34 Postoperative Cardiovascular Management
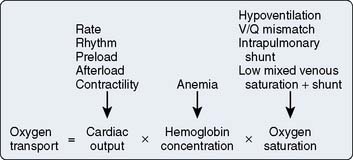
Oxygen transport
Maintaining oxygen transport (i.e., oxygen delivery [Do2]) satisfactory to meet the tissue metabolic needs is the goal of postoperative circulatory control. Oxygen transport is the product of cardiac output (CO) times arterial content of oxygen (Cao2; i.e., hemoglobin concentration × 1.34 mL oxygen per 1 g hemoglobin × oxygen saturation), and it can be affected in many ways by the cardiovascular and respiratory systems, as shown in Figure 34-1. Low CO, anemia from blood loss, and pulmonary disease can decrease Do2. Before altering the determinants of CO, including the inotropic state of the ventricles, an acceptable hemoglobin concentration and adequate oxygen saturation (Sao2) should be provided, enabling increases in CO to provide the maximum available Do2. As hemoglobin concentration increases, so does blood viscosity and, therefore, the work of the heart to eject the blood. In normal hearts (e.g., athletes), increasing hemoglobin levels to supranormal will increase performance, suggesting that in this setting the increased viscosity is less important than the increase in oxygen-carrying capacity.1 This has not been examined in patients with cardiac disease. Model analysis of data from animal investigations suggests that maintenance of the hematocrit between 30% and 33% provides the best balance between oxygen-carrying capacity and viscosity.2 This analysis also suggests that, in ischemic states, a hematocrit in this range may be desirable. Patients needing continued inotropic or mechanical support of ventricular function beyond the first few postoperative hours, especially those in need of intravascular volume expansion, should probably be transfused to a hematocrit in this range, bearing in mind that blood transfusion has been associated with decreased organ function and increased mortality in critically ill patients. A randomized trial suggested a transfusion threshold of 7 g, rather than 9 g, was associated with at least equivalent outcomes in critically ill patients who did not have acute myocardial infarction (MI) or unstable angina.3–5 Neither of these studies identified cohorts of patients who had undergone cardiac surgery. Wu et al5 found transfusion for a hematocrit of 30% or lower in elderly patients with acute MI was associated with better outcome. This study supports the concept that this is the desirable hematocrit, especially in elderly cardiac surgery patients or those experiencing a complicated course.
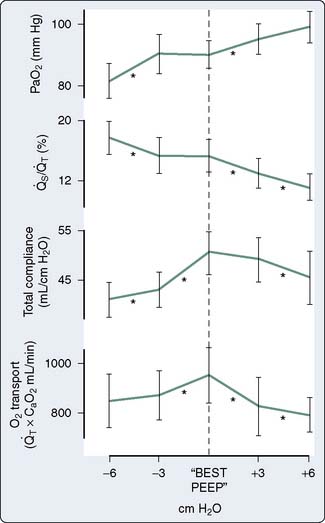
Hypoxemia from any cause reduces Do2, and acceptable arterial oxygenation (Pao2) may be achieved with the use of an increased inspired oxygen concentration (Fio2) or positive end-expiratory pressure (PEEP) in the ventilated patient. Use of PEEP or continuous positive airway pressure in the spontaneously breathing patient may improve Pao2 by reducing intrapulmonary shunt; however, venous return may be reduced, causing a decrease in CO, with DO2 decreased despite an increased Pao2 (Figure 34-2).6 It is important to measure CO as PEEP is applied. Intravascular volume expansion may be used to offset this damaging effect of PEEP7 (see Chapter 35).
In patients with marginal arterial oxygenation, pulmonary function must be monitored closely to allow prompt therapy to be undertaken for abnormalities. Measurements of airway resistance and respiratory system compliance should be made. When resistance is increased, treatment of bronchospasm may improve the Pao2 and CO, because decreases in intrathoracic pressure improve venous return. Treatment of lung overinflation may decrease pulmonary vascular resistance (PVR), benefiting right ventricular (RV) function.8 If compliance is decreased, application of PEEP or continuous positive airway pressure may help promote re-expansion of atelectatic areas and move the tidal volume to a more compliant section of the pressure-volume relation of the respiratory system9 (Figure 34-3). This will reduce the work expended by the patient during spontaneous efforts and may reduce PVR10 (see Chapter 35).
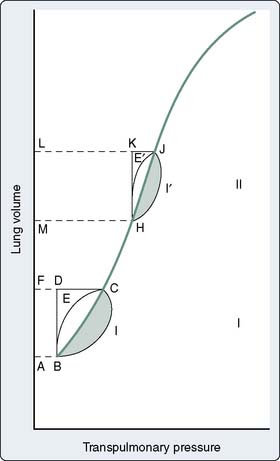
Figure 34-3 Pressure-volume diagram of elastic and resistive (nonelastic) work done on noncompliant lungs.
(From Katz JA, Marks JD: Inspiratory work with and without continuous positive airway pressure in patients with acute respiratory failure. Anesthesiology 63:598, 1985.)
Unexplained hypoxemia may be caused by right-to-left intracardiac shunting, most commonly by a patent foramen ovale. This is most likely to occur when right-sided pressures are abnormally increased; an example is the use of high levels of PEEP.11 If suspected, echocardiography should be performed, and therapy to reduce right-sided pressures should be initiated.
Patients with pulmonary disease may experience dramatic worsening of oxygenation when vasodilator therapy is started because of release of hypoxic pulmonary vasoconstriction in areas of diseased lung.12 Although CO may be increased, the worsening in Cao2 will result in a decrease in Do2. Reduced dosage of direct-acting vasodilators or trials of different agents may be indicated.
 o2) may be taken while awaiting improvement in cardiac or pulmonary function. For example, sedation and paralysis may buy time to allow reversible postoperative myocardial dysfunction to improve.
o2) may be taken while awaiting improvement in cardiac or pulmonary function. For example, sedation and paralysis may buy time to allow reversible postoperative myocardial dysfunction to improve.Temperature
Patients often are admitted to the intensive care unit (ICU) after cardiac surgery with core temperatures less than 35° C, especially after off-pump cardiac surgery. The typical pattern of temperature change during and after cardiac surgery and the hemodynamic outcomes are illustrated in Figure 34-4. Decreases in temperature after CPB occur, in part, because of redistribution of heat within the body and because of heat loss. Noback and Tinker13 found that administration of nitroprusside and the use of high flows (> 2.2 L/min/m2) during rewarming on CPB could improve the uniformity of rewarming and reduce this afterdrop from 4° C to about 2° C. Monitoring of body sites other than the blood and brain (e.g., urinary bladder, tympanic membrane temperatures) can help provide more complete rewarming, but the body temperature usually declines after bypass, especially when difficulties are encountered and the chest remains open for an extended period, and some degree of hypothermia is an almost unavoidable result.14,15 Intraoperative use of newer forced air warming blankets or cutaneous gel pads16 can help reduce the temperature loss during and after surgery.
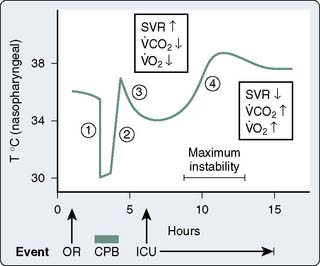
Figure 34-4 Nasopharyngeal temperature during and after cardiac surgery.
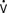 o2) are decreased on admission to the ICU because of residual hypothermia. During rapid rewarming, SVR decreases and Vco2 and
o2) are decreased on admission to the ICU because of residual hypothermia. During rapid rewarming, SVR decreases and Vco2 and 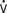 o2 increase, which can cause marked cardiac and ventilatory instability. OR, operating room.
o2 increase, which can cause marked cardiac and ventilatory instability. OR, operating room.(From Sladen RN: Management of the adult cardiac patient in the intensive care unit. In Ream AK, Fogdall RP [eds]: Acute Cardiovascular Management: Anesthesia and Intensive Care. Philadelphia, JB Lippincott, 1982, p 495.)
The normal thermoregulatory and metabolic responses to hypothermia remain intact after cardiac surgery, resulting in peripheral vasoconstriction that contributes to the hypertension commonly seen early in the ICU.17 As temperature decreases, CO is decreased because of bradycardia, whereas oxygen consumed per beat is actually increased.18 Coagulation, platelet, and immune functions are impaired by hypothermia to potentiate postoperative bleeding and infection.19–21 Other adverse consequences of postoperative hypothermia are large increases in 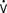 o2 and CO2 production during rewarming.22 When patients cannot increase CO (i.e., O2 delivery), the effects of this large increase in
o2 and CO2 production during rewarming.22 When patients cannot increase CO (i.e., O2 delivery), the effects of this large increase in 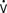 o2 include mixed venous desaturation and metabolic acidosis. Unless end-tidal carbon dioxide is monitored or arterial blood gases are analyzed often to show the increased CO2 production and guide increases in ventilation, hypercarbia will occur, causing catecholamine release, tachycardia, and pulmonary hypertension.23 These effects of rewarming are most intense when patients shiver.24 Shivering may be treated effectively with meperidine, which lowers the threshold for shivering. Muscle relaxation may provide more stable hemodynamics than meperidine but needs accompanying sedation to avoid having an awake and paralyzed patient.25,26
o2 include mixed venous desaturation and metabolic acidosis. Unless end-tidal carbon dioxide is monitored or arterial blood gases are analyzed often to show the increased CO2 production and guide increases in ventilation, hypercarbia will occur, causing catecholamine release, tachycardia, and pulmonary hypertension.23 These effects of rewarming are most intense when patients shiver.24 Shivering may be treated effectively with meperidine, which lowers the threshold for shivering. Muscle relaxation may provide more stable hemodynamics than meperidine but needs accompanying sedation to avoid having an awake and paralyzed patient.25,26
Assessment of the circulation
 Physical Examination
Physical Examination
Surgical dressings, chest tubes attached to suction, fluid in the mediastinum and pleural spaces, peripheral edema, and temperature gradients can distort or mask information obtained by the classic techniques of inspection, palpation, and auscultation in the postoperative period. However, the physician should not be deterred from applying these basic techniques in view of the potential benefit. Physical examination may be of great value in diagnosing gross or acute pathology, such as pneumothorax, hemothorax, or acute valvular insufficiency, but it is of limited value in diagnosing and managing ventricular failure. For example, in the critical care setting, experienced clinicians (e.g., internists) using only physical findings often misjudge cardiac filling pressures by a large margin.27 Low CO, in particular, is not consistently recognized by clinical signs, and systemic BP does not correlate with CO after cardiac surgery. Oliguria and metabolic acidosis, classic indicators of a low CO, are not always reliable because of the polyuria induced by hypothermia, oxygen debts induced during CPB causing acidosis, and medications or fluids given during or immediately after bypass.28
Although clinicians are taught that the adequacy of CO can be assessed by the quality of the pulses, capillary refill, and peripheral temperature, there is no relationship between these indicators of peripheral perfusion and CO or calculated systemic vascular resistance (SVR) in the postoperative period.29 By the first postoperative day, there is a crude correlation between peripheral temperature and cardiac index (CI; r = −0.60). Many patients arrive in the ICU in a hypothermic state, and residual anesthetic agents can decrease the threshold for peripheral vasoconstriction in response to this condition.30 A patient’s extremities may therefore remain warm despite a hypothermic core or a decreasing CO. Even after temperature stabilization on the first postoperative day, the relation between peripheral perfusion and CO is too crude to be used for hemodynamic management.
 Invasive Monitoring
Invasive Monitoring
Concepts regarding invasive monitoring with a pulmonary artery catheter (PAC) have been revolutionized in the past decade because of several studies in a variety of settings that fail to show a benefit from its use. In addition, there is a poor relation between filling pressures and end-diastolic volume, stroke volume (SV), or volume responsiveness. A recent review of patients admitted to medical ICUs in the United States demonstrated a reduction of more than 40% in PAC use in the 10 years before 2004.31 The same trend was evident in surgical patients, including those undergoing cardiac surgery. The PAC rarely is used in cardiac surgery patients in many other countries. Greater availability of high-quality bedside echocardiography, often performed by intensivists, has made this modality a technique of choice in the postoperative period. Measures of volume responsiveness in mechanically ventilated patients, such as pulse pressure or SV variability (from arterial waveform analysis devices), are widely recognized as more sensitive and specific indicators of the need for intravascular volume expansion than filling pressures.32
Specialized PACs have been developed that permit continuous mixed venous oxygen saturation (S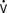 o2) monitoring, continuous CO measurement, calculation of RV volumes and ejection fraction, or have either imbedded electrodes or channels to pass atrial or ventricular pacing wires. The ability to pace through a PAC is particularly valuable in patients undergoing “minimally invasive” procedures in which the surgeon does not have adequate access to the heart to place epicardial leads. The Svo2 catheter helps evaluate the adequacy of Do2 and allows continuous assessment of the response to therapy, which may affect Do2 or
o2) monitoring, continuous CO measurement, calculation of RV volumes and ejection fraction, or have either imbedded electrodes or channels to pass atrial or ventricular pacing wires. The ability to pace through a PAC is particularly valuable in patients undergoing “minimally invasive” procedures in which the surgeon does not have adequate access to the heart to place epicardial leads. The Svo2 catheter helps evaluate the adequacy of Do2 and allows continuous assessment of the response to therapy, which may affect Do2 or 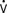 o2 (e.g., PEEP therapy). The trend in the Svo2 may function as an early warning signal of worsening in the oxygen supply/demand relation as Do2 declines or VO2 increases. In the postoperative period, the Svo2 does not correlate with CO because the latter is only one of the factors in the oxygen supply/demand relation.33 On the continuous CO catheter, a wire coil warms the blood, passing by it at time intervals determined by an algorithm, and the measured changes in temperature at the tip of the catheter are used to provide a continuous display of the CO. Although the CO displayed needs gathering of information over several minutes and is therefore not as quick as conventional thermodilution, and it does not provide beat-to-beat SV, it avoids having to give injected volumes to the patient (which can add up to a significant amount every 24 hours) and provides trends that may give earlier warning than intermittent injections. The “volumetric” PAC-computer system (REF-1; Edwards Lifesciences, Irvine, CA) uses a high-sensitivity thermistor to permit calculation of accurate right-sided volumes34 (see Chapter 14).
o2 (e.g., PEEP therapy). The trend in the Svo2 may function as an early warning signal of worsening in the oxygen supply/demand relation as Do2 declines or VO2 increases. In the postoperative period, the Svo2 does not correlate with CO because the latter is only one of the factors in the oxygen supply/demand relation.33 On the continuous CO catheter, a wire coil warms the blood, passing by it at time intervals determined by an algorithm, and the measured changes in temperature at the tip of the catheter are used to provide a continuous display of the CO. Although the CO displayed needs gathering of information over several minutes and is therefore not as quick as conventional thermodilution, and it does not provide beat-to-beat SV, it avoids having to give injected volumes to the patient (which can add up to a significant amount every 24 hours) and provides trends that may give earlier warning than intermittent injections. The “volumetric” PAC-computer system (REF-1; Edwards Lifesciences, Irvine, CA) uses a high-sensitivity thermistor to permit calculation of accurate right-sided volumes34 (see Chapter 14).
 Echocardiography
Echocardiography
Echocardiography is the technique of choice for acute assessment of cardiac function. Just as transesophageal echocardiography (TEE) has become essential for intraoperative management in various conditions, several studies document its utility in the postoperative period in the presence and absence of the PAC.35–38 It provides information that may lead to urgent surgery or prevent unnecessary surgery, gives important information about cardiac preload, and can detect acute structural and functional abnormalities. Although transthoracic echocardiography can be performed more rapidly in this setting, satisfactory images can be obtained only in about 50% of patients in the ICU39 (see Chapters 11 to 14).
Postoperative myocardial dysfunction
Studies using hemodynamic, nuclear scanning, and metabolic techniques have documented worsening in cardiac function after coronary artery bypass grafting (CABG) surgery.40–53 Although improvements in myocardial protection, surgical techniques, and operative care have been reported, similar incidences of early biventricular dysfunction (90%) were reported between 1979 and 1990. All of these studies showed significant declines in left ventricular (LV) or biventricular (when measured) function in the first postoperative hours, with gradual return to preoperative values by 8 to 24 hours. In one study, this decline was evident in only half the patients44; but in the other studies, more than 90% of patients showed at least a transient decrease in function. Decreased ventricular performance at normal or increased filling pressures occurs, suggesting decreased contractility. Similarly, “flattening” of the ventricular function curves is usually obvious, suggesting that preload expansion much greater than 10 mm Hg for CVP or 12 mm Hg for pulmonary capillary wedge pressure is of little benefit. In the classic study by Mangano,45 patients with an LV ejection fraction of less than 0.45 or ventricular dyssynergy showed more marked and prolonged dysfunction than did those patients with normal ventricles.
Satisfactory myocardial protection is important to prevent postoperative dysfunction. In off-pump surgery, the idea is to preserve coronary perfusion; but during mechanical manipulation, changes in CO and BP can occur. For CABG with CPB, most surgeons use some combination of hypothermia and crystalloid or blood cardioplegia to arrest the heart and reduce its metabolism. Although there is little consensus that any one technique is preferable in all circumstances, cold intermittent crystalloid cardioplegia with systemic hypothermia is the most widely used technique clinically and in the reported studies. Salerno et al54 recommended continuous, warm, retrograde blood cardioplegia without systemic hypothermia. Mullen et al50 suggested that blood cardioplegia had at least short-term benefit with less myocardial damage and better function; however, other studies of blood cardioplegia have showed mixed results47–52 (see Chapters 28 and 29).
Other proposed factors that contribute to postoperative ventricular dysfunction include myocardial ischemia,55 residual hypothermia,46,47 preoperative medications such as β-adrenergic antagonists,53 and ischemia/reperfusion injury (Box 34-1). Inflammatory cell activation from cytokine generation, upregulation of neutrophil adhesion molecules with neutrophil activation, oxygen free radical formation, and lipoperoxidation after ischemia/reperfusion injury may be important pathways accounting for the dysfunction. Multiple studies have showed the importance of limiting myocardial ischemia/reperfusion injury.56,57 Breisblatt et al40 observed the timing of ventricular dysfunction, and recovery after CPB for CABG was similar to what had been suggested in animal models of reperfusion injury.58–60 This nadir at 4 hours corresponds to the peak in cytokine levels. Cytokines can release nitric oxide from endothelium, which produces myocardial depression. Data evaluating complement inhibition with pexelizumab in improving outcomes represent a novel strategy61 (see Chapter 8).
Postoperative myocardial ischemia
Although intraoperative myocardial ischemia has been a focus, studies have shown that ischemia often occurs after surgery and is associated with adverse cardiac outcomes. Leung et al55 found the electrocardiographic (ECG) and segmental wall motion abnormality evidence of ischemia early after surgery in up to 40% of patients undergoing CABG surgery. Postbypass segmental wall motion abnormalities were significantly associated with adverse outcomes (e.g., MI, death; Figure 34-5). Surprisingly, these abnormalities most often appeared in the regions of the heart that had been revascularized. Hemodynamic changes rarely preceded ischemia; however, postoperative heart rates (HRs) were, as reported in other studies, significantly greater than intraoperative or preoperative values. Jain et al62 found major ECG changes in the 8 hours after cross-clamp release in 58% of CABG patients, and these changes were independent predictors of perioperative MI. Whether such changes occur because of surgery-reperfusion or events after CPB is not known. These findings do suggest that monitoring for ischemia must continue after revascularization. It may be that early recognition and treatment of ischemia or prophylactic medication can help prevent or reduce myocardial ischemia and dysfunction occurring after CABG surgery (see Chapters 6, 10, 12, 15, and 18).
Early recovery, or fast-tracking, of the cardiac surgical patient has led to some concern that ischemia will occur as patients awaken early after surgery in pain, especially because Mangano et al63 showed that sedation with a sufentanil infusion could reduce ischemia in this period. A randomized study by Cheng et al64 dispelled this concern because awakening and extubation within 6 hours of CABG were not associated with more CK-MB (isoenzyme of creatine kinase with muscle and brain subunits) release or ECG changes than overnight ventilation. Wahr et al65 showed that even with the use of propofol sedation, hemodynamic episodes (significant changes in HR and BP) were common in the 12 hours after surgery, and ST-segment changes occurred in 12% to 13% of patients.
Therapeutic interventions
 Postoperative Arrhythmias
Postoperative Arrhythmias
Patients with preoperative or newly acquired noncompliant ventricles need a correctly timed atrial contraction to provide satisfactory ventricular filling, especially when they are in sinus rhythm before surgery (see Chapters 4, 5, 10, 19, and 25). Although atrial contraction provides around 15% to 20% of ventricular filling, this may be more important in postoperative patients, when ventricular dysfunction and reduced compliance may be present. For example, in medical patients with acute MI, atrial systole contributed 35% of the SV.66 The SV is relatively fixed in patients with ventricular dysfunction, and the HR is an important determinant of CO. Rate and rhythm disorders need to be corrected when possible, using epicardial pacing wires. Approaches to postoperative rate and rhythm disturbances are listed in Table 34-1. The use of a PAC with atrial or ventricular pacing electrodes or use of lumens for pacing wires can facilitate temporary pacing if epicardial wires are not functioning. Failing that, temporary transvenous pacing wires can be placed (see Chapter 25).
| Disturbance | Usual Causes | Treatments |
|---|---|---|
| Sinus bradycardia | Preoperative/intraoperative β-blockade | Atrial pacingβ-AgonistAnticholinergic |
| Heart block (first, second, and third degree) | IschemiaSurgical trauma | Atrioventricular sequential pacingCatecholamines |
| Sinus tachycardia | Agitation/pain | Sedation/analgesia |
| Hypovolemia | Volume administration | |
| Catecholamines | Change or stop drug | |
| Atrial tachyarrhythmias | Catecholamines | Change or stop drug |
| Chamber distentionElectrolyte disorder (hypokalemia, hypomagnesemia) | Treat underlying cause (e.g., vasodilator, give K+/Mg2+)May require synchronized cardioversion or pharmacotherapy | |
| Ventricular tachycardia or fibrillation | IschemiaCatecholamines | CardioversionTreat ischemia, may require pharmacotherapy |
| Change or stop drug |
Later in the postoperative period (days 1 through 3), supraventricular tachyarrhythmias become a major problem, with atrial fibrillation (AF) predominating. The overall incidence rate is between 30% and 40%, but with increasing age and valvular surgery, the incidence rate may be in excess of 60%.67 There are many reasons for this, including genetic factors, inadequate atrial protection during surgery, electrolyte abnormalities, change in atrial size with fluid shifts, epicardial inflammation, stress, and irritation.68 Randomized trials of off-pump coronary artery bypass have found a similar incidence of postoperative AF compared with on-pump CABG.69,70
Advanced age, a history of AF, and valvular heart surgery are the most consistently identified risk factors for AF.68 Because AF is difficult to treat and potentially increases duration and cost of hospitalization, there is a great interest in effective therapy and prophylaxis.67 Many studies have showed that β-blockade significantly reduces the incidence of postoperative AF and that withdrawal of β-blockers in patients receiving them before surgery is an important risk factor. Guidelines published by the American Heart Association, American College of Cardiology, and North American Society of Pacing and Electrophysiology recommend administration of β-blockers to prevent postoperative AF if there are no contraindications.71 Sotalol, which also has some class III actions, is also effective72 and is currently available in the intravenous form in North America (see Chapters 4 and 10).
In the PAPABEAR study, oral amiodarone (10 mg/kg daily) or placebo was given 6 days before surgery through 6 days after surgery (13 days).73 Atrial tachyarrhythmias occurred in fewer amiodarone patients (48/299; 16.1%) than in placebo patients (89/302; 29.5%) overall, in patients younger than 65 years (19 [11.2%] vs. 36 [21.1%], in patients older than 65 years (28 [21.7%] vs. 54 [41.2%]), in patients who had CABG surgery only (22 [11.3%] vs. 46 [23.6%]), in patients who had valve replacement/repair surgery (25 [23.8%] vs. 44 [44.1%]), in patients who received preoperative β-blocker therapy (27 [15.3%] vs. 42 [25.0%]), and in patients who did not receive preoperative β-blocker therapy (20 [16.3%] vs. 48 [35.8%]), respectively. Postoperative sustained ventricular tachyarrhythmias occurred less frequently in amiodarone patients (1/299; 0.3%) than in placebo patients (8/302; 2.6%) (P = 0.04).73
In another study, Guarnieri et al74 evaluated 300 patients randomized in a double-blind fashion to intravenous amiodarone (1 g/day for 2 days) or to placebo immediately after cardiac surgery. The primary end points of the trial were incidence of AF and length of hospital stay. AF occurred in 67 (47%) of 142 patients on placebo versus 56 (35%) of 158 on amiodarone (P = 0.01). Length of hospital stay for the placebo group was 8.2 ± 6.2 days, and 7.6 ± 5.9 days for the amiodarone group.
After AF or other supraventricular arrhythmias develop, treatment often is urgently needed for symptomatic relief or hemodynamic benefit. The longer a patient remains in AF, the more difficult it may be to convert, and the greater the risk for thrombus formation and embolization.68,72 Treatable underlying conditions such as electrolyte disturbances or pain should be corrected while specific pharmacologic therapy is being instituted. Paroxysmal supraventricular tachycardia (uncommon in this setting) can be abolished or converted by intravenous adenosine, and atrial flutter can sometimes be converted by overdrive atrial pacing by temporary wires placed at the time of surgery. Electrical cardioversion may be necessary if hypotension is caused by the rapid rate; however, atrial arrhythmias recur in this setting.67 Rate control for AF or flutter can be achieved with various atrioventricular nodal blocking drugs, and conversion is facilitated by many of these drugs as well. Table 34-2 summarizes the various treatment modalities for supraventricular arrhythmias. If conversion to sinus rhythm does not occur, electrical cardioversion in the presence of antiarrhythmic drug therapy should be attempted or anticoagulation with warfarin started (see Chapters 4, 10, and 25).
TABLE 34-2 Treatment Modalities for Supraventricular Arrhythmias
| Treatment | Specifics* | Indications |
|---|---|---|
| Overdrive pacing by atrial wires† | Requires rapid pacer (up to 800/min); start above arrhythmia rate and slowly decrease | PAT, atrial flutter |
| Adenosine | Bolus dose of 6–12 mg; may cause 10 seconds of complete heart block | AV nodal tachycardiaBypass-tract arrhythmia |
| Atrial arrhythmia diagnosis | ||
| Amiodarone | 150 mg IV over 10 minutes, followed by infusion | Rate control/conversion to NSR in atrial fibrillation/flutter |
| β-Blockade | Esmolol, up to 0.5 mg/kg load over 1 minute, followed by infusion if tolerated | Rate control/conversion to NSR in atrial fibrillation/flutter |
| Metoprolol, 0.5–5 mg, repeat effective dose q4-6h | Rate control/conversion to NSR in atrial fibrillation/flutter | |
| Propranolol, 0.25–1 mg; repeat effective dose q4h‡ | ||
| Labetalol, 2.5–10 mg; repeat effective dose q4hr‡ | Conversion of atrial fibrillation/flutter to NSR | |
| Sotalol, 40–80 mg PO q12h | Conversion of PAT to NSR | |
| Ibutilide | 1 mg over 10 minutes; may repeat after 10 minutes | Rate control/conversion to NSR in atrial fibrillation/flutter |
| Verapamil | 2.5–5 mg IV, repeated PRN‡ | |
| Diltiazem | 0.2 mg/kg over 2 minutes, followed by 10–15 mg/hr§ | Rate control/conversion to NSR in atrial fibrillation/flutter |
| Procainamide | 50 mg/min up to 1 g, followed by 1–4 mg/min | Rate control/conversion to NSR in atrial fibrillation/flutter |
| Prevention of recurrence of arrhythmias | ||
| Treatment of wide-complex tachycardias¶¶ | ||
| Digoxin¶ | Load of 1 mg in divided doses over 4–24 hours**; may give additional 0.125-mg doses 2 hours apart (3–4 doses) | Rate control/conversion to NSR in atrial fibrillation/flutter |
| Synchronized cardioversion | 50–300 J (external); most effective with anteroposterior patches | Acute tachyarrhythmia with hemodynamic compromise (usually atrial fibrillation or flutter) |
AV, atrioventricular; IV, intravenously; NSR, normal sinus rhythm; PAT, paroxysmal atrial tachycardia; PO, orally; PRN, as needed; SVT, supraventricular tachycardia.
* See specific drug monographs for full description of indications, contraindications, and dosage. Doses are for intravenous administration; use lowest dose and administer slowly in patients with hemodynamic compromise.
† Verify pacer is not capturing ventricle.
‡ Infusion may provide better control. This drug is less useful than diltiazem because of myocardial depression.
§ Limited experience; may cause less hypotension than verapamil.
¶¶ When diagnosis is unclear (ventricular vs. supraventricular) and there is no acute hemodynamic compromise (i.e., cardioversion not indicated).
¶ Rate of administration depending on urgency of rate control.
** Less useful than other drugs because of slow onset and modest effect.
 Preload
Preload
The Frank-Starling law states that myocardial work increases as the resting length of the myocardial fiber increases.75 In vivo, this implies that SV will increase with increasing end-diastolic volume, although there is a limit at which SV reaches a plateau (and possibly decreases), with further increases in end-diastolic volume caused by excessive muscle stretch. In the normal myocardium, the Frank-Starling mechanism is the most important mechanism for increasing CO, and hypovolemia is a common cause of decreased CO and hypotension in the perioperative period. Assessment of preload is probably the single most important clinical skill for managing hemodynamic instability. Preload rapidly changes in the postoperative period because of bleeding, spontaneous diuresis, vasodilation during warming, the effects of positive-pressure ventilation and PEEP on venous return, capillary leak, and other causes.
Direct assessment of preload is clinically feasible using echocardiography. Several studies have demonstrated a fair-to-good correlation between echocardiographic and radionuclide measures of end-diastolic volume, and there is a good correlation between end-diastolic area by TEE and SV.76–79 Although the use of echocardiography to assess preload must always be tempered by the realization the clinician is viewing a two-dimensional image of a three-dimensional object, this is the most direct technique clinically available. Increased awareness of the value of TEE in the ICU and increased availability of echocardiography in general have made this modality a first choice in acute assessment of preload in the setting of unexplained or refractory hypotension. Without echocardiography, pressure measurements are used as surrogates for volume measurements. For example, in the absence of mitral valve disease, left atrial pressure is almost equal to LV end-diastolic pressure, and pulmonary artery occlusion pressure (PAOP) is almost equivalent to these two pressures. In patients without left atrial pressure catheters, the PAOP or, when shown to be equivalent to this latter number, the pulmonary artery diastolic (PAd) pressure is used (see Chapters 5 and 14).
The use of PAOP as a measure of preload may be misleading in various settings, including after cardiac surgery, when changes in pressure may not accurately reflect changes in ventricular end-diastolic volume. Studies by Ellis et al80 and Calvin et al81 suggest that fluid therapy in postoperative patients could cause a large increase in LV end-diastolic volume, with minimal or no change in PAOP. Mangano et al45,82 reported that fluid loading after CPB can uncover LV dysfunction and have also shown there is little benefit to be derived from exceeding a PAOP of about 12 mm Hg. Whether this is secondary to an open pericardium, which allows overdistention of the left ventricle, the use of PEEP causing RV distention, or other factors is unclear. Breisblatt et al83 evaluated changes in ventricular pressure-volume relations after CABG surgery while keeping a low-to-normal PAOP (10 to 15 mm Hg). The increase seen in LV end-diastolic volume was not significant enough to explain the degree of ventricular dysfunction they noted. Bouchard et al84 compared ventricular performance assessments from the PAC (i.e., LV stroke work index) with fractional area change and regional wall motion score index from TEE in 60 patients during and after cardiac surgery. They found no correlation between LV stroke work index and fractional area change and postulated that changes in ventricular compliance, loading conditions, and ventricular function alter the pressure-volume relation of the left ventricle in a manner that leads to discordant interpretations between the two techniques.
When ventricular compliance is normal and the ventricle is not distended, small changes in end-diastolic volume are usually accompanied by small changes in end-diastolic pressure. In patients with noncompliant ventricles from preexisting congestive heart failure (HF), chronic hypertrophy resulting from hypertension or valvular disease, postoperative MI, or ventricular dysfunction, small increases in ventricular volume may produce rapid increases in end-diastolic pressure, requiring therapeutic intervention.83–89 Increased intraventricular pressure will increase myocardial oxygen demand (M o2) and decrease subendocardial coronary blood flow.90 Myocardial ischemia may be the result. Increases in LV end-diastolic pressure are transmitted to the pulmonary circulation, causing congestion and, possibly, hydrostatic pulmonary edema. Although PAOP or left atrial pressure may not always show true preload, there are still good reasons to monitor them. The periods when patients are at particular risk for increases in end-diastolic pressure include awakening, endotracheal suctioning, and rapid-volume resuscitation. If myocardial ischemia or acute HF occurs, sudden increases in the end-diastolic pressure may result.
o2) and decrease subendocardial coronary blood flow.90 Myocardial ischemia may be the result. Increases in LV end-diastolic pressure are transmitted to the pulmonary circulation, causing congestion and, possibly, hydrostatic pulmonary edema. Although PAOP or left atrial pressure may not always show true preload, there are still good reasons to monitor them. The periods when patients are at particular risk for increases in end-diastolic pressure include awakening, endotracheal suctioning, and rapid-volume resuscitation. If myocardial ischemia or acute HF occurs, sudden increases in the end-diastolic pressure may result.
Many drugs may be used to reduce cardiac preload. Direct-acting vasodilators, especially nitroglycerin, increase venous capacitance, decreasing end-diastolic volume and pressure.89–91 Intravenous furosemide, besides its diuretic effect, increases venous capacitance.92 Diuretics are important to help remove the fluid that is mobilized in the days after surgery. In patients who may not tolerate the acute volume loss that is induced by the loop diuretics, a furosemide infusion allows a more gradual diuresis. Such infusions have been shown to be effective in patients with renal dysfunction.93,94 In a patient who appears refractory to diuresis, it is important to evaluate the circulatory state. Such refractoriness may suggest that a renal insult has been suffered, but it may also suggest underperfusion of the kidneys. In the latter case, the preload must be kept at the upper range of normal and CO augmented.
A new agent used in treatment of acute decompensated left-heart failure is recombinant B-type natriuretic peptide (nesiritide). The mechanism of action occurs by specific cell-surface receptors, stimulation of which increases levels of intracellular cyclic guanosine monophosphate (cGMP). The physiologic effects are mainly vasodilation, natriuresis, and renin inhibition. The result is balanced vasodilation, reducing preload and afterload, while simultaneously increasing SV and CO and promoting diuresis.95 Nesiritide has not been studied widely in the cardiac surgery population, but it has been used anecdotally in patients with poor ventricular function and high filling pressures, pulmonary hypertension, or RV failure.96 A related drug, human atrial natriuretic peptide, was shown in a small, randomized trial to reduce the need for dialysis and to improve dialysis-free survival after complicated cardiac surgery.97
Angiotensin-converting enzyme (ACE) inhibitors also can cause venodilation and reduce preload. Alternatively, opioids or benzodiazepines, or both, used to reduce endogenous catecholamine release, should be considered in the patient who needs mechanical ventilation. Morphine causes histamine release, which directly induces venodilation. In the patient with oliguria and renal failure who is fluid overloaded, peritoneal dialysis, hemodialysis, or continuous hemofiltration or dialysis may be needed.98,99
 Contractility
Contractility
Contractility is a well-defined concept in vitro, where it can be measured by the velocity of shortening of isolated muscle strips. However, it has been more complex to quantify the contractility of the intact heart because it has been difficult to find a variable to measure contractility that is also independent of preload and afterload. The pioneering work of Suga and Sagawa100,101 has shown that contractility can be measured by the end-systolic elastance of the ventricle, defined as
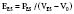
in which PES is the end-systolic pressure, VES is the end-systolic volume, and V0 is a dead-space term. EES is strictly determined by evaluating ventricular pressure-volume loops for different preloads or afterloads and by defining end-systole as the point in time at which the time-varying elastance is maximal.102 The slope of the line connecting the points at end-systole is EES. This parameter varies with changes in inotropic state but is nearly independent of preload and afterload. Routine measurement of end-systolic elastance is not clinically feasible. However, consideration of the above definition underscores the utility of TEE for the qualitative evaluation of contractility in the clinical setting. A decrease in contractility is manifested as some combination of a decrease in pressure or an increase in VES (i.e., a decrease in SV). End-systolic volume can be estimated using TEE. A large VES (implying a low ejection fraction) with a low or normal BP suggests a low value for EES and poor contractility. If BP is high, a large value of VES may be seen even if contractility is normal. By interpreting end-diastolic volume, end-systolic volume, and ejection fraction in the context of BP, an assessment of contractility is possible with TEE (see Chapters 5 and 12 to 14).
An alternative measure of contractility is the preload-recruitable stroke work, which is the slope of the line relating stroke work to preload.103 In the operating room or ICU, it often is estimated by the extent of the increase in CO that accompanies an increase in preload and is not dependent on the availability of TEE. If preload is increased by a change in patient position, BP can be a surrogate for CO because it is unlikely that SVR will change in the short time needed for the position change. A change in BP, therefore, is proportional to the change in CO.
Therapy for decreased contractility should be directed toward correcting any reversible causes, such as myocardial depressants, metabolic abnormalities, or myocardial ischemia. If the cause of depressed myocardial contractility is irreversible, positive inotropic agents may be necessary to keep a CO satisfactory to support organ function (see Chapters 5, 10, 28, and 32).
 Afterload
Afterload
Afterload is a concept that is well defined in vitro, where it refers to the added tension imposed on isolated muscle strips with contraction, but it is harder to define in vivo. In analogy with in vitro studies, afterload can be equated with ventricular wall stress, expressed as the product of cavity radius times transmural pressure divided by wall thickness, as described by the law of Laplace. However, many investigators find this definition unsatisfactory because it implies the heart generates its own afterload and because afterload would be viewed as changing during the cardiac cycle.104 If afterload is viewed as the external forces opposing ejection, possibly the best definition is the aortic input impedance, the complex ratio of pressure to flow, expressed in terms of magnitude, and the phase angle between flow and pressure for any given frequency. However, it has been difficult to quantitatively analyze the impact of impedance on overall cardiac performance (see Chapter 5). Sunagawa et al105 proposed a simplified theory of ventricular-vascular coupling within the framework of the end-systolic pressure-volume relation. Using the definition of end-systolic elastance produces the following:
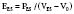
Equating SV with VE − VES (ignoring the difference between end-ejection and end-systole), it is a matter of algebra to show that
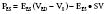
The application of an electrical law describing constant voltages and flows to the circulation, in which pulsatile flow is generated by a pump, has resulted in estimates of afterload that are questionable.104 Although SVR is a component of impedance, it cannot be equated with it. The correct downstream pressure is probably not the CVP.106 There is instead a critical opening pressure that should be used in calculating SVR that is not measurable in routine clinical care.106 Clinical use of such calculated resistances is made complex by the relation of CO to body size; the normal resistance of a small patient is much higher than that of a large one. The use of a resistance index (i.e., using CI instead of CO) partly overcomes this problem, but it is not widely used.
Calculated SVR continues to be widely used in guiding therapy or drawing conclusions about the state of the circulation. This should only be done with caution, if at all. SVR is not a complete indicator of afterload. Even if SVR were an accurate measure of impedance, the response to vasoactive agents depends on the coupling of ventricular-vascular function, not on impedance alone. Hemodynamic therapy should be guided based on the primary variables, BP and CO. If preload is appropriate, low BP and low CO are treated with an inotropic drug. If BP is acceptable (and preload appropriate) but CO is low, a vasodilator alone or in combination with an inotropic drug is used. If the patient is hypertensive (with low CO), vasodilators are indicated; if the patient is vasodilated (low BP and high CO), vasoconstrictors are used (Box 34-2).
BOX 34-2 Hemodynamic Therapy Guidelines
| Blood Pressure | Cardiac Output | Treatment |
|---|---|---|
| Low | Low | Inotrope |
| Normal | Low | Vasodilator ± Inotrope |
| High | Low | Vasodilator |
| Low | High | Vasopressor |
Postoperative hypertension
Hypertension has been a common complication of cardiac surgery, reported to occur in 30% to 80% of patients from studies in the 1970s when CABG was common in patients with normal ventricular function.107–109 The current population of older, sicker patients appears to have fewer problems with hypertension than with low-output syndromes or vasodilation. Although hypertension most commonly occurs in patients with normal preoperative ventricular function, after aortic valve replacement, or a prior history, any patient may experience development of hypertension. Multiple reasons contribute to postoperative hypertension, including preoperative hypertension, preexisting atherosclerotic vascular disease, awakening from general anesthesia, increases in endogenous catecholamines, activation of the plasma renin-angiotensin system, neural reflexes (e.g., heart, coronary arteries, great vessels), and hypothermia.110 Arterial vasoconstriction with various degrees of intravascular hypovolemia is the hallmark of perioperative hypertension.
The hazards of untreated postoperative hypertension include depressed LV performance, increased M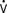 o2, cerebrovascular accidents, suture line disruption, MI, rhythm disturbances, and increased bleeding.108,111,112 Historically, therapy for hypertension in cardiac surgery was sodium nitroprusside because of its rapid onset and short duration of action.113 With multiple vasodilators available in the current era, sodium nitroprusside is no longer the drug of choice for many reasons. Nitroprusside is a potent venodilator, increasing venous capacitance (decreasing preload), and can produce arterial vasodilation, often leading to precipitous decreases in BP and a hyperdynamic cardiac state. Sodium nitroprusside can cause coronary arteriolar dilation with the potential for a steal phenomenon, resulting in myocardial ischemia.114 In patients with renal failure, the elimination of sodium nitroprusside is reduced, potentially leading to toxic effects of its metabolites cyanide or thiocyanate. This can occur if large doses are given to patients with normal renal function.
o2, cerebrovascular accidents, suture line disruption, MI, rhythm disturbances, and increased bleeding.108,111,112 Historically, therapy for hypertension in cardiac surgery was sodium nitroprusside because of its rapid onset and short duration of action.113 With multiple vasodilators available in the current era, sodium nitroprusside is no longer the drug of choice for many reasons. Nitroprusside is a potent venodilator, increasing venous capacitance (decreasing preload), and can produce arterial vasodilation, often leading to precipitous decreases in BP and a hyperdynamic cardiac state. Sodium nitroprusside can cause coronary arteriolar dilation with the potential for a steal phenomenon, resulting in myocardial ischemia.114 In patients with renal failure, the elimination of sodium nitroprusside is reduced, potentially leading to toxic effects of its metabolites cyanide or thiocyanate. This can occur if large doses are given to patients with normal renal function.
There are many alternative drugs to sodium nitroprusside for treating hypertension after cardiac surgery, including nitroglycerin,115 adrenergic-blocking agents such as β-adrenergic blockers,116 and the mixed α- and β-adrenergic blocker labetalol.117 Direct-acting vasodilators, dihydropyridine calcium channel blockers (e.g., nicardipine,118 isradipine,119 clevidipine,120–123) ACE inhibitors,113 and fenoldopam (a dopamine1 [D1] receptor agonist)124,125 also have been used. Novel therapeutic approaches are listed in Table 34-3.
| Drug | Mechanism of Action | Half-Life |
|---|---|---|
| Nicardipine | Calcium channel blocker | Intermediate |
| Clevidipine | Calcium channel blocker | Ultra-short |
| Fenoldopam | Dopamine1-receptor agonist | Ultra-short |
| Nesiritide | Brain natriuretic agonist | Short |
| Levosimendan | K+-ATP channel modulator | Intermediate |
Dihydropyridine calcium channel blockers are particularly effective in cardiac surgical patients because they relax arterial resistance vessels without negative inotropic actions or effects on atrioventricular nodal conduction and are important therapeutic options. Dihydropyridines are arterial-specific vasodilators of peripheral resistance arteries, resulting in a generalized vasodilation, including the renal, cerebral, intestinal, and coronary vascular beds. In doses that effectively reduce BP, the dihydropyridines have little or no direct negative effect on cardiac contractility or conduction. Although the dihydropyridines are more vasoselective than verapamil and diltiazem, there are also differences between dihydropyridines in this respect. Nifedipine is the least vasoselective of the dihydropyridines, isradipine and clevidipine are the most selective, and nicardipine and nimodipine are intermediately selective.120 Nicardipine is available for intravenous administration and is an important therapeutic agent to consider because of its lack of effects on vascular capacitance vessels and preload in patients after cardiac surgery.126 The pharmacokinetic profile of nicardipine suggests that effective administration requires variable rate infusions when trying to treat hypertension because of the half-life of 40 minutes. If even more rapid control is essential, a dosing strategy consisting of a loading bolus or rapid infusion dose with a constant-rate infusion may be more efficient. The effect of nicardipine may persist even though the infusion is stopped. Clevidipine, a new ultra-short-acting dihydropyridine approved in 2008 in the United States for clinical use, has a half-life of only minutes, represents a potential alternative to sodium nitroprusside, and has been studied extensively in cardiac surgical patients.121–123
Fenoldopam is a short-acting dopamine agonist approved for short-term intravenous therapy that causes arterial-specific vasodilation by stimulation of D1 receptors. Unlike sodium nitroprusside, D1-receptor stimulation also increases renal blood flow to produce diuresis and natriuresis. Fenoldopam and sodium nitroprusside were similarly effective in reducing BP in patients who experienced development of hypertension after CABG surgery.124 Fenoldopam often needs greater doses for severe hypertension that may be associated with increases in HR.
The ACE inhibitors are used in treating chronic hypertension and ventricular dysfunction. Enalaprilat is the intravenous preparation used for administration in the postoperative setting.127 These drugs are indirect vasodilators that function by inhibiting angiotensin II formation and breakdown of bradykinin. Intravenous enalaprilat has an unpredictable effect and may be long acting. A suitable role is for replacing intravenous ACE inhibitors in a patient being weaned from short-acting agents or with HF.
Postoperative vasodilation
Vasodilation and a need for vasoconstrictor support are relatively frequent complications of cardiac surgery with and without CPB. The reported incidence rate is 4% to 44%, but this wide range largely results from lack of a common definition.128–130 Vasodilation alone should be associated with a hyperdynamic circulatory state presenting as systemic hypotension, in association with an increased CO (and a low calculated SVR). More commonly, after cardiac surgery, a combination of vasodilation and myocardial dysfunction occurs, requiring vasoconstrictor and inotropic therapy. Gomez et al131–133 coined the term vasoplegia syndrome for the condition that requires high doses of vasoconstrictors, and they reported its occurrence after off-pump and on-pump surgery.
Multiple humoral and inflammatory cascades are activated by surgery alone and by CPB, aortic cross-clamping, and reperfusion, generating complement anaphylatoxins, kinins, and cytokines, many of which cause vasodilation by direct and indirect vascular mechanisms.134–136 Another potential cause of systemic vasodilation is splanchnic circulatory insufficiency resulting in endotoxemia, and this, too, has been noted after off-pump surgery137,138 (see Chapter 8). The cellular mechanisms and pathogenesis of vasodilatory shock were summarized by Landry and Oliver.135 Although the most common clinical context of vasodilatory shock is sepsis, the similarity in the cytokine response and clinical syndrome seen in sepsis with the vasodilated state seen after cardiac surgery is striking. As Landry and Oliver135 described, the stimuli of cytokines and increased tissue lactate lead to increased nitric oxide synthase and the generation of vasodilating GMP. Nitric oxide and metabolic acidosis activate potassium channels, which hyperpolarize the cell membrane, making it refractory to calcium entry and thereby refractory to norepinephrine and angiotensin II action. At this time, plasma vasopressin levels are low because of central depletion. Reports of marked vasodilatory shock after CPB responsive to vasopressin appeared when this pathophysiology was investigated.139 The ability of vasopressin to block the potassium channels and interfere with nitric oxide signaling makes it an important therapy for this syndrome. Provided there is an acceptable CO, vasopressin is a valuable agent for treating vasodilation after cardiac surgery, significantly reducing the dose requirement for norepinephrine. Systemic vasodilation also can result from hyperthermia caused by excessive warming during CPB (see Chapters 8, 28, and 29) and during warming in the ICU.
When patients experience development of acute systemic vasodilation after administration of drugs or blood products, an anaphylactic reaction should be considered. Acute anaphylaxis caused by immunoglobulin E (IgE)–mediated responses can present with systemic vasodilation and increased CO.140 Alternatively, complement-mediated transfusion reactions to any blood product can present with hypotension produced by systemic vasodilation or by thromboxane-mediated acute pulmonary vasoconstriction and RV dysfunction. Antibodies in the donor blood called leukoagglutinins, when directed against recipient white cell antigens, can actively produce white cell aggregation and thromboxane generation. These reactions have been called transfusion-related acute lung injury, which can manifest with hypotension, RV failure, and noncardiogenic pulmonary edema.140 Monitoring of RV function may, therefore, help to identify these transfusion reactions.
While underlying causes are being sought and treated, the therapeutic approach to systemic vasodilation includes intravascular volume expansion, α-adrenergic agents, and vasopressin. Administration of vasoconstrictors for more than a brief period must be guided by measures of cardiac performance because restoration of BP may camouflage a low-output state. There are no established guidelines for beginning vasoconstrictor therapy; autoregulation in vital organs is lost at mean arterial pressures (MAPs) less than 60 mm Hg, and it is reasonable to try to achieve this pressure in normotensive patients (possibly higher in hypertensive patients). A study in septic shock patients was unable to show a benefit from MAPs greater than 65 mm Hg.141
Clinicians often are concerned about the potential for constricting supply vessels or the microcirculation to vital beds (e.g., brain, kidney); although not fully evaluated in the postoperative setting, giving vasoconstrictors in septic states does not appear to have such harmful effects.142 Use of relatively low doses of vasopressin to restore responsiveness to catecholamines is physiologically sensible, but there is no clear evidence to suggest that use of vasopressin besides or instead of norepinephrine is associated with better outcome. However, dopamine was recently demonstrated to increase mortality in cardiogenic shock.143
Coronary artery spasm
Coronary artery or internal mammary artery vasospasm can occur after surgery. Mechanical manipulation and underlying atherosclerosis of the native coronary circulation and the internal mammary artery have the potential to produce transient endothelial dysfunction. The endothelium is responsible for releasing endothelium-derived relaxing factor, which is nitric oxide, a potent endogenous vasodilator substance that preserves normal endogenous vasodilation (see Chapters 6 and 8). Thromboxane can be liberated because of heparin-protamine interactions, CPB, platelet activation, or anaphylactic reactions to produce coronary vasoconstriction.144,145 Calcium administration, increased α-adrenergic tone from vasoconstrictor administration (especially in bolus doses), platelet thromboxane liberation, and calcium channel blocker withdrawal represent added reasons that may put the cardiac surgical patient at risk for spasm of native coronary vessels and arterial grafts. Engelman et al145 reported four patients who experienced development of coronary artery spasm after discontinuation of their calcium channel blockers 8 to 18 hours before surgery. In three of these patients, spasm was identified by the ECG pattern and documented as the cause of ischemia in the distribution of a nondiseased right coronary artery, with the fourth patient developing spasm in a bypassed native vessel. In two of the patients, the problem was recognized retrospectively; MIs developed, and one patient died. In the other two patients, spasm was recognized, and intravenous nitroglycerin was given (1 to 3 μg/kg/min) in combination with nifedipine, 10 mg sublingually every 5 to 6 hours, to reverse the ischemic process. The therapy of choice remains empirical. Nitroglycerin is a first-line drug, but nitrate tolerance can occur. Phosphodiesterase (PDE) inhibitors represent novel approaches to this problem and have been reported to be effective in vascular models of spasm.146 Intravenous dihydropyridine calcium channel blockers are also important therapeutic considerations.147
Reports of successful use of the radial artery as a bypass conduit have rekindled interest in this vessel.148–150 In the early days of CABG surgery, this conduit was abandoned because of its propensity to spasm. In later reports, techniques developed in the use of the internal mammary artery have been applied to the radial artery, as well as prophylactic use of diltiazem infusions.148,150 Which components of this approach are responsible for the reported success are not known, but use of a calcium channel blocking drug is recommended by many surgeons. The arterial selectivity of the dihydropyridine drugs (e.g., nicardipine) should be an advantage in this setting. However, addition of a vasodilator drug to prevent spasm of the radial artery in a patient needing a vasopressor for systemic vasodilation makes no pharmacologic sense.
Decreased contractility
Drugs that increase contractility all result in increased calcium mobilization from intracellular sites to and from the contractile proteins or sensitize these proteins to calcium. Although calcium chloride has been used to increase inotropy, evidence suggests that after CPB, its principal action is peripheral vasoconstriction.151 The same group of investigators has shown that exogenously administered calcium chloride attenuates the response to catecholamines in this setting.152 The administration of calcium salts will improve myocardial performance if there is severe hypocalcemia or hyperkalemia and may be indicated during rapid transfusion of citrated blood.153
Catecholamines, through β1-receptor stimulation in the myocardium, increase intracellular cyclic adenosine monophosphate (cAMP). This second messenger increases intracellular calcium, causing an improvement in myocardial contraction.154 Inhibition of the breakdown of cAMP by PDE inhibitors increases intracellular cAMP independent of the β receptor.155 Intracellular calcium availability can be increased by inhibiting Na+/K+-ATPase with digitalis glycosides, promoting transmembranous Na+/Ca++ exchange. However, the use of digoxin to increase myocardial contractility for postoperative ventricular dysfunction is limited by its slow onset, low potency, and narrow therapeutic safety margin. The “calcium sensitizers” constitute a new class of inotropic agents. One drug in this class, levosimendan, is being evaluated in clinical trials (Box 34-3).
BOX 34-3 Pharmacologic Approaches for Perioperative Ventricular Dysfunction
 Catecholamines
Catecholamines
The catecholamines used after surgery include dopamine, dobutamine, epinephrine, norepinephrine, and isoproterenol (Box 34-4). These drugs have various effects on α and β receptors and, therefore, various effects on HR, rhythm, and myocardial metabolism (see Chapters 10 and 32). Dosing recommendations for the catecholamines are provided in Table 34-4.
| Drug | Infusion Dose (μg/kg/min) |
|---|---|
| Dopamine*† | 2–10 |
| Dobutamine† | 2–10 |
| Epinephrine‡ | 0.03–0.20 |
| Norepinephrine‡ | 0.03–0.20 |
| Isoproterenol‡ | 0.02–0.10 |
* Less than 2 μg/kg/min predominantly “dopaminergic” (renal and mesenteric artery dilatation).
† If 10 μg/kg/min is ineffective, change to epinephrine or norepinephrine.
‡ Dose to effect; may require greater dose than indicated.
 o2 and decrease coronary perfusion pressure. In patients with bradycardias in whom pacing is not an immediate or practical option, or in those in whom an increased HR is desirable (e.g., cardiac transplant recipients, patients with regurgitant valvular lesions), isoproterenol may be used with caution bearing in mind its rather narrow therapeutic window. Although used for its β2 effects in the pulmonary vasculature, it is a weak pulmonary vasodilator.
o2 and decrease coronary perfusion pressure. In patients with bradycardias in whom pacing is not an immediate or practical option, or in those in whom an increased HR is desirable (e.g., cardiac transplant recipients, patients with regurgitant valvular lesions), isoproterenol may be used with caution bearing in mind its rather narrow therapeutic window. Although used for its β2 effects in the pulmonary vasculature, it is a weak pulmonary vasodilator.Epinephrine
Epinephrine is a potent adrenergic agonist with the desirable feature that, in low doses (< 3 μg/min), β1 – and β2-receptor effects predominate. As the dose is increased, α effects (e.g., vasoconstriction) and tachycardia occur. However, in the acutely failing heart after surgery, only drugs such as epinephrine or norepinephrine provide adequate positive inotropy and perfusion pressure. These features and its low cost make it a common first-line drug in the postoperative setting. Despite what is often stated in older literature, epinephrine causes less tachycardia than dopamine156 or dobutamine157 at equivalent inotropic doses.158 Epinephrine is a first-line therapy for anaphylaxis and, when titrated, does not produce ventricular arrhythmias. Because of the metabolic actions of β2-receptor stimulation, epinephrine infusion can cause hyperglycemia and increased serum lactate levels.159
Norepinephrine
Norepinephrine, which has potent β1– and α-receptor effects, preserves coronary perfusion pressure while not increasing HR, actions that are favorable to the ischemic, reperfused heart. When norepinephrine is used alone without a vasodilator or PDE inhibitor, the potent α1-receptor effects may have variable effects on CO. Ventricular filling pressures usually increase when this drug is given because of constriction of the capacitance vessels. Administration of a vasodilator, including the PDE inhibitors, with norepinephrine may partially oppose the vasoconstriction. Clinicians may express concern for the renal blood flow when norepinephrine is given for hypotension; however, norepinephrine has long been used as a first-line agent for hypotension and shock in ICU settings and after cardiac surgery. Despite perceived concerns, when norepinephrine is infused to increase MAP to more than 70 mm Hg in sepsis, increased urine flow and increased creatinine clearance rate occurred after 24 hours.160 Furthermore, its use in circulatory shock did not increase mortality.161 End-organ ischemia would appear to be unlikely if CO can be preserved at normal levels when norepinephrine is given. PDE inhibitors in combination with norepinephrine attenuate the arterial vasoconstrictive effects.146
Dopamine
A precursor of norepinephrine, dopamine probably achieves its therapeutic effects by releasing myocardial norepinephrine or preventing its reuptake, especially when administered in high doses.162 This indirect action may result in reduced effectiveness when given to patients with chronic HF or shock states because the myocardium becomes depleted of norepinephrine stores.163 In contrast with dobutamine, the α-agonist properties of dopamine cause increases in pulmonary artery pressure (PAP), PVR, and LV filling pressure.164–166 At low doses (< 2 μg/kg/min), dopamine stimulates renal dopaminergic receptors to increase renal perfusion more than can be explained by an increase in CO.167 Despite this action, a multicenter study demonstrated that use of low-dose dopamine in critically ill patients confers no protection from renal dysfunction.168 One review suggests there is no justification for low-dose dopamine in the ICU and that it is “bad medicine.”169 At doses greater than 10 μg/kg/min, tachycardia and vasoconstriction become the predominant actions of this drug. Tachycardia is a consistent side effect, and in patients with cardiogenic shock, dopamine was recently shown to increase mortality.142,161
Dobutamine
In contrast with dopamine, dobutamine shows mainly β1-agonist properties, with decreases in diastolic BP, and sometimes, decreased systemic BP being observed.170,171 Dobutamine is functionally similar to isoproterenol, with less tendency to induce tachycardia in the postoperative setting.172 However, Romson et al173 demonstrated that after CPB, the principal effect of dobutamine is a dose-related increase in HR. A modest effect on SV was observed in patients with poor ventricular function. Salomon et al174 showed that dobutamine increased Mvo2, which was matched by an increase in coronary blood flow, whereas dopamine increased Mvo2 but failed to increase coronary blood flow. However, the favorable actions of dobutamine may be limited if a tachycardia develops, and like dopamine, its inotropic potency is modest in comparison with that of epinephrine or norepinephrine.174
 Phosphodiesterase Inhibitors
Phosphodiesterase Inhibitors
The PDE inhibitors are nonglycosidic, nonsympathomimetic drugs that have positive inotropic effects independent of the β1-adrenergic receptor and unique vasodilatory actions independent of endothelial function or nitrovasodilators.154,155 Patients with HF have downregulation of the β1-receptor, with a decrease in receptor density and altered responses to catecholamine administration.154,175 Milrinone, amrinone, and enoximone bypass the β1 receptor, causing increases in intracellular cAMP by selective inhibition of PDE fraction III, a cAMP-specific PDE enzyme.155,176 In vascular smooth muscle, these agents cause vasodilation in the arterial and capacitance beds.177 PDE inhibitors increase CO, decrease pulmonary capillary wedge pressure, and decrease SVR and PVR in patients with biventricular dysfunction, and they are important therapeutic approaches in postoperative cardiac surgical patients. Sildenafil and other PDE5 inhibitors also are being used increasingly for pulmonary hypertension.178 The PDE5 inhibitor sildenafil, marketed with a different name from Viagra, called Revatio, was approved for the treatment of pulmonary arterial hypertension by the U.S. Food and Drug Administration and by the European Medicines Agency in 2005178 (see Chapters 10 and 24).
Effects on Vascular Responses
Any drug that increases cyclic nucleotides (e.g., cAMP, cGMP) in vascular smooth muscle will produce vasodilation.176–178 The concentration of cGMP can be increased by the release of nitric oxide produced by nitroglycerin, sodium nitroprusside, and inhaled nitric oxide, and cAMP can be increased by prostaglandin E1 (PGE1) or PGI2, or by inhibiting its breakdown by PDE inhibition. Increasing cAMP in vascular smooth muscle promotes calcium uptake by the sarcoplasmic reticulum, decreasing calcium available for contraction. The net effect of increasing calcium uptake is smooth muscle relaxation. This effect can also occur through stimulation by drugs that inhibit the breakdown of cGMP (e.g., nonspecific PDE inhibitor). Sildenafil and its congeners are PDE5 inhibitors that were originally developed for nitrate tolerance but are marketed for erectile dysfunction and pulmonary hypertension.178
PDE III inhibitors have a clinical effect as inodilators; they produce dilation of arterial and venous beds, decreasing the MAP and central filling pressures. Increases in CO are induced by multiple mechanisms, including afterload reduction and positive inotropy, but not by increasing HR.179–189 The net effect is a decrease in myocardial wall tension, representing an important contrast with most sympathomimetic agents.181 Catecholamine administration often needs the simultaneous administration of vasodilators to reduce ventricular wall tension. Milrinone and other PDE inhibitors also have unique mechanisms of vasodilation that may be favorable for coronary artery and internal mammary artery flow146 (Box 34-5).
Sildenafil inhibits PDE5, an enzyme that metabolizes cGMP, thereby increasing the cGMP-mediated relaxation.178 The current treatment modalities for pulmonary hypertension include conventional supportive therapies and more specific pharmacologic therapies that are targeted at abnormalities of endothelial function. NO and PDE5 inhibitors induce pulmonary vasodilation by increasing intracellular cGMP concentrations. Sildenafil citrate is a selective inhibitor of PDE5. Investigations in animal models and recent clinical case reports with some studies in the pediatric population suggest that sildenafil may be a promising agent in treating pulmonary hypertension. The effect of sildenafil on pulmonary vasculature appears to be independent of the underlying cause, thereby providing a role in idiopathic pulmonary arterial hypertension, pulmonary arterial hypertension associated with congenital heart disease, pulmonary hypertension secondary to lung disease, or persistent pulmonary hypertension of the newborn. It also may be beneficial in postoperative pulmonary hypertension and in patients who are difficult to wean from inhaled NO. It is administered easily and effectively and has minimal adverse systemic effects182 (see Chapter 24).
 Combination Therapy: Catecholamines and Phosphodiesterase Inhibitors
Combination Therapy: Catecholamines and Phosphodiesterase Inhibitors
Catecholamine therapy depends on the capacity of the myocardial cell to respond to β1-agonist activity. In patients with preoperative HF, the number of effective β1 receptors decreases because of downregulation, which refers to reduced density or uncoupling, such that fewer receptors are available for binding with the β1-agonist.154,175 When postoperative ventricular dysfunction is treated, a pharmacologic ceiling effect may occur with increasing doses of a single β1-agonist or even when other catecholamines are added.176 Combining PDE inhibitors with a catecholamine may significantly increase cAMP levels in patients with β1-receptor downregulation, such as patients after cardiac surgery.190 The two forms of therapy may attenuate each other’s adverse effects. Catecholamine stimulation of vascular α1 receptors induces vasoconstriction, which is attenuated by PDE inhibitors.146 Catecholamines with potent α1-agonist effects may be necessary to prevent hypotension when PDE inhibitors are given after surgery; or, alternatively, when an α agent is necessary to obtain an acceptable perfusion pressure, PDE inhibitors may be administered to augment CO. The additive improvement in hemodynamic effects of catecholamines plus amrinone, milrinone, or enoximone has been also described.187,191–195 Combined therapy may theoretically avoid dose-related adverse effects of high doses of each individual agent and is useful in RV failure196 (Box 34-6; see Chapter 10).
BOX 34-6 Acute Heart Failure: Therapeutic Goals and Treatment Summary
| Goals | Treatment |
|---|---|
| Reduce impedance to ventricular ejection | Vasodilator |
| Reduce wall stress | Vasodilator |
| Reduce filling pressures | Diuretics, venodilators |
| Increase contractility | Inotropic agents, phosphodiesterase inhibitors |
Dosage and Administration
Suggested dosing is provided in Table 34-5. Available drugs are reviewed in the following sections.
TABLE 34-5 Dosing for Phosphodiesterase Inhibitors (Cyclic Adenosine Monophosphate–Specific) Used after Surgery
| Drug | Loading Dose* | Infusion Rate |
|---|---|---|
| Amrinone | 1.5–2.0 mg/kg | 5–20 μg/kg/min |
| Milrinone | 50 μg/kg | 0.375–0.75 μg/kg/min |
| Enoximone | 0.5–1.0 mg/kg | 5–10 μg/kg/min |
* Loading doses should be administered over 5 to 10 minutes to avoid excessive vasodilation.
Amrinone
Amrinone, the first bipyridine evaluated for HF during and after cardiac surgery, has a half-life of ∼︀3.5 hours.197–201 In HF patients, an intravenous loading dose of 1.5 mg/kg and an infusion of 10 μg/kg/min resulted in a plasma concentration of 1.7 μg/mL and produced a 30% increase in CI.197 The original recommended dosing included a bolus dose of 0.75 mg/kg given intravenously over 2 to 3 minutes, followed by a maintenance infusion of 5 to 10 μg/kg/min. This dose regimen produced subtherapeutic concentrations after 5 to 10 minutes, and it failed to show any hemodynamic effect after it was given 10 minutes before termination of CPB.201,202 A loading dose of 1.5 to 2.0 mg/kg of this drug during CPB will produce therapeutic concentrations for 30 to 60 minutes, after which an infusion is required to keep therapeutic blood levels. With prolonged administration, amrinone will produce thrombocytopenia. Amrinone has been replaced with milrinone for the most part.
Milrinone
Milrinone, an analog of amrinone, is a bipyridine derivative with an inotropic activity that is almost 20 times more potent than that of amrinone, and it has a shorter half-life.183 Milrinone is an effective inodilator for patients with decompensated HF and low CO after cardiac surgery. Suggested dosing for milrinone is a loading dose of 50 μg/kg over 10 minutes, followed by an infusion of 0.5 μg/kg/min (0.375 to 0.75 μg/kg/min). By using slower loading doses, high peak concentrations can be prevented, and the vasodilation that is observed with rapid loading can be attenuated.183 A milrinone loading dose of 50 μg/kg, in combination with an infusion of 0.5 μg/kg/min, consistently maintained plasma concentrations more than 100 ng/mL. Clearance was 3.8 ± 1.7 mL/kg/min, volume of distribution was 465 ± 159 mL/kg, and terminal elimination half-time was 107 ± 77 minutes (values expressed as mean ± SD).183 Pharmacokinetic parameters were independent of dose. The relation between plasma concentration and pharmacodynamic effects produced about a 30% improvement in CI with plasma levels of 100 ng/mL, and there was a curvilinear relation between plasma levels and improvement in CI. Bailey et al183 observed that a dose of 50 μg/kg with an infusion rate of 0.5 μg/kg/min can keep plasma concentrations near the threshold of its therapeutic effects. Compared with amrinone, milrinone has a shorter context-sensitive half-time after administration is stopped, without adverse effects on platelet function.189
Kikura et al194 reported the effects of milrinone on hemodynamics and LV function in cardiac surgical patients who were already treated with catecholamines. After emergence from CPB, patients were randomly assigned to a control group (n = 10) or to one of the milrinone dosing groups: intravenous milrinone at 50 μg/kg (n = 8), 50 μg/kg + 0.5 μg/ kg/min (n = 10), or 75 μg/kg + 0.75 μg/kg/min (n = 9). Hemodynamics and TEE were recorded while constant filling pressures were maintained by volume reinfusion. In all three milrinone groups, CI and velocity of circumferential fiber shortening significantly increased from the baseline, and both were significantly greater at 5 and 10 minutes than those in the control group. The plasma concentration of milrinone with half of the maximal increase in velocity of circumferential fiber shortening was 139 ng/mL on the dose-response curve. Milrinone improves hemodynamics and LV function when constant loading conditions are maintained.194
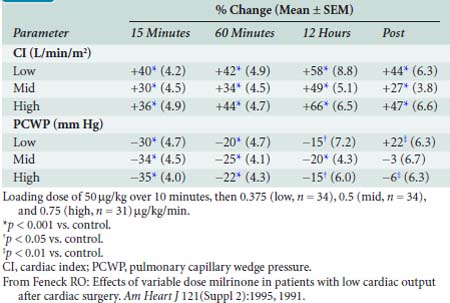
Feneck et al203 studied 99 adult patients with a low CO after elective cardiac surgery. Milrinone was administered as a loading dose of 50 μg/kg over a 10-minute period, followed by a continuous infusion of 0.375, 0.5, or 0.75 μg/kg/min (low-, middle-, and high-dose groups, respectively) for a minimum of 12 hours. They observed that milrinone therapy was associated with a rapid and well-sustained increase in CO and a decrease in PAOP in all groups. They found the increase in CI was associated with increases in SV and HR (Table 34-6).
Enoximone
Enoximone, an imidazolone derivative, is eliminated mostly by sulfoxidation, is solubilized in propylene glycol, and cannot be diluted when administered intravenously. The loading dose is 0.5 to 1.0 mg/kg, followed by an infusion of 5 to 10 μg/kg/min. Gonzalez et al187 reported using enoximone in managing a CI less than 2.2 L/min/m2 despite a pulmonary capillary wedge pressure of 15 mm Hg, catecholamine administration (e.g., dobutamine, dopamine), or IABP counterpulsation after cardiac surgery. Enoximone was administered as a 1-mg/kg loading dose over 10 minutes after a minimum of 4 hours of unsuccessful conventional therapy. An extra dose (0.5 mg/kg) was given if the increase in CO was less than 20%. A continuous infusion of the drug was administered at 3 to 10 μg/kg/min and continued for at least 8 hours. In all patients, significant increases in CI and a significant decrease in pulmonary capillary wedge pressure occurred. Naeije et al204 also reported variable effects on BP, HR, and CO of enoximone in a dose of 0.5 mg/kg after cardiac surgery. Boldt et al193 demonstrated potentiating effects of enoximone, 0.5 mg/kg, with epinephrine in a dose of 0.1 μg/kg/min.
 Levosimendan
Levosimendan
Levosimendan is a calcium-sensitizing drug that exerts positive inotropic effects through sensitization of myofilaments to calcium and vasodilation through opening of ATP-dependent potassium channels on vascular smooth muscle. These effects occur without increasing intracellular cAMP or calcium and without an increase in M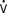 o2 at therapeutic doses. As would be expected with an inodilator, the hemodynamic effects include a decrease in PAOP in association with an increase in CO. β-Blockade does not block the hemodynamic effects of this drug. Levosimendan itself has a short elimination half-life, but it has active metabolites with elimination half-lives up to 80 hours. A study in patients with decompensated HF found hemodynamic improvements at 48 hours were similar whether patients received the drug for 24 or 48 hours. Increasing plasma levels of the active metabolite were found for 24 hours after the drug infusion was stopped.205 Currently, levosimendan is not approved for use in the United States.
o2 at therapeutic doses. As would be expected with an inodilator, the hemodynamic effects include a decrease in PAOP in association with an increase in CO. β-Blockade does not block the hemodynamic effects of this drug. Levosimendan itself has a short elimination half-life, but it has active metabolites with elimination half-lives up to 80 hours. A study in patients with decompensated HF found hemodynamic improvements at 48 hours were similar whether patients received the drug for 24 or 48 hours. Increasing plasma levels of the active metabolite were found for 24 hours after the drug infusion was stopped.205 Currently, levosimendan is not approved for use in the United States.
A randomized study enrolling 203 low-output HF patients found that levosimendan improved hemodynamics more effectively than dobutamine and was associated with a lower 6-month mortality rate.206 However, the latter finding may be caused more by adverse effects related to dobutamine than by a positive effect of levosimendan. Another study in 504 patients with LV dysfunction after acute MI demonstrated better 6-month survival with levosimendan, this time compared with placebo.207 In a small study after cardiac surgery, patients were given levosimendan; of 11 patients with severely impaired CO and hemodynamic compromise, 8 patients (73%) showed evidence of hemodynamic improvement within 3 hours after the start of levosimendan infusion. Specifically, CI and SV were increased significantly, whereas MAP, indexed SVR, mean PAP, right atrial pressure (RAP), and PAOP were decreased significantly.208 Clinical studies continue to evaluate the potential role for this new positive inotropic agent in patients with HF.
Right-heart failure
HF after cardiac surgery usually results from LV impairment. Although an isolated right-sided MI can occur perioperatively, most perioperative inferior MIs show variable involvement of the right ventricle.209 The myocardial preservation techniques that are best for the left ventricle may not offer ideal RV protection because the right ventricle is thin walled and more exposed to body and atmospheric temperature. Cardioplegic solution given through the coronary sinus (retrograde) may not reach parts of the right ventricle because of positioning of the cardioplegia cannula in relation to the venous outflow from this chamber and because the thebesian veins do not drain into the coronary sinus.210 Impairment of RV function after surgery is more severe and persistent when preoperative right coronary artery stenosis is present.211 Although depression of the ejection fraction is compensated by preload augmentation, right ventricular ejection fraction (RVEF) cannot be preserved if coronary perfusion pressure is reduced or impedance to ejection is increased.
Certain aspects of the physiology of the right ventricle make it different from the left. Normally, the RV free wall receives its blood flow during systole and diastole; however, systemic hypotension or increased RV systolic and diastolic pressures may cause supply-dependent depression of contractility when M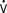 o2 is increased, whereas coronary perfusion pressure is decreased.212 The normal thin-walled right ventricle is at least twice as sensitive to increases in afterload as is the left ventricle213 (Figure 34-6). Relatively modest increases in outflow impedance from multiple causes in the postoperative period can exhaust preload reserve, causing a decrease in RVEF with ventricular dilation. RV pressure overload may be complicated by volume overload caused by functional tricuspid regurgitation.214 Decreases in RV SV will decrease LV filling, and dilation of the right ventricle can cause a leftward shift of the interventricular septum, interfering with diastolic filling of the left ventricle (i.e., ventricular interaction; Figure 34-7). A distended right ventricle limited by the pericardial cavity further decreases LV filling. RV failure has the potential to affect LV performance by decreasing pulmonary venous blood flow, decreasing diastolic distending pressure, and decreasing LV diastolic compliance. The resulting decrease in LV output will further impair RV pump function. The mechanical outcomes of RV failure in postoperative cardiac surgical patients are depicted in Figure 34-8. It can, therefore, be appreciated how, once established, RV failure is self-propagating, and aggressive treatment interventions may be needed to interrupt the vicious cycle (see Chapter 24).
o2 is increased, whereas coronary perfusion pressure is decreased.212 The normal thin-walled right ventricle is at least twice as sensitive to increases in afterload as is the left ventricle213 (Figure 34-6). Relatively modest increases in outflow impedance from multiple causes in the postoperative period can exhaust preload reserve, causing a decrease in RVEF with ventricular dilation. RV pressure overload may be complicated by volume overload caused by functional tricuspid regurgitation.214 Decreases in RV SV will decrease LV filling, and dilation of the right ventricle can cause a leftward shift of the interventricular septum, interfering with diastolic filling of the left ventricle (i.e., ventricular interaction; Figure 34-7). A distended right ventricle limited by the pericardial cavity further decreases LV filling. RV failure has the potential to affect LV performance by decreasing pulmonary venous blood flow, decreasing diastolic distending pressure, and decreasing LV diastolic compliance. The resulting decrease in LV output will further impair RV pump function. The mechanical outcomes of RV failure in postoperative cardiac surgical patients are depicted in Figure 34-8. It can, therefore, be appreciated how, once established, RV failure is self-propagating, and aggressive treatment interventions may be needed to interrupt the vicious cycle (see Chapter 24).
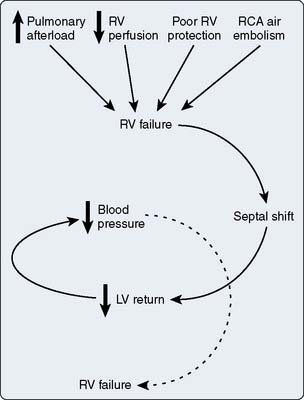
Figure 34-7 Sequence inducing right ventricular (RV) failure and causing a downward spiral of events.
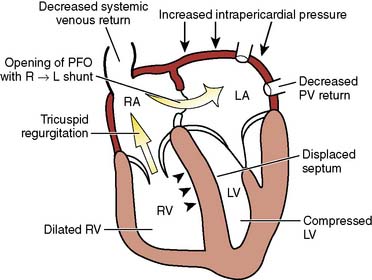
Although not consistently proved, PVR has been shown to be reversibly increased immediately after CPB and for several hours into the postoperative period.215,216 The possible mechanisms include extravascular compression by increased lung water,216 endocrine-mediated or autonomic nervous system–mediated increases in pulmonary vascular tone,217 vasoactive substances released from activated platelets and leukocytes,218 and leukocytes or platelet aggregates obstructing pulmonary vascular beds.219 Hypoxic pulmonary vasoconstriction may result in increased PVR; more commonly, hypercarbia causes an important increase in PAP.220,221 The pulmonary vascular bed has been shown to be more sensitive to the vasoconstrictor influences of respiratory acidosis after CPB as compared with the preoperative situation.222 Moderate respiratory acidosis was shown to cause depression of the RVEF and increased RV end-diastolic volume, and these changes were immediately reversed when normocarbia was reinstituted.223
 Diagnosis
Diagnosis
In the postoperative cardiac surgical patient, a low CI with RAP increased disproportionately compared with changes in left-sided filling pressures is highly suggestive of RV failure. The PAOP also may increase because of ventricular interaction, but the relation of RAP to PAOP stays close to or greater than 1.0. The absence of a step-up in pressure in going from the right atrium (RA) to the pulmonary artery (mean), provided PVR is low, suggests that RV failure is severe and the right side of the heart is acting only as a conduit. This hemodynamic presentation is typical of cardiogenic shock associated with RV infarction. The venous waveforms are accentuated with a prominent Y descent similar to constrictive pericarditis, suggesting reduced RV compliance.224 Large V waves also may be discernible and may relate to tricuspid regurgitation.
The use of a volumetric PAC to calculate right-sided volumes and ejection fraction could potentially guide management in the setting of RV failure, because increased end-diastolic volume in association with a decreased RVEF indicates decompensation. This catheter-computer system has been validated in comparison with radionuclear and ventriculographic measures, but it may not be accurate when there is tricuspid regurgitation34,225,226 (see Chapter 14). Unfortunately, tricuspid regurgitation is a common finding with RV dilation.
Echocardiography allows a qualitative interpretation of RV size, contractility, and configuration of the interventricular septum and can provide a definitive diagnosis of RV dysfunction or failure. Because of the crescent shape of the right ventricle, volume determination is not easy, but the qualitative examination and assessment for tricuspid regurgitation are valuable. TEE is also useful to determine whether the increased RAP opens a patent foramen ovale, producing a right-to-left shunt (see Chapters 12, 13, and 22). This is important because traditional methods to treat hypoxemia such as PEEP and larger tidal volumes in this setting will only increase the afterload of the right ventricle and potentially increase the shunt and hypoxemia.
 Treatment
Treatment
Treatment approaches in postoperative RV failure may differ from those used in LV failure, and they are affected by the presence of pulmonary hypertension (Table 34-7). In all cases, preload should be increased to the upper range of normal; however, the Frank–Starling relation is flat in RV failure, and to avoid ventricular dilation, the CO response to an increasing CVP should be determined. Volume loading should be stopped when the CVP exceeds 10 mm Hg and CO does not increase despite increases in this pressure.227,228 If a volumetric PAC is in use, an increase in the end-diastolic volume with unchanged or declining RVEF suggests there will be no advantage to further volume loading. The CVP should not be permitted to exceed the PAOP because if these pressures equalize, any increase obtained in pulmonary blood flow will be offset by decreased diastolic filling of the left ventricle by ventricular interdependence.229 The atrial contribution to RV filling is important when the ventricle is dilated and noncompliant. Maintenance of sinus rhythm and use of atrial pacing are important components of treating postoperative RV failure (see Chapters 4, 5, 10, 13,14, 19, 23–25 and 32).
TABLE 34-7 Treatment Approaches in Postoperative Right-Heart Failure
| Preload Augmentation |
| Volume, vasopressors, or leg elevation (CVP/PCWP < 1) |
| Decrease juxtacardiac pressures (pericardium and/or chest open) |
| Establish atrial kick and treat atrial arrhythmias (sinus rhythm, atrial pacing) |
| Afterload Reduction (pulmonary vasodilation) |
| Nitroglycerin, isosorbide dinitrate, nesiritide |
| cAMP-specific phosphodiesterase inhibitors, B2-adrenergic agonists |
| Inhaled nitric oxide |
| Nebulized PGI2 |
| Intravenous PGE1 (+left atrial norepinephrine) |
| Inotropic Support |
| cAMP-specific phosphodiesterase inhibitors, isoproterenol, dobutamine |
| Norepinephrine |
| Levosimendan |
| Ventilatory Management |
| Lower intrathoracic pressures (tidal volume < 7 mL/kg, low PEEP) |
| Attenuation of hypoxic vasoconstriction (high FIO2) |
| Avoidance of respiratory acidosis (Paco2 30–35 mm Hg, metabolic control with meperidine or relaxants) |
| Mechanical Support |
| Intra-aortic counterpulsation |
| Pulmonary artery counterpulsation |
| Right ventricular assist devices |
cAMP, cyclic adenosine monophosphate; CVP/PCWP, central venous pressure/pulmonary capillary wedge pressure; PEEP, positive end-expiratory pressure; PGE1, prostaglandin E1; PGI2, prostaglandin I2.
Although vasodilators may lead to cardiovascular collapse in RV infarction (as a result of decreases in RV filling and coronary perfusion), postoperative RV failure is often associated with increased PVR and pulmonary hypertension. In this context, attempts to decrease RV outflow impedance may be worthwhile. Intravenous vasodilators invariably reduce systemic BP, mandating the simultaneous administration of a vasoconstrictor. One way to reduce the pulmonary effects of the needed vasoconstrictor is to administer the vasoconstrictor through a left atrial catheter, treating RV dysfunction with intravenous PGs and left atrial norepinephrine.230 The PDE inhibitors commonly are used for their effect on the pulmonary vasculature and RV function. In recent years, there have been an increased interest in and availability of aerosolized pulmonary vasodilators. This route of administration reduces or even abolishes the undesirable systemic vasodilation. Delivery of the drug directly to the alveoli improves pulmonary blood flow to these alveoli, potentially improving oxygenation by better matching blood flow to ventilation. Three drugs have been used: nitric oxide, PGI2 (i.e., epoprostenol or prostacyclin), and milrinone.231,232
Nitric oxide is an important signaling molecule throughout the body. In the lung, it rapidly diffuses across the alveolar-capillary membrane and activates soluble guanylate cyclase, leading to smooth muscle relaxation by several mechanisms.231 Inhaled nitric oxide is given through a specialized delivery system in a concentration of 5 to 80 parts per million. It is available commercially in the United States, but it is costly. It has been used successfully to treat RV dysfunction associated with pulmonary hypertension after cardiac surgery,231 mitral valve replacement,233 cardiac transplantation,234 and placement of LV assist devices.235 Although it is used widely to treat the same problem in lung transplantation, a randomized trial of prophylactic inhaled nitric oxide in this population failed to show a benefit.236 This finding should not prevent its use should RV dysfunction occur in the lung transplant recipient. Potential adverse effects of inhaled nitric oxide include toxicity from forming nitrogen dioxide (NO2) and methemoglobin, rebound pulmonary hypertension from abrupt disconnection or withdrawal, and pulmonary vascular congestion because of increased pulmonary blood flow in patients with poor LV function. If administered at the recommended dosage, toxicity should not be observed; patients should have the drug gradually withdrawn, and those with poor ventricular function should be monitored closely for increases in left-sided filling pressures.
A less expensive alternative to inhaled nitric oxide is aerosolized epoprostenol (i.e., prostacyclin or PGI2). This compound binds to cell-surface PG receptors, activating adenylate cyclase, which activates protein kinase A to cause a decrease in cytosolic free calcium. It also stimulates endothelial release of nitric oxide. It is a profound vasodilator and inhibitor of platelet aggregation. Similar to inhaled nitric oxide, its delivery to ventilated alveoli can improve oxygenation by augmenting blood flow to these alveoli. Aerosolized PGI2 has been used successfully to treat pulmonary hypertension after cardiac surgery,232,237 pulmonary embolism,238 and to treat hypoxemia in patients with lung injury.239,240 Its use needs collaboration with pharmacy and respiratory therapy, as well as suitable care and monitoring of the nebulizer device. Adverse effects include the possibility of vagus-mediated bradycardia at low doses and risks similar to those of inhaled nitric oxide for abrupt withdrawal or left-sided HF.
Haraldsson et al241 studied the use of inhaled (i.e., aerosolized) milrinone in patients with mild pulmonary hypertension after CPB. They found that when delivered by this route, milrinone was an effective pulmonary vasodilator without causing systemic hypotension, and the pulmonary vasodilation was additive to that caused by inhaled PGI2.
Even a moderate increase in arterial carbon dioxide tension (Paco2) should be avoided in patients with RV failure. Although induced hypocarbia is of proven benefit in controlling PVR in neonates, the evidence in adults does not warrant this as a standard therapy because mechanical ventilation-induced changes in intrathoracic pressure have important therapeutic implications242,243 (see Chapter 35). An IABP may be of great benefit, even in patients in whom the right ventricle is mainly responsible for circulatory decompensation. This beneficial effect is mediated by increased coronary perfusion. Right-heart assist devices have a place as temporizing measures in severe intractable failure. Pulmonary artery counterpulsation is experimental, and its clinical role is uncertain.244 In cases of severe RV failure, it may be necessary to leave the sternum open or to reopen the chest if it has been closed. This decreases the tamponade-like compression of the left ventricle by the distended right ventricle, RA, and edematous mediastinal tissues.
 Effects of Mechanical Ventilation in Heart Failure
Effects of Mechanical Ventilation in Heart Failure
HF at the time of surgery has been identified as a significant predictor of postoperative respiratory complications.245 Maintenance of gas exchange in these situations usually mandates prolonged ventilatory support. Besides improving Pao2, mechanical ventilation can influence Do2 through its effects on CO. Suppression of spontaneous respiratory efforts may substantially decrease the work of breathing and improve the oxygen supply/demand relation (see Chapter 35). Traditionally, the influence of mechanical ventilation on hemodynamics has been viewed as negative. The unavoidable increase in intrathoracic pressure caused by positive-pressure ventilation or PEEP is associated with a decreased CO.246 However, in the presence of HF or myocardial ischemia, increased intrathoracic pressure has the potential to favorably affect the determinants of global cardiac performance. Understanding these heart-lung interactions is essential for the integrated management of the ventilated patient with HF after cardiac surgery. The effects of ventilation on RV and LV failure need to receive independent consideration.
Systemic venous return is proportional to the pressure gradient between the systemic veins and the RA. Changes in intrathoracic pressure imposed by positive-pressure ventilation or PEEP are transmitted to the compliant RA, causing an increase in RAP. This decreases the driving pressure for venous return, with a decrease in RV preload as the clinically most important mechanism of the decrease in RV SV caused by ventilatory support.247 The effects of mechanical ventilation on RV preload may be accentuated by hypovolemia or by an increase in venous capacitance caused by vasodilator administration.248 The effects of increased intrathoracic pressure can be overcome by fluid administration, leg elevation, or even vasopressors to increase the systemic venous pressure. Preload augmentation must be done with caution with RV failure.
Two other factors related to increased intrathoracic pressure potentially impede the diastolic filling of the left ventricle. If positive-pressure ventilation or PEEP causes an increase in PVR, RV systolic emptying may be impaired and the right ventricle will dilate. This may cause leftward displacement of the interventricular septum and decrease LV compliance.229 Independent of ventricular interaction, increased lung volume also increases juxtacardiac pressures, decreasing the transmural distending pressure.249
The end-diastolic volume and systolic BP are directly proportional to systolic wall stress or ventricular afterload. Because ventricles and the outflow vessels are surrounded by intrathoracic pressure, increases in this pressure by positive-pressure ventilation decrease the transmural pressure load (aortic or PAP relative to intrathoracic pressure) of each ventricle.250 On the right side of the heart, the hemodynamic effects of ventilatory support usually result from changes in PVR. To the extent that PEEP increases lung volume above functional residual capacity, PVR may decrease from reduced compression of extraalveolar vessels.251 Beyond that, large tidal volumes and high levels of PEEP increase PVR.252 The increase in PVR may be obvious even with normal tidal volumes in airflow-limited diseases. The effects of increased PVR in RV failure are decreased CO and further dilation.
Increased intrathoracic pressure may significantly improve LV performance as a result of the reduced transmural pressure needed to achieve an acceptable systemic BP.253 This can be viewed as afterload reduction, a favorable effect separate from the resistance to venous return that also may help such patients. Clinically significant improvements in cardiac function have been documented in patients ventilated for cardiogenic respiratory failure produced by myocardial ischemia and after CABG surgery.254,255 High LV filling pressures may help identify a subgroup benefiting from reduced afterload with increased intrathoracic pressure.256
Increased intrathoracic pressure and PEEP also have been suggested to affect the inotropic state of the ventricles. Dilation of the right ventricle in response to an increase in afterload increases the RV distending pressure, which can reduce the pressure gradient for subendocardial coronary blood flow. Decreased RV contractility related to a decreased coronary blood flow has been described with high levels of PEEP in an animal model with critical right coronary artery obstruction212 and in patients with significant right coronary artery disease.256 Left ventricular contractility does not seem to be affected by this mechanism.257 Increased intrathoracic pressure and lung distention also may modulate contractility by stimulating vagal afferents258 and releasing PGs.259 The impairment of contractility by these mechanisms appears to be minimal.
The circulatory responses to changes in ventilation always should be assessed in patients with cardiac disease; the goal of improving or maintaining Do2 must be kept in mind. This usually requires measurement of arterial oxygenation and CO. In right and biventricular failure, the increase in the airway pressure caused by ventilatory support should be kept, at a minimum, compatible with acceptable gas exchange. This means avoidance of high levels of PEEP and trials of decreased inspiratory times, flow rates, and tidal volumes. Breathing modes that emphasize spontaneous efforts such as intermittent mandatory ventilation, pressure support, or continuous positive airway pressure should be considered. Alternatively, if isolated LV failure is the reason for ventilatory therapy, improvements in cardiac performance may be achieved by positive-pressure ventilation with PEEP. In particular, patients with increased LV filling pressures, mitral regurgitation, and reversible ischemic dysfunction may improve from afterload reduction related to increased airway and intrathoracic pressures. Newer modes of ventilatory support that decrease mean airway pressure, such as cardiac cycle–specific, high-frequency jet ventilation260 and airway pressure release ventilation,261 and their roles in supporting heart function are discussed in Chapter 35.
 Effects of Ventilatory Weaning on Heart Failure
Effects of Ventilatory Weaning on Heart Failure
Traditional criteria for weaning of ventilatory support assess the adequacy of gas exchange and peak respiratory muscle strength.262 In the patient with HF, the response of global hemodynamics to spontaneous respirations must also be considered. The changes of the loading conditions of the heart brought about by resuming spontaneous ventilation can induce a vicious cycle resulting in hypoxemia and pulmonary edema.
Pulmonary congestion, often present in patients with LV dysfunction, decreases pulmonary compliance. Thus, large decreases in inspiratory intrathoracic pressure are necessary to cause satisfactory lung inflation. These negative swings of intrathoracic pressure increase venous return.263 Increased diaphragmatic movements may increase intra-abdominal pressure, further increasing the pressure gradient for venous return.264 Decreased intrathoracic pressure also increases the ventricular transmural pressures, increasing the impedance to ventricular emptying. The increased afterload causes further increases in preload, with these changes jeopardizing the myocardial oxygen balance. Accordingly, worsening of myocardial ischemia as shown by ST-segment deviations was demonstrated when ventilatory support was removed in patients ventilated after MI.265 Spontaneous ventilation episodes also precipitated ischemic dysfunction, causing LV dilation and altered thallium-201 uptake, in ventilator-dependent patients after lung injury caused by infection or complications of surgery.266
In the patient with severe ventricular dysfunction, one of the main methods of improving cardiac performance to allow separation from CPB and to maintain function in the postoperative period is to augment preload with fluid therapy. The unavoidable consequence is a positive fluid balance and a weight gain of several kilograms, even after uncomplicated surgery. Diuretic therapy to reduce this hypervolemia, as well as vasodilator therapy to reduce ventricular wall stress, should be considered before these patients are exposed to the afterload stress of ventilatory weaning.263,267
Cardiac tamponade
Cardiac tamponade is an important cause of the low CO state after cardiac surgery and occurs when the heart is compressed by an external agent, most commonly blood accumulated in the mediastinum. Hemodynamic compromise, to some degree attributable to the constraining effect of blood accumulating within the chest, is often observed in the 3% to 6% of patients needing multiple blood transfusions for hemorrhage after cardiac surgery.268 Postoperative cardiac tamponade usually manifests acutely during the first 24 hours after surgery, but delayed tamponade may develop 10 to 14 days after surgery, and it has been associated with postpericardiotomy syndrome or postoperative anticoagulation.269–271
The mechanism of hemodynamic deterioration during tamponade is discussed in Chapter 22 and mainly is the result of impaired filling of one or more of the cardiac chambers. As the external pressure on the heart increases, the distending or transmural pressure (external intracavitary pressure) is decreased. The intracavitary pressure increases in compensation lead to impaired venous return and elevation of the venous pressure. If the external pressure is high enough to exceed the ventricular pressure during diastole, diastolic ventricular collapse occurs. These changes have been documented in the right and the left hearts after cardiac surgery.272 As the end-diastolic volume and end-systolic volume decrease, there is a concomitant decrease in SV. In the most severe form of cardiac tamponade, ventricular filling occurs only during atrial systole. Adrenergic and endocrine mechanisms are activated in an effort to maintain venous return and perfusion pressure.273,274 Intense sympathoadrenergic activation increases venous return by constricting venous capacitance vessels. Tachycardia helps to maintain CO in the face of a reduced SV. Adrenergic mechanisms may also explain decreased urinary output and sodium excretion, but these phenomena also may be caused by reduced CO or a reduction in atrial natriuretic factor from decreased distending pressure of the atria.273
The diagnosis of cardiac tamponade depends on a high degree of suspicion. Tamponade after heart surgery is a clinical entity distinct from the tamponade typically seen in medical patients in whom the pericardium is intact and the heart is surrounded by a compressing fluid. In the setting of cardiac surgery, the pericardial space is often left open and in communication with one or both of the pleural spaces, and the compressing blood is, at least in part, in a clotted, nonfluid state and able to cause localized compression of the heart. Serious consideration should be given to the possibility of tamponade after cardiac surgery in any patient with inadequate or worsening hemodynamics, as evidenced by hypotension, tachycardia, increased filling pressures, or low CO, especially when there has been excessive chest tube drainage. A more subtle presentation of postoperative tamponade is gradually increasing needs for inotropic and pressor support. Many of the classic signs of tamponade may not be present in these patients, partly because they are usually sedated and ventilated, but also because the pericardium is usually left open, resulting in a more gradual increase in the restraining effects of blood accumulation. There may be localized accumulations that affect one chamber more than another.275 The classic findings of increased CVP or equalization of CVP, PAd, and PAOP may not occur.276,277 It may, therefore, be difficult in the face of a declining CO and increased filling pressures to distinguish tamponade from biventricular failure. A useful clue may be pronounced respiratory variation in BP with mechanical ventilation in association with high filling pressures and low CO because the additional external pressure applied to the heart by positive-pressure ventilation may further impair the already compromised ventricular filling in the presence of tamponade.
Echocardiography may provide strong evidence for the diagnosis of tamponade.277–280 Echolucent crescents between the RV wall and the pericardium or the posterior LV wall and the pericardium are visible with transthoracic imaging or TEE. Echogenicity of grossly bloody pericardial effusions, especially when clots have been formed, may sometimes make delineation of the borders of the pericardium and the ventricular wall difficult, compromising the sensitivity of this technique. A classic echocardiographic sign of tamponade is diastolic collapse of the RA or right ventricle, with the duration of collapse bearing a relation to the severity of the hemodynamic alteration, but such findings are often absent in the postcardiac surgery patient.277,281,282 Often, transthoracic imaging is difficult because of mechanical ventilation, and TEE is required for satisfactory imagining (see Chapters 12 and 13).
The definitive treatment of tamponade is surgical exploration with evacuation of hematoma. The chest may have to be opened in the ICU if tamponade proceeds to hemodynamic collapse. For delayed tamponade, pericardiocentesis may be acceptable. Medical palliation in anticipation of re-exploration consists of reinforcing the physiologic responses that are already occurring while preparing for definitive treatment. Venous return can be increased by volume administration and leg elevation. The lowest tidal volume and PEEP compatible with adequate gas exchange should be used.283 Epinephrine in high doses gives the needed chronotropic and inotropic boost to the ventricle and increases systemic venous pressures. Sedatives and opioids should be given with caution because they may interfere with adrenergic discharge and precipitate abrupt hemodynamic collapse. Occasionally, patients experience development of significant cardiac tamponade without accumulation of blood in the chest. Edema of the heart, lungs, and other tissues in the chest after CPB may not allow chest closure at the first operation and need staged chest closure after the edema has subsided.284 Similarly, it was found that some patients with inadequate hemodynamics after cardiac surgery despite maximum support in the ICU improved with opening of the chest because of relief of this tamponade effect. Reclosure of the chest in the operating room often is possible after a few days of continued cardiovascular support and diuresis.
Transplanted heart
Postoperative circulatory control in the heart transplant recipient differs from that of the nontransplant population in three major respects: The transplanted heart is noncompliant with a relatively fixed SV, acute rejection must be considered when cardiac performance is poor or suddenly deteriorates, and these patients are at risk for acute RV failure if pulmonary hypertension develops.285
The fixed SV combined with denervation of the donor heart means that maintenance of CO often is dependent on therapy to maintain an increased HR (110 to 120 beats/min). The drug most commonly used is isoproterenol because it is a potent inotropic agent and because it causes a dose-related increase in HR. Its vasodilating β2-receptor effect on the pulmonary vasculature may be of benefit if PVR is above normal. Alternatively, atrial pacing may be used to maintain HR if contractility appears normal. Pacing is often used to allow the withdrawal of isoproterenol in the first postoperative days. Parasympatholytic drugs, such as atropine, do not have any effect on the transplanted heart (see Chapter 23).
The adequacy of immunosuppression is monitored by percutaneous myocardial biopsy, usually performed at weekly intervals in the first month. Less invasive techniques, such as echocardiographic evaluation of diastolic function and sophisticated ECG analysis, are being evaluated.286 Although acute rejection is diagnosed histologically, if suspected clinically (i.e., acute deterioration in cardiac function), it must be treated with intense immunosuppressive therapy. The agents and doses used vary from institution to institution, but they usually include high-dose steroids and monoclonal antibody to T3 lymphocytes (OKT3).287 Pharmacologic management and sometimes mechanical support of biventricular function are required because severe impairment of contractility, ventricular dilation, and even cardiovascular collapse may occur.
Preoperative evaluation helps screen patients with fixed pulmonary hypertension because the normal donor right ventricle may fail acutely if presented with an increased PAP in the recipient.285 However, patients may have progression of disease between the time of evaluation and surgery, or the right ventricle may be inadequately protected during harvest or transport. When separation from CPB is attempted, acute RV dilation and failure occur, and such patients may emerge from the operating room on multiple drug therapy, including the inhaled agents nitric oxide and prostacyclin, as described earlier. Gradual withdrawal of these drugs occurs in the first postoperative days, with close monitoring of PAPs and oxygenation.
1 Jones M., Tunstall-Pedow D.S. Blood doping—a literature review. Br J Sports Med. 1989;23:84.
2 Mirhashemi S., Ertefai S., Merrmer K., Intaglietta M. Model analysis of the enhancement of tissue oxygenation by hemodilution due to increased microvascular flow velocity. Microvasc Res. 1987;34:290.
3 Vincent J.L., Baron J.F., Reinhart K., et al. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288:1499.
4 Hebert P.C., Wells G., Blajchman M.A., et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med. 1999;340:409.
5 Wu W.C., Rathore S.S., Wang Y., et al. Blood transfusions in elderly patients with acute myocardial infarction. N Engl J Med. 2001;345:1230.
6 Suter P.M., Fairley H.B., Isenbery M.D. Optimum end-expiratory pressure in patients with acute pulmonary failure. N Engl J Med. 1975;292:284.
7 Dhainaut J.F., Devaux J.Y., Monsallier J.E., et al. Mechanisms of decreased left ventricular preload during continuous positive-pressure ventilation in ARDS. Chest. 1986;90:74.
8 Robotham J.L., Scharf S.M. Effects of positive and negative pressure ventilation on cardiac performance. Clin Chest Med. 1983;4:161.
9 Katz J.A., Marks J.D. Inspiratory work with and without continuous positive airway pressure in patients with acute respiratory failure. Anesthesiology. 1985;63:598.
10 Rasanen J., Vaisanen I.T., Heikkil J., et al. Acute myocardial infarction complicated by left ventricular dysfunction and respiratory failure. The effects of continuous airway pressure. Chest. 1987;87:158.
11 Sukernik M.R., Mets B., Bennett-Guerrero E. Patent foramen ovale and its significance in the perioperative period. Anesth Analg. 2001;93:137.
12 Radermacher P., Santak B., Becker H., et al. Prostaglandin E1 and nitroglycerin reduce pulmonary capillary pressure but worsen Va/Q distribution in patients with ARDS. Anesthesiology. 1989;70:601.
13 Noback C.R., Tinker J.H. Hypothermia after cardiopulmonary bypass in man; amelioration by nitroprusside-induced vasodilation during rewarming. Anesthesiology. 1980;53:277.
14 Ramsay J.G., Ralley F.E., Whalley D.G., et al. Site of temperature monitoring and prediction of afterdrop after open heart surgery. Can Anaesth Soc J. 1985;32:607.
15 Pujol A., Fusciardi J., Ingrand P., et al. Afterdrop after hypothermic cardiopulmonary bypass: The value of tympanic membrane temperature monitoring. J Cardiothorac Vasc Anesth. 1996;10:336.
16 Grocott H.P., Mathew J.P., Carver E.H., et al. A randomized controlled trial of the Arctic Sun temperature management system versus conventional methods for preventing hypothermia during off-pump cardiac surgery. Anesth Analg. 2004;98:298.
17 Licker M., Schweizer A., Ralley F.E. Thermoregulatory and metabolic responses following cardiac surgery. Eur J Anaesthesiol. 1996;13:502.
18 Buckberg G.D., Brazier J.R., Nelson R.L., et al. Studies of the effects of hypothermia on regional myocardial blood flow and metabolism during cardiopulmonary bypass. 1. The adequately perfused beating, fibrillating, and arrested heart. J Thorac Cardiovasc Surg. 1977;73:87.
19 Reed L.R.II, Bracey A.W.Jr, Hudson J.D., et al. Hypothermia and blood coagulation: Dissociation between enzyme activity and clotting factor levels. Circ Shock. 1990;32:141.
20 Valeri C.R., Khabbaz K., Khuri S.F., et al. Effect of skin temperature on platelet function in patients undergoing extracorporeal bypass. J Thorac Cardiovasc Surg. 1992;104:108.
21 Valeri C.R., Feingold H., Cassidy G., et al. Hypothermia-induced reversible platelet dysfunction. Ann Surg. 1987;205:175.
22 Donati F., Maille J.G., Blain R., et al. End-tidal carbon dioxide tension and temperature changes after coronary artery bypass surgery. Can Anaesth Soc J. 1985;32:272.
23 Sladen R.N. Temperature and ventilation after hypothermic cardiopulmonary bypass. Anesth Analg. 1985;64:816.
24 Ralley F.E., Wynands J.E., Ramsay J.G., et al. The effects of shivering on oxygen consumption and carbon dioxide production in patients re-warming from hypothermic cardiopulmonary bypass. Can J Anaesth. 1988;35:332.
25 Kurz A., Ikeda T., Sessler D.I., et al. Meperidine decreases the shivering threshold twice as much as the vasoconstriction threshold. Anesthesiology. 1997;86:1046.
26 Sladen R.N., Berend J.Z., Fassero J.S., Zehnder E.B. Comparison of vecuronium and meperidine on the clinical and metabolic effects of shivering after hypothermic cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 1995;9:147.
27 Connors A.F., McCaffree D.R., Gray B.A. Evaluation of right heart catheterization in the critically ill patient with acute myocardial infarction. N Engl J Med. 1983;308:263.
28 Ariza M., Gothard J.W., Macnaughton P., et al. Blood lactate and mixed venous-arterial Pco2 gradient as indices of poor peripheral perfusion following cardiopulmonary bypass surgery. Intensive Care Med. 1991;17:320.
29 Bailey J.M., Levy J.H., Kopel M.A., et al. Relationship between clinical evaluation of peripheral perfusion and global hemodynamics in adults after cardiac surgery. Crit Care Med. 1990;18:1353.
30 Sessler D. Mild perioperative hypothermia. N Engl J Med. 1997;336:1730.
31 Wiener R.S., Welch H.G. Trends in the use of pulmonary artery catheters in the US, 1993-2004. JAMA. 2007;298:423.
32 Mayer J., Boldt J., Poland R., et al. Continuous arterial pressure waveform-based cardiac outputs using the FloTrac/Vigeleo: Review & meta-analysis. J Cardiothorac Vasc Anesth. 2009;23:401.
33 Magilligan D.J., Teasdall R., Eisinminger R., Peterson E. Mixed venous oxygen saturation as a predictor of cardiac output in the postoperative cardiac surgical patient. Ann Thorac Surg. 1987;44:260.
34 Spinale F.G., Smith A.C., Crawford F.A. Right ventricular function using thermodilution. Surg Forum. 1988;39:242.
35 Reichert C.L.A., Visser C.A., Doolen J.J., et al. Transesophageal echocardiography in hypotensive patients after cardiac operations. Comparison with hemodynamic parameters. J Thorac Cardiovasc Surg. 1992;104:321.
36 Kochar G.S., Jacobs L.E., Kotler M.N. Right atrial compression in post-operative cardiac patients: Detection by transesophageal echocardiography. J Am Coll Cardiol. 1990;16:511.
37 Chan K-L. Transesophageal echocardiography for assessing cause of hypotension after cardiac surgery. Am J Cardiol. 1988;62:1142.
38 Costachescu T., Denault A., Guimond J.G., et al. The hemodynamically unstable patient in the intensive care unit: Hemodynamic vs TEE monitoring. Crit Care Med. 2002;30:1214.
39 Hwang J.J., Shyu K.G., Chen J.J., et al. Usefulness of transesophageal echocardiography in the treatment of critically ill patients. Chest. 1993;104:861.
40 Breisblatt W.M., Stein K.L., Wolfe C.J., et al. Acute myocardial dysfunction and recovery: A common occurrence after cardiopulmonary bypass surgery. J Am Coll Cardiol. 1990;15:1261.
41 Roberts A.J., Spies M., Meyers S.N., et al. Early and long-term improvement in left ventricular performance following coronary bypass surgery. Surgery. 1980;88:467.
42 Roberts A.J., Spies M., Sanders J.H., et al. Serial assessment of left ventricular performance following coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1981;81:69.
43 Gray R., Maddahi J., Berman D., et al. Scintigraphic and hemodynamic demonstration of transient left ventricular dysfunction immediately after uncomplicated coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1979;77:504.
44 Reduto L.A., Lawrie G.M., Reid J.W., et al. Sequential postoperative assessment of left ventricular performance with gated cardiac blood pool imaging following aortocoronary bypass surgery. Am Heart J. 1981;101:59.
45 Mangano D.T. Biventricular function after myocardial revascularization in humans: Deterioration and recovery patterns during the first 24 hours. Anesthesiology. 1985;62:571.
46 Czer L., Hamer A., Murthey F., et al. Transient hemodynamic dysfunction after myocardial revascularization. J Thorac Cardiovasc Surg. 1983;86:226.
47 Fremes S.E., Weisel R.D., Mickle D.G., et al. Myocardial metabolism and ventricular function following cold potassium cardioplegia. J Thorac Cardiovasc Surg. 1985;89:531.
48 Codd J.E., Barner H.B., Pennington D.G., et al. Intraoperative myocardial protection: Comparison of blood and asanguineous cardioplegia. Ann Thorac Surg. 1985;39:125.
49 Roberts A.J., Woodhall D.D., Knauf D.G., Alexander J.A. Coronary artery bypass graft surgery: Clinical comparison of cold blood potassium cardioplegia, warm cardioplegic induction, and secondary cardioplegia. Ann Thorac Surg. 1985;40:483.
50 Mullen J.C., Christakis G.T., Weisel R.D., et al. Late postoperative ventricular function after blood and crystalloid cardioplegia. Circulation. 1986;74(Suppl III):89.
51 Rousou J.A., Engleman R.M., Breyer R.H., et al. The effect of temperature and hematocrit level of oxygenated cardioplegic solutions of myocardial preservation. J Thorac Cardiovasc Surg. 1988;95:625.
52 Khuri S.F., Warner K.G., Josa M., et al. The superiority of continuous cold blood cardioplegia in the metabolic protection of the hypertrophied human heart. J Thorac Cardiovasc Surg. 1988;95:442.
53 Phillips H.R., Carter J.E., Okada R.D., et al. Serial changes in left ventricular ejection in the early hours after aortocoronary bypass grafting. Chest. 1983;83:28.
54 Salerno T.A., Houck J.P., Barrozo C.A.M., et al. Retrograde continuous warm blood cardioplegia: A new concept in myocardial protection. Ann Thorac Surg. 1991;51:245.
55 Leung J.M., O’Kelly B., Browner W.S., et al. Prognostic importance of postbypass regional wall-motion abnormalities in patients undergoing coronary artery bypass graft surgery. Anesthesiology. 1989;71:16.
56 Shernan S.K. Perioperative myocardial ischemia reperfusion injury. Anesthesiol Clin North America. 2003;21:465.
57 Verrier E.D., Shernan S.K., Taylor K.M. Terminal complement blockade with pexelizumab during coronary artery bypass graft surgery requiring cardiopulmonary bypass: A randomized trial. JAMA. 2004;291:2319.
58 Bolli R. Oxygen-derived free radicals and postischemic myocardial dysfunction. J Am Coll Cardiol. 1988;12:239.
59 Entman M., Michael M., Rossen R., et al. Inflammation in the course of early myocardial ischemia. FASEB J. 1991;5:2529.
60 Maxwell S., Lip G. Reperfusion injury: A review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol. 1997;58:95.
61 Shernan S.K., Fitch J.C., Nussmeier N.A., et al. Impact of pexelizumab, an anti-C5 complement antibody, on total mortality and adverse cardiovascular outcomes in cardiac surgical patients undergoing cardiopulmonary bypass. Ann Thorac Surg. 2004;77:942.
62 Jain U., Laflamme C.J.A., Aggarwal A., et al. Electrocardiographic and hemodynamic changes and their association with myocardial infarction during coronary artery bypass surgery. Anesthesiology. 1997;86:576.
63 Mangano D.T., Siciliano D., Hollenberg M., et al. Postoperative myocardial ischemia. Therapeutic trials using intensive analgesia following surgery. Anesthesiology. 1992;76:342.
64 Cheng D.C.H., Karski J., Peniston C., et al. Morbidity outcome in early versus conventional tracheal extubation after coronary artery bypass grafting: A prospective randomized controlled trial. J Thorac Cardiovasc Surg. 1996;112:755.
65 Wahr J.A., Plunkett J.J., Ramsay J.G., et al. Cardiovascular responses during sedation after coronary revascularization. Anesthesiology. 1996;84:1350.
66 Rahimtoola S.H., Ehsani A., Sinno M.Z., et al. Left atrial transport function in myocardial infarction: Importance of its booster function. Am J Med. 1975;59:686.
67 Mathew J.P., Fontes M.L., Tudor I.C., et al. A multicenter risk index for atrial fibrillation after cardiac surgery. JAMA. 2004;291:1720.
68 Hill L.L., Kattapuram M., Hogue C.W. Management of atrial fibrillation after cardiac surgery. Part 1. Pathophysiology and risks. J Cardiothorac Vasc Anesth. 2002;16:483.
69 Van Dijk D., Nierich A.P., Jansen E.W.L., et al. Early outcome after off-pump versus on-pump coronary bypass surgery. Circulation. 2001;104:1761.
70 Puskas J.D., Williams W.H., Mahoney E.M., et al. Off-pump versus conventional coronary artery bypass grafting: Early and 1-year graft patency, cost, and quality of life outcomes. JAMA. 2004;291:1841.
71 Fuster V., Ryden L.E., Asinger R.W., et al. ACC/AHA/ECC guidelines for the management of patients with atrial fibrillation (executive summary). J Am Coll Cardiol. 2001;38:1231.
72 Hill L.L., De Wet C., Hogue C.W. Management of atrial fibrillation after cardiac surgery. Part II. Prevention and treatment. J Cardiothorac Vasc Anesth. 2002;16:626.
73 Mitchell L.B., Exner D.V., Wyse D.G., et al. Prophylactic Oral Amiodarone for the Prevention of Arrhythmias that Begin Early After Revascularization, Valve Replacement, or Repair: PAPABEAR: A randomized controlled trial. JAMA. 2005;294:3093.
74 Guarnieri T., Nolan S., Gottlieb S.O., et al. Intravenous amiodarone for the prevention of atrial fibrillation after open heart surgery: The Amiodarone Reduction in Coronary Heart (ARCH) trial. J Am Coll Cardiol. 1999;34:343.
75 Sarnoff S.T., Berglund E. Ventricular function. I. Starling’s law of the heart studied by means of simultaneous right and left ventricular function curves in the dog. Circulation. 1954;9:706.
76 Urbanowicz J.H., Shaaban M.J., Cohen N.H., et al. Comparison of transesophageal echocardiographic and scintigraphic estimates of left ventricular end-diastolic volume index and ejection fraction in patients following coronary artery bypass grafting. Anesthesiology. 1990;72:607.
77 Harpole D.H., Clements F.M., Quill T., et al. Right and left ventricular performance during and after abdominal aortic aneurysm repair. Ann Surg. 1989;209:356.
78 Thys D.M., Hillel Z., Goldman M.E., et al. A comparison of hemodynamic indices derived by invasive monitoring and two-dimensional echocardiography. Anesthesiology. 1987;67:630.
79 Smith M.D., MacPhail B., Harrison M.R., et al. Value and limitations of transesophageal echocardiography in determination of left ventricular volumes and ejection fraction. J Am Coll Cardiol. 1992;19:1213.
80 Ellis R.J., Mangano D.T., Van Dyke D.C. Relationship of wedge pressure to end-diastolic volume in patients undergoing myocardial revascularization. J Thorac Cardiovasc Surg. 1974;78:605.
81 Calvin J.E., Driedger A.A., Sibbald W.J. Does the pulmonary capillary wedge pressure predict left ventricular preload in critically ill patients? Crit Care Med. 1981;9:437.
82 Mangano D.T., Van Dyke D.C., Ellis R.J. The effect of increasing preload on ventricular output and ejection in man. Circulation. 1980;62:535.
83 Breisblatt W.M., Vita N., Armuchastegui M., et al. Usefulness of serial radionuclide monitoring during graded nitroglycerin infusion for unstable angina pectoris for determining left ventricular function and individualized therapeutic dose. Am J Cardiol. 1988;61:685.
84 Bouchard M.J., Denault A., Couture P., et al. Poor correlation between hemodynamic and echocardiographic indexes of left ventricular performance in the operating room and intensive care unit. Crit Care Med. 2004;32:644.
85 Breisblatt W.M., Navratil D.L., Burns M.J., Spaccavento L.J. Comparable effects of intravenous nitroglycerin and intravenous nitroprusside in acute ischemia. Am Heart J. 1988;116:465.
86 Ghani M.F., Parker B.M., Smith J.R. Recognition of myocardial infarction after cardiac surgery and its relation to cardiopulmonary bypass. Am Heart J. 1974;88:18.
87 Chiarello M., Gold W.K., Leinbach R.C., et al. Comparison between the effects of nitroprusside and nitroglycerin on ischemic injury during acute myocardial infarction. Circulation. 1976;54:766.
88 Marchionni N., Schneeweiss A., Di Bari M., et al. Age-related hemodynamic effects of intravenous nitroglycerin for acute myocardial infarction and left ventricular failure. Am J Cardiol. 1988;61:81E.
89 Natarajan D., Khurana T.R., Karnade V., et al. Sustained hemodynamic effects with therapeutic doses of intravenous nitroglycerin in congestive heart failure. Am J Cardiol. 1988;62:319.
90 Katz A.M. Cardiomyopathy of overload: A major determinant of prognosis in congestive heart failure. N Engl J Med. 1990;322:100.
91 Muir A.L., Nolan J. Modulation of venous tone in heart failure. Am Heart J. 1991;6:1948.
92 Dikshit K., Vyden J.K., Forrester J.S., et al. Renal and extrarenal hemodynamic effects of furosemide in congestive heart failure after acute myocardial infarction. N Engl J Med. 1973;288:1087.
93 Copeland J.G., Campbell D.W., Plachetka J.R., et al. Diuresis with continuous infusion of furosemide after cardiac surgery. Am J Surg. 1983;146:796.
94 Krasna M.J., Scott G.E., Scholz P.M., et al. Postoperative enhancement of urinary output in patients with acute renal failure using continuous furosemide therapy. Chest. 1986;89:294.
95 Adams K.F., Mathur V.S., Gheorghiade M. B-type natriuretic peptide: From bench to bedside. Am Heart J. 2003;145:S34.
96 Moazami N., Damiano D.J., Bailey M.S., et al. Nesiritide (BNP) in the management of postoperative cardiac patients. Ann Thorac Surg. 2003;75:1974.
97 Sward K., Valsson F., Odencrants P., et al. Recombinant human atrial natriuretic peptide in ischemic acute renal failure: A randomized placebo-controlled trial. Crit Care Med. 2004;32:1310.
98 Caver A., Saccaggi A., Ronco C., et al. Continuous arteriovenous hemofiltration in the critically ill patient. Clinical use and operational characteristics. Ann Intern Med. 1983;99:455.
99 Lamer C., Valleaux T., Plaisance P., et al. Continuous arteriovenous hemodialysis for acute renal failure after cardiac operations [Letter]. J Thorac Cardiovasc Surg. 1990;99:175.
100 Sagawa K. The end-systolic pressure-volume relation of the ventricle: Definition, modifications, and clinical use. Circulation. 1981;63:1223.
101 Suga H., Sagawa K., Shoukas A.A. Load independence of the instantaneous pressure-volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res. 1973;32:314.
102 Kass D.A., Maughan W.L. From “Emax” to pressure-volume relations: A broader view. Circulation. 1988;77:1203.
103 Glower D.D., Spratt J.A., Snow N.D., et al. Linearity of the Frank-Starling relationship in the intact heart: The concept of preload recruitable stroke work. Circulation. 1985;71:994.
104 Hettrick D.A., Warltier D.C. Ventriculoarterial coupling. In: Warltier D.C., editor. Ventricular Function. Baltimore: Williams & Wilkins; 1995:153-179.
105 Sunagawa K., Maughan W.L., Sagawa K. Optimal arterial resistance for the maximal stroke work studied in isolated canine left ventricle. Circ Res. 1985;56:586.
106 Urzua J., Meneses G., Fajardo C., et al. Arterial pressure-flow relationships in patients undergoing cardiopulmonary bypass. Anesth Analg. 1997;84:958.
107 Estafanous F.G., Tarazi R.C. Systemic arterial hypertension associated with cardiac surgery. Am J Cardiol. 1980;46:685.
108 Roberts A.J., Niarchos A.P., Subramanian V.A., et al. Systemic hypertension associated with coronary artery bypass surgery. Predisposing factors, hemodynamic characteristics, humoral profile, and treatment. J Thorac Cardiovasc Surg. 1977;74:846.
109 Vuylsteke A., Feneck R.O., Jolin-Mellgard A., et al. Perioperative blood pressure control: A prospective survey of patient management in cardiac surgery. J Cardiothorac Vasc Anesth. 2000;14:269.
110 Wallach R., Karp R.B., Reves J.G., et al. Pathogenesis of paroxysmal hypertension developing during and after coronary bypass surgery: A study of hemodynamic and humoral factors. Am J Cardiol. 1980;46:559.
111 Stinson E.B., Holloway E.L., Derby G.C., et al. Control of myocardial performance early after open-heart operations by vasodilator treatment. J Thorac Cardiovasc Surg. 1977;73:523.
112 Aronson S., Boisvert D., Lapp W. Isolated systolic hypertension is associated with adverse outcomes from coronary artery bypass grafting surgery. Anesth Analg. 2002;94:1079.
113 Roberts A.J., Niarchos A.P., Subramanian V.A., et al. Hypertension following coronary artery bypass surgery. Comparison of hemodynamic responses to nitroprusside, phentolamine and converting enzyme inhibitor. Circulation. 1978;58(Suppl I):49.
114 Becker L.C. Conditions of vasodilator-induced coronary steal in experimental myocardial ischemia. Circulation. 1978;57:1103.
115 Flaherty J.T., MaGee P.A., Gardner T.L., et al. Comparison of intravenous nitroglycerin and sodium nitroprusside for treatment of acute hypertension developing after coronary artery bypass surgery. Circulation. 1982;65:1072.
116 Gray R.J., Bateman J.M., Czer L.S.C., et al. Comparison of esmolol and nitroprusside for acute postcardiac surgical hypertension. Am J Cardiol. 1987;59:887.
117 Morel D.R., Forster A., Suter P.M. IV labetalol in the treatment of hypertension following coronary artery surgery. Br J Anaesth. 1982;54:1191.
118 David D., Dubois C., Loria Y. Comparison of nicardipine and sodium nitroprusside in the treatment of paroxysmal hypertension following aortocoronary bypass surgery. J Cardiothorac Vasc Anesth. 1991;5:357.
119 Leslie J., Brister N.W., Levy J.H., et al. Treatment of postoperative hypertension following coronary artery bypass surgery: Double-blind comparison of intravenous isradipine and sodium nitroprusside. Circulation. 1994;90(Suppl II):256.
120 Bailey J.M., Lu W., Levy J.H., et al. Clevidipine in adult cardiac surgical patients: A dose-finding study. Anesthesiology. 2002;96:1086.
121 Levy J.H., Mancao M.Y., Gitter R. Clevidipine effectively and rapidly controls blood pressure preoperatively in cardiac surgery patients: Results of the efficacy study of clevidipine assessing its preoperative antihypertensive effect in cardiac surgery-1 (ESCAPE-1) Trial. Anesth Analg. 2007;105:918-925.
122 Aronson S., Dyke C.M., Stierer K.A., et al. The ECLIPSE Trials: Comparative studies of clevidipine to nitroglycerin, sodium nitroprusside, and nicardipine for acute hypertension treatment in cardiac surgery patients. Anesth Analg. 2008;107:1110-1121.
123 Singla H., Wartier D.C., Gandhi S.D., et al. Treatment of Acute Postoperative Hypertension in Cardiac Surgical Patients (ESCAPE 2). Anesth Analg. 2008;107:59.
124 Hill A.J., Feneck R.O., Walesby R.K. A comparison of fenoldopam and nitroprusside in the control of hypertension following coronary artery surgery. J Cardiothorac Vasc Anesth. 1993;7:279.
125 Goldberg M.E., Cantillo J., Nemiroff M.S., et al. Fenoldopam infusion for the treatment of postoperative hypertension. J Clin Anesth. 1993;5:386.
126 Levy J.H., Huraux C., Nordlander M. Treatment of perioperative hypertension. In: Epstein M., editor. Calcium Antagonists in Clinical Medicine. Philadelphia: Hanley & Belfus, 1997.
127 Demarco T., Daly P.A., Liv N., et al. Enalaprilat, a new parenteral angiotensin-converting enzyme inhibitor: Rapid changes in systemic and coronary hemodynamics and humoral profile in chronic heart failure. J Am Coll Cardiol. 1987;9:1131.
128 Mekontso-Dessap A., Houel R., Soustelle C., et al. Risk factors for post-cardiopulmonary bypass vasoplegia in patients with preserved left ventricular function. Ann Thorac Surg. 2001;71:1428.
129 Tuman K.J., McCarthy R.J., O’Connor C.J., et al. Angiotensin-converting enzyme inhibitors increase vasoconstrictor requirements after cardiopulmonary bypass. Anesth Analg. 1995;80:473.
130 Kristoff A.S., Magder S. Low systemic vascular resistance state in patients undergoing cardiopulmonary bypass. Crit Care Med. 1999;27:1121.
131 Gomez W.J., Erlichman M.R., Batista-Filho M.L., et al. Vasoplegic syndrome after off-pump coronary artery bypass surgery. Eur J Cardiothorac Surg. 2003;23:165.
132 Biglioli P., Cannata A., Alamanni F., et al. Biological effects of off pump vs on pump coronary artery surgery: Focus on inflammation, hemostasis and oxidative stress. Eur J Cardiothorac Surg. 2003;24:260.
133 Gomez W.J., Carvalho A.C., Palma J.H., et al. Vasoplegic syndrome: A new dilemma. J Thorac Cardiovasc Surg. 1994;107:942.
134 Chenoweth D.E., Cooper S.W., Hugli T.E., et al. Complement activation during cardiopulmonary bypass: Evidence of generation of C3a and C5a anaphylatoxins. N Engl J Med. 1981;304:497.
135 Landry D.W., Oliver J.A. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345:588.
136 Haeffner-Cavaillon N., Roussellier N., Ponzio O., et al. Induction of interleukin-1 production in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1989;98:1100.
137 Bennett-Guerrero E., Barclay G.R., Weng P.L., et al. Endotoxin-neutralizing capacity of serum from cardiac surgical patients. J Cardiothorac Vasc Anesth. 2001;15:451.
138 Aydin N.B., Gercekoglu H., Aksu B., et al. Endotoxemia in coronary artery bypass surgery: A comparison of the off-pump technique and conventional cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2003;125:843.
139 Argenziano M., Chen J.M., Choudhri A.F., et al. Management of vasodilatory shock after cardiac surgery: Identification of predisposing factors and use of a novel pressor agent. J Thorac Cardiovasc Surg. 1998;116:973.
140 Levy J.H., Adkinson N.F. Anaphylaxis during cardiac surgery: Implications for clinicians. Anesth Analg. 2008;106:392.
141 LeDoux D., Astiz M.E., Carpati C.M., et al. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med. 2000;28:2729.
142 Desjars P., Pinaud M., Potel G., et al. A reappraisal of norepinephrine therapy in human septic shock. Crit Care Med. 1987;15:134.
143 Levy J.H. Treating shock—old drugs, new ideas. N Engl J Med. 2010;362:841-843.
144 Addonizio V.P., Smith J.B., Strauss J.F., et al. Thromboxane synthesis and platelet secretion during cardiopulmonary bypass with bubble oxygenator. J Thorac Cardiovasc Surg. 1980;79:91.
145 Engelman R.M., Harji-Rovsov I., Breyer R.H., et al. Rebound vasospasm after coronary revascularization. Ann Thorac Surg. 1984;37:469.
146 Salmenpera M.T., Levy J.H. Effects of phosphodiesterase inhibitors on the human internal mammary artery. Anesth Analg. 1996;82:954.
147 Huraux C., Makita T., Szlam F., et al. Vasodilator effects of clevidipine on human internal mammary artery. Anesth Analg. 1997;85:1000.
148 Acar C., Jebara V.A., Portoghese M., et al. Revival of the radial artery for coronary artery bypass grafting. Ann Thorac Surg. 1992;54:652.
149 da Costa F.D.A., da Costa I.A., Poffo R., et al. Myocardial revascularization with the radial artery: A clinical and angiographic study. Ann Thorac Surg. 1996;62:475.
150 Dietl C.A., Benoit C.H. Radial artery graft for coronary revascularization: Technical considerations. Ann Thorac Surg. 1995;60:102.
151 Royster R.L., Butterworth J.F.I.V., Prielipp R.C., et al. A randomized, blinded, placebo-controlled evaluation of calcium chloride and epinephrine for inotropic support after emergence from cardiopulmonary bypass. Anesth Analg. 1992;74:3.
152 Zaloga G.P., Strickland R.A., Butterworth J.F.I.V., et al. Calcium attenuated epinephrine’s beta-adrenergic effects in postoperative heart surgery patients. Circulation. 1991;81:196.
153 Drop L.J. Ionized calcium, the heart, and hemodynamic function. Anesth Analg. 1985;64:432.
154 Bristow M.R., Ginsburg R., Umans V., et al. β1– and β2-adrenergic receptor subpopulations in normal and failing human ventricular myocardium. Coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circ Res. 1986;59:297.
155 Levy J.H., Bailey J.M., Deeb M. Intravenous milrinone in cardiac surgery. Ann Thorac Surg. 2002;73:325.
156 Stephenson L.W., Blackstone E.H., Kouchoukos N.T. Dopamine vs. epinephrine in patients following cardiac surgery: Randomized study. Surg Forum. 1976;27:272.
157 Butterworth J.F., Prielipp R.C., Zaloga G.P., et al. Is dobutamine less chronotropic than epinephrine after coronary bypass surgery? Anesthesiology. 1990;73(Suppl 3A):A61.
158 Sung B.H., Robinson C., Thadani U., et al. Effects of L-epinephrine on hemodynamics and cardiac function in coronary disease: Dose-response studies. Clin Pharmacol Ther. 1988;43:308.
159 Totaro R.J., Raper R.F. Epinephrine-induced lactic acidosis following cardiopulmonary bypass. Crit Care Med. 1997;25:1693.
160 Albanese J., Leone M., Garnier F., et al. Renal effects of norepinephrine in septic and nonseptic patients. Chest. 2004;126:534.
161 De Backer D., Biston P., Devriendt J., et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779-789.
162 Leier C.V., Heran P.T., Huss P., et al. Comparative systemic and regional hemodynamic effects of dopamine and dobutamine in patients with cardiomyopathic heart failure. Circulation. 1978;58:466.
163 Port J.D., Gilbert E.M., Larrabee P., et al. Neurotransmitter depletion compromises the ability of indirect acting amines to provide inotropic support in the failing human heart. Circulation. 1990;81:929.
164 Lehmann A., Boldt J. New pharmacologic approaches for the perioperative treatment of ischemic cardiogenic shock. J Cardiothorac Vasc Anesth. 2005;19:97.
165 DiSesa V., Gold J., Shemin R., et al. Comparison of dopamine and dobutamine in patients requiring postoperative circulatory support. Clin Cardiol. 1986;9:253.
166 Royster R.L. Intraoperative administration of inotropes in cardiac surgery patients. J Cardiothorac Anesth. 1990;4:17.
167 Davis R.F., Cappas D.G., Kirklin J.K., et al. Acute oliguria after cardiopulmonary bypass: Renal functional improvement with low-dose dopamine infusion. Crit Care Med. 1982;10:852.
168 Bellomo R., Chapman M., Finfer S., et al. Low-dose dopamine in patients with early renal dysfunction: A placebo-controlled randomized trial: Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet. 2000;356:2139.
169 Holmes C., Walley K.R. Bad medicine: Low-dose dopamine in the ICU. Crit Care Med. 2003;123:1266.
170 Ward H.B., Einzig S., Wang T., et al. Enhanced cardiac efficiency with dobutamine after global ischemia. J Surg Res. 1982;33:32.
171 Stephens J., Ead H., Spurrell R. Haemodynamic effects of dobutamine with special reference to myocardial blood flow: A comparison with dopamine and isoprenaline. Br Heart J. 1981;42:269.
172 Fowler M.B., Alderman E.L., Oesterle S.N., et al. Dobutamine and dopamine after cardiac surgery: Greater augmentation of myocardial blood flow with dobutamine. Circulation. 1984;70(Suppl I):103.
173 Romson J.L., Leung J.M., Bellows W.H., et al. Effects of dobutamine on hemodynamics and left ventricular performance after cardiopulmonary bypass in cardiac surgical patients. Anesthesiology. 1999;91:1318.
174 Salomon N.W., Plachetka J.R., Copeland J.G. Comparison of dopamine and dobutamine following coronary artery bypass grafting. Ann Thorac Surg. 1982;33:48.
175 Bristow M.R., Ginsburg R., Minobe W., et al. Decreased catecholamine sensitivity and beta-adrenergic receptor density in failing human hearts. N Engl J Med. 1982;307:205.
176 Gain K.R., Appleman M.M. Distribution and regulation of the phosphodiesterases of muscle tissues. Adv Cyclic Nucleotide Res. 1978;10:221.
177 Weishaar R.E., Burrows S.D., Kobylarz D.C., et al. Multiple molecular forms of cyclic nucleotide phosphodiesterase in cardiac and smooth muscle and in platelets. Isolation, characterization, and effects of various reference phosphodiesterase inhibitors and cardiotonic agents. Biochem Pharmacol. 1986;35:787.
178 Archer S.L., Michelakis E.D. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009;361:1864.
179 Benotti J.R., Grossman W., Braunwald E., et al. Hemodynamic assessment of amrinone. N Engl J Med. 1987;299:1373.
180 Levy J.L., Bailey J.M. Amrinone: Its effects on vascular resistance and capacitance in human subjects. Chest. 1994;105:62.
181 Benotti J.R., Grossman W., Braunwald E., et al. Effects of amrinone on myocardial energy metabolism and hemodynamics in patients with severe congestive heart failure due to coronary artery disease. Circulation. 1980;62:28.
182 Galie N., Ghofrani H.A., Torbicki A., et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148.
183 Bailey J.M., Levy J.H., Kikura M., et al. Pharmacokinetics of milrinone during cardiac surgery. Anesthesiology. 1994;81:616.
184 Konstam M.A., Cohen S.R., Weiland D.S., et al. Relative contribution of inotropic and vasodilator effects to amrinone-induced hemodynamic improvement in congestive heart failure. Am J Cardiol. 1986;57:242.
185 Firth B.G., Ratner A.V., Grassman E.D., et al. Assessment of the inotropic and vasodilator effects of amrinone versus isoproterenol. Am J Cardiol. 1984;54:1331.
186 Baim D.S., McDowell A.V., Cherniles J., et al. Evaluation of a new bipyridine inotropic agent—Milrinone—in patients with severe congestive heart failure. N Engl J Med. 1983;309:748.
187 Gonzalez M., Desager J-P, Jacquemart J-L, et al. Efficacy of enoximone in the management of refractory low-output states following cardiac surgery. J Cardiothorac Anesth. 1988;2:409.
188 Boldt J., Kling D., Zickmann B., et al. Efficacy of the phosphodiesterase inhibitor enoximone in complicated cardiac surgery. Chest. 1990;98:53.
189 Kikura M., Lee M.K., Safon R., et al. Effect of milrinone on platelets in patients undergoing cardiac surgery. Anesth Analg. 1995;81:44.
190 Gilbert E.M., Mealey P., Volkman K., et al. Combination therapy with enoximone and dobutamine is superior to nitroprusside and dobutamine in heart failure. Circulation. 1988;78:109.
191 Gage J., Rutman H., Lucido D., et al. Additive effects of dobutamine and amrinone on myocardial contractility and ventricular performance in patients with severe heart failure. Circulation. 1986;74:367.
192 Prielipp R.C., MacGregor D.A., Butterworth J.F., et al. Pharmacodynamics and pharmacokinetics of milrinone administration to increase oxygen delivery in critically ill patients. Chest. 1996;109:1291.
193 Boldt J., Kling D., Moosdorf R., Hempelmann G. Enoximone treatment of impaired myocardial function during cardiac surgery: Combined effects with epinephrine. J Cardiothorac Anesth. 1990;4:462.
194 Kikura M., Levy J.H., Bailey J.M., et al. Effects of milrinone on ventricular function after emergence from cardiopulmonary bypass. Anesth Analg. 1997;85:16.
195 Kikura M., Levy J.H., Bailey J.M., et al. A bolus dose of 1.5mg/kg amrinone effectively improves low cardiac output state following separation from cardiopulmonary bypass in cardiac surgical patients. Acta Anaesthesiol Scand. 1998;42:825.
196 Bondy R., Ramsay J.G. Reversal of refractory right ventricular failure with amrinone. J Cardiothorac Anesth. 1991;5:255.
197 Edelson J., LeJemtel T.H., Alousi A.A., et al. Relationship between amrinone plasma concentration and cardiac index. Clin Pharmacol Ther. 1981;29:723.
198 Park G.B., Kershner R.P., Angelotti J., et al. Oral bioavailability and intravenous pharmacokinetics of amrinone in humans. J Pharm Sci. 1983;72:817.
199 Kikura M., Levy J.H. New cardiac drugs. Int Anesthesiol Clin. 1995;33:21.
200 Rocci M.L., Wilson H., Likoff M., et al. Amrinone pharmacokinetics after single and steady-state doses in patients with chronic cardiac failure. Clin Pharmacol Ther. 1983;33:260.
201 Bailey J.M., Levy J.H., Rogers G., et al. Pharmacokinetics of amrinone during cardiac surgery. Anesthesiology. 1991;75:961.
202 Ramsay J.G., DeJesus J.M., Wynands J.E., et al. Amrinone before termination of cardiopulmonary bypass: Hemodynamic variables and oxygen utilization in the postbypass period. Can J Anaesth. 1992;39:342.
203 Feneck R.O. Effects of variable dose milrinone in patients with low cardiac output after cardiac surgery. Am Heart J. 1991;121(Suppl 2):1995.
204 Naeije R., Carlier E., DeSmet J.M., et al. Enoximone in low-output states following cardiac surgery. J Crit Care. 1989;4:112.
205 Kivikko M., Lehtonen L., Colucci W.S., et al. Sustained hemodynamic effects of intravenous levosimendan. Circulation. 2003;107:81.
206 Follath F., Cleland J.G., Just H., et al. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): A randomized double-blind trial. Lancet. 2002;360:196.
207 Moiseyev V.S., Poder P., Anderjevs N., et al. Safety and efficacy of a novel calcium sensitizer, levosimendan, in patients with left ventricular failure due to an acute myocardial infarction. A randomized, placebo-controlled, double-blind study (RUSSLAN). Eur Heart J. 2002;23:1422.
208 Labriola C., Siro-Brigiani M., Carrata F., et al. Hemodynamic effects of levosimendan in patients with low-output heart failure after cardiac surgery. Int J Clin Pharmacol Ther. 2004;42:204.
209 Jacobs A., Leopold J., Bates E., et al. Cardiogenic shock caused by right ventricular infarction: A report from the SHOCK registry. J Am Coll Cardiol. 2003;41:1273.
210 Hayashida N., Ikonomidis J.S., Weisel R.D., et al. Adequate distribution of warm cardioplegic solution. J Thorac Cardiovasc Surg. 1995;110:800.
211 Boldt J., Kling D., Thiel A., et al. Revascularization of the right coronary artery: Influence on thermodilution right ventricular ejection fraction. J Cardiothorac Anesth. 1988;2:140.
212 Schulman D.S., Biondi J.W., Zohgbi S., et al. Coronary flow and right ventricular performance during positive end-expiratory pressure. Am Rev Respir Dis. 1990;141:1531.
213 Matthay R.A., Ellis J.H., Steele P.P. Methoxamine-induced increase in afterload. Effect on left ventricular performance in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1978;117:871.
214 Mikami T., Kudo T., Sakurai N., et al. Mechanisms for development of functional tricuspid regurgitation determined by pulsed Doppler and two-dimensional echocardiography. Am J Cardiol. 1984;53:160.
215 Heinonen J., Salmenpera M., Takkunen O. Increased pulmonary artery diastolic-pulmonary wedge pressure gradient after cardiopulmonary bypass. Can Anaesth Soc J. 1985;32:165.
216 Byrick R.J., Kay J.C., Noble W.H. Extravascular lung water accumulation in patients following coronary artery surgery. Can Anaesth Soc J. 1977;24:332.
217 Chernow B., Rainey T.G., Lake C.R. Endogenous and exogenous catecholamines in critical care medicine. Crit Care Med. 1982;10:407.
218 Colman R.W. Platelet and neutrophil activation in cardiopulmonary bypass. Ann Thorac Surg. 1990;49:32.
219 Chenoweth D.E., Cooper S.W., Hugli T.E., et al. Complement activation during cardiopulmonary bypass. Evidence for generation of C3a and C5a anaphylatoxins. N Engl J Med. 1991;304:497.
220 Anjoyu-Lindskog E., Broman L., Broman M., Holmgren A. Effects of oxygen on central hemodynamics and VA/Q distribution after coronary bypass surgery. Acta Anaesthesiol Scand. 1983;27:378.
221 Salmenpera M., Heinonen J. Pulmonary vascular responses to moderate changes in Paco2 after cardiopulmonary bypass. Anesthesiology. 1986;64:311.
222 Viitanen A., Salmenpera M., Hynynen M., Heinonen J. Pulmonary vascular resistance before and after cardiopulmonary bypass. The effect of Paco2. Chest. 1989;95:773.
223 Viitanen A., Salmenpera M., Heinonen J. Right ventricular response to hypercarbia after cardiac surgery. Anesthesiology. 1990;73:393.
224 Lloyd E.A., Gersh B.J., Kennelly B.M. Hemodynamic spectrum of “dominant” right ventricular infarction in 19 patients. Am J Cardiol. 1981;48:1016.
225 Kay H., Afshari M., Barash P., et al. Measurement of ejection fraction by thermal dilution techniques. J Surg Res. 1983;34:337.
226 Spinale F.G., Smith A.C., Carabello B.A., Crawford F.A. Right ventricular function computed by thermodilution and ventriculography; a comparison of methods. J Thorac Cardiovasc Surg. 1990;99:141.
227 Dell’Italia L.J., Starling M.R., Blumhardt R., et al. Comparative effects of volume loading, dobutamine, and nitroprusside in patients with predominant right ventricular infarction. Circulation. 1985;72:1327.
228 Berisha S., Kastrati A., Goda A., Popa Y. Optimal value of filling pressure in the right side of the heart in acute myocardial infarction. Br Heart J. 1990;63:98.
229 Brinker J.A., Weiss J.L., Lapp D.L., et al. Leftward septal displacement during right ventricular loading in man. Circulation. 1980;61:626.
230 D’Ambra M.N., LaRaia P.J., Philbin D.M., et al. Prostaglandin E1—a new therapy for refractory right heart failure and pulmonary hypertension after mitral valve replacement. J Thorac Cardiovasc Surg. 1985;89:567.
231 Ichinose F., Roberts J.D., Zapol W.M. Inhaled nitric oxide: A selective pulmonary vasodilator; current uses and therapeutic potential. Circulation. 2004;109:3106.
232 De Wet C.J., Affleck D.G., Jacobsohn E., et al. Inhaled prostacyclin is safe, effective, and affordable in patients with pulmonary hypertension, right heart dysfunction, and refractory hypoxemia after cardiothoracic surgery. J Thorac Cardiovasc Surg. 2004;127:1058.
233 Girard C., Lehot J-J, Pannetier J-C, et al. Inhaled nitric oxide after mitral valve replacement in patients with chronic pulmonary artery hypertension. Anesthesiology. 1992;77:880.
234 Ardehali A., Hughes K., Sadeghi A., et al. Inhaled nitric oxide for pulmonary hypertension after cardiac transplantation. Transplantation. 2001;72:638.
235 Argenziano M., Choudhri A.F., Moazami N., et al. Randomized, double-blind trial of inhaled nitric oxide in LVAD recipients with pulmonary hypertension. Ann Thorac Surg. 1998;65:340.
236 Meade M.O., Granton J.T., Matte-Martyn A., et al. A randomized trial of inhaled nitric oxide to prevent ischemia-reperfusion injury after lung transplantation. Am J Respir Crit Care Med. 2003;167:1483.
237 Haraldsson A., Kieler-Jensen N., Ricksten S-E. Inhaled prostacyclin for treatment of pulmonary hypertension after cardiac surgery or heart transplantation: A pharmacodynamic study. J Cardiothorac Vasc Anesth. 1996;10:864.
238 Webb S.A., Scott S., van Heerden P.V. The use of inhaled aerosolized prostacyclin (IAP) in the treatment of pulmonary hypertension secondary to pulmonary embolism. Intensive Care Med. 1996;22:353.
239 Heerden P.V., Barden A., Michalopoulos N., et al. Dose-response to inhaled aerosolized prostacyclin for hypoxemia due to ARDS. Chest. 2000;117:819.
240 Domenighetti G., Stricker H., Waldispuehl B. Nebulized prostacyclin (PGI2) in acute respiratory distress syndrome: Impact of primary (pulmonary injury) and secondary (extrapulmonary injury) on gas exchange response. Crit Care Med. 2001;29:57.
241 Haraldsson A., Kieler-Jensen N., Ricksten S.E. The additive pulmonary vasodilatory effects of inhaled prostacyclin and inhaled milrinone in postcardiac surgical patients with pulmonary hypertension. Anesth Analg. 2001;93:1439.
242 Drummond W.H., Gregory G.A., Heyman M.A., Phibbs R.A. The independent effects of hyperventilation, tolazoline, and dopamine on infants with persistent pulmonary hypertension. J Pediatr. 1981;98:603.
243 Mahdi M., Salem M.R., Joseph N.J., et al. Influence of moderate hypocapnia on pulmonary vascular tone following mitral valve replacement. Anesthesiology. 1991;75:A166.
244 Miller D.C., Moreno-Cabral R.J., Stinson E.B., et al. Pulmonary artery balloon counterpulsation for acute right ventricular infarction. J Thorac Cardiovasc Surg. 1980;80:760.
245 Higgins T.L., Yared J.P., Paranandi L., et al. Risk factors for respiratory complications after cardiac surgery. Anesthesiology. 1991;75:A258.
246 Vuori A., Jalonen J., Laaksonen V. Continuous positive airway pressure during mechanical ventilation and spontaneous ventilation: Effects on central hemodynamics and oxygen transport. Acta Anaesthesiol Scand. 1979;23:453.
247 Morgan B.C., Abel F.L., Mullins G.L., Guntheroth W.G. Flow patterns in cavae, pulmonary artery, pulmonary vein and aorta in intact dogs. Am J Physiol. 1966;210:903.
248 Harken H.A., Brennan M.F., Smith B., Barsamian E.M. The hemodynamic response to positive end-expiratory pressure ventilation in hypovolemic patients. Surgery. 1974;76:786.
249 Wallis T.W., Robotham J.L., Compean R., Kindred M.K. Mechanical heart-lung interaction with positive end-expiratory pressure. J Appl Physiol. 1983;54:1039.
250 Pinsky M.R., Matuschak G.M., Klain M. Determinants of cardiac augmentation by elevations in intrathoracic pressure. J Appl Physiol. 1985;58:1189.
251 Canada E., Benumof J.L., Tousdale F.R. Pulmonary vascular resistance correlates in intact normal and abnormal canine lungs. Crit Care Med. 1982;10:719.
252 Permutt S., Howell J.B.L., Proctor D.F., Riley R.L. Effect of lung inflation on static pressure-volume characteristics of pulmonary vessels. J Appl Physiol. 1961;16:64.
253 McGregor M. Pulsus paradoxus. N Engl J Med. 1979;301:480.
254 Grace M.P., Greenbaum D.M. Cardiac performance in response to PEEP in patients with cardiac dysfunction. Crit Care Med. 1982;10:358.
255 Mathru M., Rao T.L.K., El-Etr A.A., Pifarre R. Hemodynamic response to changes in ventilatory patterns in patients with normal and poor left ventricular reserve. Crit Care Med. 1982;10:423.
256 Schulman D.S., Biondi J.W., Matthay R.A., et al. Effect of positive end-expiratory pressure on right ventricular performance-importance of baseline right ventricular function. Am J Med. 1988;84:57.
257 Calvin J.E., Driedger A.A., Sibbald W.J. Positive end-expiratory pressure (PEEP) does not depress left ventricular function in patients with pulmonary edema. Am Rev Respir Dis. 1981;124:121.
258 Glick G., Wechsler A.S., Epstein S.E. Reflex cardiovascular depression produced by stimulation of pulmonary stretch receptors in the dog. J Clin Invest. 1979;48:467.
259 Dunham B.M., Grindlinger G.A., Utsunomiya T., et al. Role of prostaglandins in positive end-expiratory pressure-induced negative inotropism. Am J Physiol. 1981;241:783.
260 Pinsky M.R., Marquez J., Martin D., Klain M. Ventricular assist by cardiac cycle–specific increases in intrathoracic pressure. Chest. 1987;91:709.
261 Garner W., Downs J.B., Stock M.C., Rasanen J. Airway pressure release ventilation (APRV)—a human trial. Chest. 1988;94:779.
262 Cane R.D., Shapiro B.A. Ventilator discontinuance and weaning. Anesthesiol Clin North America. 1987;5:749.
263 Lemaire F., Teboul J-L, Cinotti L., et al. Acute left ventricular dysfunction during unsuccessful weaning from mechanical ventilation. Anesthesiology. 1988;69:171.
264 Permutt S. Circulatory effects of weaning from mechanical ventilation: The importance of transdiaphragmatic pressure. Anesthesiology. 1988;69:157.
265 Rasanen J., Nikki P., Heikkila J. Acute myocardial infarction complicated by respiratory failure. The effects of mechanical ventilation. Chest. 1984;85:21.
266 Hurford W.E., Lynch K.E., Strauss W., et al. Myocardial perfusion as assessed by thallium 201 scintigraphy during the discontinuation of mechanical ventilation in ventilator-dependent patients. Anesthesiology. 1991;74:1007.
267 Tahvanainen J., Salmenpera M., Nikki P. Extubation criteria after weaning from intermittent mandatory ventilation and continuous positive airway pressure. Crit Care Med. 1983;11:702.
268 Russo A., O’Connor W., Waxman H. Atypical presentations and echocardiographic findings in patients with cardiac tamponade occurring early and late after cardiac surgery. Chest. 1993;104:71.
269 Yilmaz A.T., Arslan M., Demirklic U., et al. Late posterior cardiac tamponade after open heart surgery. J Cardiovasc Surg. 1996;37:615.
270 Borkon A.M., Schaff H.V., Gardner T.J., et al. Diagnosis and management of postoperative pericardial effusions and late cardiac tamponade following open heart surgery. Ann Thorac Surg. 1981;31:512.
271 King T.E., Stelzner T.J., Steven A., Sahn S.A. Cardiac tamponade complicating the postpericardiotomy syndrome. Chest. 1983;83:500.
272 Chuttani K., Pandian N.G., Mohanty P.K., et al. Left ventricular diastolic collapse. An echocardiographic sign of regional cardiac tamponade. Circulation. 1991;83:1999.
273 Fowler N.O. Physiology of cardiac tamponade and pulsus paradoxus. Physiological, circulatory, and pharmacologic responses in cardiac tamponade. Mod Concepts Cardiovasc Dis. 1978;47:115.
274 Hynynen M., Salmenpera M., Harjula A.L.J., et al. Atrial pressure and hormonal and renal responses to acute cardiac tamponade. Ann Thorac Surg. 1990;49:632.
275 Kochar G.S., Jacobs L.E., Kotler M.N. Right atrial compression in postoperative cardiac patients: Detection by transesophageal echocardiography. J Am Coll Cardiol. 1990;16:511.
276 Bommer W.J., Follette D., Pollock M., et al. Tamponade in patients undergoing cardiac surgery: A clinical-echocardiographic diagnosis. Am Heart J. 1995;130:1216.
277 Chuttani K., Tischler M.D., Pandian N.G., et al. Diagnosis of cardiac tamponade after cardiac surgery: Relative value of clinical, echocardiographic, and hemodynamic signs. Am Heart J. 1994;127:913.
278 D’Cruz I.A., Kensey K., Campbell C., et al. Two-dimensional echocardiography in cardiac tamponade occurring after cardiac surgery. J Am Coll Cardiol. 1985;5:1250.
279 Reichert C.L., Visser C.A., Koolen J.J., et al. Transesophageal echocardiography in hypotensive patients after cardiac operations. Comparison with hemodynamic parameters. J Thorac Cardiovasc Surg. 1992;104:321.
280 Settle H.P., Adolph R.J., Fowler N.O., et al. Echocardiographic study of cardiac tamponade. Circulation. 1977;56:951.
281 Singh S.M., Wann L.S., Schuchard G.H., et al. Right ventricular and right atrial collapse in patients with cardiac tamponade—a combined echocardiographic and hemodynamic study. Circulation. 1984;70:966.
282 Russo A.M., O’Connor W.H., Waxman H.L. Atypical presentations and echocardiographic findings in patients with cardiac tamponade occurring early and late after cardiac surgery. Chest. 1993;104:71.
283 Mattila I., Takkunen O., Mattila P., et al. Cardiac tamponade and different modes of artificial ventilation. Acta Anaesthesiol Scand. 1984;23:236.
284 Ziemer G., Karck M., Muller H., Luhmer I. Staged chest closure in pediatric cardiac surgery preventing typical and atypical cardiac tamponade. Eur J Cardiothorac Surg. 1992;6:91.
285 Addonizio L.J., Gersony W.M., Robbins R.C., et al. Elevated pulmonary vascular resistance and cardiac transplantation. Circulation. 1987;76(Suppl V):52.
286 Valantine H.A., Schroeder J.S. Cardiac transplantation. Intensive Care Med. 1989;15:283.
287 O’Connell J.B., Renlund D.G., Bristow M.R. Murine monoclonal CD3 antibody (OKT3) in cardiac transplantation: Three-year experience. Transplant Proc. 1989;21(Suppl 2):31.

