5 Prediction of Radiation Response
The prescription for clinical radiotherapy is usually based on such factors as tumor site, stage, and grade. All patients with tumors falling in the same category with regard to these clinical parameters receive the same radiation schedule, conforming to the institute’s current policy. An increasing body of evidence now shows that even for similar histologic type and extent of tumor, wide variations exist in the response to irradiation. For example, the fractionation scheme (e.g., conventional, accelerated, hyper- or hypo-fractionation) has been shown to influence outcome in clinical trials.1–5 Total dose also influences outcome. It is therefore likely that a single radiotherapy prescription is not optimum for all patients, even those within the same clinical category. Some patients, but not all, may benefit from accelerated fractionation (depending on proliferation rate of the tumor), some but not all may benefit from hyperfractionation (depending on the shape of the survival curve of the tumor cells), some may require higher doses (radioresistant tumors), others may be overtreated with conventional doses (radiosensitive tumors), while for others, conventional radiotherapy of 1.8 to 2 Gy per day for 6 to 7 weeks will be the best choice.
Genetic Assays: Data-Driven Approach
Tumors
The clinical value of microarrays was shown by studies in breast cancer,6,7 lymphoma,8 lung adenocarcinoma,9 glioma,10 and others that showed tumors could be subdivided into groups based on their gene expression profiles—and that these subdivisions have clinical relevance, since the different groups have different prognoses (Fig. 5-1). In addition, the prognostic potential of microarrays was shown by van’t Veer and colleagues,11 who defined a 70-gene signature that predicted the chance of distant metastases in young women with breast cancer. Several other signatures with the same utility have been found in subsequent studies, often containing different genes.12,13 Of note is that these different signatures usually select the same patients as being at high or low risk, suggesting that many different gene sets may have similar prognostic potential for the same clinical situation and may represent different genes on common deregulated pathways.14–16 In addition to distant metastases, it also appears to be possible to predict the chance of developing regional lymph node metastases in sites such as head and neck, breast, and others.17–19
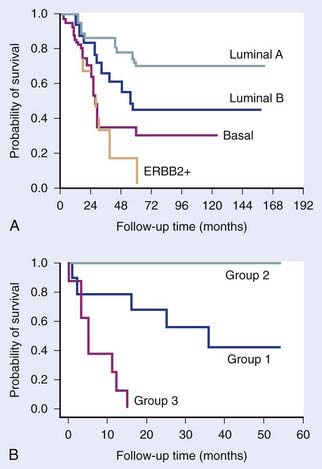
FIGURE 5-1 • Examples of the clinical value of gene expression profiling by microarrays. (A) Study by Sorlie and colleagues7 on breast tumors showing that clusters defined by differences in expression patterns have significantly different outcomes. (B) Study by Garber et al.9 showing a similar result for lung tumors.
(Redrawn from Sorlie T, Tibshirani R, Parker J, et al: Repeated observation of breast tumor subtypes in independent gene expression data sets, Proc Natl Acad Sci USA 100:8418–8423, 2003; Garber ME, Troyanskaya OG, Schluens K, et al: Diversity of gene expression in adenocarcinoma of the lung, Proc Natl Acad Sci USA 98:13784–13789, 2001.)
It is useful at this stage to distinguish between the terms prognostic and predictive. Several gene signatures have been described that appear to discriminate between good and poor outcome, but in a variety of disease sites and in patients given a variety of different treatments. Such signatures can be described as prognostic and probably reflect degree of malignancy. Examples are the “wound” signature,20 hypoxic signatures,21 genetic instability signatures,22 and stem cell signatures.23 By contrast, a signature that discriminates between good and bad responders to a specific treatment can be described as predictive. Such signatures can help in treatment selection, whereas prognostic signatures are inherently less useful. A predictive signature can also, in principle, help unravel causes of resistance, leading to potential new intervention strategies.
To date, very few predictive signatures have been described. Chung and colleagues24 defined a 75-gene high risk signature for patients with head and neck cancer treated with primary surgery followed by radiation and/or chemotherapy. Pramana and colleagues25 subsequently showed that this signature also predicted locoregional control in an independent series of head and neck cancer patients treated with radiotherapy and concomitant cisplatin (Fig. 5-2). However, the specificity of this signature for radiotherapy, with or without chemotherapy, remains to be determined. Nuyten and colleagues26 described a signature for predicting local recurrence after breast conserving therapy (local excision plus radiotherapy). Signatures have also been described that predict tamoxifen resistance in breast cancer.27,28 Validated signatures predicting response to radiotherapy alone in cancer patients have not yet been reported.
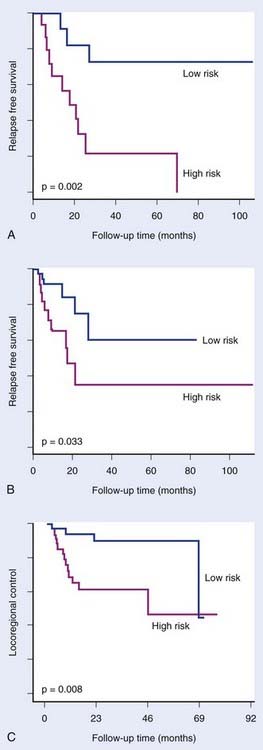
FIGURE 5-2 • Chung et al.24 defined an expression signature comprising 75 genes associated with high risk of relapse in head and neck cancer treated with primary surgery with or without subsequent treatment with radiation and/or chemotherapy. The signature significantly distinguished good and poor prognosis groups in an initial group of 28 patients (A) and in a subsequent group of 60 patients (B). Pramana et al.25 found this signature to predict locoregional control after concurrent radiotherapy and cisplatin in an independent group of 70 head and neck tumors (C).
(Redrawn from Chung CH, Parker JS, Ely K, et al: Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor–kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma, Cancer Res 66:8210–8218, 2006; Pramana J, Van den Brekel MW, van Velthuysen ML, et al: Gene expression profiling to predict outcome after chemoradiation in head and neck cancer, Int J Radiat Oncol Biol Phys 69:1544–1552, 2007.)
MicroRNAs (miRNA) also have been shown to have predictive or prognostic potential. These are small 18- to 22-nucleotide single-stranded RNAs. They are transcribed from genomic DNA like messenger RNA (mRNA), but do not code for proteins. Instead, they bind to partially complementary sequences on target messenger RNAs, thereby inhibiting translation and sometimes causing mRNA degradation. It is estimated that the expression of up to half of all genes are regulated by miRNAs. It is therefore not surprising that they have been shown to be involved in carcinogenesis and treatment response. Reports are now appearing of the predictive potential of miRNAs,29,30 which may ultimately prove to be at least as powerful as mRNA profiling for prediction (Fig. 5-3). To date, no study testing miRNA as a predictor of outcome after radiotherapy has been reported, although specific miRNAs have been shown to be induced by hypoxia and to correlate with outcome in breast cancer patients treated by surgery and adjuvant therapies.29
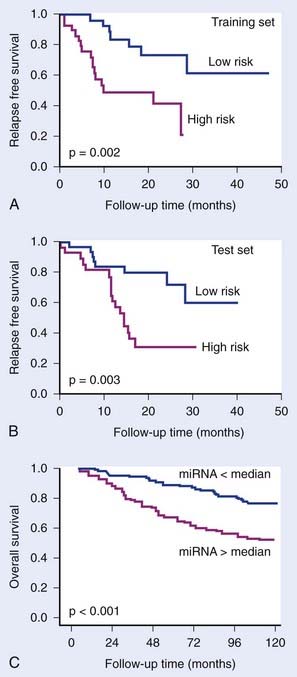
FIGURE 5-3 • The prognostic potential of microRNAs (miRNA). Yu et al.30 showed that a 5-gene miRNA signature distinguished good and poor outcome groups in non–small cell lung cancer in a training group of 56 patients (A) and in a test group of 56 patients (B). Camps et al.29 showed that a single miRNA, hsa-miR-210, which is induced under hypoxia, could distinguish good and poor outcome in 219 breast cancer patients. Camps et al. showed that a single miRNA, hsa-miR-210, which is induced under hypoxia, could distinguish good and poor outcome in 219 breast cancer patients (C).
(Redrawn from Yu SL, Chen HY, Chang GC, et al: MicroRNA signature predicts survival and relapse in lung cancer, Cancer Cell13:48-57, 2008, and Camps C, Buffa FM, Colella S, et al: hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer, Clin Cancer Res 14:1340-1348, 2008.)
Methylation of DNA in the so-called CpG islands in gene promoter regions can silence expression of those genes. Conversely, demethylation can activate transcription. Deregulation of methylation can thus lead to silencing of tumor suppressors or activation of oncogenes. Deregulation of genes involved in drug metabolism, the DNA damage response, and others can affect response to chemotherapy and radiotherapy. The most consistent result concerning prediction is in glioblastoma, where methylation of the O-6-methylguanine-DNA methyltransferase (MGMT) promoter is associated with improved survival in patients treated with radiotherapy plus alkylating agents, particularly temozolamide.31,32 The MGMT enzyme can reverse drug-induced DNA alkylation, thus reversing its cytotoxic effects. Gene methylation will reduce expression of this DNA repair gene, leading to increased tumor responses. Studies in other tumors have also shown correlations between methylation status of specific genes and outcome in colorectal cancer,33 neuroblastoma,34 and others. No clinical studies on radiotherapy alone have reported on methylation status. Several methods have been developed to measure DNA methylation, some of them approaching a genome-wide scale, and are now being increasingly applied to test their predictive potential.
Comparative genomic hybridization (CGH) measures copy number variations (CNV; amplifications and deletions) in regions of genomic DNA, currently at a 5 to 10 kilobase resolution. This method allows characterization of recurrent chromosome changes in tumors, which can pinpoint relevant known or novel oncogenes and suppressor genes. In addition, CNVs can be correlated with outcome to define CNV predictors, analogous to gene expression predictors. The resolution of current arrays is such that candidate genes in affected loci can be rapidly traced and tested further for their involvement if desired or necessary. Reports have appeared showing the prognostic potential of CNVs in several cancer types, including breast,35 gliomas,36 colorectal cancers,37 Wilms’ tumor38 and others (Fig. 5-4). Few studies have been done on radiotherapy patients, although van den Broek and colleagues39 reported finding specific gains and losses in head and neck cancers that correlated with outcome after treatment with combined chemoradiotherapy. It is likely that CGH will complement gene expression and other genome wide assays in defining robust predictors.
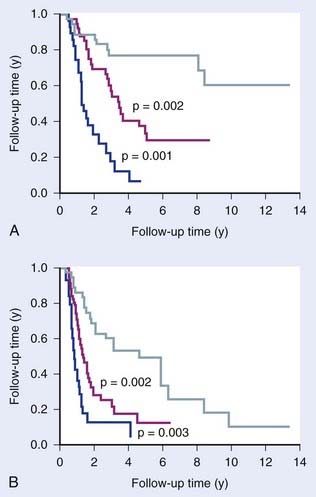
FIGURE 5-4 • The prognostic potential of comparative genomic hybridization (CGH). Idbaih et al.36 defined three groups of gliomas based on their patterns of genomic amplifications and deletions that showed prognostic significance for overall survival (A) and progression free survival (B).
(Redrawn from Chin SF, Teschendorff AE, Marioni JC, et al: High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer, Genome Biol 8:R215, 2007.)
Normal Tissues
Predicting the chance of adverse normal tissue reactions after radiation would also be a valuable aid, allowing dose adjustments and/or protective measures (radioprotective or ameliorating drugs) to be prescribed to patients at risk. It further opens the possibility of increasing tumor radiation doses for the remainder (the majority) of patients, leading to potential increases in cure rates for the patient population as a whole. Several factors are known to affect the response of normal tissues to irradiation. The extent of cell kill of parenchymal cells in the organ at risk is clearly important but is not the only factor. Cytokines are known to play an important role,40,41 TGF-β being a key cytokine in fibrosis development, influencing fibroblast proliferation and differentiation.
Attempts have been made to predict the chance of adverse reactions from the transcriptional response of patient’s lymphocytes irradiated ex vivo.42 The underlying assumption is that the genetic profile of the patient will affect radiation response in all tissues and can therefore be monitored in easily accessible peripheral blood lymphocytes. The further assumption is that sensitivity is best monitored not necessarily by the basal level of gene expression but by transcriptional response to radiation. In the Svensson study,42 an expression signature in ex vivo irradiated lymphocytes was identified that discriminated prostate cancer patients with severe late complications following radiotherapy (over-responders) from patients without such complications (nonresponders). Lymphocytes have also been used with some success for predicting radiation induced complications using colony survival43 and apoptosis assays,44 supporting the use of these cells for predictive purposes.
Gene polymorphisms have also been studied in relation to adverse radiotherapy induced reactions. Single-nucleotide polymorphisms (SNP) in several genes have been found to correlate with radiation induced fibrosis,45 particularly in the TGF-β gene. Lymphocytes represent ideal test material, since they are easy to obtain and SNPs will be the same in all cells (in contrast to gene expression). SNP predictors of adverse reactions are not yet robust, because the studies carried out to date have been small and only a few candidate genes have been investigated. The method appears promising, but evaluation will have to await the results of several ongoing larger studies looking at genome-wide SNPs.
Genetic Assays: Hypothesis-Driven Approach
Radiosensitivity
Many radiosensitivity genes have been discovered using knockout, knock-down or chemical inhibitor strategies. Many of these genes are involved in DNA repair. However, in recent years it has become clear that the DNA damage response is highly complex, so that predicting the extent of radiation induced killing for any random cell line or tumor has proven to be difficult. One approach has therefore been to use a genome-wide strategy, not dependent on known data or pathways, in which genetic signatures are sought that correlate with intrinsic radiosensitivity in a series of cell lines or tumors. Torres-Roca et al.46 described such an approach on selected cell lines from the National Cancer Institute (NCI) panel and found a small set of genes correlating with radiosensitivity. More recently, Amundson and colleagues47 extended the studies to the complete NCI panel of cell lines and found a larger set of genes correlating with intrinsic radiosensitivity, with some overlap with the Torres-Roca set. Khodarev and colleagues48 used a different approach combining animal tumor and cell line studies, creating a radioresistant and radiosensitive pair of cell lines, to define a radiosensitivity signature. Little has so far been reported on testing radiosensitivity signatures. Pramana et al.25 tested one of these signatures (Torres-Roca) on a group of head and neck cancer patients treated with radiation plus cisplatin, but no significant correlation was found. Recently, this signature has been independently refined, and the updated signature showed a strong trend (P = .06) that patients with tumors predicted to be radiosensitive had a better outcome after chemoradiotherapy (Torres-Roca J, et al., presented at ASTRO, Boston, 2008).
Hypoxia
It has been known for more than half a century that hypoxic cells are up to three times more radioresistant than normoxic cells. In addition, hypoxic cells in tumors are often more slowly proliferating and harder to reach with drugs, because they often are at a distance from blood vessels, making them more chemoresistant. Studies using glass electrodes to measure oxygen tension in human tumors have shown, firstly, how ubiquitous hypoxia is, and secondly, that it is a negative prognostic factor for all the three current major treatment modalities of surgery, radiotherapy, and chemotherapy.49–54
Cells react to hypoxia by reducing the expression of many genes. However, in contrast, there are a smaller number of genes that are up-regulated. Many of these are dependent on HIF-1α (hypoxia inducible factor), a protein that is stabilized under hypoxic conditions, leading to accumulation in the cell. HIF-1α, a transcription factor, then switches on transcription of a plethora of other genes that have an HRE (HIF responsive element, a specific short DNA sequence) in their promoter region. This allows the cell to adapt to hypoxic stress by, among others, increasing glucose uptake and stimulating angiogenesis. Such up-regulated genes represent a hypoxic signature. Several such signatures have been defined by growing cells under hypoxic conditions and subsequently measuring their expression profiles. Chi et al.21 showed that such in vitro–defined signatures have prognostic significance and are more predictive of outcome (overall and relapse-free survival) in breast and ovarian cancer than present clinical parameters. As expected, patients with tumors showing high expression of hypoxia genes, indicating high tumor hypoxia, did worse.
The cell’s response to hypoxia also depends on the degree and duration of hypoxia. More severe hypoxia and longer times under hypoxia are reflected by altered gene expression patterns. Signatures have therefore been described for both acute and chronic hypoxia, and for different degrees of hypoxia.21,55 These are potentially important distinctions, since there is accumulating evidence that acute hypoxia is more dangerous than chronic hypoxia (equally radioresistant, more viable, more DNA repair–proficient).56,57 Indeed, Seigneuric and colleagues55 showed that in vitro signatures derived from short exposures to hypoxia (acute) predicted outcome in breast cancers whereas those derived from longer exposures (chronic) did not (Fig. 5-5).
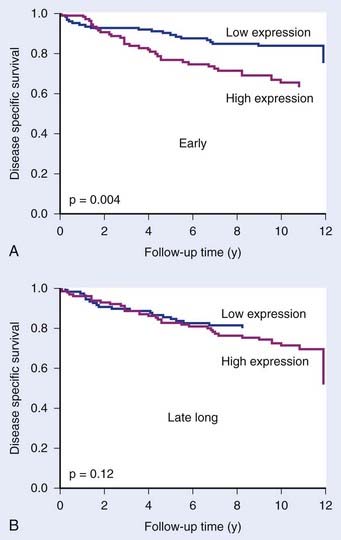
FIGURE 5-5 • The prognostic potential of gene expression signatures for hypoxia. Seigneuric et al.55 analyzed cell line data to define signatures for genes up-regulated at early times during hypoxic incubation (“early”; acute hypoxia) or after long times (“late long”) chronic hypoxia. Acute signatures (A) had greater potential to distinguish good and poor prognosis in breast cancer patients than chronic signatures (B).
(From Seigneuric R, Starmans MH, Fung G, et al: Impact of supervised gene signatures of early hypoxia on patient survival, Radiother Oncol 83:374-382, 2007.)
An alternative method of deriving hypoxic signatures was described by Winter et al.58 They first chose 10 genes known to be HIF-1α (and thus hypoxia)–dependent. They then looked for other genes whose expression correlated with expression of these 10 “seed” genes across a series of human head and neck carcinomas. In this way they defined a hypoxic metagene of 99 genes. This metagene had prognostic value in both head and neck and breast cancer, supporting the notion that hypoxia is an important negative prognostic factor and that gene signatures can be used to monitor it.
Repopulation
During the course of fractionated radiotherapy, tumor cells that have survived (remained clonogenic) up to that point can begin to divide and will increase the number of cells to be killed with the remaining dose fractions. Such tumor cell repopulation is a particular risk during weekends and other gaps in therapy. Cell kill by radiotherapy is thus counteracted by cell production (repopulation) in the treatment gaps. The greater the capacity of a tumor for repopulation, the greater the effective resistance of that tumor will be. Further, the more gaps in treatment there are, the greater the opportunity will be for repopulation. Evidence for this is strongest in head and neck cancer where a number of studies on the influence of treatment time, including split dose and accelerated fractionation, have consistently shown either worse local control if the treatment time is longer, or that higher radiation doses are needed for a given level of local control.1–4 This is most likely to be due to repopulation, which can therefore limit cure in some cancers. The influence of repopulation will vary between cancers of the same type, and between different types of cancer. Cancers that are in general more slowly growing, (e.g., breast and prostate), may be at less risk from repopulation, although some tumors within these types may be capable of rapid repopulation. It is therefore important to be able to predict repopulation in individual tumors.
Great strides have been made in understanding the molecular events driving and accompanying cell proliferation. Cell culture studies have employed cell populations synchronized in different cell cycle phases, as well as resting cells stimulated to proliferate by serum addition. These have defined a host of cycle phase–specific and proliferation-specific genes. Such proliferation-associated gene signatures include genes that regulate cell cycle progression, such as the cyclin-dependent kinases, and the many external and signal transduction pathways that control entry into and exit from the cell cycle, including cytokines, hormones, growth and antigrowth factors, checkpoints, cell adhesion, and others. Some of these signatures have been shown to have prognostic potential for outcome of breast cancer.58a In addition, using data-driven approaches, several expression-profiling studies on clinical material have found gene signatures correlating with outcome that contain a preponderance of proliferation-associated genes. Such studies include those on lymphoma,59,60 breast cancer,61 hepatocellular carcinoma,62 and others.63 These signatures, and those derived from cell culture studies, have thus been shown to have prognostic potential. These have not yet been tested on patients receiving curative radiotherapy as the primary treatment.
Validation of Genetic Signatures
Genetic signatures must be validated before clinical application. This is especially so for genome-wide measurements, where the possibility of false positives is high. During development of a signature on a clinical series (the training set), internal validation is usually performed (e.g., leave-one-out cross-validation). This is the first of several steps. In the case of the van’t Veer et al. 70-gene signature for predicting metastasis in women with breast cancer,11 this was defined on a test series of 78 patients, was subsequently validated on a larger series of patients,64 and is currently being tested in a large randomized trial.65–68 The progression from definition in a training set to validation in a separate clinical series followed by testing in a randomized trial is a route now being followed for several promising gene signatures in different diseases and therapies.69 These are necessary steps before routine application of such signatures in the clinic.
Genetic Assay Considerations
Combining Assays
Given the potential deficiencies of individual assays, a combination of assays may prove superior. Indeed, combining CGH data with expression profiling allowed Adler and colleagues to separate relevant from irrelevant genetic changes and discover the most important genes driving the “wound” signature, a set of genes with prognostic significance in breast cancer.70 Other studies have also shown the added value of combining expression profiling with CGH35,71 for defining better predictors and better subclassifications of tumors. Combining three approaches (CGH, methylation, and mRNA expression) is now possible, as shown by Sadikovic and colleagues,72 providing a more complete picture of genetic and epigenetic changes.
Cell Based and Functional Assays
Radiosensitivity
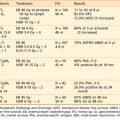
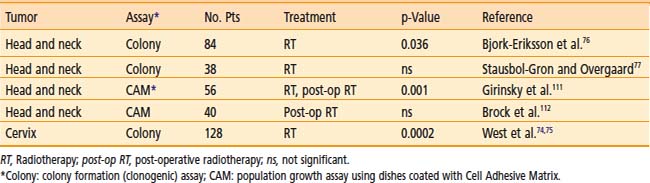
The most relevant measure of radiosensitivity is based on the fraction of cells surviving a particular radiation dose, defined as the ability of a cell to undergo at least six doublings, thus forming a clone of at least 50 cells. This is termed the colony-forming, or clonogenic, assay. A summary of predictive assays studies for radiosensitivity is shown in Table 5-1. The most convincing study is that of West and colleagues73,74 on cervix carcinomas treated by radiotherapy alone. Explanted tumor cells irradiated in vitro had SF2 values that correlated with outcome. Patients with tumors exhibiting SF2 values higher than the median value (radioresistant) had significantly worse local control and significantly worse survival rates than did those with tumors with SF2 values below the median. This trend was the same for all tumor stages. Two of the larger studies on head and neck tumors also showed a positive correlation of in vitro radiosensitivity with local control.75,76 These clinical studies support the notion that in vitro measurements of radiosensitivity, with all their potential limitations, have relevance to the response of tumors in situ.
Table 5-1 Clinical Studies Correlating Outcome After Radiotherapy with Intrinsic Radiosensitivity Measured on Cells Taken from Primary Tumor Material Before Treatment and Irradiated and Assayed in Vitro
Ionizing radiation also induces chromosome aberrations, including fragments, translocations (dicentrics or reciprocal), rings, chromatid exchanges, gaps, complex types, and micronuclei, all being dose related. Many studies have shown a good correlation between chromosome damage and cell kill.77–79 Chromosome aberrations can also be measured in a matter of days and can be detected in cells after repair with doses less than 1 Gy, making it as sensitive as γH2AX detection of DSBs. However, although chromosome damage assays have a lot of attractive features as radiosensitivity predictors, ex vivo culturing and irradiations would be required, making it unlikely to prove robust enough for routine clinical use.
Repopulation
Here the goal is to predict, before treatment begins, which tumors are capable of rapid proliferation during treatment. These could be then selected for adjusted radiotherapy schedules or alternative or extra-treatment modalities. Several methods have been tried, including simply counting the frequency of mitoses. Flow cytometry has advantages over counting cells under a microscope, allowing the quantitative measurements of many thousands of cells per minute. By using fluorescent dyes that bind to DNA, DNA histograms can be generated and analyzed for the fraction of cells in each cycle phase (G1, S, and G2/M). A more functional assessment of proliferation can be obtained using analogs of thymidine, bromodeoxyuridine (BrdU), and iododeoxyuridine (IdU), which are incorporated into DNA during the S phase. Fluorescent conjugated antibodies allow the degree of analog incorporation per cell to be rapidly measured by flow cytometry. Cell kinetics can be measured in patients with this method by injection or infusion of thymidine analogs at nontoxic tracer doses. The combination of thymidine analogs and flow cytometry allows rapid measurement of the proportion of labeled cells (labeling index [LI]). In addition, by taking samples a few hours after analog administration, one can determine both the LI and the rate of movement through the S phase (TS).80 The ratio of the two (TS/LI) approximates the potential doubling time, Tpot, a parameter describing the cell number doubling time of a tumor population in the absence of cell loss. Staining and measuring can be accomplished in 1 day. Disadvantages include the necessity of administering a drug (the thymidine analog) and the inability to reliably distinguish malignant from non-malignant cells in a biopsy.
A multicenter study from 11 different centers was carried out for head and neck tumor patients receiving radiotherapy alone given in an overall time of at least 6 weeks, with a total of 476 patients.81 All patients received the thymidine analog prior to treatment. A univariate analysis showed that only LI was significantly associated with local control (P < .03), higher values correlating with worse outcome. Tpot showed no trend. In a multivariate analysis of local control, LI lost its significance (P < .16). Two potential confounding factors in this study were that each center carried out its own flow cytometry and analysis (rather than a standard reference center), and in none of these analyses was an adequate distinction made between normal and malignant cells. This study suggests that LI but not Tpot may predict repopulation during radiotherapy, but not strongly.
Finding a proliferation marker that does not require administration of a potentially toxic substance remains a worthwhile goal. Such markers include antibodies to Ki67 (cycle-specific), PCNA (S phase–specific),82 cyclin A (S/G2 phase–specific)83 and DNA polymerase alpha (cycle-specific).84 These all provide static parameters and can be measured by either immunohistochemistry or flow cytometry. The rapidly increasing knowledge of cell cycle control gives hope that expression profiles will be found that can predict repopulation capacity.
Ideally, measurements of proliferation during, and not before, treatment are desired, since this is when the dangerous repopulation takes place. Labeling measurements can be made during treatment, but the data will be strongly dominated by doomed and dying cells that constitute the vast majority of cells after the first few 2 Gy fractions. Such measurements are therefore likely to be misleading, as shown by animal studies.85 Until ways can be found to distinguish doomed but intact cells from surviving cells, measurements during treatment will remain unreliable at best, and often come too late to change treatment.
Hypoxia
The most direct method to date for measuring tumor hypoxia is the use of glass oxygen electrodes inserted into the tumor. Multiple measurements can then be taken along several tracks, allowing the distribution of oxygen tension to be assessed. The mean or median oxygen tension can be calculated, as well as the fraction of values below a cut-off, usually 5 or 10 mm Hg, giving an estimate of the hypoxic fraction. Several studies have correlated such measurements with outcome after radiotherapy.49–53 These studies have shown remarkable uniformity in that most, but not all, found that pretreatment oxygen tension was a significant prognostic indicator. These included different tumor sites and all three major treatment modalities, although no sufficiently large series have been published for surgery alone or chemotherapy alone. Hypoxia could affect chemotherapy outcome through lower drug concentrations at hypoxic sites, and the fact that hypoxic cells tend to proliferate slower, reducing the effectiveness of many drugs. Exposure to hypoxia can also lead to selection of apoptosis-resistant cells,86 and consequently to malignant progression and an increase in metastatic capacity.87,88 These may be contributing reasons why hypoxia is also a bad prognostic indicator for surgery. A major disadvantage of electrode methods is its invasive nature and its restriction to accessible tumors.
One of the current most widely used alternative methods to electrodes is the administration of a bioreductive drug, in particular, the nitroimidazoles. Such drugs have been shown to be selectively reduced in and bind to hypoxic cells and can be detected with a labeled drug or by antibodies developed against bound products.89,90 Two of these nitroimadazoles, pimonidazole and EF5, are approved for human use as hypoxic markers.90,91 Of interest is that the pimonidazole staining fraction in head and neck tumors does not appear to correlate with electrode oxygen measurements in the same tumors.92 Possible reasons include the influence of stroma and necrosis on the polarographic measurements, or that one method may be more influenced than the other by acute (fluctuating) hypoxia. Of further interest is that the only study measuring both pimonidazole staining fraction and oxygen tension with Eppendorf electrodes found that neither parameter correlated with outcome in cervix cancer patients treated with radiotherapy alone.93
Several other methods, both direct and indirect, have also been applied in the clinic for measuring tumor hypoxia.54 These include noninvasive assessment of hypoxia using the imaging techniques of PET or SPECT or with MRI, and measuring expression of endogenous markers associated with hypoxia, such as HIF-1α and CA9 (see discussion of genetic assays above). Few studies with sufficient statistical power have yet been carried out to test the predictive potential of these techniques for radiotherapy patients. Finally, it should be noted that none of the methods can distinguish between clonogenic and nonclonogenic cells. Extrapolation from changes in the measured hypoxia parameter occurring during treatment to reoxygenation patterns of the hypoxic, clonogenic cells therefore cannot be made with any degree of certainty.
Normal Tissues
Several studies have tested the relationship between the in vitro radiosensitivity of either fibroblasts or lymphocytes and the severity of normal tissue reactions. Geara et al.94 and Johansen and colleagues95 found a significant correlation between fibroblast radiosensitivity and late reactions. These and other studies indicated that colony survival of fibroblasts after in vitro irradiation may predict for late normal tissue damage, primarily fibrosis. However, two subsequent larger studies could not confirm these results.96,97 In vitro radiosensitivity of lymphocytes, measured either by colony, cytogenetic or apoptosis assays, have been reported to predict normal tissue morbidity in some studies.43,44,98
Problems with cell-based assays include their technical difficulty and the long assay times. Clinical confounding factors include an often inaccurate estimate of dose in the target tissue.99 While cell based assays have been useful in showing that intrinsic radiosensitivity of somatic cells is probably a contributing cause to differences observed between patients in their reactions to radiotherapy, it is unlikely that they will be routinely useful as predictors for reasons stated above. In addition, intrinsic radiosensitivity is not the only determinant of radiation morbidity. There are a number of biologic factors that influence treatment response. For several tissues (e.g., lung, skin, and intestinal mucosa), the involvement of cytokine-mediated multicellular interactions are implicated, including those mediated by interleukins 2 and 6 (IL-2, IL-6), and interferon alpha (IFN-α).100 Transforming growth factor beta (TGF-β) clearly also plays an important role in generating and modulating tissue fibrosis in many tissues and organs.40,41,101 Understanding the mechanisms of normal tissue radiation response other than the conventional radiobiologic paradigm of target cell death will ultimately lead to better prediction.
Action Based on Assay Results
Several options are available for high hypoxic fraction tumors. The main current ones are the use of a chemical hypoxic cell radiosensitizer such as nimorazole,102 or applying carbogen (increases blood oxygen) with or without nicotinamide (counteracts acute hypoxia), which is also undergoing clinical testing.103 An alternative approach would be to selectively kill the hypoxic cells using a bioreductive agent such as tirapazamine. This promising approach is also undergoing clinical testing.104 Future possibilities include the delivery of gene-encoded toxins coupled to hypoxia-specific promoters and the use of anaerobic bacteria as tumor (hypoxia)-specific delivery vectors.105–107
The Future
Finally, accurate prediction will only be really useful when it not only indicates whether the tumor in an individual patient will be resistant to radiotherapy or other therapies, but when it can also guide the physician in choosing the best therapy. This will depend on the availability of pathway-specific drugs. An optimum predictor should then indicate which radioresistance pathway is activated or deregulated so that the appropriate drug inhibiting that pathway can be chosen for combining with radiotherapy. Indications from studies on hypoxia, DNA repair, growth receptor signaling, proliferation, and others suggest that this will be possible in the medium-term future. At the time of writing, there are as yet no validated markers or signatures for predicting response to available pathway specific agents in combination with radiotherapy,108 and this must come through integrating marker assays into clinical trials, preferably those where the effectiveness of such agents are specifically being tested within the trial.
Summary
The four main factors likely to be relevant for predicting outcome after radiation therapy are intrinsic tumor cell radiosensitivity, normal tissue radiosensitivity, tumor hypoxia, and tumor cell proliferation. Positive correlations with outcome have been reported for all four parameters, measured by clonogenic assays, oxygen electrodes, and BrdU/IdU-flow cytometry, although variability in results have been seen between studies for all parameters. Problems with these functional assays include reproducibility difficulties, long assays times, the necessity for ex vivo cell cultures, the need for invasive procedures, addition of tracer drugs, and the difficulty of carrying out more than one assay per patient. These have led to the use of genome-wide assays, in principle allowing multiple factors to be assessed simultaneously, as well as indicating which genes and pathways cause resistance. Gene expression, SNP, CGH, microRNA, methylation, and others have all shown promise as predictors, as well as some assay combinations. None have yet been validated fully in randomized trials, although several are in this phase of testing. Many prognostic signatures have been described, but few predictive signatures have been reported that are specific for a particular therapy, such as radiotherapy. Such therapy-specific predictors will be the most useful for choosing optimum treatments for individuals and are achieving increasingly more attention for both chemotherapy and radiotherapy. On a final note, it should not be forgotten that one of the most robust predictors of outcome of radiotherapy is the size of the tumor (Fig. 5-6). Measuring tumor volume dose not indicate the way ahead for biologists or clinicians in the future, but it should always be taken into account, as with other clinical factors of proven importance, when assessing potential predictive assays.
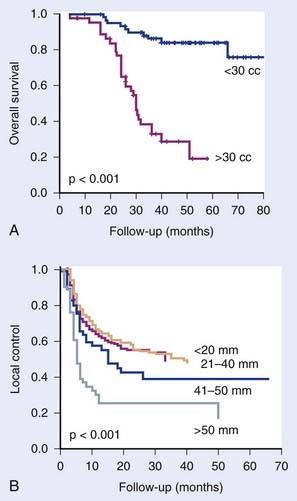
FIGURE 5-6 • Examples of studies showing that one of the most important clinical predictors is tumor volume. (A) Study of Kim et al.109 on 106 cervix cancer patients treated with concurrent chemotherapy and radiotherapy. (B) Study of Begg et al.80 on head and neck cancer patients treated with radiotherapy alone (numbers against curves are tumor diameters).
(A, Redrawn from Coco Martin JM, Mooren E, Ottenheim C, et al: Potential of radiation-induced chromosome aberrations to predict radiosensitivity in human tumour cells, Int J Radiat Biol 75:1161–1168, 1999; Liu SC, Minton NP, Giaccia AJ, et al: Anticancer efficacy of systemically delivered anaerobic bacteria as gene therapy vectors targeting tumor hypoxia/necrosis, Gene Ther 9:291–296, 2002. B, Reprinted by permission from Macmillan Publishers Ltd, Gene Ther 9:291–296, 2002.)
1 Overgaard J, Hjelm-Hansen M, Johansen LV, et al. Comparison of conventional and split-course radiotherapy as primary treatment in carcinoma of the larynx. Acta Oncol. 1988;27:147-152.
2 Overgaard J, Hansen HS, Specht L, et al. Five compared with six fractions per week of conventional radiotherapy of squamous-cell carcinoma of head and neck: DAHANCA 6 and 7 randomised controlled trial. Lancet. 2003;362:933-940.
3 Fu KK, Pajak TF, Trotti A, et al. A Radiation Therapy Oncology Group (RTOG) phase III randomized study to compare hyperfractionation and two variants of accelerated fractionation to standard fractionation radiotherapy for head and neck squamous cell carcinomas: first report of RTOG 9003. Int J Radiat Oncol Biol Phys. 2000;48:7-16.
4 Bourhis J, Overgaard J, Audry H, et al. Hyperfractionated or accelerated radiotherapy in head and neck cancer: a meta-analysis. Lancet. 2006;368:843-854.
5 Skladowski K, Maciejewski B, Golen M, et al. Continuous accelerated 7-days-a-week radiotherapy for head-and-neck cancer: long-term results of phase III clinical trial. Int J Radiat Oncol Biol Phys. 2006;66:706-713.
6 Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-752.
7 Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418-8423.
8 Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503-511.
9 Garber ME, Troyanskaya OG, Schluens K, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784-13789.
10 Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157-173.
11 van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530-536.
12 Wang HP, Long XH, Sun ZZ, et al. Identification of differentially transcribed genes in human lymphoblastoid cells irradiated with 0.5 Gy of gamma-ray and the involvement of low dose radiation inducible CHD6 gene in cell proliferation and radiosensitivity. Int J Radiat Biol. 2006;82:181-190.
13 Foekens JA, Atkins D, Zhang Y, et al. Multicenter validation of a gene expression-based prognostic signature in lymph node-negative primary breast cancer. J Clin Oncol. 2006;24:1665-1671.
14 Fan C, Oh DS, Wessels L, et al. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med. 2006;355:560-569.
15 Yu JX, Sieuwerts AM, Zhang Y, et al. Pathway analysis of gene signatures predicting metastasis of node-negative primary breast cancer. BMC Cancer. 2007;7:182.
16 Roepman P, Kemmeren P, Wessels LF, et al. Multiple robust signatures for detecting lymph node metastasis in head and neck cancer. Cancer Res. 2006;66:2361-2366.
17 Roepman P, Wessels LF, Kettelarij N, et al. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet. 2005;37:182-186.
18 Chung CH, Levy S, Yarbrough WG. Clinical applications of genomics in head and neck cancer. Head Neck. 2006;28:360-368.
19 O’Donnell RK, Kupferman M, Wei SJ, et al. Gene expression signature predicts lymphatic metastasis in squamous cell carcinoma of the oral cavity. Oncogene. 2005;24:1244-1251.
20 Chang HY, Nuyten DS, Sneddon JB, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005;102:3738-3743.
21 Chi JT, Wang Z, Nuyten DS, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 2006;3:e47.
22 Carter SL, Eklund AC, Kohane IS, et al. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043-1048.
23 Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503-1521.
24 Chung CH, Parker JS, Ely K, et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006;66:8210-8218.
25 Pramana J, Van den Brekel MW, van Velthuysen ML, et al. Gene expression profiling to predict outcome after chemoradiation in head and neck cancer. Int J Radiat Oncol Biol Phys. 2007;69:1544-1552.
26 Nuyten DS, Kreike B, Hart AA, et al. Predicting a local recurrence after breast-conserving therapy by gene expression profiling. Breast Cancer Res. 2006;8:R62.
27 Loi S, Haibe-Kains B, Desmedt C, et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics. 2008;9:239.
28 Chanrion M, Negre V, Fontaine H, et al. A gene expression signature that can predict the recurrence of tamoxifen-treated primary breast cancer. Clin Cancer Res. 2008;14:1744-1752.
29 Camps C, Buffa FM, Colella S, et al. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14:1340-1348.
30 Yu SL, Chen HY, Chang GC, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13:48-57.
31 Brandes AA, Franceschi E, Tosoni A, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008;26:2192-2197.
32 Eoli M, Menghi F, Bruzzone MG, et al. Methylation of O6-methylguanine DNA methyltransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin Cancer Res. 2007;13:2606-2613.
33 Shen L, Catalano PJ, Benson ABIII, et al. Association between DNA methylation and shortened survival in patients with advanced colorectal cancer treated with 5-fluorouracil based chemotherapy. Clin Cancer Res. 2007;13:6093-6098.
34 Yang Q, Kiernan CM, Tian Y, et al. Methylation of CASP8, DCR2, and HIN-1 in neuroblastoma is associated with poor outcome. Clin Cancer Res. 2007;13:3191-3197.
35 Chin SF, Teschendorff AE, Marioni JC, et al. High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007;8:R215.
36 Idbaih A, Marie Y, Lucchesi C, et al. BAC array CGH distinguishes mutually exclusive alterations that define clinicogenetic subtypes of gliomas. Int J Cancer. 2008;122:1778-1786.
37 Kim MY, Yim SH, Kwon MS, et al. Recurrent genomic alterations with impact on survival in colorectal cancer identified by genome-wide array comparative genomic hybridization. Gastroenterology. 2006;131:1913-1924.
38 Natrajan R, Little SE, Sodha N, et al. Analysis by array CGH of genomic changes associated with the progression or relapse of Wilms’ tumour. J Pathol. 2007;211:52-59.
39 van den Broek GB, Wreesmann VB, Van den Brekel MW, et al. Genetic abnormalities associated with chemoradiation resistance of head and neck squamous cell carcinoma. Clin Cancer Res. 2007;13:4386-4391.
40 Rodemann HP, Blaese MA. Responses of normal cells to ionizing radiation. Semin Radiat Oncol. 2007;17:81-88.
41 Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6:702-713.
42 Svensson JP, Stalpers LJ, Esveldt-van Lange RE, et al. Analysis of gene expression using gene sets discriminates cancer patients with and without late radiation toxicity. PLoS Med. 2006;3:e422.
43 West CM, Davidson SE, Elyan SA, et al. Lymphocyte radiosensitivity is a significant prognostic factor for morbidity in carcinoma of the cervix. Int J Radiat Oncol Biol Phys. 2001;51:10-15.
44 Ozsahin M, Crompton NE, Gourgou S, et al. CD4 and CD8 T-lymphocyte apoptosis can predict radiation-induced late toxicity: a prospective study in 399 patients. Clin Cancer Res. 2005;11:7426-7433.
45 Alsner J, Andreassen CN, Overgaard J. Genetic markers for prediction of normal tissue toxicity after radiotherapy. Semin Radiat Oncol. 2008;18:126-135.
46 Torres-Roca JF, Eschrich S, Zhao H, et al. Prediction of radiation sensitivity using a gene expression classifier. Cancer Res. 2005;65:7169-7176.
47 Amundson SA, Do KT, Vinikoor LC, et al. Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 2008;68:415-424.
48 Khodarev NN, Beckett M, Labay E, et al. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci USA. 2004;101:1714-1719.
49 Hockel M, Knoop C, Schlenger K, et al. Intratumoral pO2 histography as predictive assay in advanced cancer of the uterine cervix. Adv Exp Med Biol. 1994;345:445-450.
50 Nordsmark M, Overgaard J. A confirmatory prognostic study on oxygenation status and loco-regional control in advanced head and neck squamous cell carcinoma treated by radiation therapy. Radiother Oncol. 2000;57:39-43.
51 Brizel DM, Dodge RK, Clough RW, et al. Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radiother Oncol. 1999;53:113-117.
52 Fyles A, Milosevic M, Hedley D, et al. Tumor hypoxia has independent predictor impact only in patients with node-negative cervix cancer. J Clin Oncol. 2002;20:680-687.
53 Janssen HL, Haustermans KM, Balm AJ, et al. Hypoxia in head and neck cancer: how much, how important? Head Neck. 2005;27:622-638.
54 Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225-239.
55 Seigneuric R, Starmans MH, Fung G, et al. Impact of supervised gene signatures of early hypoxia on patient survival. Radiother Oncol. 2007;83:374-382.
56 Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180-192.
57 Martinive P, Defresne F, Bouzin C, et al. Preconditioning of the tumor vasculature and tumor cells by intermittent hypoxia: implications for anticancer therapies. Cancer Res. 2006;66:11736-11744.
58 Winter SC, Buffa FM, Silva P, et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007;67:3441-3449.
58a Starmans MH, Krishnapuram B, Steck H, et al. Robust prognostic value of a knowledge-based proliferation signature across large patient microarray studies spanning different cancer types. Br J Cancer. 2008;99:1884-1890.
59 Cuadros M, Dave SS, Jaffe ES, et al. Identification of a proliferation signature related to survival in nodal peripheral T-cell lymphomas. J Clin Oncol. 2007;25:3321-3329.
60 Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3:185-197.
61 Dai H, Van’T Veer L, Lamb J, et al. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005;65:4059-4066.
62 Villanueva A, Toffanin S, Llovet JM. Linking molecular classification of hepatocellular carcinoma and personalized medicine: preliminary steps. Curr Opin Oncol. 2008;20:444-453.
63 Whitfield ML, George LK, Grant GD, et al. Common markers of proliferation. Nat Rev Cancer. 2006;6:99-106.
64 van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999-2009. 19
65 Mook S, Schmidt MK, Viale G, et al. The 70-gene prognosis-signature predicts disease outcome in breast cancer patients with 1–3 positive lymph nodes in an independent validation study. Breast Cancer Res Treat. 2009;116:295-302.
66 Desmedt C, Piette F, Loi S, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13:3207-3214.
67 Bogaerts J, Cardoso F, Buyse M, et al. Gene signature evaluation as a prognostic tool: challenges in the design of the MINDACT trial. Nat Clin Pract Oncol. 2006;3:540-551.
68 Buyse M, Loi S, Van’T Veer L, et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98:1183-1192.
69 Bonnefoi H, Potti A, Delorenzi M, et al. Validation of gene signatures that predict the response of breast cancer to neoadjuvant chemotherapy: a substudy of the EORTC 10994/BIG 00-01 clinical trial. Lancet Oncol. 2007;8:1071-1078.
70 Adler AS, Lin M, Horlings H, et al. Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet. 2006;38:421-430.
71 Blenk S, Engelmann JC, Pinkert S, et al. Explorative data analysis of MCL reveals gene expression networks implicated in survival and prognosis supported by explorative CGH analysis. BMC Cancer. 2008;8:106.
72 Sadikovic B, Yoshimoto M, Al Romaih K, et al. In vitro analysis of integrated global high-resolution DNA methylation profiling with genomic imbalance and gene expression in osteosarcoma. PLoS ONE. 2008;3:e2834.
73 West CM, Davidson SE, Roberts SA, et al. Intrinsic radiosensitivity and prediction of patient response to radiotherapy for carcinoma of the cervix. Br J Cancer. 1993;68:819-823.
74 West CM, Davidson SE, Roberts SA, et al. The independence of intrinsic radiosensitivity as a prognostic factor for patient response to radiotherapy of carcinoma of the cervix. Br J Cancer. 1997;76:1184-1190.
75 Bjork-Eriksson T, West C, Karlsson E, et al. Tumor radiosensitivity (SF2) is a prognostic factor for local control in head and neck cancers. Int J Radiat Oncol Biol Phys. 2000;46:13-19.
76 Stausbol-Gron B, Overgaard J. Relationship between tumour cell in vitro radiosensitivity and clinical outcome after curative radiotherapy for squamous cell carcinoma of the head and neck. Radiother Oncol. 1999;50:47-55.
77 Carrano AV. Chromosome aberrations and radiation-induced cell death. II. Predicted and observed cell survival. Mutat Res. 1973;17:355-366.
78 Cornforth MN, Bedford JS. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat Res. 1987;111:385-405.
79 Coco Martin JM, Mooren E, Ottenheim C, et al. Potential of radiation-induced chromosome aberrations to predict radiosensitivity in human tumour cells. Int J Radiat Biol. 1999;75:1161-1168.
80 Begg AC, McNally NJ, Shrieve DC, et al. A method to measure the duration of DNA synthesis and the potential doubling time from a single sample. Cytometry. 1985;6:620-626.
81 Begg AC, Haustermans K, Hart AA, et al. The value of pretreatment cell kinetic parameters as predictors for radiotherapy outcome in head and neck cancer: a multicenter analysis. Radiother Oncol. 1999;50:13-23.
82 Celis JE, Bravo R, Larsen PM, et al. Cyclin: a nuclear protein whose level correlates directly with the proliferative state of normal as well as transformed cells. Leuk Res. 1984;8:143-157.
83 Juan G, Li X, Darzynkiewicz Z. Correlation between DNA replication and expression of cyclins A and B1 in individual MOLT-4 cells. Cancer Res. 1997;57:803-807.
84 Wahl AF, Geis AM, Spain BH, et al. Gene expression of human DNA polymerase alpha during cell proliferation and the cell cycle. Mol Cell Biol. 1988;8:5016-5025.
85 Begg AC, Hofland I, Kummermehr J. Tumour cell repopulation during fractionated radiotherapy: correlation between flow cytometric and radiobiological data in three murine tumours. Eur J Cancer. 1991;27:537-543.
86 Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88-91.
87 Cairns RA, Kalliomaki T, Hill RP. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001;61:8903-8908.
88 Hasan NM, Adams GE, Joiner MC, et al. Hypoxia facilitates tumour cell detachment by reducing expression of surface adhesion molecules and adhesion to extracellular matrices without loss of cell viability. Br J Cancer. 1998;77:1799-1805.
89 Hodgkiss RJ, Jones G, Long A, et al. Flow cytometric evaluation of hypoxic cells in solid experimental tumours using fluorescence immunodetection. Br J Cancer. 1991;63:119-125.
90 Evans SM, Hahn S, Pook DR, et al. Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res. 2000;60:2018-2024.
91 Raleigh JA, Chou SC, Calkins-Adams DP, et al. A clinical study of hypoxia and metallothionein protein expression in squamous cell carcinomas. Clin Cancer Res. 2000;6:855-862.
92 Nordsmark M, Loncaster J, Chou SC, et al. Invasive oxygen measurements and pimonidazole labeling in human cervix carcinoma. Int J Radiat Oncol Biol Phys. 2001;49:581-586.
93 Nordsmark M, Loncaster J, Aquino-Parsons C, et al. The prognostic value of pimonidazole and tumour pO2 in human cervix carcinomas after radiation therapy: a prospective international multi-center study. Radiother Oncol. 2006;80:123-131.
94 Geara FB, Peters LJ, Ang KK, et al. Prospective comparison of in vitro normal cell radiosensitivity and normal tissue reactions in radiotherapy patients. Int J Radiat Oncol Biol Phys. 1993;27:1173-1179.
95 Johansen J, Bentzen SM, Overgaard J, et al. Evidence for a positive correlation between in vitro radiosensitivity of normal human skin fibroblasts and the occurrence of subcutaneous fibrosis after radiotherapy. Int J Radiat Biol. 1994;66:407-412.
96 Russell NS, Grummels A, Hart AA, et al. Low predictive value of intrinsic fibroblast radiosensitivity for fibrosis development following radiotherapy for breast cancer. Int J Radiat Biol. 1998;73:661-670.
97 Peacock J, Ashton A, Bliss J, et al. Cellular radiosensitivity and complication risk after curative radiotherapy. Radiother Oncol. 2000;55:173-178.
98 Barber JB, Burrill W, Spreadborough AR, et al. Relationship between in vitro chromosomal radiosensitivity of peripheral blood lymphocytes and the expression of normal tissue damage following radiotherapy for breast cancer. Radiother Oncol. 2000;55:179-186.
99 Russell NS, Begg AC. Editorial radiotherapy and oncology 2002: predictive assays for normal tissue damage. Radiother Oncol. 2002;64:125-129.
100 Okunieff P, Cornelison T, Mester M, et al. Mechanism and modification of gastrointestinal soft tissue response to radiation: role of growth factors. Int J Radiat Oncol Biol Phys. 2005;62:273-278.
101 Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys. 2000;47:277-290.
102 Overgaard J, Hansen HS, Overgaard M, et al. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother Oncol. 1998;46:135-146.
103 Kaanders JH, Pop LA, Marres HA, et al. ARCON: experience in 215 patients with advanced head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2002;52:769-778.
104 Rischin D, Peters L, Fisher R, et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Radiation in patients with locally advanced head and neck cancer: a randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02). J Clin Oncol. 2005;23:79-87.
105 Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-447.
106 Nuyts S, Van Mellaert L, Theys J, et al. Clostridium spores for tumor-specific drug delivery. Anticancer Drugs. 2002;13:115-125.
107 Liu SC, Minton NP, Giaccia AJ, et al. Anticancer efficacy of systemically delivered anaerobic bacteria as gene therapy vectors targeting tumor hypoxia/necrosis. Gene Ther. 2002;9:291-296.
108 Riesterer O, Milas L, Ang KK. Use of molecular biomarkers for predicting the response to radiotherapy with or without chemotherapy. J Clin Oncol. 2007;25:4075-4083.
109 Kim H, Kim W, Lee M, et al. Tumor volume and uterine body invasion assessed by MRI for prediction of outcome in cervical carcinoma treated with concurrent chemotherapy and radiotherapy. Jpn J Clin Oncol. 2007;37:858-866.