annual incidence of PBC range from 0.7 to 49 per 1 million population. Both the prevalence and incidence of PBC seem to have increased over time.2 The increase in prevalence possibly reflects an increase in survival time in patients with PBC.
In the United States,3 the age-adjusted reported incidence of PBC per 1 million person-years is 45 for women and 7 for men (27 overall). The reported prevalence per 1 million population is 654 for women and 121 for men (402 overall); these figures represent the highest prevalence rates for PBC ever reported.
PATHOGENESIS
Although the cause of PBC remains unknown, several lines of evidence suggest an autoimmune pathogenesis. The evidence includes the intense humoral and cellular response to an intracytoplasmic antigen, presence of highly specific antimitochondrial antibodies (AMA), involvement of T lymphocytes in the destruction of bile ducts, and numerous defects in immunologic regulation. Like other autoimmune diseases, PBC has a clear female predominance. Both PBC and the presence of AMA occur more frequently in close relatives of patients who have PBC than in controls. PBC is associated with an increased incidence of autoimmune disease of other types in both patients with PBC and their first-degree relatives.4,5 PBC seems to be triggered by an immune-mediated response to one or more allo- or autoantigens, which leads to progressive destruction of bile ducts, chronic cholestasis, and eventual biliary cirrhosis. Immunohistochemical phenotyping of inflammatory cells surrounding the bile ducts shows a combination of CD4+ and CD8+ T lymphocytes, accompanied by B lymphocytes and natural killer cells. Bile duct destruction is induced directly by the cytotoxicity of CD4+ and CD8+ T cells in contact with biliary epithelium. B lymphocytes are relatively uncommon in the inflammatory reaction but sometimes can be seen in clusters. Intracellular adhesion molecules (e.g., intracellular adhesion molecule-1 [ICAM-1]) are strongly expressed on many epithelial cells, particularly in areas of lymphocyte damage; these molecules may facilitate the interaction between destructive lymphocytes and their targets. In the early biliary lesions of PBC, eosinophilic infiltration and granulomas often are seen. PBC is principally a disease of the small intrahepatic bile ducts, with loss of biliary epithelial cells that line these ducts and resulting cholestatic damage. PBC is not restricted to the liver; abnormalities of salivary and lacrimal glands with an associated cellular phenotypic change similar to that seen in the biliary epithelial cells also occur. Three spontaneous autoimmune biliary disease mouse models6–8 and two induced models of PBC9,10 have been reported.
AUTOANTIBODIES
The mechanisms by which AMA directed to proteins located on the inner surface of mitochondrial membranes develop are unknown; PDC-E2 or a cross-reactive molecule is overexpressed on biliary epithelial cells in PBC, predominantly at the luminal domain, and PDC-E2–specific CD4+ T cells are present in portal inflammatory infiltrates of affected persons. Although AMA are predominantly of the IgG1 and IgG3 classes, most patients who have PBC exhibit polyclonal elevation of serum IgM levels; the IgM is not directed at mitochondrial or nuclear antigens. This phenomenon is suggestive of polyclonal activation of the B-cell compartment with an associated failure of isotype switching, representing aberrant B-cell activation.11 AMA do not appear to be cytotoxic: (1) They persist after liver transplantation without evidence of disease recurrence; (2) disease severity is unrelated to antibody titer; (3) they are not always present in PBC; and (4) they develop in animal models after the injection of recombinant PDC-E2 protein, but without resulting bile duct destruction or inflammation. Further, the different types and numbers of mitochondrial antigens recognized by Western immunoblot analysis at the time of the patient’s presentation is independent of the stage of the liver disease and not associated with specific clinical, biochemical, histologic, and immunologic features or with the Mayo risk score (see later).12
Antinuclear antibodies (ANA) are present in nearly one half of patients with PBC and in up to 85% of patients with AMA-negative PBC (see later). The most relevant immunofluorescent reactivities of ANA in patients with PBC are anti-multiple nuclear dots antibodies ([anti-MND], with the molecular target being a 100-kd soluble protein called Sp100), anticentromere antibodies, and antinuclear envelope antibodies. The immunofluorescence pattern of the antinuclear envelope antibodies is characterized as being rim-like and membranous; its molecular targets are structural components of the nuclear pore complex, such as gp210 and nucleoprotein p62, and of the nuclear membrane, such as lamin B receptors. Antibodies against the nuclear pore protein gp210 (anti-gp210) are found in 25% of patients with AMA-positive PBC and in up to 50% of those with AMA-negative PBC. ANA with the MND and rim-like and membranous patterns, which are relatively rare or absent in normal and pathologic controls, are strongly associated with PBC and can be considered to be surrogate markers of PBC in AMA-negative patients.13–15 The specificity of anti-gp210 for PBC when detected by immunoblotting is greater than 99%, whereas antibodies to p62 (anti-p62) are found in approximately 25% of patients and are highly specific for PBC. Anti-p62 antibodies seem to be mutually exclusive with anti-gp210 antibodies. Further, anti-gp210 and possibly anti-p62 also offer prognostic information in that they seem to be associated with aggressive disease with a poor prognosis.13,16,17 In Japanese patients with PBC, anticentromere antibodies are associated with the development of portal hypertension.18
GENETIC FACTORS
The occurrence of PBC in relatives of affected persons plus abnormalities of cell-mediated immunity in first-degree relatives of patients with PBC suggests a genetic association. This association is further supported by the finding that PBC exhibits a higher concordance rate in monozygotic than dizygotic twin pairs, suggesting a genetic component to disease susceptibility and expression of the susceptibility through genes that regulate the immune response.19 Many of the familial risk and genetic studies of PBC have failed to distinguish, however, between true genetic risk and shared environmental exposure.
Although no link between PBC and a specific human leukocyte antigen (HLA) class I phenotype has been found, HLA class II molecules may contribute to the development of this condition. The HLA associations with PBC detected most commonly have been with the DRB1*0801 allele in European and North American white populations and DRB1*0803 in Japanese populations.20,21 Although the DRB1*08 allele seems to impart a significant risk for PBC, with odds ratios of 3 or higher in white and Japanese populations, a study from China reported that the frequencies of the DRB1*0701 and DRB1*03 alleles were increased significantly in patients with PBC compared with controls, with no difference in the frequencies of the DRB1*08 allele.22 A few class II HLA alleles demonstrate protective associations against PBC. These alleles include DQA1*0102 in United States and Japanese studies and DQB1*0602 in a United States study. More recently, DRB*13 was found to be protective against PBC in patients from the United Kingdom and Italy, and DRB1*11 was protective in patients from Italy but not those from the United Kingdom.20,23 An association with HLA class III genes, which code for complement components C2 and C4, cytokines, and complement factor B, has been studied less extensively. Earlier studies reported an increased frequency of haplotype C4B2 and haplotype C4A*Q0 in patients with PBC. Smaller, older studies of the association between PBC and alleles affecting the expression of tumor necrosis factor-α have been reported, but an association remains unresolved.
MOLECULAR MIMICRY
Molecular mimicry between host autoantigens and unrelated exogenous proteins is one of the hypotheses to explain how autoantibodies to self-proteins arise, break tolerance, and lead to autoimmune disease. Molecular mimicry of an extrinsic protein produced by an infectious agent has long been suggested as a possible initiating event in PBC. Infectious agents incriminated in the immune response in PBC include various bacteria and viruses, most recently Chlamydia pneumoniae,24 Novospingobium aromaticivorans,25 and human betaretrovirus.26 Microorganisms produce a multitude of foreign antigens that collectively constitute the major determinants recognized by the immune system. These antigens potentially include a variety of carbohydrates, lipids, and proteins that can be recognized by specific receptors on inflammatory cells. In PBC, PDC-E2 appears to be an ideal candidate for foreign antigens to mimic. PDC-E2, particularly its inner lipoyl domain, is highly conserved among bacteria, yeasts, and mammals. Autoimmune phenomena in PBC could result from peptides that mimic T-cell epitopes of microbial proteins and that are derived from, and presented by, abnormally expressed HLA class II molecules. Molecular mimicry has been invoked to explain the breaking of tolerance against mitochondrial antigens. Definitive evidence for this theory is still lacking, however.
XENOBIOTICS AND OTHER IMPLICATED AGENTS
Xenobiotics are foreign compounds that may alter self-proteins by inducing a change in the molecular structure of the native protein sufficient to induce an immune response. The immune response may then result in the recognition of both the modified and the native proteins.27 The continued presence of the self-protein may perpetuate the immune response initiated by the xenobiotic-induced adduct, thereby leading to chronic autoimmunity. Because many xenobiotics are metabolized in the liver, the potential for liver-specific alteration of proteins is substantial. To address the hypothesis that PBC is induced by xenobiotic exposure, Long and colleagues28 synthesized the inner lipoylated domain of PDC-E2, replaced the lipoic acid moiety with synthetic structures, and quantified the reactivity of these structures with sera from patients with PBC. AMA from all patients reacted more strongly to 3 of the 18 modified organic autoepitopes than to the native domain. Defective sulfoxidation of certain compounds, such as bile acids, estrogen, or drugs, and selenium deficiency have been proposed as underlying mechanisms that may lead to this process. These hypotheses remain unproved.
CLINICAL FEATURES
SYMPTOMATIC DISEASE
The typical patient with symptomatic disease (Table 89-1) is a middle-aged woman with a complaint of fatigue or pruritus. Other symptoms include right upper quadrant abdominal pain, anorexia, and jaundice. Fatigue, although relatively nonspecific, is considered to be the most disabling symptom by many patients, and it worsens in some patients as the disease progresses.29,30 Fatigue in patients with PBC does not correlate with several markers of disease severity, or with the patient’s age or thyroid status, but does correlate with sleep disturbance and depression.29 In a four-year follow-up study, the severity of fatigue was quite stable overall; only liver transplant recipients experienced a significant improvement in fatigue.31 In that study, fatigue was also found on multivariate analysis to be an independent predictor of mortality, particularly cardiac death.31
Table 89-1 Symptoms and Signs of Primary Biliary Cirrhosis at Presentation
| SYMPTOM OR SIGN | FREQUENCY (%) |
|---|---|
| Fatigue | 21-85 |
| Pruritus | 19-55 |
| Hyperpigmentation | 25 |
| Hepatomegaly | 25 |
| Splenomegaly | 15 |
| Xanthelasma | 10 |
| Jaundice | 3-10 |
| Right upper quadrant pain | 8 |
| None | 25-61 |
Pruritus may occur at any point, early or late, in the course of the disease, or intermittently throughout the course. Pruritus generally is intermittent during the day and is most troublesome in the evening and at night. Pruritus often resolves as the disease progresses, but in some patients, severe, intractable pruritus can develop in earlier stages of the disease and may require liver transplantation for effective management. In a population-based study of 770 patients with PBC from England, the cumulative risk of developing pruritus was 19%, 45%, and 57% at 1, 5, and 10 years, respectively.30
Most patients with PBC do not have jaundice at the time of diagnosis. Jaundice occurs later in the course of the disease and usually is persistent and associated with a worse prognosis. Symptoms also may relate to fat-soluble vitamin deficiency, bone pain with or without spontaneous fractures, or an associated autoimmune disease that may occur in patients with PBC (Table 89-2). Symptoms and signs of advanced liver disease, such as ascites, bleeding from gastroesophageal varices, and encephalopathy, usually occur late in the course of PBC.
Table 89-2 Diseases Associated with Primary Biliary Cirrhosis
| DISEASE | FREQUENCY (%) |
|---|---|
| Keratoconjunctivitis sicca (Sjögren’s syndrome) | 72-100 |
| Renal tubular acidosis | 50-60 |
| Arthritis/arthropathy | 4-42 |
| Gallstones | 33 |
| Autoimmune thyroiditis | 15-20 |
| Scleroderma and its variants | 15-19 |
| Raynaud’s disease | 8 |
| CREST or any of its components | 7 |
| Scleroderma | 3-4 |
| Cutaneous disorders—lichen planus, discoid lupus, pemphigoid | 11 |
| Hepatocellular carcinoma | 1-2 |
| Pulmonary fibrosis | Rare |
| Celiac disease | Rare |
CREST, calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia.
ASSOCIATED DISEASES
Many of the diseases found frequently in patients with PBC (see Table 89-2) are thought to be related to disturbances in immune mechanisms. These associated disorders include Sjögren’s syndrome (characterized by dry eyes [keratoconjunctivitis sicca] and dry mouth), scleroderma and its variants, rheumatoid arthritis, some cutaneous disorders, renal tubular acidosis, and thyroiditis.
DIAGNOSIS
The diagnosis of PBC is established by liver biochemical test results consistent with chronic cholestasis plus the presence in serum of AMA. Liver biopsy helps to confirm the diagnosis of PBC but may not be necessary for establishing the diagnosis in patients with characteristic chronic cholestasis and AMA.31
BIOCHEMICAL FEATURES
Liver biochemical test results show a cholestatic picture. Almost all patients have increased serum levels of alkaline phosphatase (three to four times the upper limit of normal) and gamma glutamyl transpeptidase. Serum aminotransferase (aspartate aminotransferase [AST], alanine aminotransferase [ALT]) levels are mildly elevated (usually less than three times normal); marked elevations (more than five times normal) are distinctly unusual and may suggest PBC-autoimmune hepatitis overlap syndrome (see Chapter 88) or coexisting viral hepatitis. Serum bilirubin levels usually are normal in early stages and increase slowly over the course of the disease; levels ultimately may exceed 20 mg/dL. A high serum bilirubin level, low serum albumin, and prolonged prothrombin time indicate a poor prognosis and advanced disease. Serum immunoglobulin levels, especially IgM, are increased, as are serum levels of bile acids, in particular cholic and chenodeoxycholic acids, and cholesterol.
HISTOPATHOLOGIC FEATURES
The initial lesion on a liver biopsy specimen in PBC (Figs. 89-1 and 89-2A and B) is damage to epithelial cells of the small bile ducts. The most important and only diagnostic clue in many cases is ductopenia, defined as the absence of interlobular bile ducts in greater than 50% of portal tracts. The florid duct lesion, in which the epithelium of the interlobular and segmental bile ducts degenerates segmentally, with formation of poorly defined, noncaseating epithelioid granulomas, is nearly diagnostic of PBC but is found in a relatively small number of cases, mainly in early stages.
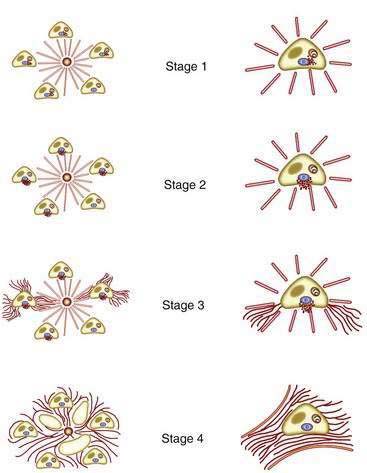
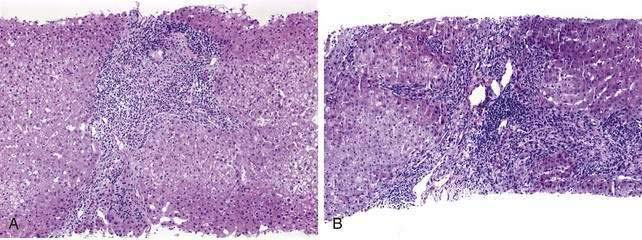
The two most popular histologic staging systems are those proposed by Ludwig and colleagues and Scheuer, which classify the disease in four stages. Both systems describe progressive pathologic changes, beginning initially in the portal areas surrounding the bile ducts and culminating in cirrhosis. Ludwig stage 1 disease is characterized by inflammatory destruction of the intrahepatic septal and interlobular bile ducts that range up to 100 µm in diameter. These lesions often are focal and described as florid duct lesions, characterized by marked inflammation and necrosis around a bile duct. The portal tracts usually are expanded by lymphocytes, with only sparse neutrophils or eosinophils seen. In stage 2 disease (see Fig. 89-2A), the inflammation extends from the portal tract into the hepatic parenchyma, a lesion called interface hepatitis, or formerly, piecemeal necrosis. Destruction of bile ducts with proliferation of bile ductules can be seen. Stage 3 disease is characterized by scarring and fibrosis. Lymphocytic involvement of the portal and periportal areas, as well as the hepatic parenchyma, can be seen, but the hallmark of this stage is the presence of fibrosis without regenerative (or regenerating) nodules. Stage 4 disease is characterized by cirrhosis with fibrous septa and regenerative nodules (see Fig. 89-2B).
Most patients with PBC demonstrate progression of the liver disease; a few patients have a prolonged course of histologic stability, and only rare patients have sustained regression. A time course Markov model has been used to describe the rate of histologic progression over time (Table 89-3).
Table 89-3 Time Course of Histologic Progression to a Higher Stage in Patients with Primary Biliary Cirrhosis

Liver biopsy has been considered necessary for confirming the diagnosis of PBC and excluding other liver diseases. This routine indication for diagnostic purposes has been questioned, however.32 In a patient with AMA in serum, the combination of a serum alkaline phosphatase level greater than 1.5 times the upper limit of normal plus a serum AST level less than 5 times the upper limit of normal yields a 98.2% positive predictive value for a diagnosis of PBC. Therefore, a liver biopsy is not necessary to confirm the diagnosis in most patients with PBC and should be performed in only the minority of AMA-positive patients with a serum alkaline phosphatase level less than 1.5 times normal or a serum AST level greater than 5 times normal.32
NATURAL HISTORY
ASYMPTOMATIC PRIMARY BILIARY CIRRHOSIS
Several reports have described the natural history of asymptomatic patients who have AMA, abnormal liver biochemical test levels consistent with cholestasis, and liver histologic features diagnostic of or compatible with PBC. Asymptomatic patients have less advanced disease than that typically seen in symptomatic patients.33 Patients who are asymptomatic at presentation may survive longer, but a majority will eventually have progressive disease. A median survival of approximately 10 years has been reported for this group of patients. Patients who remain asymptomatic for several years may have a significantly longer survival than that in symptomatic patients, but their life expectancy is still less than that of an age- and gender-matched population. Symptoms of PBC will develop in approximately 40% of the initially asymptomatic patients within five to seven years of follow-up, and most asymptomatic patients ultimately will become symptomatic if the follow-up period is long enough (95% after 20 years).33 When symptoms develop, life expectancy falls significantly and is the same as that for other symptomatic patients. The mortality rate for liver-related causes is significantly higher in initially symptomatic patients than in initially asymptomatic patients; however, an excess rate of non–liver-related mortality in initially asymptomatic patients has been reported to decrease the median survival in these patients to that in initially symptomatic patients.33
SYMPTOMATIC PRIMARY BILIARY CIRRHOSIS
When compared with asymptomatic patients, patients with PBC who have symptoms of chronic cholestasis show a more rapid progression to end-stage liver disease and have a worse prognosis. Several independent predictors of a poor prognosis have been identified in this group of patients (Table 89-4).
Table 89-4 Independent Predictors of Survival in Patients with Primary Biliary Cirrhosis in Various Clinical Studies
| Clinical |
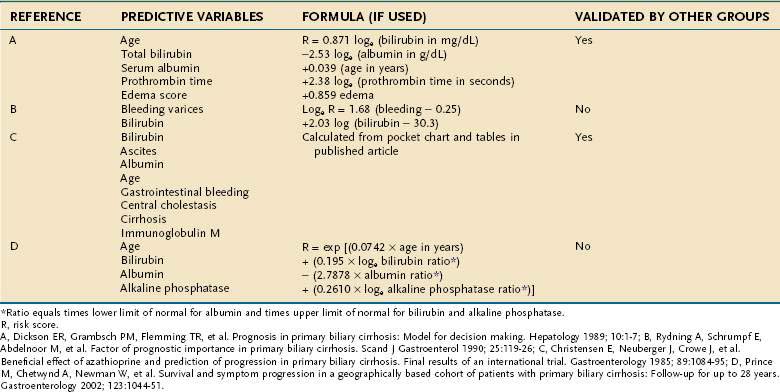
PREDICTING SURVIVAL
When untreated, PBC may follow a course that extends over a 15- to 20-year period. In patients with a serum bilirubin level greater than 10 mg/dL, however, the average life expectancy is reduced to two years. In order to predict survival in patients with PBC, prognostic models, some of which rely on Cox’s proportional hazard analysis, have been developed (Table 89-5). Among these models, the Mayo risk score has been cross-validated and is widely used in predicting survival and in guiding referral of patients for liver transplantation. These prognostic models also can be used for monitoring the efficacy of experimental drugs in clinical trials. Although all the prognostic models are of help in clinical decision-making, they should not replace clinical judgment in determination of the optimal timing of liver transplantation in an individual patient (see Chapter 95).
TREATMENT
URSODEOXYCHOLIC ACID
Because of its safety and patient adherence to treatment with the drug, UDCA has received the most attention of any drug used to treat PBC. Treatment with UDCA leads to rapid improvement in liver biochemical test levels and a decrease in the histologic severity of interface hepatitis, inflammation, cholestasis, bile duct paucity, and bile duct proliferation.34,35 UDCA significantly decreases the risk of development of gastroesophageal varices and ascites and delays progression to cirrhosis.36,37 The predicted probability that cirrhosis will develop after 5 years of therapy with UDCA for patients with stage 1, 2, or 3 disease at diagnosis is 4%, 12%, and 59%, respectively; at 10 years of therapy with UDCA, the probability of cirrhosis is 17%, 27%, and 76%, respectively.37 These figures confirm the beneficial effect of UDCA on delaying progression to cirrhosis, as compared with disease progression in the absence of treatment. Moreover, UDCA reduces proliferation of colonic epithelial cells, and its long-term use in patients with PBC significantly reduces the probability that colorectal adenomas will recur following removal.38
The beneficial effect of UDCA on long-term survival in patients with PBC was questioned in the past on the basis of results of two meta-analyses.39 These meta-analyses had serious methodologic flaws derived primarily from mixing different patient populations, including some in which the duration of treatment—with suboptimal doses of UDCA—was too short to demonstrate an effect.39 When an effective dose of UDCA (13 to 15 mg/kg/day) has been used and an appropriate number of patients received treatment for an appropriate period of time, UDCA has been shown clearly to improve survival free of liver transplantation (Fig. 89-3).40 On the contrary, a more recent meta-analysis that included only randomized, controlled trials in which the doses of UDCA administered and the duration of follow-up were adequate concluded that the frequency of liver transplantation was reduced significantly, with a marginally significant reduction in the rate of death or liver transplantation, but that the rate of death alone was not reduced in the group of patients who received UDCA.41 In addition, in the sensitivity analyses that included studies that administered placebo as control, long-term studies (>48 months), and large-sized studies (>100 patients), long-term treatment with UDCA reduced the frequencies of liver transplantation and death or liver transplantation significantly.41
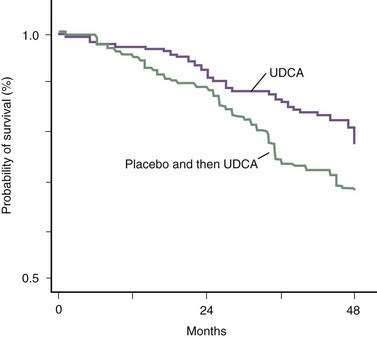
Four reports have provided further data on the long-term effects of UDCA in PBC.42–45 A study of 262 patients who received the standard dose of UDCA daily for a mean of eight years found that survival without liver transplantation was only slightly lower than survival of an age- and gender-matched healthy control population and that treatment with UDCA could normalize the survival rate of patients with PBC when given at early stages.42 Another study of 192 patients with PBC treated with UDCA for a mean of 6.7 years found that patients with PBC who have a biochemical response (decrease of serum alkaline phosphatase levels to less that 40% of the baseline or to normal) after 1 year of treatment had a survival rate better than that predicted by the Mayo risk score and similar to that of a historical control population.43 A study of 297 patients treated with UDCA for a median of 5.7 years showed that survival without liver transplantation in low-risk patients was significantly better than survival predicted by the Mayo risk score and slightly lower than that for an age- and gender-matched control group of the general population.44 Another report evaluated the effect of UDCA on hepatocellular carcinoma and found that the increased risk in UDCA-treated patients with PBC was three-fold, in contrast to an eight-fold increase in patients with PBC not treated with UDCA.45 A few studies have failed to demonstrate a survival benefit in UDCA-treated patients with PBC,8,46,47 but several flaws in study design, most notably small numbers of patients enrolled46 and use of suboptimal doses of UDCA,8,47 preclude meaningful conclusions.48 The experience in the United Kingdom8 is different from that of other European countries in that lower doses (6 to 8 mg/kg/day) of UDCA are used and therapy is started later in the course of the disease.
OTHER DRUGS
Budesonide
Budesonide is a newer glucocorticoid structurally related to 16α-hydroxyprednisolone, with extensive first-pass hepatic metabolism and minimal systemic availability. In a randomized, multicenter trial,49 79 patients with noncirrhotic (stage 1 to 3) PBC were enrolled, and 41 were randomized to treatment with oral budesonide (6 mg daily) in combination with UDCA (15 mg/kg/day), whereas 36 received UDCA alone. At three years of treatment, the combination of budesonide and UDCA led to greater histologic improvement compared with UDCA alone. Side effects of glucocorticoids led to discontinuation of treatment in only one patient, and seven other patients reported mild glucocorticoid-related side effects. In an earlier, open-label study,50 22 patients with PBC who had experienced a suboptimal response to UDCA for a number of years were treated with oral budesonide (3 mg three times daily) for one year. The addition of budesonide to treatment with UDCA was associated with improvement in liver enzyme levels, without affecting other important prognostic markers such as the bilirubin level and Mayo risk score. In that study,50 the addition of budesonide was associated with a significant worsening of osteoporosis and cosmetic effects, particularly in those patients with more advanced (stage 3 to 4) PBC. A pharmacokinetic study showed that the hepatic metabolism of budesonide is markedly reduced in patients with stage 4 PBC compared with those who have stage 1 to 2 disease.51 In that study,51 portal vein thrombosis, possibly related to budesonide, developed in two of the seven patients with stage 4 PBC. Collectively, the data suggest that budesonide may be of potential benefit for patients with early-stage PBC but is associated with important systemic glucocorticoid-related adverse events in patients with more advanced-stage disease. Therefore, before budesonide can be recommended for the treatment of PBC, appropriately designed controlled trials of long-term duration are necessary.
Methotrexate
Patients with PBC who demonstrated clinical, biochemical, and histologic improvement with methotrexate therapy have been described in anecdotal reports. In a placebo-controlled trial of methotrexate for PBC, methotrexate in a dose of 7.5 mg per week for up to six years was not only of no benefit, but also was associated with more unfavorable outcomes than was observed with placebo. A large randomized trial evaluating UDCA (15 mg/kg/day) plus methotrexate (15 mg/m2 of body surface area weekly, maximal dose of 20 mg/week) versus UDCA plus placebo has been reported.52 In that study, 265 patients with PBC and a serum bilirubin level below 3 mg/dL were assigned to one of the two treatment groups; the mean period of study was 7.5 years. The hazard ratio for death with or without liver transplantation was no better in the methotrexate-UDCA combination group than in the UDCA-placebo group. Therefore, methotrexate should not be recommended routinely as monotherapy or as an adjuvant to UDCA.
Other Medications and Combination Therapy
Small pilot, open-label studies of short duration have been reported for mycophenolate mofetil, rituximab, bezafibrate, rifampin, lamivudine plus zidovudine (Combivir), and sulindac.53–56 Although some improvement in liver biochemical test levels was observed with these medications, none of these agents can be recommended outside of clinical trials.
The use of combination therapy with drugs that have different properties has been evaluated in open and controlled trials. Combinations studied include UDCA and methotrexate, UDCA and colchicine, cyclosporine and prednisone, chlorambucil and prednisolone, UDCA and prednisone or prednisolone, UDCA and sulindac, and UDCA, prednisone, and azathioprine. Although some liver biochemical improvement in the short term has been reported with some of these combinations, the small numbers of patients enrolled, short follow-up period, and risk of drug-related side effects do not allow recommendation of any of these combinations for the treatment of PBC. Furthermore, none of these combinations seems to be more effective than UDCA alone. The encouraging results of the combination of UDCA with budesonide in patients with early PBC49 require confirmation in larger, controlled trials.
COMPLICATIONS OF CHRONIC CHOLESTASIS
BONE DISEASE
Osteopenic bone disease with a predisposition to spontaneous fracturing is a common complication of chronic cholestatic liver disease. In North America, most patients with osteopenia from cholestasis have osteoporosis rather than osteomalacia. Osteoporosis is defined as defective bone formation, whereas osteomalacia is defective bone mineralization resulting from vitamin D deficiency. Women with PBC lose bone mass at a rate approximately twice that seen in age-matched controls, and this accelerated bone loss is the result of decreased formation rather than increased resorption of bone. The cause of osteoporosis associated with PBC is poorly understood, but the pathogenesis seems to be related to cholestasis itself. In one study,57 patients with PBC and a rate of bone loss higher than 2% per year were identified as those with more severe cholestasis, whereas in another study,58 bone mass was significantly lower in patients with stage 3 to 4 PBC than in the general population matched for age and gender, but such a difference was not seen for patients with stage 1 to 2 PBC.58 Genetic susceptibility for the development of osteoporosis in PBC has been suggested, including vitamin D receptor gene polymorphisms,57,59–61 collagen type Ia1 gene polymorphisms,57 and insulin-like growth factor-1 gene polymorphisms,62 but contradictory results have been reported and no definitive genetic susceptibility and bone disease in PBC has been confirmed.
The severity and progression of bone disease can be assessed by measurement of bone mineral density in different sites, in particular the lumbar spine and femur. Dual-energy x-ray absorptiometry and dual-photon absorptiometry are noninvasive techniques that quantify bone mass accurately. At the time of referral for or diagnosis of PBC, approximately 20% of patients have osteoporosis, as defined by a T-score below −2.5 in either the lumbar spine or the femoral neck, and approximately 10% have severe bone disease, as defined by a Z-score below −2.58,59 (The T-score is the number of standard deviations below the mean peak value in young gender-matched normal subjects, whereas the Z-score is the number of standard deviations below mean normal values corrected for age and gender.) The risk of osteoporosis (T-score below −2.5) is eight times higher in patients with PBC than in a gender-matched population, whereas the risk of severe bone disease (Z-score below −2) is four times higher in patients with PBC than in a healthy gender- and age-matched population.58
In patients with PBC, as in the general population, older age, postmenopausal status, and lower body-mass index are independent risk factors for the development of osteoporosis. In patients with PBC, however, the severity of osteoporosis increases as liver disease advances; bone mass in patients with stage 1 or 2 PBC is similar to that in a normal age- and gender-matched population, but bone mass is significantly lower in patients with stage 3 or 4 disease.58 Higher serum bilirubin levels, possibly as an indication of more advanced PBC, correlate significantly with a higher rate of bone loss.58 The reported cumulative frequency of fragility fractures in patients with PBC ranges from 10% to 26%, with a cumulative frequency of vertebral fractures of 10% to 20%. One half of patients with PBC who undergo liver transplantation have severe bone disease and one half of patients with PBC (almost exclusively those with preexisting osteopenia) experience a pathologic fracture during the first months after liver transplantation.
Treatment of the bone disease includes adequate exercise and supplemental calcium (1200 to 1500 mg daily orally) and vitamin D (600 to 800 IU daily orally, or if deficiency is present, 25,000 to 50,000 IU orally once or twice per week). Treatment with estrogens significantly prevents loss of bone mass in postmenopausal patients with PBC63 and was not associated with worsening cholestasis in a series of 46 patients with PBC who received estrogen treatment for a mean period of almost five years.63 Because of the carcinogenic properties of estrogens, their lack of protective cardiovascular effects, a possible increased risk of dementia, and resumption of menses, however, postmenopausal women are not enthusiastic about taking estrogens. Raloxifene, a selective estrogen receptor modulator, looks promising as an alternative to estrogen replacement therapy for postmenopausal osteoporosis. Raloxifene was evaluated in a pilot study of nine postmenopausal women with PBC who showed a significant increase in bone mass after one year of treatment with raloxifene; this improvement in bone mass was not seen in an age- and menopausal status-matched control group.64 Raloxifene deserves evaluation in a large controlled trial in patients with PBC. Bisphosphonates also hold promise in the treatment of osteoporosis in patients with PBC. Although etidronate was no better than placebo in one randomized controlled study,65 alendronate (70 mg/wk) was found to improve bone mass significantly after two years of treatment in 13 patients with PBC when compared with etidronate66 and in 15 patients with PBC when compared with 13 patients treated with placebo.67 Other bisphosphonates, including risedronate and pamidronate, deserve evaluation, as do parathormone derivatives.68
HYPERLIPIDEMIA
Lipid abnormalities are found in up to 85% of patients with PBC. High-density lipoprotein (HDL) levels usually are most prominently elevated in the early stages of PBC; as the disease progresses, HDL levels decrease and low-density lipoprotein (LDL) levels increase (see Chapter 72). The risk of atherosclerosis in these patients with hyperlipidemia does not appear to be increased.69 Xanthelasmas (deposits of cholesterol in the skin) may develop in some patients with hyperlipidemia and can be troublesome. Therapy with UDCA has been shown to lower the LDL levels in patients with PBC and has been useful in some patients with xanthelasmas. Surgical removal of xanthelasmas is seldom successful, and such attempts should be avoided. The use of simvastatin has been evaluated in six patients with PBC and hypercholesterolemia.70 In all patients, serum LDL levels decreased, whereas HDL levels remained largely unchanged after two months of therapy; of interest, the serum alkaline phosphatase and gamma glutamyl transpeptidase levels also improved. Further evaluation of simvastatin for PBC is warranted.
PRURITUS
The cause of pruritus in patients with PBC remains an enigma. Various agents may provide symptomatic relief (Table 89-6). The bile acid-binding resin cholestyramine was the first medication described to alleviate this symptom. Therapy with cholestyramine is successful in a majority of patients who can tolerate the unpleasant side effects of bad taste, bloating, and occasional constipation. The recommended total dose is 3 to 12 g/day orally, and the drug is most effective when one half of the dose is given 30 minutes before and one half is given 30 minutes after breakfast, to permit maximal bile acid binding as the gallbladder empties. All drugs that can potentially bind to cholestyramine should be taken several hours before or after the cholestyramine. Other bile acid sequestrants such as colestipol and colesevelam have not been studied.
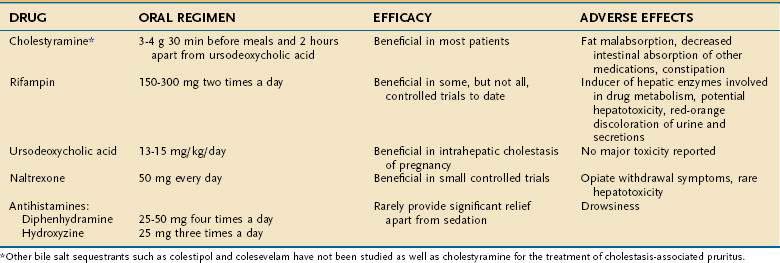
Occasionally, treatment with UDCA alleviates pruritus, although on occasion pruritus may worsen with initiation of UDCA. In warm countries, exposure to ultraviolet light without sun block can alleviate pruritus, and not surprisingly, the pruritus of PBC subsides during the summer months. The hypothesis has been proposed that pruritus may be related to the release of endogenous opioids. Intravenous infusion of the opiate receptor antagonist naloxone has shown a clear benefit in a double-blind trial. Oral opiate receptor antagonists such as nalmefene and naltrexone have led to amelioration of pruritus in patients with PBC, although further trials are needed to evaluate their safety. The serotonin antagonist ondansetron decreased the pruritus associated with cholestasis in studies using subjective methodology but not in a study of patients with PBC that applied behavior methodology (i.e., measurement of scratching activity).71 The serotonin reuptake inhibitor sertraline (75 to 100 mg orally) was associated with relief of pruritus as assessed by a visual analog scale and healing of excoriations as assessed by physical examination in a randomized, placebo-controlled trial.72 Because of their sedative effects, antihistamines such as diphenhydramine and hydroxyzine are helpful for treating the insomnia associated with pruritus, which is always more troublesome at night. Phenobarbital may have the same effect. The pruritus of PBC is almost always cured by liver transplantation, which is a viable option for patients with severe intractable pruritus (see Table 89-6).
STEATORRHEA
Steatorrhea can occur in patients with advanced PBC. Several causes have been described. The most important cause is decreased bile acid delivery with insufficient micellar concentration of bile acids in the small intestine (see Chapters 64 and 101). Occasionally, exocrine pancreatic insufficiency can be found as part of a widespread glandular dysfunction seen in some patients with PBC. Coexisting celiac disease has been reported in a small number of patients with PBC, and small intestinal bacterial overgrowth may be the cause of steatorrhea in some patients with PBC and scleroderma. Because each of these causes has specific and different treatments, determining the exact cause of steatorrhea is important. Patients with decreased intestinal bile acid concentrations usually benefit from substitution of medium-chain triglycerides for long-chain triglycerides in their diets and a decrease in total fat intake. Patients with exocrine pancreatic insufficiency will benefit from pancreatic replacement therapy; patients with celiac disease require gluten withdrawal from the diet; and patients with small intestinal bacterial overgrowth should receive intermittent broad-spectrum oral antibiotic therapy.
LIVER TRANSPLANTATION
The best therapeutic alternative for patients with end-stage PBC is liver transplantation (see Chapter 95). The major manifestations of chronic liver disease that should prompt an evaluation for liver transplantation in patients with other causes of chronic liver disease apply to patients with PBC. These indications include complications related to portal hypertension, including bleeding from gastroesophageal varices, diuretic-resistant ascites, hepatorenal syndrome, and hepatic encephalopathy. In patients with PBC, the development of complications associated with chronic cholestasis, such as a poor quality of life secondary to disabling fatigue, intractable pruritus, and severe muscle wasting, as well as persistent increases in the serum bilirubin level in the absence of hepatic malignancy, should prompt clinicians to consider referral for liver transplantation, even in patients without cirrhosis on a liver biopsy specimen.
Data from the United Network for Organ Sharing show a clear trend toward decreased rates of liver transplantation for PBC.73 From 1995 to 2006, the absolute number of liver transplants in the United States increased an average of 249 cases per year, but the absolute number of transplants performed for PBC decreased by an average of 5.4 cases per year,73 despite the steady increase in the incidence and prevalence of PBC.2 A similar trend has been observed in Europe, where the proportion of liver transplants performed in patients with PBC decreased from 55% in the early years of transplantation (late 1980s) to 11% in 2006.73 Because UDCA is now prescribed nearly universally to patients with PBC, the decline in the number of liver transplants for PBC is consistent with improved survival resulting from treatment with UDCA.
Liver transplantation clearly improves survival, as well as quality of life, for patients with PBC. One-year survival rates after liver transplantation are currently higher than 90%, with five-year survival rates higher than 80% in most transplant centers. The advent of prognostic models has helped identify factors that predict survival (see Table 89-5). The Mayo risk score (http://www.mayoclinic.org/gi-rst/mayomodel2.html), which is based on the patient’s age, serum bilirubin level, serum albumin level, prothrombin time, presence of edema, and need for treatment with diuretics is superior to the Child-Turcotte-Pugh score in predicting survival. The Model for End-stage Liver Disease (MELD) score (http://www.mayoclinic.org/meld/mayomodel6.html) is also a reliable measure of mortality risk in patients with end-stage liver disease, including PBC, and is used as a disease severity index to determine organ allocation priorities (see Chapter 95).75
PBC-specific autoantibodies against mitochondria (AMA) and gp210 protein generally persist in a patient’s serum after liver transplantation. PBC may recur in the allograft, with a frequency of recurrence ranging from 11% to 34% and a median time to diagnosis of recurrence of 36 to 61 months after transplantation.76–81 The risk of recurrence increases with time, so that by 10 years, histologic recurrence may be found in 30% to 50% of patients. Tacrolimus-based immunosuppression has been the most consistently identified risk factor for disease recurrence, although older recipient age and male gender of the recipient also have been identified as risk factors for recurrence. One study found that the use of UDCA in patients with recurrent PBC improves serum alkaline phosphatase and aminotransferase levels at three years post-transplantation but has no effect on progression of histologic stage.81 Recurrent PBC following liver transplantation does not seem to decrease survival significantly, although in some studies a small proportion of patients had graft failure.
AMA-NEGATIVE PRIMARY BILIARY CIRRHOSIS
As discussed earlier, AMA are detected in the serum of 90% of patients with PBC by the indirect immunofluorescence technique and in 95% of patients by the immunoblotting technique. AMA-negative PBC is the designation for those patients who clinically, biochemically, and histologically appear to have the classic features of PBC but are found not to have AMA in serum by indirect immunofluorescence or immunoblotting techniques. Of patients who have PBC by all other criteria, 5% are confirmed AMA negative.82 Terms such as “autoimmune cholangitis” and “autoimmune cholangiopathy” have been used in the past to describe this entity, but the current consensus holds that there is no practical difference between AMA-positive and AMA-negative PBC and, therefore, no need for an alternative label to describe AMA-negative PBC.83
Most patients with AMA-negative PBC have antinuclear (perinuclear/rim-like or multiple nuclear dot pattern) or smooth muscle antibodies (or both). Although these patients may be distinguished by the lack of AMA in serum, the specific AMA antigen PDC-E2 is expressed on the apical region of their biliary epithelium, as occurs in AMA-positive patients—an observation suggesting that the pathogenesis of both conditions may be identical. Whether a different genetic susceptibility exists for the development of AMA-positive PBC and AMA-negative PBC is still uncertain. In one study, the frequencies of HLA DRβ1*08 and DQβ1*04 were reduced significantly in patients with AMA-negative PBC as compared with AMA-positive patients.84 When regulatory T cells and the subgroup of T cells suggested to have a role in the genesis of autoimmune disease were examined in patients with PBC, however, no difference was found between those who had and those who did not have AMA.85
Patents with AMA-negative PBC tend to follow a clinical course, and to demonstrate a therapeutic response to UDCA, similar to those in AMA-positive patients.83,86 When a patient with AMA-negative PBC is evaluated, other diseases that may manifest in a similar manner should be excluded. The absence of AMA makes liver biopsy mandatory to look for features of PBC and rule out other liver diseases. Also, imaging by magnetic resonance cholangiopancreatography (MRCP) is essential to identify other cholangiopathies such as primary sclerosing cholangitis. Liver biopsy and MRCP, along with select laboratory tests, will allow the exclusion of conditions that should be considered in the differential diagnosis, such as celiac disease, hepatitis C, sarcoidosis, small-duct primary sclerosing cholangitis, and IgG4-associated autoimmune cholangitis (see Chapter 68).
Patients with AMA-negative PBC should be treated with UDCA in a dose of 13 to 15 mg/kg/day. When histologic features of superimposed autoimmune hepatitis are detected, the combination of glucocorticoids and UDCA should be considered (see Chapter 88).86
Invernizzi P, Selmi C, Mackay IR, et al. From bases to basis: Linking genetics to causation in primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2005;3:401-10. (Ref 19.)
Jackson H, Solaymani-Dodaran M, Card TR, et al. Influence of ursodeoxycholic acid on the mortality and malignancy associated with primary biliary cirrhosis: A population-based cohort study. Hepatology. 2007;46:1131-7. (Ref 45.)
Jacob DA, Neumann UP, Bahra M, et al. Long-term follow-up after recurrence of primary biliary cirrhosis after liver transplantation in 100 patients. Clin Transplant. 2006;20:211-20. (Ref 80.)
Jones DE, Bhala N, Burt J, et al. Four year follow-up of fatigue in a geographically defined primary biliary cirrhosis cohort. Gut. 2006;55:536-41. (Ref 31.)
Lazaridis KN, Juran BD, Boe GM, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785-92. (Ref 5.)
Mayo MJ, Handem I, Saldana S, et al. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology. 2007;45:666-74. (Ref 72.)
Menon KV, Angulo P, Weston S, et al. Bone disease in patients with primary biliary cirrhosis: Independent predictors and rate of progression. J Hepatol. 2001;35:316-23. (Ref 58.)
Pares A, Caballeria J, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130:715-20. (Ref 43.)
Poupon RE, Lindor KD, Pares A, et al. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol. 2003;39:12-16. (Ref 35.)
Prince M, James OFW. The epidemiology of primary biliary cirrhosis. Clin Liver Dis. 2003;7:795-819. (Ref 2.)
Zein CO, Angulo P, Lindor KD. When is liver biopsy needed in the diagnosis of primary biliary cirrhosis? Clin Gastroenterol Hepatol. 2003;1:89-95. (Ref 32.)
Zein C, Jorgensen RA, Clarke B, et al. Alendronate improves bone mineral density in patients with PBC: A randomized placebo-controlled trial. Hepatology. 2005;42:762-71. (Ref 67.)
1. Dahlan Y, Smith L, Simmonds D, et al. Pediatric-onset primary biliary cirrhosis. Gastroenterology. 2003;125:1476-9.
2. Prince M, James OFW. The epidemiology of primary biliary cirrhosis. Clin Liver Dis. 2003;7:795-819.
3. Kim WR, Lindor KD, Locke GRIII, et al. Epidemiology and natural history of primary biliary cirrhosis in a U.S. community. Gastroenterology. 2000;119:1631-6.
4. Watt FE, James OF, Jones DE. Patterns of autoimmunity in primary biliary cirrhosis patients and their families: A population-based cohort study. QJM. 2004;97:397-406.
5. Lazaridis KN, Juran BD, Boe GM, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785-92.
6. Koarada S, Wu Y, Fertig N, et al. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol. 2004;173:2315-23.
7. Oertelt S, Lian ZX, Cheng CM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655-60.
8. Wakabayashi K, Lian ZX, Moritoki Y, et al. IL-2 receptor alpha(-/-) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240-9.
9. Leung PS, Quan C, Park O, et al. Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol. 2003;170:5326-32.
10. Rieger R, Gershwin ME. The X and why of xenobiotics in primary biliary cirrhosis. J Autoimmun. 2007;28:76-84.
11. Kikuchi K, Lian ZX, Yang GX, et al. Bacterial CpG induces hyper-IgM production in CD27(+) memory B cells in primary biliary cirrhosis. Gastroenterology. 2005;128:304-12.
12. Muratori L, Muratori P, Granito A, et al. The Western immunoblotting pattern of anti-mitochondrial antibodies is independent of the clinical expression of primary biliary cirrhosis. Dig Liver Dis. 2005;37:108-12.
13. Muratori P, Muratori L, Ferrari R, et al. Characterization and clinical impact of antinuclear antibodies in primary biliary cirrhosis. Am J Gastroenterol. 2003;98:431-7.
14. Vergani D, Bogdanos DP. Positive markers in AMA-negative PBC. [comment, editorial]. Am J Gastroenterol. 2003;98:241-3.
15. Granito A, Muratori P, Muratori L, et al. Antinuclear antibodies giving the “multiple nuclear dots” or the “rim-like/membranous” patterns: Diagnostic accuracy for primary biliary cirrhosis. Aliment Pharmacol Therap. 2006;24:1575-83.
16. Invernizzi P, Podda M, Battezzati PM, et al. Autoantibodies against nuclear pore complexes are associated with more active and severe liver disease in primary biliary cirrhosis. J Hepatol. 2001;34:366-72.
17. Miyachi K, Hankins RW, Matsushima H, et al. Profile and clinical significance of anti-nuclear envelope antibodies found in patients with primary biliary cirrhosis: A multicenter study. J Autoimmun. 2003;20:247-54.
18. Nakamura M, Kondo H, Mori T, et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology. 2007;45:118-27.
19. Invernizzi P, Selmi C, Mackay IR, et al. From bases to basis: Linking genetics to causation in primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2005;3:401-10.
20. Donaldson PT, Baragiotta A, Heneghan MA, et al. HLA class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: A large-scale study. Hepatology. 2006;44:667-74.
21. Mullarkey ME, Stevens AM, McDonnell WM, et al. Human leukocyte antigen class II alleles in Caucasian women with primary biliary cirrhosis. Tissue Antigens. 2005;65:199-205.
22. Liu HY, Deng AM, Zhou Y, et al. Analysis of HLA alleles polymorphism in Chinese patients with primary biliary cirrhosis. Hepatobiliary Pancreat Dis Int. 2006;5:129-32.
23. Invernizzi P, Battezzati PM, Crosignani A, et al. Peculiar HLA polymorphisms in Italian patients with primary biliary cirrhosis. J Hepatol. 2003;38:401-6.
24. Abdulkarim AS, Petrovic LM, Kim WR, et al. Primary biliary cirrhosis: An infectious disease caused by Chlamydia pneumoniae? J Hepatol. 2004;40:380-4.
25. Selmi C, Balkwill DL, Invernizzi P, et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology. 2003;38:1250-7.
26. Xu L, Sakalian M, Shen Z, et al. Cloning the human beta retrovirus proviral genome from patients with primary biliary cirrhosis. Hepatology. 2004;39:151-6.
27. Medzhitov R, Janeway CAJr. How does the immune system distinguish self from nonself? Semin Immunol. 2000;12:185-8.
28. Long SA, Quan C, van de Water J, et al. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: Connecting xenobiotics with primary biliary cirrhosis. J Immunol. 2001;167:2956-63.
29. Goldblatt J, Taylor PJS, Lipman T, et al. The true impact of fatigue in primary biliary cirrhosis: A population study. Gastroenterology. 2002;122:1235-41.
30. Prince M, Chetwynd A, Newman W, et al. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: Follow-up for up to 28 years. Gastroenterology. 2002;123:1044-51.
31. Jones DE, Bhala N, Burt J, et al. Four year follow-up of fatigue in a geographically defined primary biliary cirrhosis cohort. Gut. 2006;55:536-41.
32. Zein CO, Angulo P, Lindor KD. When is liver biopsy needed in the diagnosis of primary biliary cirrhosis? Clin Gastroenterol Hepatol. 2003;1:89-95.
33. Prince M, Chetwynd A, Craig JV, et al. Asymptomatic primary biliary cirrhosis: Clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53:865-70.
34. Pares A, Caballeria L, Rodes J, et al. Long-term effects of ursodeoxycholic acid in primary biliary cirrhosis: Results of a double-blind controlled multicentric trial. J Hepatol. 2000;32:561-6.
35. Poupon RE, Lindor KD, Pares A, et al. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol. 2003;39:12-6.
36. Lindor KD, Jorgensen RA, Therneau TM, et al. Ursodeoxycholic acid delays the onset of esophageal varices in primary biliary cirrhosis. Mayo Clin Proc. 1997;72:1137-40.
37. Corpechot C, Carrat F, Poupon R, et al. Primary biliary cirrhosis: Incidence and predictive factors of cirrhosis development in ursodiol-treated patients. Gastroenterology. 2002;122:652-8.
38. Serfaty L, De Leusse A, Rosmorduc O, et al. Ursodeoxycholic acid therapy and the risk of colorectal adenoma in patients with primary biliary cirrhosis: An observational study. Hepatology. 2003;38:203-9.
39. Lindor KD, Poupon R, Poupon RE, et al. Ursodeoxycholic acid for primary biliary cirrhosis. Lancet. 2000;355:657-8.
40. Poupon R, Lindor KD, Cauch-Dudek K, et al. Combined analysis of French, American and Canadian randomized controlled trials of ursodeoxycholic acid therapy in primary biliary cirrhosis. Gastroenterology. 1997;113:884-90.
41. Shi J, Wu C, Lin Y, et al. Long-term effects of mid-dose ursodeoxycholic acid in primary biliary cirrhosis: A meta-analysis of randomized controlled trials. Am J Gastroenterol. 2006;101:1529-38.
42. Corpechot C, Carrat F, Bahr A, et al. The effect of ursodeoxycholic acid therapy on the natural history of primary biliary cirrhosis. Gastroenterology. 2005;128:297-03.
43. Pares A, Caballeria J, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130:715-20.
44. ter Borg PC, Schalm SW, Hansen BE, et al. Prognosis of ursodeoxycholic acid-treated patients with primary biliary cirrhosis. Results of a 10-yr cohort study involving 297 patients. Am J Gastroenterol. 2006;l:2044-50.
45. Jackson H, Solaymani-Dodaran M, Card TR, et al. Influence of ursodeoxycholic acid on the mortality and malignancy associated with primary biliary cirrhosis: A population-based cohort study. Hepatology. 2007;46:1131-37.
46. Papatheodoridis GV, Hadziyannis ES, Deutsch M, et al. Ursodeoxycholic acid for primary biliary cirrhosis: Final results of a 12-year, prospective, randomized, controlled trial. Am J Gastroenterol. 2002;97:2063-70.
47. Combes B, Luketic VA, Peters MG, et al. Prolonged follow-up of patients in the U.S. multicenter trial of ursodeoxycholic acid for primary biliary cirrhosis. Am J Gastroenterol. 2004;99:264-8.
48. Corpechot C, Poupon R. Geotherapeutics of primary biliary cirrhosis: Bright and sunny around the Mediterranean but still cloudy and foggy in the United Kingdom. Hepatology. 2007;46:963-5.
49. Rautiainen H, Karkkainen P, Karvonen AL, et al. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: A three-year randomized trial. Hepatology. 2005;41:747-52.
50. Angulo P, Jorgensen RA, Keach JC, et al. Oral budesonide in the treatment of patients with primary biliary cirrhosis with a suboptimal response to ursodeoxycholic acid. Hepatology. 2000;31:318-23.
51. Hempfling W, Grunhage F, Dilger K, et al. Pharmacokinetics and pharmacodynamic action of budesonide in early- and late-stage primary biliary cirrhosis. Hepatology. 2003;38:196-02.
52. Combes B, Emerson SS, Flye NL, et al. Methotrexate (MX) plus ursodeoxycholic acid (UDCA) in the treatment of primary biliary cirrhosis. Hepatology. 2005;42:1184-93.
53. Talwalkar JA, Angulo P, Keach JC, et al. Mycophenolate mofetil for the treatment of primary biliary cirrhosis in patients with an incomplete response to ursodeoxycholic acid. J Clin Gastroenterol. 2005;39:168-71.
54. Meyers RP, Shaheen AA, Swain MG, et al. Rituximab for primary biliary cirrhosis (PBC) refractory to ursodeoxycholic acid (UDCA). Hepatology. 2007;46:550A.
55. Leuschner M, Holtmeier J, Ackermann H, et al. The influence of sulindac on patients with primary biliary cirrhosis that respond incompletely to ursodeoxycholic acid: A pilot study. Eur J Gastroenterol Hepatol. 2002;14:1369-76.
56. Mason AL, Farr GH, Xu L, et al. Pilot studies of single and combination of retroviral therapy in patients with primary biliary cirrhosis. Am J Gastroenterol. 2004;99:2348-55.
57. Pares A, Guanabens N, Alvarez L, et al. Collagen type Ia1 and vitamin D receptor gene polymorphisms and bone mass in primary biliary cirrhosis. Hepatology. 2001;33:554-60.
58. Menon KV, Angulo P, Weston S, et al. Bone disease in patients with primary biliary cirrhosis: Independent predictors and rate of progression. J Hepatol. 2001;35:316-23.
59. Springer JE, Cole DE, Rubin LA, et al. Vitamin D-receptor genotypes as independent genetic predictors of decreased bone mineral density in primary biliary cirrhosis. Gastroenterology. 2000;118:145-51.
60. Lakatos LP, Bajnok E, Hegedus D, et al. Vitamin D receptor Bsm I, estrogen receptor alpha gene Pvu and Xba I and IL-1 receptor-antagonist gene VNRT polymorphism and bone mineral density in Hungarian patients with primary biliary cirrhosis. Eur J Gastroenterol Hepatol. 2002;14:733-40.
61. Vogel A, Strassburg GP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis. Hepatology. 2002;35:126-31.
62. Lakatos LP, Bajnok E, Tornai I, et al. Insulin-like growth factor I (IGF-I) gene microsatellite repeat, collagen type Ia1 gene (COLIA 1) Sp1 polymorphism and bone disease in primary biliary cirrhosis. Eur J Gastroenterol Hepatol. 2004;16:753-9.
63. Menon KV, Angulo P, Boe G, et al. Safety and efficacy of estrogen therapy in preventing bone loss in primary biliary cirrhosis. Am J Gastroenterol. 2003;98:889-92.
64. Levy C, Harnois DM, Angulo P, et al. Raloxifene improves bone mass in osteopenic women with primary biliary cirrhosis: Results of a pilot study. Liver Int. 2003;25:117-21.
65. Lindor KD, Jorgensen RA, Tiegs RD, et al. Etidronate for osteoporosis in primary biliary cirrhosis. A randomized trial. J Hepatol. 2000;33:878-82.
66. Guanabens N, Pares A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol. 2003;98:2268-74.
67. Zein C, Jorgensen RA, Clarke B, et al. Alendronate improves bone mineral density in patients with PBC: A randomized placebo-controlled trial. Hepatology. 2005;42:762-71.
68. Greenspan SL, Bone HG, Ettinger MP, et al. Effect of recombinant human parathyroid hormone (1-84) on vertebral fracture and bone mineral density in postmenopausal women with osteoporosis: A randomized trial. Ann Intern Med. 2007;146:326-39.
69. Longo M, Crosignani A, Battezzati PM, et al. Hyperlipidaemic state and cardiovascular risk in primary biliary cirrhosis. Gut. 2002;51:265-9.
70. Ritzel U, Leonhardt U, Nather M, et al. Simvastatin in primary biliary cirrhosis: Effects on serum lipids and distinct disease markers. J Hepatol. 2002;36:454-8.
71. Jones EA, Molenaar HA, Oosting J. Ondansetron in the treatment of pruritus of cholestasis: A randomized controlled trial. Hepatogastroenterology. 2007;54:1196-9.
72. Mayo MJ, Handem I, Saldana S, et al. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology. 2007;45:666-74.
73. Lee J, Belanger A, Doucette JT, et al. Transplantation trends in primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2007;5:1313-15.
74. European liver transplant registry. http://www.eltr.org/publi/index_rv.php3.
75. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33:464-70.
76. Liermann-Garcia RF, Evangelista Garcia C, McMaster P, et al. Transplantation for primary biliary cirrhosis: Retrospective analysis of 400 patients in a single center. Hepatology. 2001;33:22-7.
77. Sanchez EQ, Levy MF, Goldstein RM, et al. The changing clinical presentation of recurrent primary biliary cirrhosis after liver transplantation. Transplantation. 2003;76:1583-88.
78. Sylvestre PB, Batts KP, Burgart LJ, et al. Recurrence of primary biliary cirrhosis after liver transplantation: Histologic estimate of incidence and natural history. Liver Transpl. 2003;9:1086-93.
79. Neuberger J, Gunson B, Hubscher S, et al. Immunosuppression affects the rate of recurrent primary biliary cirrhosis after liver transplantation. Liver Transpl. 2004;10:488-91.
80. Jacob DA, Neumann UP, Bahra M, et al. Long-term follow-up after recurrence of primary biliary cirrhosis after liver transplantation in 100 patients. Clin Transplant. 2006;20:211-20.
81. Charatcharoenwitthaya P, Pimentel S, Talwalkar JA, et al. Long-term survival and impact of ursodeoxycholic acid treatment for recurrent primary biliary cirrhosis after liver transplantation. Liver Transpl. 2007;13:1236-45.
82. Oertelt S, Rieger R, Selmin C, et al. A sensitive bead assay for antimitochondrial antibodies chipping away at AMA-negative primary biliary cirrhosis. Hepatology. 2007;45:659-65.
83. Heathcote EJ. Management of primary biliary cirrhosis. The American Association for the Study of Liver Diseases practice guidelines. Hepatology. 2000;31:1005-13.
84. Stone J, Wade JA, Cauch-Dudek K, et al. Human leukocyte antigen class II associations in serum antimitochondrial antibodies (AMA)-positive and AMA-negative primary biliary cirrhosis. J Hepatol. 2002;36:8-13.
85. Lan RY, Cheng C, Lian ZX, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology. 2006;43:729-37.
86. Gisbert JP, Jones EA, Pajares JM, et al. Review article. Is there an optimal therapeutic regimen for antimitochondrial antibody-negative primary biliary cirrhosis (autoimmune cholangitis)? Aliment Pharmacol Ther. 2003;17:17-27.