[level-membership-for-pediatrics-category]Chapter 42
Prenatal Imaging
Imaging Techniques
The accuracy of identifying spinal anomalies during nontargeted screening ultrasonography varies, depending on the skill and experience of the operator. The accuracy of a referral center performing detailed targeting studies for a suspected neural tube defect (elevated maternal serum α-fetoprotein) is close to 100%. A detailed protocol should be performed. Because fetuses with open neural tube defects typically have Chiari II malformations, the fetal brain should be scanned initially. A small cisterna magna with rounded small cerebellum is termed the “banana sign” and is 99% sensitive in the diagnosis of a Chiari II malformation (Fig. 42-1). The frontal bones may be concave, and this is termed the “lemon sign.” This sign is less specific and may be present in 1% to 2% of normal fetuses and may resolve by the third trimester. Ventriculomegaly may be present. Axial and longitudinal views of the spine should be obtained. Spine ossification progresses from 10 to 22 weeks’ gestation. By 16 weeks’ gestation, neural arch ossification is complete to L5. By 19 weeks, S1 is completely ossified, and by 22 weeks, S2 is ossified. Splayed pedicles are best visualized in the transverse plane. An overlying sac may be imaged in transverse and longitudinal planes, with higher-frequency transducers showing cord tethering and placode contents (Fig. 42-2).
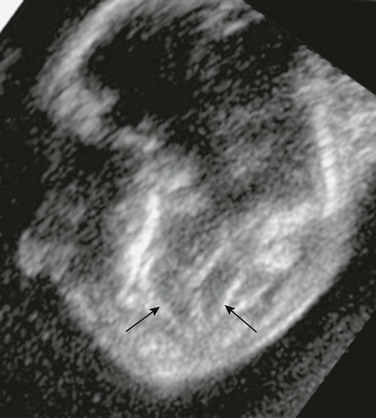
Figure 42-1 Banana sign.
An axial ultrasonographic image through the posterior fossa in a 21-week fetus with a Chiari II malformation shows the crowding of the cerebellum around the brainstem that has been termed the “banana sign” (demarcated by arrows).
Open Spinal Dysraphism
Abnormalities in neurulation may result from defects in disjunction, the process by which the neural tube separates from the overlying ectoderm. A large site of complete failure of disjunction may result in MMC. Prenatally, the findings of MMC are clearly detectable with ultrasonography and MRI. In the presence of elevation of the neural placode, secondary to expansion of the subarachnoid space, the lesion is referred to as MMC (Fig. 42-3). This is distinguished from myeloschisis, also known as myelocele, where an open neural tube defect exists, but the subarachnoid space is not expanded, and the neural placode remains within the confines of the dysraphic spinal canal (Fig. 42-4). MMC occurs during the formation of the primitive neural tube (neurulation) in the third week of gestation, when a localized failure of neural tube closure occurs. This failure may occur anywhere along the length of the spinal cord, but it is most common in the lumbar region. The resulting lesion is an open spinal canal with a flat neural placode instead of a cylindrical spinal cord. Imaging reveals the failure of neurulation in the MMC as a posterior osseocutaneous defect. When present, the expansion of the subarachnoid space is readily apparent (see Fig. 42-3). The neurologic deficits sustained by the fetus are postulated to occur in stages—a “two-hit” hypothesis. The first “hit” is the original defect in neurulation that creates the dysraphism and any associated myelodysplasia. The second “hit” is the secondary chemical or physical trauma (or both) to the neural tissue as a result of its exposure to the intrauterine environment.
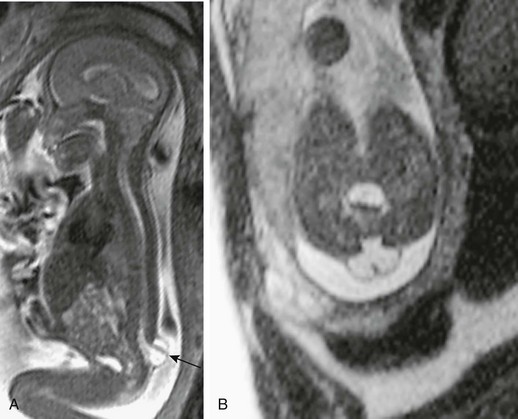
Figure 42-3 Myelomeningocele.
A, A sagittal half-Fourier acquisition single-shot turbo spin echo (HASTE) magnetic resonance image in a 21-week fetus with a small myelomeningocele sac protruding at the lumbosacral junction level. The neural tissue can be readily identified traversing the sac (arrow). Note the Chiari II malformation findings. B, An axial HASTE image at the lumbosacral level shows the expanded subarachnoid space protruding through the wide posterior element deficiency. Note the neural placode outside the confines of the canal, traversing the sac.
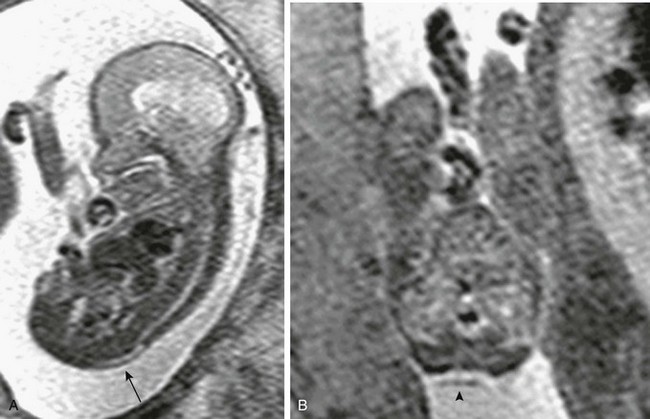
Figure 42-4 Myeloschisis.
A, A sagittal half-Fourier acquisition single-shot turbo spin echo (HASTE) image of the spine in a 19-week fetus with a posterior spinal defect. The placode remains within the spinal canal (arrow). B, An axial HASTE image at the lumbosacral level shows the wide posterior element deficiency and neural placode within the confines of the broad canal (arrowhead).
A unified theory regarding the pathogenesis of the associated Chiari II malformation was postulated by McLone and Knepper, who suggested that the open spinal canal and associated free drainage of cerebrospinal fluid (CSF) promote collapse of the primitive ventricular system and cause lack of expansion of the rhombencephalic vesicle, from which the posterior fossa develops. This lack of distention leads to an abnormally small posterior fossa and subsequent herniation and other associated malformations in the brain. The Chiari II malformation is a pancerebral anomaly, affecting broad areas of the brain. Abnormalities include herniation of the medulla, cerebellar tonsils, and vermis through the foramen magnum; a small posterior fossa; “beaking” of the tectum of the midbrain; an enlarged massa intermedia of the thalami; partial or complete callosal dysgenesis; and structural changes in the skull (Fig. 42-5). Migrational abnormalities, particularly subependymal gray matter heterotopia, also are commonly seen. The cause of hydrocephalus in patients with MMC is frequently debated. Current theories include mechanical obstruction secondary to anatomic changes associated with the Chiari II malformation and dysfunctional CSF absorption. Clinically, hydrocephalus may not be present at birth but may become apparent after early postnatal closure of the defect.
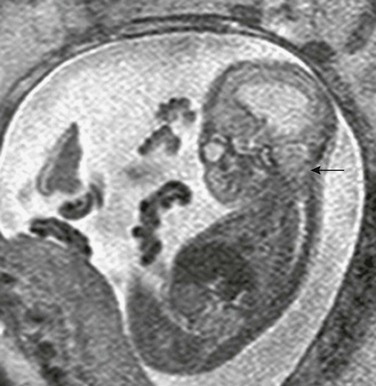
Figure 42-5 Chiari II malformation.
A sagittal half-Fourier acquisition single-shot turbo spin echo image at the craniocervical junction in a 19-week fetus shows the hindbrain herniation and funneling of the posterior fossa of the Chiari II malformation (arrow). Note the ventriculomegaly and loss of the supratentorial subarachnoid spaces.
Treatment
The Management of Myelomeningocele Study trial found that fetuses who underwent in utero repair of MMC demonstrated superior standardized test scores for motor skills and that twice as many children were walking independently at 30 months of age compared with those randomized to postnatal surgery. Additionally, prenatal repair led to a reduction in hindbrain herniation (Fig. 42-6), and these children were half as likely to require ventricular shunting.
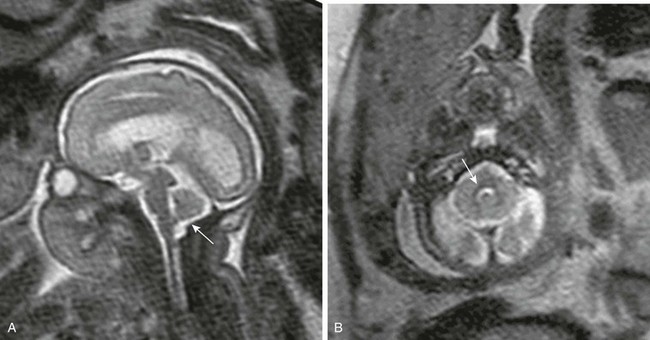
Figure 42-6 Reversal of hindbrain herniation after fetal myelomeningocele repair.
A, A sagittal half-Fourier acquisition single-shot turbo spin echo (HASTE) magnetic resonance image of the same fetus as in Figure 42-5 reveals the dramatic improvement in the appearance of the brain 6 weeks after surgery. The hindbrain herniation has resolved, with return of the posterior fossa subarachnoid spaces (arrow). The supratentorial subarachnoid spaces are now normal, but the ventricles remain enlarged. B, An axial HASTE image through the posterior fossa shows the now normal appearance of the fourth ventricle (arrow).
Closed Spinal Dysraphism
Lipomyelomeningocele
Premature disjunction of the cutaneous ectoderm from the neuroectoderm allows mesenchyme to contact the inner portion of the developing neural tube. As the tube begins to close, the mesenchyme is induced to become fat, the presence of which may interfere with neurulation. This may result in lipomyelomeningocele–lipomyeloschisis. These lesions are skin covered and consequently not associated with the Chiari II malformation or abnormal elevation of maternal serum or amniotic fluid α-fetoprotein and acetylcholinesterase (markers of an open neural tube). A small lipomyelomeningocele may be harder to detect with screening ultrasonography but should be apparent with MRI, particularly later in the second trimester and in the third trimester (Fig. 42-7). As in the case of MMC, the distinction between lipomyelomeningocele and lipomyeloschisis lies in the location of the placode–lipoma interface with respect to the plane of the back. The expansion of the subarachnoid space characterizes the lipomyelomeningocele, but in contrast to MMC, it is much less common than its flat counterpart. The placode in the lipomyelomeningocele is much more likely to be deformed, being rotated toward the lipoma and away from the protrusion of the meninges. This poses an additional problem for the pediatric neurosurgeon performing postnatal repair because the spinal nerve roots are similarly deformed, with shortened roots on the side of the lipoma, which tether the cord. The elongated roots on the side of the meninges must be carefully negotiated as the surgeon attempts to access the placode–lipoma interface.
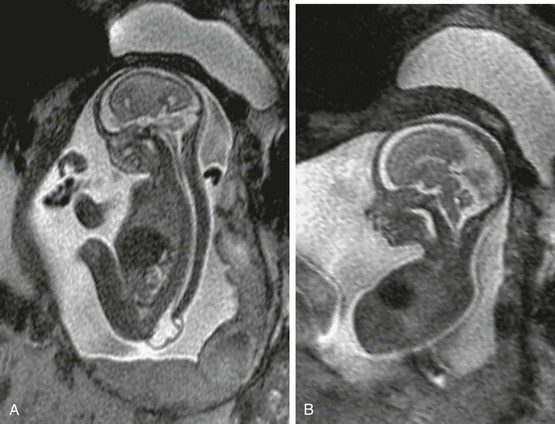
Figure 42-7 Lipomyelomeningocele.
A, A sagittal half-Fourier acquisition single-shot turbo spin echo (HASTE) magnetic resonance image of a 21-week fetus shows a large lumbosacral defect with neural tissue traversing the sac. The sac appears slightly thicker walled than that of a myelomeningocele, although correlation with open neural tube defect markers is required. B, An axial HASTE image through the midline of the brain shows normal morphology, including a normal corpus callosum. No Chiari II malformation is present.
Split Cord Malformation
The spinal cord is focally split into two hemicords, often asymmetrically (Fig. 42-8). This may involve the entire anteroposterior aspect of the cord or a portion of the spinal cord, the latter being quite rare. Each hemicord contains a central canal, and at least one dorsal horn and one ventral horn, from which nerve roots arise. An osseous septum between dual dural tubes (type I) or fibrous septum within a single dural tube (type II) separates the hemicords. Occasionally, no intervening septum exists in the type II lesion. SCM most commonly occurs in the lumbar region, and the type I is frequently associated with vertebral body anomalies. Cervicothoracic junction lesions may be more common than is currently reported because they are often asymptomatic owing to the absence of spinal cord tethering.
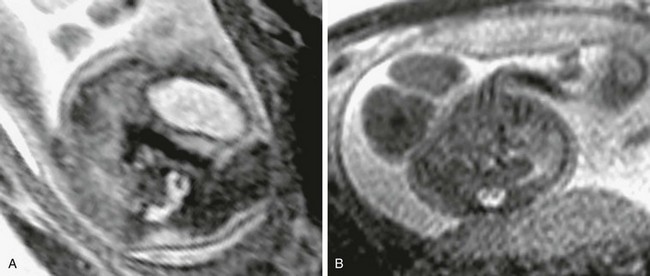
Figure 42-8 Split cord malformation.
A, An axial half-Fourier acquisition single-shot turbo spin echo (HASTE) image through a type I split cord malformation. The osseous septum traversing the two canals is visible. B, An axial HASTE image through a type II split cord malformation. No septum is visible between the two hemicords.
Type I SCM is readily recognized with fetal imaging (see Fig. 42-8). An often subtle alteration in spinal alignment from the associated anomalous vertebral bodies should be a clue that a spinal anomaly is present. When present, the osseous septum may appear on ultrasonography as an echogenic structure traversing the spinal canal. Echo planar MRI techniques may be valuable in assessing for the osseous septum, which commonly tethers the spinal cord, and is significant in determining the extent of postnatal surgical intervention. The simple fibrous septum or cord duplication without intervening septum (type II) may be difficult to visualize prenatally, particularly when imaging is performed in the second trimester. SCM is present in 40% of MMC, although this most commonly involves only duplication or splitting of the placode, which may be impossible to detect prenatally. It is imperative to search for SCM when MMC is encountered because the spinal cord may remain tethered by the septum after MMC repair.
Terminal Myelocystocele
This rare malformation may represent a severe manifestation of the persistent terminal ventricle, resulting from an inability of CSF to escape from the neural tube during its formation. The marked dilation of the distal central canal is also referred to as the terminal ventricle of the spinal cord, which herniates through a posterior lumbosacral spinal defect. The leptomeninges herniate around the bulbous distal spinal cord (Fig. 42-9). With prenatal imaging, terminal myelocystocele may sometimes be distinguished from the MMC with close attention to the morphology and wall thickness of the protruding sac, the absence of Chiari II malformation, and the lack of elevation of maternal serum and amniotic fluid markers of an open neural tube defect. A degree of downward displacement of the cerebellar tonsils through the foramen magnum and reduction of the infratentorial and supratentorial subarachnoid spaces may develop late in gestation with a large terminal myelocystocele, but this should not be mistaken for the Chiari II malformation.
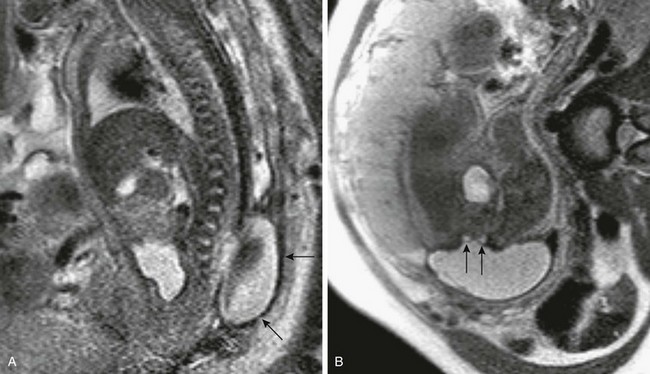
Figure 42-9 Terminal myelocystocele.
A, A sagittal half-Fourier acquisition single-shot turbo spin echo (HASTE) magnetic resonance image in a 32-week fetus shows the thick-walled posterior sac (arrows) protruding through a focal lower lumbar osseous defect. B, An axial HASTE image through the osseous defect and large sac shows the distal spinal cord (arrows) splitting around the dilated central canal.
Meningocele
The simple posterior meningocele is characterized by a meningeal-lined CSF sac, which protrudes through a posterior osseous spinal defect (Fig. 42-10). The spinal cord does not enter the sac, although it may be associated with hypertrophy of the filum terminale or spinal cord tethering. Its etiology is not well understood, but some postulate that CSF pulsations cause the meninges to herniate through a focal posterior osseous defect. Most commonly encountered in the thoracic spine, these anomalies rarely may be present in utero and must be differentiated from thoracic MMC. Similar to the terminal myelocystocele and lipomyelomeningocele, these skin-covered (closed) lesions do not have an associated Chiari II malformation, and the maternal serum or amniotic fluid does not contain markers of an open neural tube defect.
Adzick, NS, Thom, EA, Spong, CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364(11):993–1004.
Glenn, OA, Barkovich, AJ. Magnetic resonance imaging of the fetal brain and spine: an increasingly important tool in prenatal diagnosis, part 1. AJNR Am J Neuroradiol. 2006;27(8):1604–1611.
Glenn, OA, Barkovich, AJ. Magnetic resonance imaging of the fetal brain and spine: an increasingly important tool in prenatal diagnosis: part 2. AJNR Am J Neuroradiol. 2006;27(9):1807–1814.
Griffiths, PD, Paley, MN, Widjaja, E, et al. In utero magnetic resonance imaging for brain and spinal abnormalities in fetuses. BMJ. 2005;331(7516):562–565.
Rossi, A, Gandolfo, C, Morana, G, et al. Current classification and imaging of congenital spinal abnormalities. Semin Roentgenol. 2006;41(4):250–273.
Adzick, NS, Sutton, LN, Crombleholme, TM, et al. Successful fetal surgery for spina bifida [letter]. Lancet. 1998;352:1666–1675.
Bruner, JP, Tulipan, N, Paschall, RL, et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819–1825.
Dias, MS, McLone, DG. Hydrocephalus in the child with dysraphism. Neurosurg Clin North Am. 1993;4:715–726.
Hasan, SJ, Keirstead, HS, Muir, GD, et al. Axonal regeneration contributes to repair of injured brainstem-spinal neurons in embryonic chick. J Neurosci. 1993;13:492–507.
Heffez, DS, Aryanpur, J, Rotellini, NA, et al. Intrauterine repair of experimental surgically created dysraphism. Neurosurgery. 1993;32:1005–1010.
Inagaki, T, Schoenwoif, GC, Walker, ML. Experimental model: change in the posterior fossa with surgically induced spina bifida aperta in mouse. Pediatr Neurosurg. 1997;26:185–189.
Johnson, MP, Sutton, LN, Rintoul, N, et al. Fetal myelomeningocele repair: short-term clinical outcomes. Am J Obstet Gynecol. 2003;189:482–487.
Korenromp, MJ, van Gool, JD, Bruinese, HW, et al. Early fetal leg movements in myelomeningocele [letter]. Lancet. 1986;1:917–918.
Larsen, WJ. Human embryology, 2nd ed. New York: Churchill Livingstone; 1997.
Levine, D, Barnes, PD, Madsen, JR, et al. Central nervous system abnormalities assessed with prenatal magnetic resonance imaging. Obstet Gynecol. 1999;94:1011–1019.
McLone, DG, Knepper, PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1–12.
Meuli, M, Meuli-Simmen, C, Hutchins, GM, et al. The spinal cord lesion in human fetuses with myelomeningocele: implications for fetal surgery. J Pediatr Surg. 1997;32:448–452.
Paek, BW, Farmer, DL, Wilkinson, CC, et al. Hindbrain herniation develops in surgically created myelomeningocele but is absent after repair in fetal lambs. Am J Obstet Gynecol. 2000;183:1119–1123.
Pilu, G, Falco, P, Perolo, A, et al. Ultrasound evaluation of the fetal neural axis. In: Callen P, ed. Ultrasonography in obstetrics and gynecology. 4th ed. Philadelphia, PA: Saunders; 2000:277–306.
Sutton, LN, Adzick, NS, Bilaniuk, LT, et al. Improvement in hindbrain herniation demonstrated by serial fetal magnetic resonance imaging following fetal surgery for myelomeningocele. JAMA. 1999;282:1826–1831.
Tulipan, N, Bruner, JP, Hernanz-Schulman, M, et al. Effect of intrauterine myelomeningocele repair on central nervous system structure and function. Pediatr Neurosurg. 1999;31:183–188.
Tulipan, N, Hernanz-Schulman, M, Lowe, LH, et al. Intrauterine myelomeningocele repair reverses preexisting hindbrain herniation. Pediatr Neurosurg. 1999;31:137–142.
Walsh, DS, Adzick, NS, Sutton, LN, et al. The rationale for in utero repair of myelomeningocele. Fetal Diagn Ther. 2001;16:312–322.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 42
Prenatal Imaging
Imaging Techniques
The accuracy of identifying spinal anomalies during nontargeted screening ultrasonography varies, depending on the skill and experience of the operator. The accuracy of a referral center performing detailed targeting studies for a suspected neural tube defect (elevated maternal serum α-fetoprotein) is close to 100%. A detailed protocol should be performed. Because fetuses with open neural tube defects typically have Chiari II malformations, the fetal brain should be scanned initially. A small cisterna magna with rounded small cerebellum is termed the “banana sign” and is 99% sensitive in the diagnosis of a Chiari II malformation (Fig. 42-1). The frontal bones may be concave, and this is termed the “lemon sign.” This sign is less specific and may be present in 1% to 2% of normal fetuses and may resolve by the third trimester. Ventriculomegaly may be present. Axial and longitudinal views of the spine should be obtained. Spine ossification progresses from 10 to 22 weeks’ gestation. By 16 weeks’ gestation, neural arch ossification is complete to L5. By 19 weeks, S1 is completely ossified, and by 22 weeks, S2 is ossified. Splayed pedicles are best visualized in the transverse plane. An overlying sac may be imaged in transverse and longitudinal planes, with higher-frequency transducers showing cord tethering and placode contents (Fig. 42-2).
Figure 42-1 Banana sign.
An axial ultrasonographic image through the posterior fossa in a 21-week fetus with a Chiari II malformation shows the crowding of the cerebellum around the brainstem that has been termed the “banana sign” (demarcated by arrows).
Open Spinal Dysraphism
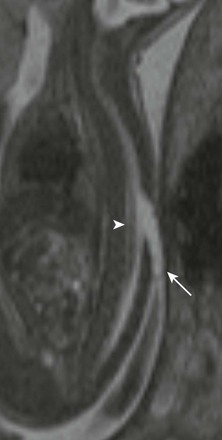
Abnormalities in neurulation may result from defects in disjunction, the process by which the neural tube separates from the overlying ectoderm. A large site of complete failure of disjunction may result in MMC. Prenatally, the findings of MMC are clearly detectable with ultrasonography and MRI. In the presence of elevation of the neural placode, secondary to expansion of the subarachnoid space, the lesion is referred to as MMC (Fig. 42-3). This is distinguished from myeloschisis, also known as myelocele, where an open neural tube defect exists, but the subarachnoid space is not expanded, and the neural placode remains within the confines of the dysraphic spinal canal (Fig. 42-4). MMC occurs during the formation of the primitive neural tube (neurulation) in the third week of gestation, when a localized failure of neural tube closure occurs. This failure may occur anywhere along the length of the spinal cord, but it is most common in the lumbar region. The resulting lesion is an open spinal canal with a flat neural placode instead of a cylindrical spinal cord. Imaging reveals the failure of neurulation in the MMC as a posterior osseocutaneous defect. When present, the expansion of the subarachnoid space is readily apparent (see Fig. 42-3). The neurologic deficits sustained by the fetus are postulated to occur in stages—a “two-hit” hypothesis. The first “hit” is the original defect in neurulation that creates the dysraphism and any associated myelodysplasia. The second “hit” is the secondary chemical or physical trauma (or both) to the neural tissue as a result of its exposure to the intrauterine environment.
Figure 42-3 Myelomeningocele.
A, A sagittal half-Fourier acquisition single-shot turbo spin echo (HASTE) magnetic resonance image in a 21-week fetus with a small myelomeningocele sac protruding at the lumbosacral junction level. The neural tissue can be readily identified traversing the sac (arrow). Note the Chiari II malformation findings. B, An axial HASTE image at the lumbosacral level shows the expanded subarachnoid space protruding through the wide posterior element deficiency. Note the neural placode outside the confines of the canal, traversing the sac.
Figure 42-4 Myeloschisis.
A, A sagittal half-Fourier acquisition single-shot turbo spin echo (HASTE) image of the spine in a 19-week fetus with a posterior spinal defect. The placode remains within the spinal canal (arrow). B, An axial HASTE image at the lumbosacral level shows the wide posterior element deficiency and neural placode within the confines of the broad canal (arrowhead).
A unified theory regarding the pathogenesis of the associated Chiari II malformation was postulated by McLone and Knepper, who suggested that the open spinal canal and associated free drainage of cerebrospinal fluid (CSF) promote collapse of the primitive ventricular system and cause lack of expansion of the rhombencephalic vesicle, from which the posterior fossa develops. This lack of distention leads to an abnormally small posterior fossa and subsequent herniation and other associated malformations in the brain. The Chiari II malformation is a pancerebral anomaly, affecting broad areas of the brain. Abnormalities include herniation of the medulla, cerebellar tonsils, and vermis through the foramen magnum; a small posterior fossa; “beaking” of the tectum of the midbrain; an enlarged massa intermedia of the thalami; partial or complete callosal dysgenesis; and structural changes in the skull (Fig. 42-5). Migrational abnormalities, particularly subependymal gray matter heterotopia, also are commonly seen. The cause of hydrocephalus in patients with MMC is frequently debated. Current theories include mechanical obstruction secondary to anatomic changes associated with the Chiari II malformation and dysfunctional CSF absorption. Clinically, hydrocephalus may not be present at birth but may become apparent after early postnatal closure of the defect.
Figure 42-5 Chiari II malformation.
A sagittal half-Fourier acquisition single-shot turbo spin echo image at the craniocervical junction in a 19-week fetus shows the hindbrain herniation and funneling of the posterior fossa of the Chiari II malformation (arrow
[/not-level-membership-for-pediatrics-category]
