[level-membership-for-basic-science-category]
CHAPTER 21 Prenatal Testing and Reproductive Genetics
Until recently, couples at high risk of having a child with a genetic disorder had to choose between taking the risk or considering other reproductive options, such as long-term contraception, sterilization, and termination of pregnancy. Other alternatives included adoption, long-term fostering, and donor insemination (DI).
The ethical issues surrounding prenatal diagnosis and selective termination of pregnancy are both complex and emotive, and are considered more fully in Chapter 24 (p. 363). In this chapter, we focus on the practical aspects of prenatal testing and diagnosis, including prenatal screening, as well as some aspects of reproductive genetics.
Techniques Used in Prenatal Diagnosis
There are several techniques that can be used for the prenatal diagnosis of hereditary disorders and structural abnormalities (Table 21.1).
| Technique | Optimal Time (Weeks) | Disorders Diagnosed |
|---|---|---|
| Non-Invasive | ||
| Maternal serum screening | ||
| α-Fetoprotein | 16 | Neural tube defects |
| Triple test | 16 | Down syndrome |
| Ultrasound | 18 | Structural abnormalities (e.g., central nervous system, heart, kidneys, limbs) |
| Invasive | ||
| Amniocentesis | 16 | |
| Fluid | Neural tube defects | |
| Cells | Chromosome abnormalities, metabolic disorders, molecular defects | |
| Chorionic villus sampling | 10–12 | Chromosome abnormalities, metabolic disorders, molecular defects |
| Fetoscopy | ||
| Blood (cordocentesis) | Chromosome abnormalities, hematological disorders, congenital infection | |
| Liver | Metabolic disorders (e.g., ornithine transcarbamylase deficiency) | |
| Skin | Hereditary skin disorders (e.g., epidermolysis bullosa) | |
Amniocentesis
Amniocentesis involves the aspiration of 10 to 20 ml of amniotic fluid through the abdominal wall under ultrasonographic guidance (Figure 21.1). This is usually performed around the 16th week of gestation. The sample is spun down to yield a pellet of cells and supernatant fluid. The fluid can be used in the prenatal diagnosis of neural tube defects by assay of α-fetoprotein (p. 328). The cell pellet is resuspended in culture medium with fetal calf serum, which stimulates cell growth. Most of these cells in the amniotic fluid, that have been shed from the amnion, fetal skin, and urinary tract epithelium, are non-viable, but a small proportion will grow. After approximately 14 days, there are usually sufficient cells for chromosome and DNA analysis, although a longer period may be required before enough cells are obtained for biochemical assays. Increasingly, sensitive polymerase chain reaction (PCR) techniques make direct DNA analysis possible without the need for culture.
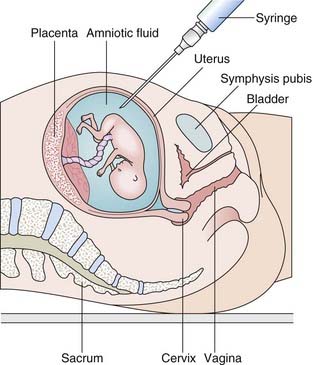
Chorionic Villus Sampling
In contrast to amniocentesis, chorionic villus sampling (CVS), first developed in China, enables prenatal diagnosis to be undertaken during the first trimester. This procedure is usually carried out at 11 to 12 weeks’ gestation under ultrasonographic guidance by either transcervical or, more usually, transabdominal aspiration of chorionic villus (CV) tissue (Figure 21.2). This tissue is fetal in origin, being derived from the outer cell layer of the blastocyst (i.e., the trophoblast). Maternal decidua, normally present in the biopsy sample, must be removed before the sample is analyzed. Placental biopsy is the term used when the procedure is carried out at later stages of pregnancy.
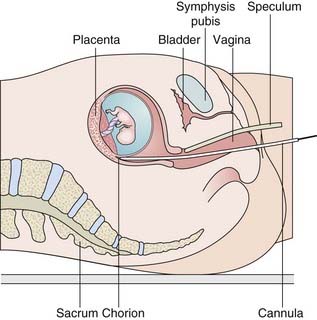
Chromosome analysis can be undertaken on CV tissue either directly, looking at metaphase spreads from actively dividing cells, or after culture. Direct chromosomal analysis of CV tissue usually allows a provisional result to be given within 24 hours. Nowadays rapid, direct fluorescent in-situ hybridization (FISH) probing (p. 34), or DNA analysis by the multiplex ligation-dependent probe amplification technique (p. 66), is used to test for common chromosome aneuploidies prior to a standard karyotype following culture of CV tissue; this may also detect other chromosome abnormalities and balanced rearrangements. For single-gene disorders, sufficient CV tissue is usually obtained to allow prenatal diagnosis by immediate biochemical assay or DNA analysis using uncultured CV tissue.
Ultrasonography
Ultrasonography offers a valuable means of prenatal diagnosis. It can be used not only for obstetric indications, such as placental localization and the diagnosis of multiple pregnancies, but also for prenatal diagnosis of structural abnormalities not associated with known chromosomal, biochemical or molecular defects. Ultrasonography is particularly valuable because it is non-invasive and conveys no known risk to the fetus or mother. It does, however, require expensive equipment and a skilled, experienced operator. For example, a search can be made for polydactyly as a diagnostic feature of a multiple abnormality syndrome, such as one of the autosomal recessive short-limb polydactyly syndromes that are associated with severe pulmonary hypoplasia—invariably lethal (Figure 21.3). Similarly, a scan can reveal that the fetus has a small jaw, which can be associated with a posterior cleft palate and other more serious abnormalities in several single-gene syndromes (Figure 21.4).
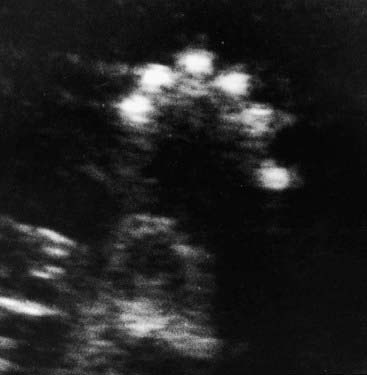
FIGURE 21.3 Ultrasonographic image of a transverse section of the hand of a fetus showing polydactyly.
Nuchal Translucency
In addition, the observation that increased nuchal translucency (NT) is seen in fetuses who are subsequently born with Down syndrome has resulted in the introduction of measurements of nuchal pad thickness (Figure 21.5) in the first and second trimesters as part of screening for Down syndrome (p. 273). In fact, the finding is not specific and may be seen in various chromosomal anomalies as well as isolated congenital heart disease.
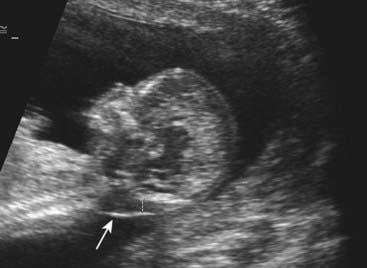
Fetoscopy
Fetoscopy has also been used to obtain samples of tissue from the fetus that can be analyzed as a means of achieving the prenatal diagnosis of several rare disorders. These have included inherited skin disorders such as epidermolysis bullosa and, before DNA testing became available, metabolic disorders in which the enzyme is expressed only in certain tissues or organs, such as the liver—e.g., ornithine transcarbamylase deficiency (p. 172).
Cordocentesis
Although fetoscopy can also be used to obtain a small sample of fetal blood from one of the umbilical cord vessels in the procedure known as cordocentesis, improvements in ultrasonography have enabled visualization of the vessels in the umbilical cord, allowing transabdominal percutaneous fetal blood sampling. Fetal blood sampling is used routinely in the management of rhesus iso-immunization (p. 205) and can be used to obtain samples for chromosome analysis to resolve problems associated with possible chromosomal mosaicism in CV or amniocentesis samples.
Radiography
The fetal skeleton can be visualized by radiography from 10 weeks onwards, and this technique has been used in the past to diagnose inherited skeletal dysplasias. It is now employed only occasionally because of the dangers of radiography to the fetus (p. 26) and the widespread availability of detailed ultrasonography.
Prenatal Screening
The history of widespread prenatal (antenatal) screening really began with the finding, in the early 1970s, of an association between raised maternal serum α-fetoprotein (AFP) and neural tube defects (NTDs). Estimation of AFP levels was gradually introduced into clinical service, and the next significant development was ultrasonography, followed, in the 1980s, by the identification of maternal serum biochemical markers for Down syndrome. These are discussed in more detail below. Where the incidence of a genetic condition was high, for instance thalassemia in Cyprus, prenatal screening came into practice, as described in Chapter 20 (p. 321). However, molecular genetic advances, rather than biochemical, mean that the range of prenatal screening is continuing to evolve.
Testing for cystic fibrosis and fragile X syndrome are available in the UK, mainly for those willing to pay privately, and in Israel, for example, a wide range of relatively rare diseases can be screened for on the basis that they are more common in specific population groups that were originally isolates with multiple inbreeding, and therefore certain mutations are prevalent. Besides Tay-Sachs disease (carrier testing in this case is biochemical; see Chapter 20), familial dysautonomia, Canavan disease, Bloom syndrome, ataxia telangiectasia (North African Jews), limb-girdle muscular dystrophy (Libyan Jews) and Costeff syndrome (Iraqi Jews) are among the conditions for which screening is available. It does not come free of charge but the level of uptake of this screening is high, revealing the lengths to which some societies will go in order to avoid having children with serious genetic conditions. As DNA testing becomes more automated, rapid, and affordable, there will be pressure from some quarters to screen for many conditions, even though they are individually very rare. This challenge is already emerging with the potential use of microarray CGH in prenatal testing. If the use of microarray becomes routine there may be great difficulty in interpreting the consequences of rare or unique copy number variants. This ethical challenge is discussed more fully in Chapter 24.
Neural Tube Defects
In 1972 it was recognized that many pregnancies in which the baby had an open NTD (p. 258) could be detected at 16 weeks’ gestation by assay of AFP in maternal serum. AFP is the fetal equivalent of albumin and is the major protein in fetal blood. If the fetus has an open NTD, the level of AFP is raised in both the amniotic fluid and maternal serum as a result of leakage from the open defect. Open NTDs fulfil the criterion of being serious disorders, as anencephaly is invariably fatal, and between 80% and 90% of the small proportion of babies who survive with an open lumbosacral lesion are severely handicapped.
Unfortunately maternal serum AFP screening for NTDs is neither 100% sensitive nor 100% specific (p. 319). The curves for the levels of maternal serum AFP in normal and affected pregnancies overlap (Figure 21.6), so that in practice an arbitrary cut-off level has to be introduced below which no further action is taken. This is usually either the 95th centile, or 2.5 multiples of the median (MoM); as a result around 75% of screened open spina bifida cases are detected. Those pregnant women with results that lie above this arbitrary cut-off level are offered detailed ultrasonography; which is usually sufficient to diagnose NTD. In fact, ultrasonography has more or less superceded maternal serum screening as a means of diagnosing NTD. Anencephaly shows a dramatic deficiency in the cranium (Figure 21.7) and an open myelomeningocele is almost invariably associated with herniation of the cerebellar tonsils through the foramen magnum. This deforms the cerebellar hemispheres, which then have a curved appearance known as the ‘banana sign’; the forehead is also distorted, giving rise to a shape referred to as the ‘lemon sign’ (Figure 21.8). A posterior encephalocele is readily visualized as a sac in the occipital region (Figure 21.9) and always prompts a search for additional anomalies that might help diagnose a recognizable condition such as Meckel-Gruber syndrome.
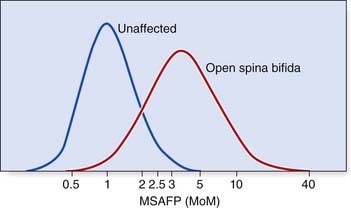
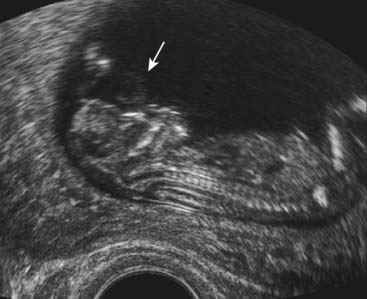
FIGURE 21.7 Anencephaly (arrow). There is no cranium and this form of neural tube defect is incompatible with life.
(Courtesy Dr. Helen Liversedge, Exeter, UK.)
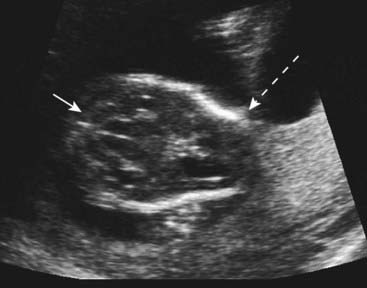
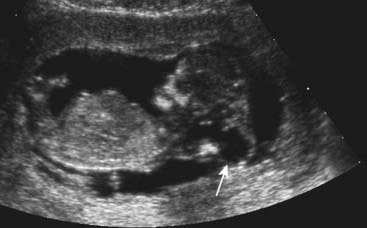
A raised maternal serum AFP concentration is not specific for open NTDs (Box 21.1). Other causes include threatened miscarriage, twin pregnancy and a fetal abnormality such as exomphalos, in which there is a protrusion of abdominal contents through the umbilicus.
As a result of these screening modalities there has been a striking decline in the incidence of open NTDs in liveborn and stillborn babies. Other contributory factors are a general improvement in diet and the introduction of periconceptional folic acid supplementation (p. 258). In England and Wales the combined incidence of anencephaly and spina bifida in liveborn and stillborn babies fell from 1 in 250 in 1973 to 1 in 6250 in 1993.
Down Syndrome and Other Chromosome Abnormalities
The Triple Test
Confirmation of a chromosome abnormality in an unborn baby requires cytogenetic or molecular studies using material obtained by an invasive procedure such as CVS or amniocentesis (p. 325). However, chromosome abnormalities, and in particular Down syndrome, can be screened for in pregnancy by taking into account risk factors such as maternal age and the levels of three biochemical markers in maternal serum (Table 21.2).
| Advanced age (35 years or older) Maternal serum |
MoM* |
| α-Fetoprotein | (0.75) |
| Unconjugated estriol | (0.73) |
| Human chorionic gonadotrophin | (2.05) |
| Inhibin-A | (2.10) |
* Values in parentheses refer to the mean values in affected pregnancies, expressed as multiples of the median (MoMs) in normal pregnancies.
Using age alone as a screening parameter, if all pregnant women aged 35 years and over opt for fetal chromosome analysis approximately 35% of all Down syndrome pregnancies will be detected (Table 21.3). If three biochemical markers are also included (this being the so-called triple test), 60% of all Down syndrome pregnancies will be detected when a risk of 1 in 250 or greater is the cut-off for offering amniocentesis. This approach will also result in the detection of approximately 50% of all cases of trisomy 18 (p. 275). In the latter condition all the biochemical parameters are low, including hCG.
Table 21.3 Detection Rates Using Different down Syndrome Screening Strategies
| Screening Modality | Percent of All Pregnancies Tested | Percent of Down Syndrome Cases Detected |
|---|---|---|
| Age alone | ||
| 40 years and older | 1.5 | 15 |
| 35 years and older | 7 | 35 |
| Age + AFP | 5 | 34 |
| Age + AFP, µE3 + hCG | 5 | 61 |
| Age + AFP, µE3, hCG + inhibin-A | 5 | 75 |
| NT alone | 5 | 61 |
| NT + age | 5 | 69 |
| hCG, AFP + age | 5 | 73 |
| NT + AFP, hCG + age | 5 | 86 |
AFP, α-fetoprotein; µE3, unconjugated estriol; hCG, human chorionic gonadotrophin; NT, nuchal translucency.
Ultrasonography
Almost all pregnant women are routinely offered a ‘dating’ scan at around 12 weeks’ gestation. At around this time there is a strong association between chromosome abnormalities and the abnormal accumulation of fluid behind the baby’s neck—increased fetal nuchal translucency (NT) (see Figure 21.5). This applies to Down syndrome, the other autosomal trisomy syndromes (trisomies 13 and 18; p. 275), Turner syndrome, and triploidy, as well as a wide range of other fetal abnormalities and rare syndromes. The risk for Down syndrome correlates with absolute values of NT as well as maternal age (Figure 21.10) but, because NT also increases with gestational age, it is more usual now to relate the risk to the percentile value for any given gestational age. In one study, for example, 80% of Down syndrome fetuses had NT above the 95th percentile. By combining information on maternal age with the results of fetal NT thickness measurements, together with maternal serum markers, it is possible to detect more than 80% of fetuses with trisomy 21 if invasive testing is offered to the 5% of pregnant women with the highest risk (see Table 21.3). Some babies with Down syndrome have duodenal atresia, which shows up as a ‘double bubble sign’ on ultrasonography of the fetal abdomen (Figure 21.11).
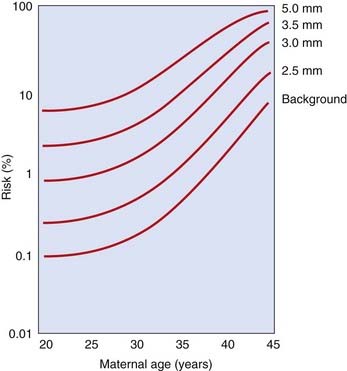
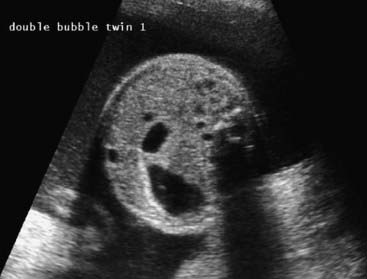
FIGURE 21.11 The ‘double bubble sign’, suggestive of duodenal atresia, sometimes associated with Down syndrome.
(Courtesy Dr. Helen Liversedge, Exeter, UK.)
In many centers, it is also standard practice to offer a detailed ‘fetal anomaly’ scan to all pregnant women at 18 weeks. Although chromosome abnormalities cannot be diagnosed directly, their presence can be suspected by the detection of an abnormality, such as exomphalos (Figure 21.12) or a rocker-bottom foot (Figure 21.13) (Table 21.4). A chromosome abnormality is found in 50% of fetuses with exomphalos identified at 18 weeks, and a rocker-bottom foot is a very characteristic, though not specific, finding in babies with trisomy 18 (p. 275), who are invariably growth retarded. The use of other ultrasonographic ‘soft markers’ in identifying chromosome abnormalities in pregnancy is discussed in the following section (p. 335).
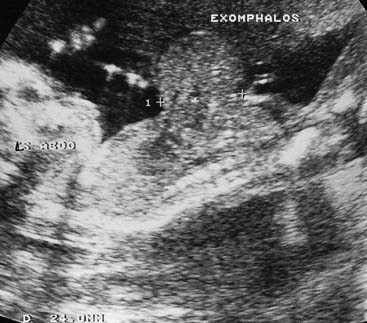
FIGURE 21.12 Ultrasonogram at 18 weeks showing exomphalos.
(Courtesy Dr. D. Rose, City Hospital, Nottingham, UK.)
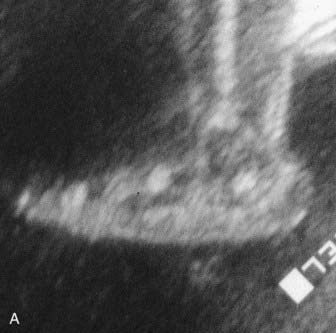
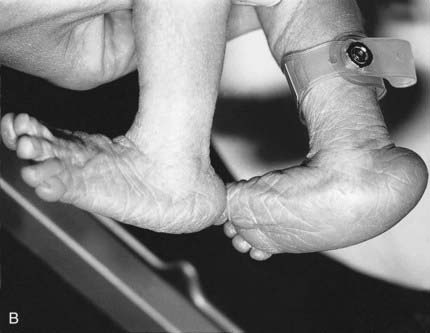
Table 21.4 Prenatal Ultrasonographic Findings Suggestive of a Chromosome Abnormality
| Feature | Chromosome Abnormality |
|---|---|
| Cardiac defect (especially common atrioventricular canal) | Trisomy 13, 18, 21 |
| Clenched overlapping fingers | Trisomy 18 |
| Cystic hygroma or fetal hydrops | Trisomy 13, 18, 21 |
| Duodenal atresia | 45,X (Turner syndrome) Trisomy 21 |
| Exomphalos | Trisomy 13, 18 |
| Rocker-bottom foot | Trisomy 18 |
Indications for Prenatal Diagnosis
There are numerous indications for offering prenatal diagnosis. Ideally, couples at high prior risk of having a baby with an abnormality should be identified and assessed before embarking on a pregnancy so that, in an unrushed manner, they can be counseled and come to a decision about which option they wish to pursue. Certain orthodox Jewish communities are extremely well organized in this respect vis-à-vis Tay-Sachs disease, as described in Chapter 20 (pp. 313–314). A less satisfactory alternative is that couples are identified early in pregnancy so that they still have an opportunity to consider prenatal diagnostic options. Unfortunately, many couples at increased risk because of their family history or previous reproductive history are still not referred until mid-pregnancy, when it may be too late to undertake the most thorough clinical and laboratory work-up in preparation for prenatal diagnosis.
Advanced Maternal Age
This has been the most common indication for offering prenatal diagnosis. There is a well-recognized association of advanced maternal age with increased risk of having a child with Down syndrome (see Table 18.4; p. 274) and the other autosomal trisomy syndromes. No standard criterion exists for determining at what age a mother should be offered the option of an invasive prenatal diagnostic procedure for fetal chromosome analysis. Most centers routinely offer amniocentesis or CVS to women age 37 years or older, and the option is often discussed with women from the age of 35 years onward. These risk figures relate to the maternal age at the expected date of delivery. The risk figures for Down syndrome at the time of CVS, amniocentesis, and delivery differ (see Figure 18.1; p. 274) because a proportion of pregnancies with trisomy 21 are lost spontaneously during the first and second trimesters. Interestingly, despite industrial-scale efforts to screen for Down syndrome, there has been a slight rise in the numbers of live births in the United Kingdom since 2000, following a steady decline from the widespread introduction in screening in 1989 (National Down Syndrome Cytogenetic Register). However, the numbers of prenatal diagnoses and terminations for Down syndrome has also increased over this period. Both observations are attributed to the slightly older age at which women are now having children, and there may also be an increasing willingness to raise a child with the condition.
Previous Child with a Chromosome Abnormality
Although there are a number of series with slightly different recurrence risk figures, for couples who have had a child with Down syndrome because of non-disjunction, or a de novo unbalanced Robertsonian translocation, the risk in a subsequent pregnancy is usually given as the mother’s age-related risk plus approximately 1%. If one of the parents has been found to carry a balanced chromosomal rearrangement, such as a chromosomal translocation (p. 44) or pericentric inversion (p. 48), that has caused a previous child to be born with serious problems due to an unbalanced chromosome abnormality, the recurrence risk is likely to be between 1% to 2% and 15% to 20%. The precise risk will depend on the nature of the parental rearrangement and the specific segments of the individual chromosomes involved (p. 48).
Family History of a Chromosome Abnormality
Couples may be referred because of a family history of a chromosome abnormality, most commonly Down syndrome. For most couples, there will usually be no increase in risk compared with the general population, as most cases of trisomy 21 and other chromosomal disorders will have arisen as a result of non-disjunction rather than as a result of a familial translocation, or other rearrangement. However, each situation should be evaluated carefully, either by confirming the nature of the chromosome abnormality in the affected individual or, if this is not possible, by urgent chromosome analysis of blood from the relevant parent at risk. The results of parental chromosomal analysis can usually be obtained within 3 to 4 days; if normal, an invasive prenatal diagnostic procedure is not then appropriate as the risk is no greater than that for the general population.
Family History of a Neural Tube Defect
Careful evaluation of the pedigree is necessary to determine the risk that applies to each pregnancy. Risks can be determined based on empiric data (p. 346). In high-risk situations, ultrasonographic examination of the fetus, possibly in conjunction with assay of maternal serum AFP, can be offered. However, even with good equipment and an experienced ultrasonographer, small closed NTDs can still be missed. Fortunately, the latter types of NTD are not usually associated with the serious problems seen with large open NTDs (p. 258).
Family History of Undiagnosed Learning Difficulty
An increasingly common scenario is the urgent referral of a pregnant couple who already has a child, or close relative, with an undiagnosed learning difficulty, with or without dysmorphic features. Whereas in the past a standard karyotype and fragile X syndrome test might be carried out, today it is incumbent on geneticists to use the latest technique of microarray-CGH (p. 281). This must first be undertaken on the affected child or individual, of course, and producing an urgent report (2 weeks) is a challenge for the laboratories.
Abnormalities Identified In Pregnancy
The widespread introduction of prenatal diagnostic screening procedures, such as triple testing and fetal anomaly scanning, has meant that many couples unexpectedly present with diagnostic uncertainty during the pregnancy that can be resolved only by an invasive procedure such as amniocentesis or CVS. Other factors, such as poor fetal growth, can also be an indication for prenatal chromosome analysis, as confirmation of a serious and non-viable chromosome abnormality, such as trisomy 18 or triploidy (p. 276), can influence subsequent management of the pregnancy and mode of delivery.
Other High-Risk Factors
These factors include parental consanguinity, a poor obstetric history, and certain maternal illnesses. Parental consanguinity increases the risk that a child will have a hereditary disorder or congenital abnormality (pp. 113). Consequently, if the parents are concerned, it is appropriate to offer detailed ultrasonography to try to exclude a serious structural abnormality. It may also be appropriate to offer to test the couple for cystic fibrosis and spinal muscular atrophy carrier status, and possibly other conditions depending on ethnicity. A poor obstetric history, such as recurrent miscarriages or a previous unexplained stillbirth, could indicate an increased risk of problems in a future pregnancy and detailed ultrasonographic monitoring. A history of three or more unexplained miscarriages should be investigated by parental chromosome studies to exclude a chromosomal rearrangement such as a translocation or inversion (pp. 44, 48). Maternal illnesses, such as poorly controlled diabetes mellitus (p. 235) or epilepsy treated with anticonvulsant medications such as sodium valproate (p. 261), would also be indications for detailed ultrasonography. Both of these factors convey an increased risk of structural abnormality in a fetus.
Special Problems in Prenatal Diagnosis
An Ambiguous Chromosome Result
In approximately 1% of cases, CVS shows evidence of apparent chromosome mosaicism—i.e., the presence of two or more cell lines with different chromosome constitutions (p. 50). This can occur for several reasons:
An Unexpected Chromosome Result
The Presence of a Marker Chromosome
A third difficult situation is the finding of a small additional chromosome known as a marker chromosome, that is, a small chromosomal fragment the specific identity of which cannot be determined by conventional cytogenetic techniques (p. 33). If this is found to be present in one of the parents, then it is unlikely to be of any significance to the fetus. If, on the other hand, it is a de novo finding, there is up to a 15% chance that the fetus will be phenotypically abnormal. The risk is lower when the marker chromosome contains satellite material (p. 17), or is made up largely of heterochromatin (p. 32), than when it does not have satellites and is mostly made up of euchromatin (p. 32). The availability of FISH (p. 34) means that the origin of the marker chromosome can often be determined more specifically, so that it is possible to give more precise prognostic information. The most common single abnormality of this kind is a marker chromosome 15.
Ultrasonographic ‘Soft’ Markers
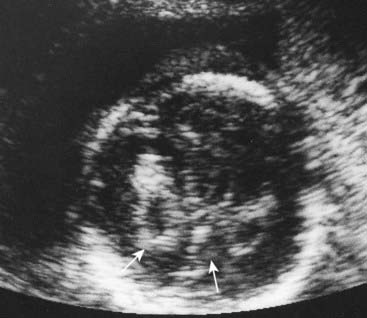
Sophisticated ultrasonography has resulted in the identification of subtle anomalies in the fetus, the significance of which is not always clear. For example, choroid plexus cysts are sometimes seen in the developing cerebral ventricles in mid-trimester (Figure 21.14). Initially, it was thought that these were invariably associated with the fetus having trisomy 18 but in fact they occur frequently in normal fetuses, although if they are very large and do not disappear spontaneously they can be indicative of a chromosome abnormality.
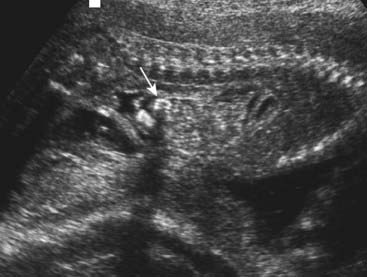
Increased echogenicity of the fetal bowel (Figure 21.15) has been reported in association with cystic fibrosis—the prenatal equivalent of meconium ileus (p. 301). Initial reports suggested this finding could convey a risk as high as 10% for the fetus having cystic fibrosis, but it is now clear that this risk is probably no greater than 1% to 2%. Novel ultrasonographic findings of this kind are often called soft markers, and their interpretation must be approached cautiously in the effort to distinguish normal from abnormal variation.
Preimplantation Genetic Diagnosis
At the eight-cell stage, the early embryo is biopsied and one, or sometimes two, cells are removed for analysis. Whatever genetic analysis is undertaken, it is essential that this is a practical possibility on genomic material from a single cell. From the embryos tested, two that are both healthy and unaffected by the disorder from which they are at risk are reintroduced into the mother’s uterus. Implantation must then occur for a successful pregnancy and this is a major hurdle—the success rate for the procedure is only about 25% to 30% per cycle of treatment, even in the best centers. A variation of the technique is removal of the first, and often second, polar bodies from the unfertilized oocyte, which lie under the zona pellucida. Because the first polar body degenerates quite rapidly, analysis is necessary within 6 hours of retrieval. Analysis of polar bodies is an indirect method of genotyping because the oocyte and first polar body divide from each other during meiosis I and therefore contain different members of each pair of homologous chromosomes.
In the United Kingdom, centers must be licensed to practice PGD and are regulated by the Human Fertilization and Embryology Authority (HFEA), though this body is due to be abolished. In numerical terms, the impact of PGD has been small to date, but a wide and increasing range of genetic conditions has now been tested (Table 21.5). The most common referral reasons for single-gene disorders are cystic fibrosis, myotonic dystrophy, Huntington disease, β-thalassemia, spinal muscular atrophy, and fragile X syndrome. The technique for identifying normal and abnormal alleles in these conditions, and DNA linkage analysis where appropriate, is PCR (p. 56). Sex selection in the case of serious X-linked conditions is available where single-gene analysis is not possible. The biggest group of referrals for PGD, however, is chromosome abnormalities—reciprocal and robertsonian translocations in particular (pp. 45, 47). Genetic analysis in these cases uses FISH technology (p. 34) and substantial work has to be undertaken for the couple prior to treatment because of the unique nature of many translocations.
Table 21.5 Some of the Conditions for Which Preimplantation Genetic Diagnosis has been Used and is Available
| Mode of Inheritance | Disease |
|---|---|
| Autosomal dominant |
Ornithine transcarbamylase deficiency
Incontinentia pigmenti
Other serious disorders
MELAS, mitochondrial myopathy encephalopathy, lactic acidosis, stroke.
In recent years, PGD has occasionally been used not only to select embryos unaffected for the genetic disorder for which the pregnancy is at risk, but also to provide a human leukocyte antigen tissue-type match so that the new child can act as a bone marrow donor for an older sibling affected by, for example, Fanconi anemia. The ethical debate surrounding these so-called savior sibling cases is discussed further in Chapter 24.
A further development using micromanipulation methods has attracted a lot of attention. To circumvent the problem of genetic disease resulting from mutation in the mitochondrial genome, the nucleus of the oocyte from the genetic mother (who carried the mitochondrial mutation) was removed and inserted into a donor oocyte from which the nucleus had been removed. This is cell nuclear replacement technology, similar to that used in reproductive cloning experiments in animals (’Dolly’ the sheep; see p. 369). The resulting fertilization led to the headline that the fetus had three genetic parents. The technique has also been used in other situations where the oocytes are generally of poor quality, but its use is extremely limited.
Assisted Conception and Implications for Genetic Disease
In Vitro Fertilization
Many thousands of babies worldwide have been born by IVF over the past 30 years, when the technique was first successful. The indication for the treatment in most cases is subfertility, which now affects one in seven couples. In some Western countries, 1% to 3% of all births are the result of assisted reproductive technologies (ARTs). The cohort of offspring conceived in this way is therefore very large, and evidence is gathering that the risk of birth defects is increased by 30% to 40% compared with the general population conceived in the normal way and about 50% more children are likely to be small for gestational age (SGA). Specifically, a small increase in certain epigenetic conditions due to defective genomic imprinting (p. 121) has been observed—Beckwith-Wiedemann (p. 124) and Angelman (p. 123) syndromes, and ‘hypomethylation’ syndrome, though the possible mechanisms are unclear. In cases studied, loss of imprinting (LOI) was observed at the KCNQ1OT1 locus (see Figure 7.27; p. 125) in the case of Beckwith-Wiedemann syndrome, and at the SNRPN locus (Figure 7.23; p. 123) in the case of Angelman syndrome. No apparent imprinting differences explain the increase in SGA babies conceived by ICSI.
Epigenetic events around the time of fertilization and implantation are crucial for normal development (p. 103). If there is a definite increased risk of conditions from abnormal imprinting after ARTs, this may relate, in part, to the extended culture time of embryos, which has become a trend in infertility clinics. Instead of transferring cleavage-stage embryos, it is now more routine to transfer blastocysts, which allows the healthier looking embryos to be selected. However, in animal models it has been shown that in vitro culture affects the extent of imprinting, gene expression, and therefore the potential for normal development.
Donor Insemination
Naturally, all of these issues apply in an equivalent way to women who wish to be egg donors.
Assisted Conception and the Law
In the United States, no federal law exists to regulate the practice of assisted conception other than the requirement that outcomes of IVF and ICSI must be reported. In the United Kingdom, strict regulation operates through the HFEA based on the Human Fertilization and Embryology Act of 1990. The HFEA reports to the Secretary of State for Health, issues licences, and arranges inspections of registered centers. The different licences granted are for treatment (Box 21.2), storage (gametes and embryos), and research (on human embryos in vitro). A register of all treatment cycles, the children born by IVF, and the use of donated gametes, must be kept. The research permitted under licence covers treatment of infertility, increase in knowledge regarding birth defects, miscarriage, genetic testing in embryos, the development of the early embryo, and potential treatment of serious disease. At the time of writing it is not clear what arrangements will be put in place when the HFEA is abolished.
Non-Invasive Prenatal Diagnosis
At the turn of the 19th century, it was discovered that fetal cells reach the maternal circulation, but confirmation that cell-free DNA of fetal origin (placentally derived) is present in the plasma of pregnant women was not made until 1997. This fact has now been exploited in clinical practice as early as 6 to 7 weeks of pregnancy to determine fetal sex by detection of Y-chromosome DNA, as well as fetal Rhesus D gene. Early determination of fetal sex is clinically useful in a pregnancy at risk of an X-linked recessive disorder, and also in congenital adrenal hyperplasia (see the following section). The problem with analyzing cell-free fetal DNA is one of isolation because maternal cell-free DNA constitutes about 95% of all the cell-free DNA in the maternal circulation. The absence of Y-chromosome DNA might indicate the fetus is female, or that the quantity of fetal DNA is very low. This is resolved by using real-time PCR to quantify the amount of fetal or total DNA present in plasma.
Prenatal Treatment
A possible model for successful prenatal treatment is provided by the autosomal recessive disorder congenital adrenal hyperplasia (CAH) (p. 174). Affected female infants are born with virilization of the external genitalia. There is evidence that in a proportion of cases the virilization can be prevented if the mother takes a powerful steroid known as dexamethasone in a very small dose from 4 to 5 weeks’ gestation onward. Specific prenatal diagnosis of CAH can be achieved by DNA analysis of CV tissue. If this procedure confirms that the fetus is both female and affected, the mother continues to take low doses of dexamethasone throughout pregnancy, which suppresses the fetal pituitary–adrenal axis and can prevent virilization of the female fetus. If the fetus is male and either affected or unaffected, the mother ceases to take dexamethasone and the pregnancy can proceed uneventfully.
Treatment of a fetus affected with severe combined immunodeficiency (p. 203) has also been reported. The immunological tolerance of the fetus to foreign antigens introduced in utero means that the transfused stem cells are recognized as ‘self’, with the prospect of good long-term results.
When gene therapy (p. 350) has been proved to be both safe and effective, the immunological tolerance of the fetus should make it easier to commence such therapy before birth rather than afterward. This will have the added advantage of reducing the period in which irreversible damage can occur in organs such as the central nervous system, which can be affected by progressive neurodegenerative disorders.
Abramsky L, Chapple J, editors. Prenatal diagnosis: the human side. Cheltenham. UK: Nelson Thornes, 2003.
Brock DJH, Rodeck CH, Ferguson Smith MA, editors. Prenatal diagnosis and screening. Edinburgh, UK: Churchill Livingstone, 1992.
A comprehensive multiauthor textbook covering all aspects of prenatal diagnosis.
Drife JO, Donnai D, editors. Antenatal diagnosis of fetal abnormalities. London, UK: Springer, 1991.
The proceedings of a workshop on the practical aspects of prenatal diagnosis.
European Society for Human Reproduction and Embryology PGD Steering Committee. ESHRE Preimplantation Genetic Diagnosis Consortium: data collection III (May 2001). Hum Reprod. 2002;17:233-246.
An up-to-date appraisal of the use of PGD.
Lilford RJ, editor. Prenatal diagnosis and prognosis. Oxford, UK: Butterworth-Heinemann, 1990.
Stranc LC, Evans JA, Hamerton JL. Chorionic villus sampling and amniocentesis for prenatal diagnosis. Lancet. 1997;349:711-714.
Whittle MJ, Connor JM, editors. Prenatal diagnosis in obstetric practice. Oxford, UK: Blackwell, 1989.
Describes prenatal diagnostic techniques and the types of abnormalities identified.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 21 Prenatal Testing and Reproductive Genetics
Until recently, couples at high risk of having a child with a genetic disorder had to choose between taking the risk or considering other reproductive options, such as long-term contraception, sterilization, and termination of pregnancy. Other alternatives included adoption, long-term fostering, and donor insemination (DI).
The ethical issues surrounding prenatal diagnosis and selective termination of pregnancy are both complex and emotive, and are considered more fully in Chapter 24 (p. 363). In this chapter, we focus on the practical aspects of prenatal testing and diagnosis, including prenatal screening, as well as some aspects of reproductive genetics.
Techniques Used in Prenatal Diagnosis
There are several techniques that can be used for the prenatal diagnosis of hereditary disorders and structural abnormalities (Table 21.1).
| Technique | Optimal Time (Weeks) | Disorders Diagnosed |
|---|---|---|
| Non-Invasive | ||
| Maternal serum screening | ||
| α-Fetoprotein | 16 | Neural tube defects |
| Triple test | 16 | Down syndrome |
| Ultrasound | 18 | Structural abnormalities (e.g., central nervous system, heart, kidneys, limbs) |
| Invasive | ||
| Amniocentesis | 16 | |
| Fluid | Neural tube defects | |
| Cells | Chromosome abnormalities, metabolic disorders, molecular defects | |
| Chorionic villus sampling | 10–12 | Chromosome abnormalities, metabolic disorders, molecular defects |
| Fetoscopy | ||
| Blood (cordocentesis) | Chromosome abnormalities, hematological disorders, congenital infection | |
| Liver | Metabolic disorders (e.g., ornithine transcarbamylase deficiency) | |
| Skin | Hereditary skin disorders (e.g., epidermolysis bullosa) | |
Amniocentesis
Amniocentesis involves the aspiration of 10 to 20 ml of amniotic fluid through the abdominal wall under ultrasonographic guidance (Figure 21.1). This is usually performed around the 16th week of gestation. The sample is spun down to yield a pellet of cells and supernatant fluid. The fluid can be used in the prenatal diagnosis of neural tube defects by assay of α-fetoprotein (p. 328). The cell pellet is resuspended in culture medium with fetal calf serum, which stimulates cell growth. Most of these cells in the amniotic fluid, that have been shed from the amnion, fetal skin, and urinary tract epithelium, are non-viable, but a small proportion will grow. After approximately 14 days, there are usually sufficient cells for chromosome and DNA analysis, although a longer period may be required before enough cells are obtained for biochemical assays. Increasingly, sensitive polymerase chain reaction (PCR) techniques make direct DNA analysis possible without the need for culture.
Chorionic Villus Sampling
In contrast to amniocentesis, chorionic villus sampling (CVS), first developed in China, enables prenatal diagnosis to be undertaken during the first trimester. This procedure is usually carried out at 11 to 12 weeks’ gestation under ultrasonographic guidance by either transcervical or, more usually, transabdominal aspiration of chorionic villus (CV) tissue (Figure 21.2). This tissue is fetal in origin, being derived from the outer cell layer of the blastocyst (i.e., the trophoblast). Maternal decidua, normally present in the biopsy sample, must be removed before the sample is analyzed. Placental biopsy is the term used when the procedure is carried out at later stages of pregnancy.
Chromosome analysis can be undertaken on CV tissue either directly, looking at metaphase spreads from actively dividing cells, or after culture. Direct chromosomal analysis of CV tissue usually allows a provisional result to be given within 24 hours. Nowadays rapid, direct fluorescent in-situ hybridization (FISH) probing (p. 34), or DNA analysis by the multiplex ligation-dependent probe amplification technique (p. 66), is used to test for common chromosome aneuploidies prior to a standard karyotype following culture of CV tissue; this may also detect other chromosome abnormalities and balanced rearrangements. For single-gene disorders, sufficient CV tissue is usually obtained to allow prenatal diagnosis by immediate biochemical assay or DNA analysis using uncultured CV tissue.
Ultrasonography
Ultrasonography offers a valuable means of prenatal diagnosis. It can be used not only for obstetric indications, such as placental localization and the diagnosis of multiple pregnancies, but also for prenatal diagnosis of structural abnormalities not associated with known chromosomal, biochemical or molecular defects. Ultrasonography is particularly valuable because it is non-invasive and conveys no known risk to the fetus or mother. It does, however, require expensive equipment and a skilled, experienced operator. For example, a search can be made for polydactyly as a diagnostic feature of a multiple abnormality syndrome, such as one of the autosomal recessive short-limb polydactyly syndromes that are associated with severe pulmonary hypoplasia—invariably lethal (Figure 21.3). Similarly, a scan can reveal that the fetus has a small jaw, which can be associated with a posterior cleft palate and other more serious abnormalities in several single-gene syndromes (Figure 21.4).
FIGURE 21.3 Ultrasonographic image of a transverse section of the hand of a fetus showing polydactyly.
Nuchal Translucency
In addition, the observation that increased nuchal translucency (NT) is seen in fetuses who are subsequently born with Down syndrome has resulted in the introduction of measurements of nuchal pad thickness (Figure 21.5) in the first and second trimesters as part of screening for Down syndrome (p. 273). In fact, the finding is not specific and may be seen in various chromosomal anomalies as well as isolated congenital heart disease.
Fetoscopy
Fetoscopy has also been used to obtain samples of tissue from the fetus that can be analyzed as a means of achieving the prenatal diagnosis of several rare disorders. These have included inherited skin disorders such as epidermolysis bullosa and, before DNA testing became available, metabolic disorders in which the enzyme is expressed only in certain tissues or organs, such as the liver—e.g., ornithine transcarbamylase deficiency (p. 172).
Cordocentesis
Although fetoscopy can also be used to obtain a small sample of fetal blood from one of the umbilical cord vessels in the procedure known as cordocentesis, improvements in ultrasonography have enabled visualization of the vessels in the umbilical cord, allowing transabdominal percutaneous fetal blood sampling. Fetal blood sampling is used routinely in the management of rhesus iso-immunization (p. 205) and can be used to obtain samples for chromosome analysis to resolve problems associated with possible chromosomal mosaicism in CV or amniocentesis samples.
Radiography
The fetal skeleton can be visualized by radiography from 10 weeks onwards, and this technique has been used in the past to diagnose inherited skeletal dysplasias. It is now employed only occasionally because of the dangers of radiography to the fetus (p. 26) and the widespread availability of detailed ultrasonography.
Prenatal Screening
The history of widespread prenatal (antenatal) screening really began with the finding, in the early 1970s, of an association between raised maternal serum α-fetoprotein (AFP) and neural tube defects (NTDs). Estimation of AFP levels was gradually introduced into clinical service, and the next significant development was ultrasonography, followed, in the 1980s, by the identification of maternal serum biochemical markers for Down syndrome. These are discussed in more detail below. Where the incidence of a genetic condition was high, for instance thalassemia in Cyprus, prenatal screening came into practice, as described in Chapter 20 (p. 321). However, molecular genetic advances, rather than biochemical, mean that the range of prenatal screening is continuing to evolve.
Testing for cystic fibrosis and fragile X syndrome are available in the UK, mainly for those willing to pay privately, and in Israel, for example, a wide range of relatively rare diseases can be screened for on the basis that they are more common in specific population groups that were originally isolates with multiple inbreeding, and therefore certain mutations are prevalent. Besides Tay-Sachs disease (carrier testing in this case is biochemical; see Chapter 20), familial dysautonomia, Canavan disease, Bloom syndrome, ataxia telangiectasia (North African Jews), limb-girdle muscular dystrophy (Libyan Jews) and Costeff syndrome (Iraqi Jews) are among the conditions for which screening is available. It does not come free of charge but the level of uptake of this screening is high, revealing the lengths to which some societies will go in order to avoid having children with serious genetic conditions. As DNA testing becomes more automated, rapid, and affordable, there will be pressure from some quarters to screen for many conditions, even though they are individually very rare. This challenge is already emerging with the potential use of microarray CGH in prenatal testing. If the use of microarray becomes routine there may be great difficulty in interpreting the consequences of rare or unique copy number variants. This ethical challenge is discussed more fully in Chapter 24.
Neural Tube Defects
In 1972 it was recognized that many pregnancies in which the baby had an open NTD (p. 258) could be detected at 16 weeks’ gestation by assay of AFP in maternal serum. AFP is the fetal equivalent of albumin and is the major protein in fetal blood. If the fetus has an open NTD, the level of AFP is raised in both the amniotic fluid and maternal serum as a result of leakage from the open defect. Open NTDs fulfil the criterion of being serious disorders, as anencephaly is invariably fatal, and between 80% and 90% of the small proportion of babies who survive with an open lumbosacral lesion are severely handicapped.
Unfortunately maternal serum AFP screening for NTDs is neither 100% sensitive nor 100% specific (p. 319). The curves for the levels of maternal serum AFP in normal and affected pregnancies overlap (Figure 21.6), so that in practice an arbitrary cut-off level has to be introduced below which no further action is taken. This is usually either the 95th centile, or 2.5 multiples of the median (MoM); as a result around 75% of screened open spina bifida cases are detected. Those pregnant women with results that lie above this arbitrary cut-off level are offered detailed ultrasonography; which is usually sufficient to diagnose NTD. In fact, ultrasonography has more or less superceded maternal serum screening as a means of diagnosing NTD. Anencephaly shows a dramatic deficiency in the cranium (Figure 21.7) and an open myelomeningocele is almost invariably associated with herniation of the cerebellar tonsils through the foramen magnum. This deforms the cerebellar hemispheres, which then have a curved appearance known as the ‘banana sign’; the forehead is also distorted, giving rise to a shape referred to as the ‘lemon sign’ (Figure 21.8). A posterior encephalocele is readily visualized as a sac in the occipital region (Figure 21.9) and always prompts a search for additional anomalies that might help diagnose a recognizable condition such as Meckel-Gruber syndrome.
FIGURE 21.7 Anencephaly (arrow). There is no cranium and this form of neural tube defect is incompatible with life.
(Courtesy Dr. Helen Liversedge, Exeter, UK.)
A raised maternal serum AFP concentration is not specific for open NTDs (Box 21.1). Other causes include threatened miscarriage, twin pregnancy and a fetal abnormality such as exomphalos, in which there is a protrusion of abdominal contents through the umbilicus.
As a result of these screening modalities there has been a striking decline in the incidence of open NTDs in liveborn and stillborn babies. Other contributory factors are a general improvement in diet and the introduction of periconceptional folic acid supplementation (p. 258). In England and Wales the combined incidence of anencephaly and spina bifida in liveborn and stillborn babies fell from 1 in 250 in 1973 to 1 in 6250 in 1993.
Down Syndrome and Other Chromosome Abnormalities
The Triple Test
Confirmation of a chromosome abnormality in an unborn baby requires cytogenetic or molecular studies using material obtained by an invasive procedure such as CVS or amniocentesis (p. 325). However, chromosome abnormalities, and in particular Down syndrome, can be screened for in pregnancy by taking into account risk factors such as maternal age and the levels of three biochemical markers in maternal serum (Table 21.2).
| Advanced age (35 years or older) Maternal serum |
MoM* |
| α-Fetoprotein | (0.75) |
| Unconjugated estriol | (0.73) |
| Human chorionic gonadotrophin | (2.05) |
| Inhibin-A | (2.10) |
* Values in parentheses refer to the mean values in affected pregnancies, expressed as multiples of the median (MoMs) in normal pregnancies.
[/not-level-membership-for-basic-science-category]
