[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 90 Portal Hypertension and Gastrointestinal Bleeding
Variceal hemorrhage, hepatic encephalopathy, and ascites—the major complications of cirrhosis of the liver—result from portal hypertension, defined as an increase in hepatic sinusoidal pressure to 6 mm Hg or greater. Portosystemic collaterals decompress the hypertensive hepatic sinusoids and give rise to varices at the gastroesophageal junction and elsewhere. These portosystemic collaterals also may allow ammonia derived from the intestine to reach the brain, thereby resulting in hepatic encephalopathy through a pathologic process of several intermediary steps involving the peripheral benzodiazepine-type receptors, neurosteroids, and γ-aminobutyric acid (GABA) receptors (see Chapter 92). Additionally, portal hypertension is associated with renal retention of sodium and water and the formation of ascites (see Chapter 91). Indeed, portal hypertension and its complications remain important clinical problems despite advances in treatment and improved understanding of both the molecular basis and pathophysiology of portal hypertension.
NORMAL PORTAL CIRCULATION
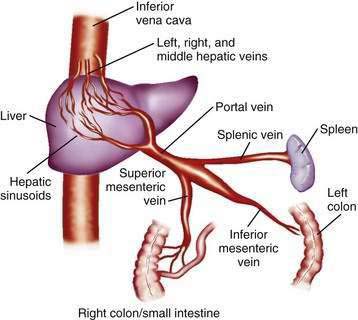
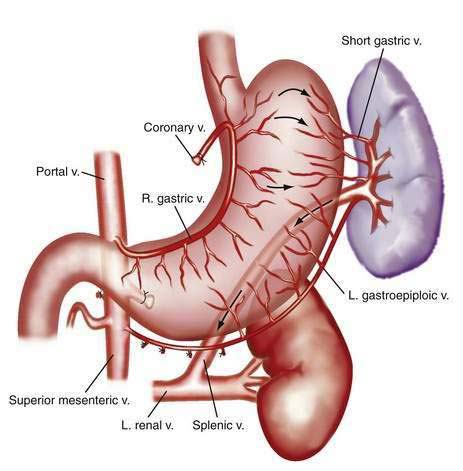
The portal venous system carries capillary blood from the esophagus, stomach, small and large intestine, pancreas, gallbladder, and spleen to the liver. The portal vein is formed by the confluence of the splenic vein and the superior mesenteric vein behind the neck of the pancreas.1 The inferior mesenteric vein usually drains into the splenic vein. The left gastric vein, also called the left coronary vein, usually drains into the portal vein at the confluence of the splenic vein and superior mesenteric vein (Fig. 90-1). The portal vein is approximately 7.5 cm in length and runs dorsal to the hepatic artery and bile duct into the hilum of the liver. The uppermost 5 cm of the portal vein does not receive any tributaries.2 In the hilum of the liver, the portal vein divides into the left and right portal vein branches, which supply the left and right sides of the liver, respectively. The umbilical vein drains into the left portal vein. The cystic vein from the gallbladder drains into the right portal vein, whereas the portal venules drain into hepatic sinusoids that, in turn, are drained by the hepatic veins into the inferior vena cava. The left and middle hepatic veins usually join and drain into the inferior vena cava separately but adjacent to the confluence of the right hepatic vein with the inferior vena cava. The caudate lobe drains separately into the inferior vena cava (see Chapter 71).
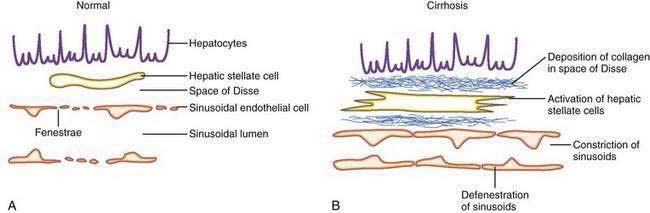
The sinusoids are highly permeable and thus facilitate the transport of macromolecules to the parenchymal hepatocytes that reside on the extraluminal side of the endothelial cells. The hepatic sinusoids are highly permeable because they lack a proper basement membrane and because the endothelial cells that line the sinusoids contain fenestrae. Other unique aspects of the hepatic sinusoids are the space of Disse, a virtual space located extraluminal to the endothelial cell and adjacent to the hepatocyte, and its cellular constituents, the hepatic stellate cell (HSC) and the Kupffer cell (Fig. 90-2; see also Chapters 71 and 72). These two cell types probably play an important role, in concert with the endothelial cell, in regulating sinusoidal hemodynamics and homeostasis and may contribute to the sinusoidal derangements that occur in portal hypertension. Under basal conditions, HSCs maintain a quiescent phenotype and accumulate vitamin A. On activation, however, as occurs in cirrhosis and portal hypertension, these cells are postulated to develop contractile abilities that permit them to function as sinusoidal pericytes. Kupffer cells contribute to vascular homeostasis by generating cytokines with potent cellular and vasoregulatory actions, including tumor necrosis factor. Endothelial cells and smooth muscle cells in nonsinusoidal hepatic vessels such as the portal venule and the terminal hepatic venule are important in hepatic vasoregulation, particularly in the normal liver, where HSCs are quiescent, unactivated, and presumably less contractile.
HEMODYNAMIC PRINCIPLES OF PORTAL HYPERTENSION
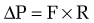
in which the pressure gradient in the portal circulation (ΔP) is a function of portal flow (F) and resistance to flow (R). Increases in portal resistance or portal flow can contribute to increased pressure. Portal hypertension almost always results from increases in both portal resistance and portal flow (Fig. 90-3). One exception is that of an arteriovenous fistula, which in the initial stages causes portal hypertension largely through an increase in portal flow in the absence of an increase in resistance. The mechanism of the increase in portal resistance depends on the site and cause of portal hypertension; in the Western world, the most common cause is liver cirrhosis (see later). Because of the increase in hepatic resistance and the decrease in hepatic compliance, small changes in flow that do not increase pressure in the normal liver can have a prominent stimulatory effect on portal pressure in the cirrhotic liver. The increase in portal venous inflow is part of a generalized systemic derangement termed the hyperdynamic circulatory state. Collateral vessels that dilate and new vascular sprouts that form connect the high-pressure portal venous system with lower-pressure systemic veins. Unfortunately, this process of angiogenesis and collateralization is insufficient for normalizing portal pressure and actually causes complications of portal hypertension, such as esophageal varices.3 Approaches to block this angiogenic process are a compelling target for drug development.
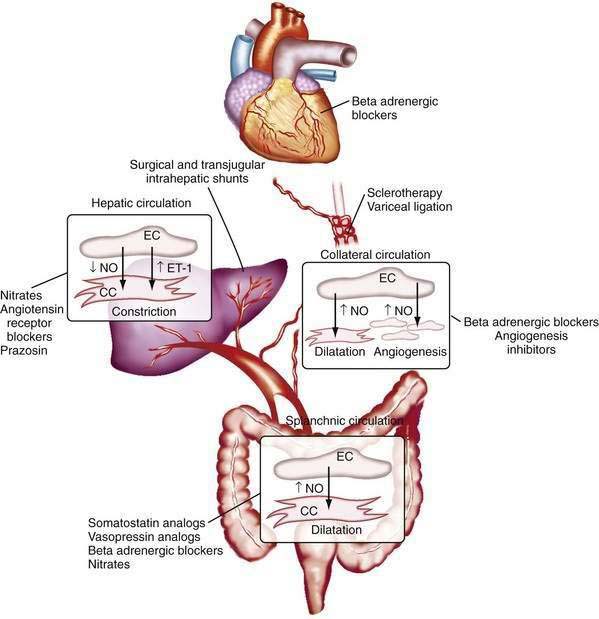
The changes in portal flow and resistance also can be viewed as originating from mechanical and vascular factors. Mechanical factors include the fibrosis and nodularity of the cirrhotic liver with distortion of the vascular architecture and the remodeling that is recognized to occur in the systemic and splanchnic vasculature in response to the chronic increases in flow and shear stress that characterize the hyperdynamic circulatory state. Vascular factors include intrahepatic vasoconstriction, which contributes to increased intrahepatic resistance, and the splanchnic and systemic vasodilatation that accompanies the hyperdynamic circulatory state. The vascular factors that contribute to portal hypertension are particularly important because they are reversible and dynamic and therefore compelling targets for experimental therapies (Fig. 90-4). Conversely, effective therapies for the fixed, mechanical component of portal hypertension caused by scar, regenerative nodules, and vascular remodeling are currently lacking. Indeed, most available therapies for portal hypertension focus on correction of hemodynamic alterations in the portal circulation. Approaches include use of nonselective β-adrenergic blocking agents, octreotide, and vasopressin to reduce the hyperdynamic circulation, portal venous inflow, and splanchnic vasodilatation.4,5 Alternative agents reduce the increased intrahepatic resistance and include angiotensin receptor blockers and mononitrates.
INCREASED INTRAHEPATIC RESISTANCE
In cirrhosis, increased portal resistance occurs in great part as a result of mechanical factors that reduce vessel diameter. In addition to regenerative nodules and fibrotic bands, these mechanical factors include capillarization of the sinusoids and swelling of cells, including hepatocytes and Kupffer cells. As discussed earlier, however, reduced hepatic vessel diameter resulting in increased portal resistance, even when caused by cirrhosis, is not a purely mechanical phenomenon.6 Hemodynamic changes in the hepatic circulation also contribute to increased intrahepatic resistance.7,8 These changes are characterized by hepatic vasoconstriction and impaired responses to vasodilatory stimuli. The increase in intrahepatic resistance is determined largely by changes in vessel radius, with small reductions in vessel radius causing prominent increases in resistance. Blood viscosity and vessel length also can influence resistance, albeit to a much smaller extent. The factors that regulate resistance can be viewed in the context of the law of Poiseuille:
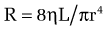
in which R is resistance, ηL is the product of blood viscosity and vessel length, and r is vessel radius.
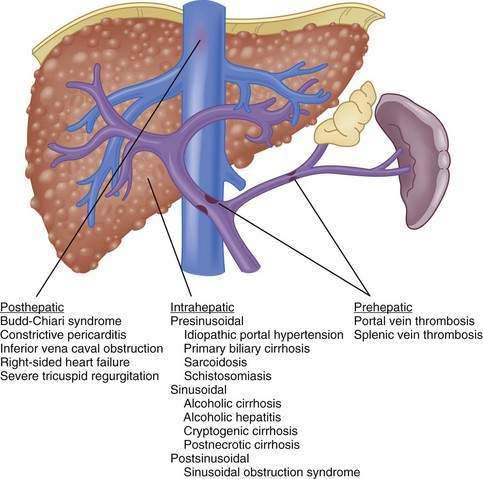
Although vasoactive changes were estimated initially to account for 10% to 30% of the increase in portal resistance in cirrhosis, subsequent studies have suggested that these figures actually may underestimate the contribution of hepatic vasoconstriction to the increased resistance observed in the cirrhotic liver. In noncirrhotic causes of portal hypertension, the increase in resistance may occur at sites upstream (prehepatic) or downstream (posthepatic) of the liver, as in portal vein thrombosis and hepatic vein thrombosis, respectively (Fig. 90-5). Furthermore, the site of increased intrahepatic resistance can be further delineated as the sinusoids (sinusoidal), upstream from the sinusoids within the portal venules (presinusoidal), or downstream from the sinusoids in the hepatic venules (postsinusoidal), as in alcoholic cirrhosis, schistosomiasis, and sinusoidal obstruction syndrome, respectively. Pressure is increased only in the portal circulation behind the site of increased resistance, and in isolated portal vein thrombosis, hepatic function frequently remains largely preserved despite prominent portal hypertension.
Most evidence suggests that a decrease in the production of the vasodilator NO and an increase in the production of the vasoconstrictor ET-1 jointly contribute to the increase in hepatic vascular resistance. In experimental models of cirrhosis, the bioavailability of hepatic NO is diminished because of a reduction in the production of NO by endothelial cells.7,9,10 A similar paradigm is observed in the human cirrhotic liver.11 Most studies indicate that the reduction in NO production occurs not through a reduction in hepatic eNOS protein levels9,10 but through defects in the steps necessary to activate existing eNOS protein. For example, increases in the production of the eNOS-inhibiting protein caveolin-1 have been observed in experimental models of cirrhosis10 and in human cirrhosis. Another pathway that contributes to deficient generation of NO by eNOS is a reduction in the level of AKT (protein kinase B) phosphorylation of eNOS and upregulation of the eNOS inhibiting protein, GRK (G protein-coupled receptor kinase), in the cirrhotic liver.12 Irrespective of the mechanism of deficiency, the lack of availability of NO is thought to allow HSCs, which are activated and highly contractile in liver cirrhosis, to constrict the sinusoids that they envelop, thereby increasing portal pressure. The role of the HSCs in this process remains controversial, however, because evidence is mixed regarding whether the site of the increase in intrahepatic resistance in cirrhosis is the sinusoids, where stellate cells reside, or the pre- or postsinusoidal venules (or both), which are devoid of stellate cells and in which endothelial cells signal smooth muscle cells. Furthermore, increasing evidence points toward diverse origins of these myofibroblastic cells within the cirrhotic sinusoids, with portal myofibroblasts as well as HSC postulated to play important roles.13 In this regard, therapies that target myofibroblast migration may be a compelling therapeutic target by limiting the density of these contractile cells within the hepatic sinusoids.14 Finally, the contribution of HSCs to hepatic angiogenesis may also be an important target for treating fibrosis and portal hypertension.15
In clinical practice, NO can be delivered by NO donor agents such as mononitrates. NO donor agents exert their beneficial effects in part by relaxing the actively contractile stellate cells.16,17 The systemic actions of these agents, however, tend to cause side effects and exacerbate the hyperdynamic circulatory state. In studies utilizing a liver-specific NO donor compound, the increased intrahepatic vascular resistance could be corrected by the generation of additional NO and consequent relaxation of HSCs.18 In cirrhosis, however, deficient endothelial cell NO generation may be accompanied by impaired stellate cell relaxation in response to NO,19 perhaps because of diminished response of the NO second messenger cyclic guanosine monophosphate (cGMP) in activated cells.16 In this situation, a prominent beneficial effect of NO donors is less predictable.
Excessive ET-1 also contributes to increased intrahepatic vasoconstriction in portal hypertension through vasoconstrictive effects in the liver, presumably by enhancing HSC contractility.20,21 In experimental models, ET-1 protein and receptor expression are increased, most notably in HSCs and endothelial cells.20,22,23 In humans with portal hypertension, plasma and liver ET-1 levels also are increased.24 The reason for activation of the ET-1 system in portal hypertension is not known, but this effect may be secondary to transforming growth factor-β (TGF-β), a key fibrogenic growth factor.23 Clinical trials of ET antagonists in patients with portal hypertension are in progress; however, the variable effects of ET modulation in experimental models of portal hypertension, as well as the possible hepatotoxicity of these compounds, have limited enthusiasm for studies in humans.25 Other therapies for portal hypertension may provide benefit through the ET pathway. For example, somatostatin, which reduces portal pressure by constricting the splanchnic circulation, also may act by inhibiting ET-1–dependent HSC contraction.26
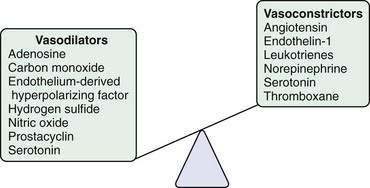
Other vasoactive mediators, including cysteinyl leukotrienes, thromboxane, angiotensin, and hydrogen sulfide, also have been implicated in the development of increased intrahepatic resistance in cirrhosis.27,28 Some of these mediators, particularly angiotensin, which causes contraction of HSCs, have been studied in humans. Attempts to reduce portal pressure using pharmacologic agents that inhibit angiotensin activation of HSC contraction have met with mixed results thus far.29
HYPERDYNAMIC CIRCULATION
In addition to the increases in portal resistance discussed earlier, a major factor in the development and perpetuation of portal hypertension is an increase in portal venous flow, or the hyperdynamic circulation. The term portal venous inflow indicates the total blood that drains into the portal circulation, not the blood flow in the portal vein itself, which may actually be diminished in portal hypertension because of portosystemic collateral shunts. The hyperdynamic circulation is characterized by peripheral and splanchnic vasodilatation, reduced mean arterial pressure, and increased cardiac output. Vasodilatation, particularly in the splanchnic bed, permits an increase in inflow of systemic blood into the portal circulation.30
Splanchnic vasodilatation is caused in large part by relaxation of splanchnic arterioles and ensuing splanchnic hyperemia. Studies of experimental portal hypertension have demonstrated that splanchnic vascular endothelial cells are primarily responsible for mediating splanchnic vasodilatation and enhanced portal venous inflow through excess generation of NO.31–39 This excess generation of NO and ensuing vasodilatation, hyperdynamic circulation, and hyperemia in the splanchnic and systemic circulation contrasts with the hepatic circulation, in which NO deficiency contributes to increased intrahepatic resistance.
The mechanism of excess NO production from the endothelial cells of the systemic and splanchnic arterial circulation is an area of active investigation. Some of the increase in NO production probably occurs from shear stress–dependent and shear stress–independent increases in the expression of eNOS, which can be corrected in part by beta blockers.37,40–45 Activation of existing eNOS by cytokines or mechanical factors also seems to contribute to excess systemic and splanchnic NO generation through pathways that include eNOS phosphorylation and protein interactions.42–46 The physiologic stimuli that mediate this process are not well understood but may include ET-1, which is increased in the serum of patients with portal hypertension, and the cytokine tumor necrosis factor-α (TNF-α) because inhibitors of TNF improve portal pressure and the splanchnic circulatory disturbances in both human and experimental portal hypertension. TNF-α may be derived from intestinal endotoxin, and intestinal decontamination appears to correct the hyperdynamic circulation in humans, suggesting a link with intestinal inflammation.47 Vascular endothelial growth factor (VEGF) has also been implicated in this process by excessively activating eNOS.48 In humans with portal hypertension, therapeutic inhibition of NOS has met with mixed clinical results.
In one study, inhibition of NOS corrected altered systemic hemodynamics,49 but other studies have not demonstrated significant portal pressure–reducing effects of systemic NOS inhibition.50 Other mediators that may contribute to systemic and splanchnic vasodilatation include anandamide, an endogenous vasodilatory cannabinoid,51–53 heme oxygenase,17,54–56 and cyclooxygenase.57 Compelling evidence also supports a primary defect in smooth muscle cells in portal hypertension, perhaps because of defects in potassium channels.58–62 In fact, many pharmacologic therapies for portal hypertension target the splanchnic arteriolar smooth muscle cells, rather than endothelial cells, to reduce splanchnic vasodilatation. For example, octreotide, a synthetic analog of somatostatin, causes marked but transient reductions in portal pressure by contracting splanchnic smooth muscle cells, thereby limiting portal venous inflow, especially after meals. Nonselective beta blockers and vasopressin also reduce portal pressure by constricting splanchnic arterioles and thereby reducing portal venous inflow. Because intrahepatic resistance persists, therapies targeted toward the increase in portal venous inflow usually do not normalize portal pressure entirely but often blunt the prominent increases in portal venous inflow that occur in response to a meal. Combination therapy with an agent that reduces increased intrahepatic resistance, such as a nitrate, and an agent that reduces portal venous inflow, such as a beta blocker, are more effective in reducing portal pressure than is either agent alone.
COLLATERAL CIRCULATION AND VARICES
The portal vein–systemic collateral circulation develops and expands in response to elevation of the portal pressure.63 Blood flow in the low volumes that normally perfuse these collaterals and flow toward the portal circulation is reversed in portal hypertension because the increased portal pressure exceeds systemic venous pressure. Therefore, flow is reversed in these collateral vessels, and blood flows out of the portal circulation toward the systemic venous circulation.
Four distinct zones of venous drainage at the gastroesophageal junction are particularly relevant to the formation of esophageal varices.64 The gastric zone, which extends for 2 to 3 cm below the gastroesophageal junction, comprises veins that are longitudinal and located in the submucosa and lamina propria. They come together at the upper end of the cardia of the stomach and drain into short gastric and left gastric veins. The palisade zone extends 2 to 3 cm proximal to the gastric zone into the lower esophagus. Veins in this zone run longitudinally and in parallel in four groups corresponding to the esophageal mucosal folds. These veins anastomose with veins in the lamina propria. The perforating veins in the palisade zone do not communicate with extrinsic (periesophageal) veins in the distal esophagus. The palisade zone is the dominant watershed area between the portal and systemic circulations. More proximal to the palisade zone in the esophagus is the perforating zone, where there is a network of veins. These veins are less likely to be longitudinal and are termed perforating veins because they connect the veins in the esophageal submucosa and the external veins. The truncal zone, the longest zone, is approximately 10 cm in length, located proximally to the perforating zone in the esophagus, and usually characterized by four longitudinal veins in the lamina propria.
The fundus of the stomach drains through short gastric veins into the splenic vein. In the presence of portal hypertension, varices may therefore form in the fundus of the stomach. Splenic vein thrombosis usually results in isolated gastric fundal varices. Because of the proximity of the splenic vein to the renal vein, spontaneous splenorenal shunts may develop and are more common in patients with gastric varices than in those with esophageal varices.65,66
The predominant collateral flow pattern in intrahepatic portal hypertension is through the right and left coronary veins, with only a small portion of flow through the short gastric veins. Therefore, most patients with intrahepatic causes of portal hypertension have esophageal varices or gastric varices in continuity with esophageal varices. Unfortunately, portal hypertension caused by cirrhosis generally persists and progresses despite the development of even an extensive collateral circulation. Progression of portal hypertension results from (1) the prominent obstructive resistance in the liver; (2) resistance within the collaterals themselves; and (3) continued increase in portal vein inflow. The collateral circulatory bed develops through a combination of angiogenesis, the development of new blood vessels, and dilatation and increased flow through preexisting collaterals.3,67 Experimental evidence suggests that VEGF, a key NO stimulatory growth factor, may contribute to both the angiogenic and collateral vessel responses.55,68 Inhibition of VEGF or NO may attenuate the collateral vessel propagation by inhibiting angiogenic responses in experimental models of portal hypertension and collateralization.67–72 Some pharmacologic agents used in the management of portal hypertension, such as beta blockers and octreotide, may act in part by constricting collateral vessels.73–76 Approaches to inhibiting VEGF and angiogenesis are worth studying therapeutically.48
The development of gastroesophageal varices requires a portal pressure gradient of at least 10 mm Hg. Furthermore, a portal pressure gradient of at least 12 mm Hg is thought to be required for varices to bleed; other local factors that increase variceal wall tension also are needed77 because all patients with a portal pressure gradient of greater than 12 mm Hg do not necessarily bleed. Factors that influence variceal wall tension can be viewed in the context of the law of Laplace:
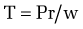
where T is variceal wall tension, P is the transmural pressure gradient between the variceal lumen and esophageal lumen, r is the variceal radius, and w is the variceal wall thickness. When the variceal wall thins and the varix increases in diameter and pressure, the tolerated wall tension is exceeded and the varix will rupture. These physiologic observations are manifested clinically by the observation that patients with larger varices (r) in sites of limited soft tissue support (w), with elevated portal pressure (P), tend to be at greatest risk for variceal rupture from variceal wall tension (T) that becomes excessive. One notable site in which soft tissue support is limited is at the gastroesophageal junction. The lack of tissue support and high vessel density may contribute to the greater frequency of bleeding from varices at the gastroesophageal junction. The law of Laplace also has implications for the relevance of pharmacologic therapies aimed at reducing portal pressure. Reductions in portal pressure will reduce the variceal transmural pressure gradient, thereby reducing the risk that variceal wall tension will become excessive and varices will rupture. Clinically, a reduction in the hepatic venous pressure gradient to less than 12 mm Hg almost negates the risk of variceal hemorrhage. The changes in portal pressure and local variceal factors, however, are dynamic and influenced by a number of physiologic (an increase in intra-abdominal pressure, meal-induced increases in portal pressure), diurnal (circadian changes in portal pressure), and pathophysiologic (acute alcohol use) factors, and portal pressure and esophageal variceal pressure may vary at different times.
MEASUREMENT OF PORTAL PRESSURE
HEPATIC VEIN PRESSURE GRADIENT
The HVPG is the difference between the wedged hepatic venous pressure (WHVP) and free hepatic vein pressure (FHVP). The HVPG has been used to assess portal hypertension since its first description in 1951,78 and has been validated as the best predictor for the development of complications of portal hypertension.
Measurement of the HVPG requires passage of a catheter into the hepatic vein under radiologic guidance until the catheter can be passed no further, that is, until the catheter has been “wedged” in the hepatic vein. The catheter can be passed into the hepatic vein through the femoral vein or using a transjugular venous approach. The purpose of wedging the catheter is to form a column of fluid that is continuous between the hepatic sinusoids and the catheter. Therefore, the measured pressure of fluid within the catheter reflects hepatic sinusoidal pressure. One of the drawbacks of using a catheter that is wedged in the hepatic vein is that the WHVP measured in a more fibrotic area of liver may be higher than the pressure measured in a less fibrotic area because of regional variation in the degree of fibrosis. Using a balloon-occluding catheter in the right hepatic vein to create a stagnant column of fluid in continuity with the hepatic sinusoids eliminates this variation in measurement of WHVP because the balloon catheter measures the WHVP averaged over a wide segment of the liver.79 HVPG is not effective for detecting presinusoidal causes of portal hypertension. For example, in portal hypertension secondary to portal vein thrombosis, the HVPG is normal. Moreover, the HVPG may underestimate sinusoidal pressure in primary biliary cirrhosis and other presinusoidal causes of portal hypertension.80 Therefore, HVPG is accurate for detecting only sinusoidal and postsinusoidal causes of portal hypertension.
The HVPG represents the gradient between the pressure in the portal vein and the intra-abdominal inferior vena caval pressure. An elevation in intra-abdominal pressure increases both WHVP and FHVP equally, so that the HVPG is unchanged. The advantage of the HVPG is that variations in the “zero” reference point have no impact on the HVPG.81 The HVPG is measured at least three times to demonstrate that the values are reproducible. Total occlusion of the hepatic vein by the inflated balloon to confirm that the balloon is in a wedged position is demonstrated by injecting contrast into the hepatic vein. A sinusoidal pattern should be seen, with no collateral circulation to other hepatic veins. The contrast washes out promptly with deflation of the balloon. Correct positioning of the balloon also is demonstrated by a sharp increase in the recorded pressure on inflation of the balloon. The pressure then becomes steady until the balloon is deflated, when the pressure drops sharply. In experienced hands, measurement of the HVPG is highly reproducible, accurate, and safe.
Measurement of the HVPG has been proposed for the following indications: (1) to monitor portal pressure in patients taking drugs used to prevent variceal bleeding; (2) as a prognostic marker82; (3) as an end-point in trials using pharmacologic agents for the treatment of portal hypertension83; (4) to assess the risk of hepatic resection in patients with cirrhosis; and (5) to delineate the cause of portal hypertension (i.e., presinusoidal, sinusoidal, or postsinusoidal (Table 90-1), usually in combination with venography, right-sided heart pressure measurements, and transjugular liver biopsy. Although the indication for HVPG measurement with the most potential for widespread use is monitoring the efficacy of therapies to reduce portal pressure, HVPG monitoring is not done routinely in clinical practice because no controlled trials have yet demonstrated its usefulness.84
PORTAL VEIN PRESSURE
Direct measurement of the pressure in the portal vein is a rarely used method that can be carried out through a percutaneous transhepatic route, transvenous approach, or, rarely, intraoperatively (although anesthesia can affect portal pressure). The transhepatic route requires portal vein puncture performed under ultrasound guidance. A catheter is then threaded over a guidewire into the main portal vein. With increasing use of the transjugular intrahepatic portosystemic shunt (TIPS) (see later), radiologists have gained expertise in puncturing the portal vein and measuring portal vein pressure by a transjugular route. Direct portal pressure measurements are carried out when HVPG cannot be measured, as in patients with occluded hepatic veins caused by the Budd-Chiari syndrome, in whom a surgical portosystemic shunt is being contemplated,85 or in patients with intrahepatic, presinusoidal causes of portal hypertension, such as idiopathic portal hypertension, in which the HVPG may be normal.
ENDOSCOPIC VARICEAL PRESSURE
Varices rupture and bleed when the expanding force of intravariceal pressure exceeds variceal wall tension. Measurement of the difference between intravariceal pressure and pressure within the esophageal lumen (the transmural pressure gradient across the varices) is potentially a more important indicator of bleeding risk than measurement of HVPG,86,87 especially in patients with portal vein thrombosis and other causes of portal hypertension associated with a normal HVPG.
A miniature pneumatic pressure sensitivity gauge attached to the tip of an endoscope (Varipres Solid Components, Barcelona, Spain) allows noninvasive measurement of variceal pressure. Patients with previous variceal bleeding have been demonstrated to have higher variceal pressures than those in patients without previous bleeding.88 A variceal pressure greater than 18 mm Hg during a bleeding episode is associated with failure to control bleeding and predicts early rebleeding.89 Moreover, patients on pharmacologic therapy who show a decrease in variceal pressure of greater than 20% from baseline have a low probability of bleeding, as compared with patients who do not demonstrate a greater than 20% decrease in variceal pressure, in whom the risk of variceal bleeding is 46%.88 Variceal pressure measurements determined with use of Varipres are considered satisfactory when they meet the following criteria: (1) a stable intraesophageal pressure; (2) absence of artifacts caused by esophageal peristalsis; and (3) correct placement of the gauge over the varix, as shown by fine fluctuations in the pressure tracing that correspond to the cardiac cycle and respirations. Therefore, measurement of variceal pressure requires both a skilled endoscopist and a cooperative patient, and, even in expert hands, accurate variceal pressure measurements cannot be obtained in 25% of patients.
Manometry using an endoscopic balloon to measure variceal pressure is subject to observer bias because it relies on visual appearance to determine whether the varices have collapsed.90–92 With this technique, a balloon is inserted into the esophagus and inflated until the varices are noted on endoscopy to collapse. The pressure in the balloon required to collapse the varices represents the variceal pressure. In general, techniques of measuring variceal pressure are still considered experimental and not suitable for routine clinical use.
DETECTION OF VARICES
UPPER GASTROINTESTINAL ENDOSCOPY
Upper gastrointestinal endoscopy is the most commonly used method to detect varices. The current consensus is that all patients with cirrhosis of the liver should be screened for esophageal varices by endoscopy. In patients in whom no varices are detected on initial endoscopy, endoscopy to look for varices should be repeated in 2 to 3 years. If small varices are detected on the initial endoscopy, endoscopy should be repeated in 1 to 2 years.93,94 None of the various noninvasive methods of determining which patients benefit most from endoscopic screening are accurate enough to recommend for routine use in clinical practice.95 The role of noninvasive markers in predicting the risk of large esophageal varices requires study in large multicenter trials. Preliminary data suggest that wireless video capsule endoscopy (see Chapter 19)96 and computed tomography (CT) imaging are alternative screening modalities in patients who are not candidates for upper endoscopy. Moreover, CT screening may be more cost-effective than endoscopy.97
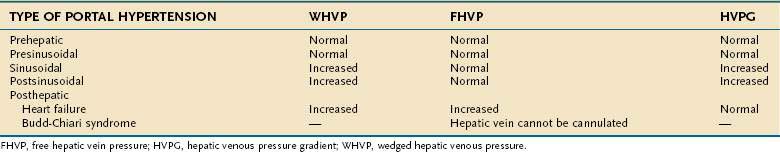
Endoscopic grading of esophageal varices is subjective. Various criteria have been used to try to standardize the reporting of esophageal varices. The best known of these criteria are those compiled by the Japanese Research Society for Portal Hypertension. The descriptors include red color signs, color of the varix, form (size) of the varix, and location of the varix.98 Red color signs include red “wale” markings, which are longitudinal whip-like marks on the varix; cherry-red spots, which usually are 2 to 3 mm or less in diameter; hematocystic spots, which are blood-filled blisters 4 mm or greater in diameter; and diffuse redness. The color of the varix can be white or blue. The form of the varix at endoscopy is described most commonly. Esophageal varices may be small and straight (grade I); tortuous and occupying less than one third of the esophageal lumen (grade II); or large and occupying more than one third of the esophageal lumen (grade III). Varices can be in the lower third, middle third, or upper third of the esophagus. Of all of the aforementioned descriptors, the size of the varices in the lower third of the esophagus is the most important. The size of the varices in the lower third of the esophagus is determined during withdrawal of the endoscope (Fig. 90-6). As much air as possible should be aspirated from the stomach while the esophageal lumen is fully inflated. Small varices, that is, those occupying less than one third of the lumen, are less than 5 mm in diameter, whereas large varices are greater than 5 mm in diameter.98,99 As a point of reference, any varix larger in diameter than an open pinch biopsy forceps is likely to be greater than 5 mm in diameter. Patients with large esophageal varices, Child (or Child-Pugh) class C cirrhosis (see later), and red color signs on varices have the highest risk of variceal bleeding within 1 year.100 The increase in bleeding risk attributable to the presence of red color signs, however, is not independent of the risk associated with large variceal size. Therefore, prophylactic treatment to prevent variceal bleeding is recommended in all patients with large esophageal varices irrespective of the presence or absence of red color signs (see later).
ULTRASONOGRAPHY
Ultrasound examination of the liver with Doppler study of the vessels has been used widely to assess patients with portal hypertension. Features suggestive of portal hypertension on ultrasonography include splenomegaly, portosystemic collateral vessels, and reversal of the direction of flow in the portal vein (hepatofugal flow). Some studies have demonstrated that a portal vein diameter greater than 13 mm and the absence of respiratory variations in the splenic and mesenteric veins are sensitive but nonspecific markers of portal hypertension.101,102 These criteria are not used routinely in clinical practice in most centers. Ultrasound examination can detect thrombosis of the portal vein, which appears as nonvisualization or cavernous transformation (a cavernoma) of the portal vein; the latter finding indicates an extensive collateral network in place of the portal vein.103 Splenic vein thrombosis also can be demonstrated. Portal blood flow can be measured by Doppler ultrasonography, which is the easiest research method for detecting postprandial increases in splanchnic blood flow.104 Although Doppler ultrasonography is clinically useful in the initial evaluation of portal hypertension, the technique is not widely used to provide quantitative assessments of the degree of portal hypertension. Transient elastography may be useful in detecting portal hypertension but is not sufficiently sensitive to recommend as a modality to monitor decreases in portal pressure in patients on pharmacotherapy (see Chapter 73).105
COMPUTED TOMOGRAPHY
Computed tomography (CT) is useful for demonstrating many features of portal hypertension, including abnormal configuration of the liver, ascites, splenomegaly, and collateral vessels (Fig. 90-7). Detection of varices may be an emerging indication for CT. Diagnosis of fundal varices by multidetector row CT (MDCT) is at least as accurate as endoscopic ultrasonography (see later). CT is especially helpful in distinguishing submucosal from perigastric fundal varices106 and is considered a less invasive alternative to conventional angiographic portography in assessing portosystemic collaterals. At present, however, CT is not a recommended screening method for detecting large esophageal varices, but it may be a cost-effective method of screening for varices and preferred to endoscopy by patients.97
MAGNETIC RESONANCE IMAGING
Gadolinium-enhanced magnetic resonance imaging (MRI) is becoming recognized as a potentially useful method of detecting esophageal varices.107 In addition, MRI can be used to measure portal and azygous blood flow, which is increased in patients with portal hypertension.108 MRI provides excellent detail of the vascular structures of the liver and can detect portal venous thrombosis and spleen stiffness in patients with portal hypertension, but the role of MRI in the assessment of portal hypertension requires further study. Unlike transient elastography using ultrasound, MRI can accurately assess the stiffness of even fatty livers.109
ENDOSCOPIC ULTRASONOGRAPHY
Endoscopic ultrasound examination (endosonography) using radial or linear array echo-endoscopes or endoscopic ultrasound mini-probes passed through the working channel of a diagnostic endoscope has been applied as an investigational tool in the evaluation of patients with varices. Endoscopic ultrasonography has been used to study several aspects of esophageal varices, including the cross-sectional area of varices to identify patients at increased risk of bleeding77; size of and flow in the left gastric vein, azygous vein, and paraesophageal collaterals; changes after endoscopic therapy; and recurrence of esophageal varices following variceal ligation (see later).110 Endosonography can be combined with endoscopic measurement of transmural variceal pressure to allow estimation of variceal wall tension, which is a predictor of variceal bleeding (see earlier).111–113
CAUSES OF PORTAL HYPERTENSION
The usual classification of causes of portal hypertension is based on the site of increased resistance to portal blood flow—namely, prehepatic, intrahepatic, and posthepatic—and is outlined in Figure 90-5. Intrahepatic sites of increased resistance can be presinusoidal, sinusoidal, or postsinusoidal. Many causes of portal hypertension are associated with an increase in resistance at more than one site. For example, alcoholic cirrhosis may be associated with increased resistance at the presinusoidal, sinusoidal, and postsinusoidal levels. Therefore, classification based on the site of resistance may not be possible for all diseases that cause portal hypertension. A more useful classification is clinically based and considers common and less common causes of portal hypertension (Table 90-2).
COMMON CAUSES
Cirrhosis
Complications related to portal hypertension are the usual clinical manifestations of cirrhosis of the liver. Although all causes of cirrhosis are associated with portal hypertension, some features are disease specific. In alcoholic liver disease, elevation of the portal pressure is accurately reflected by the HVPG; moreover, portal hypertension may occur in the absence of cirrhosis but is more marked when cirrhosis is present. Perivenular lesions implicated in the pathogenesis in noncirrhotic alcoholic liver injury account for the presinusoidal component of portal hypertension in these patients.114 Autoimmune hepatitis also may be associated with portal hypertension in the absence of cirrhosis115; however, the risk of variceal bleeding is low in patients with autoimmune hepatitis. In patients with hemochromatosis, portal hypertension may be seen even before cirrhosis; the severity of portal hypertension increases with increasing fibrosis. Patients with hemochromatosis may bleed from varices despite an HVPG less than 12 mm Hg, indicating a presinusoidal component of portal hypertension. Phlebotomy therapy in patients with hemochromatosis may result in a decrease in portal hypertension.116 In patients with primary biliary cirrhosis, portal hypertension also may occur before cirrhosis has developed. The risk of variceal bleeding increases with an increase in the histologic stage of the disease.117 In earlier stages of primary biliary cirrhosis, portal hypertension is predominantly presinusoidal, but as the disease progresses, a sinusoidal component develops. Therefore, the HVPG may underestimate portal pressure in patients with primary biliary cirrhosis.80 Portal hypertension occurs in patients with primary sclerosing cholangitis and in those with biliary strictures. A long duration of biliary obstruction usually is required, although portal hypertension has been known to develop in a few months in patients with chronic bile duct obstruction caused by chronic alcoholic pancreatitis.118 Portal hypertension in patients with biliary obstruction regresses following relief of the biliary obstruction.
Schistosomiasis
Schistosomiasis may be the most common cause of portal hypertension worldwide (see Chapter 82). Bleeding from esophageal varices is a major cause of death in patients with hepatosplenic schistosomiasis. Portal hypertension results from presinusoidal obstruction caused by deposition of eggs of Schistosoma mansoni and Schistosoma japonicum in the presinusoidal portal venules. The host reaction results in granulomatous inflammation, which causes presinusoidal and periportal fibrosis.119 The fibrosis that results is sometimes called “clay pipestem” or simply “pipestem” fibrosis and usually is associated with sustained heavy infection. The periportal collagen deposition leads to progressive obstruction of portal blood flow, portal hypertension, and variceal bleeding, along with splenomegaly and hypersplenism. Lobular architecture usually is preserved. Coinfection with hepatitis B or C virus in patients with hepatic schistosomiasis can result in more rapid progression of fibrosis, hepatic failure, and an increased risk of hepatocellular carcinoma.120
Extrahepatic Portal Vein Thrombosis
Extrahepatic portal vein thrombosis is a prehepatic, presinusoidal cause of portal hypertension and a common cause of portal hypertension in children (see Chapter 83). The most common causes of portal vein thrombosis include hematologic disorders such as polycythemia vera or other myeloproliferative disorders. Other causes include a prothrombotic state, such as antithrombin, protein C, or protein S deficiency; antiphospholipid syndrome (or antiphospholipid antibody syndrome); paroxysmal nocturnal hemoglobinuria; oral contraceptive use; a neoplasm, usually intra-abdominal; an inflammatory disease, such as pancreatitis, inflammatory bowel disease, or diverticulitis; abdominal trauma; and postoperative states, especially postsplenectomy. Cirrhosis is a cause of portal vein thrombosis.121 Older studies suggested that portal vein thrombosis occurs in approximately 6% of patients with cirrhosis and in up to 25% of those with cirrhosis and hepatocellular carcinoma.122 With improved imaging, portal vein thrombosis is now known to be a more common complication of cirrhosis, and the association with hepatocellular carcinoma may not be as strong as previously thought. Isolated splenic vein thrombosis caused by a pancreatic neoplasm or pancreatitis usually is not associated with a thrombophilia. Umbilical vein sepsis may be an etiologic factor in children with portal vein thrombosis, but even in these cases, an associated prothrombotic state may predispose the patient to portal vein thrombosis.
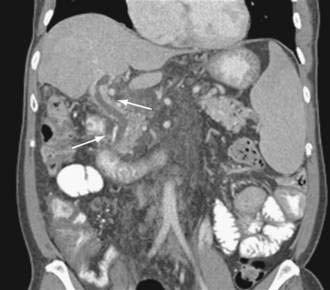
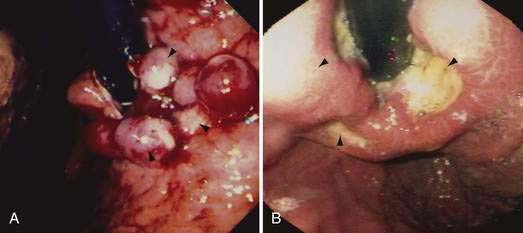
Acute and subacute portal vein thrombosis usually does not manifest with variceal bleeding.1 Chronic portal vein thrombosis is suggested by nonvisualization of the portal or splenic vein and an extensive collateral circulation. Patients may present with nonspecific symptoms or with variceal bleeding and hypersplenism. Bleeding usually is from gastroesophageal varices but may be from duodenal varices and, rarely, other ectopic sites. Gallbladder varices (Fig. 90-8) also have been described in patients with portal vein thrombosis.123
The treatment of portal vein thrombosis is symptomatic, with the aim of controlling variceal bleeding or preventing recurrent variceal bleeding. Patients in whom esophageal varices are not large, and a thrombophilia is detected, are best managed with anticoagulation because in these patients, the benefits of anticoagulation outweigh the risks.124 Local or systemic thrombolytic therapy is seldom required and is generally reserved for patients in whom a portal vein thrombus extends into the superior mesenteric vein, with danger of impending intestinal ischemia. Endoscopic therapy is used to control acute variceal bleeding and to prevent recurrent bleeding. Use of pharmacologic agents such as beta blockers to prevent variceal bleeding is probably also effective in patients with portal vein thrombosis, but this approach has not been well studied. Patients with portal vein thrombosis have lower mortality and morbidity rates from variceal bleeding than those reported in patients with cirrhosis and variceal bleeding, owing to the lack of coagulopathy and synthetic liver dysfunction. Surgical portosystemic shunt procedures are carried out in patients in whom bleeding cannot be controlled by conservative measures. If a suitable vein is not available for anastomosis, a large collateral vein may be anastomosed to a systemic vein.125 Placement of a TIPS is possible in some patients with chronic portal vein thrombosis.
Idiopathic Portal Hypertension
Idiopathic portal hypertension is uncommon in Western countries but is common in parts of Asia such as India and Japan. This disorder is diagnosed when the portal pressure is elevated in the absence of significant histologic changes in the liver or extrahepatic portal vein obstruction.126 A liver biopsy specimen from affected patients may be entirely normal,122 although increased concentrations of ET-1 have been noted in the periportal hepatocytes, portal venules, and hepatic sinusoids of patients with idiopathic portal hypertension.127 Various terms used (rather loosely) to describe idiopathic portal hypertension include hepatoportal sclerosis, noncirrhotic portal fibrosis, and Banti’s syndrome.128,129 Use of the term idiopathic portal hypertension probably is best restricted to portal hypertension in patients in whom no hepatic lesion is found on light microscopy. The term hepatoportal sclerosis suggests obliterative portal venopathy with subendothelial thickening of the intrahepatic portal veins; thrombosis and recanalization of these veins may follow. Fibrosis of the portal tracts is prominent later in the course.
The cause of idiopathic portal hypertension is unclear in a majority of patients, although chronic arsenic intoxication, exposure to vinyl chloride, and hypervitaminosis A have been implicated (see Chapter 87). These etiologic factors are present in only a minority of patients. The dominant clinical features of the condition are variceal bleeding and hypersplenism related to a markedly enlarged spleen. Liver biochemical test levels are usually normal, although the serum alkaline phosphatase level may be mildly elevated. Ascites is uncommon. The HVPG in this disorder usually is normal because the site of increased resistance is presinusoidal.130 Surgical portosystemic shunts are well tolerated in these patients, although hepatic encephalopathy may occur on long-term follow-up evaluation.122 Liver transplantation is rarely required in these patients.
Idiopathic portal hypertension may be confused with incomplete septal cirrhosis, which probably is an unrelated condition characterized by incomplete septa and liver nodularity.131 Patients with incomplete septal cirrhosis are clinically similar to patients with cirrhosis and may progress to end-stage liver disease and require liver transplantation.
LESS COMMON CAUSES
Nodular Regenerative Hyperplasia
Nodular regenerative hyperplasia is a histopathologic diagnosis characterized by atrophy of zone 3 hepatocytes and hypertrophy of zone 1 hepatocytes without significant fibrosis (see Chapters 35 and 94).132 This disorder has been recognized increasingly as a cause of portal hypertension and may even occur after liver transplantation.133 Similar histologic changes may be seen in well-established Budd-Chiari syndrome.134 The nodular hyperplasia may not be apparent on histologic examination unless a reticulin stain is carried out to demonstrate the micronodules. These regenerative nodules are believed to result from an imbalance between hyperperfused areas of the liver, with resulting regenerative nodules, and poorly perfused areas, with resulting atrophy. Nodular regenerative hyperplasia is associated with a variety of conditions, predominantly hematologic and rheumatologic in nature. Liver biochemical abnormalities include mild elevation of the serum aminotransferase levels. Portal hypertension manifesting as variceal bleeding is the predominant clinical presentation. Ascites also may develop in these patients, suggesting that an increase in sinusoidal pressure occurs.135 Hepatocellular carcinoma does not occur, but liver transplantation may be required in some patients.
Partial Nodular Transformation of the Liver
Partial nodular transformation of the liver is an uncommon lesion that is characterized by large nodules in the perihilar region.136 These nodules may be visible on imaging studies of the liver. The rest of the liver may be normal or may show changes of nodular regenerative hyperplasia. Liver biochemical test levels usually are normal. Like nodular regenerative hyperplasia, partial nodular transformation of the liver is believed to be related to an imbalance in portal perfusion of the liver, but the abnormality is restricted to the hilar branches, whereas in nodular regenerative hyperplasia the abnormality is more diffuse. Variceal bleeding is the predominant presentation in partial nodular transformation of the liver, although patients with large nodules may experience abdominal pain. Hepatocellular carcinoma may rarely develop in these regenerating nodules. Treatment with a surgical portosystemic shunt is associated with good long-term results.
Fibropolycystic Liver Disease
Fibropolycystic liver disease is a term which encompasses Caroli’s disease, Caroli’s complex (Caroli’s disease with congenital hepatic fibrosis), congenital hepatic fibrosis, and polycystic liver disease. Congenital hepatic fibrosis usually occurs in association with Caroli’s disease of the liver, polycystic disease of the kidney, and medullary sponge kidney (see Chapter 62). The major manifestation of congenital hepatic fibrosis is variceal bleeding.137 A portosystemic shunt may be placed in these patients to treat refractory variceal bleeding, with a low long-term risk of hepatic encephalopathy. Patients with polycystic liver disease, whether associated with polycystic kidney disease or not, rarely present with portal hypertension (see Chapter 94).138 Portal hypertension may decrease after treatment of the cysts.
Sarcoidosis
Portal hypertension is an uncommon manifestation of hepatic sarcoidosis (see Chapter 35).139 The site of increased intrahepatic resistance in patients with sarcoidosis seems to be postsinusoidal, in view of the elevated HVPG. In early disease, however, the resistance is predominantly at a presinusoidal level. Treatment with glucocorticoids may decrease portal hypertension in some patients with hepatic sarcoidosis.
Malignancy
Portal hypertension has been associated with leukemias, lymphomas, and systemic mastocytosis (see Chapters 34 and 35).140 Portal hypertension also may occur in patients with hepatocellular carcinoma independent of the presence of cirrhosis (see Chapter 94). The pathogenesis of portal hypertension in patients with hepatocellular carcinoma is thought to be multifactorial; contributing factors include portal vein thrombosis, pressure by the tumor on the portal vein, and, in some cases, a hepatic artery–portal vein fistula. Esophageal varices may be seen in patients with hepatic metastases, although variceal bleeding is unusual.141
Splanchnic Arteriovenous Fistula
Splanchnic arteriovenous fistula should be suspected when the onset of ascites and variceal bleeding is acute, especially in the presence of an abdominal bruit. When a splanchnic artery ruptures into a mesenteric vein, the portal pressure increases acutely, reaching levels of systemic arterial pressure.142 The result is acute portal hypertension with development of ascites and variceal bleeding. A bruit may be heard in the left upper quadrant of the abdomen with a splenic arteriovenous fistula and in the right upper quadrant with a hepatic artery–portal vein fistula. With a long-standing fistula, secondary hepatic changes of perisinusoidal fibrosis related to an increase in portal venous inflow may be present. In the early stages, embolization or ligation of the fistula will ameliorate the portal hypertension. In late stages, however, portal fibrosis may be advanced, and the portal hypertension may not correct completely with embolization of the fistula.
Hereditary Hemorrhagic Telangiectasia
Hereditary hemorrhagic telangiectasia (HHT), or Osler-Weber-Rendu disease, is an unusual cause of portal hypertension (see also Chapters 19 and 36). Diagnostic criteria include mucocutaneous telangiectasias, epistaxis, arteriovenous fistulas of the viscera (usually lung or liver), and a family history of the disorder. Manifestations of HHT depend on the site of fistula formation. A fistula between the hepatic artery and hepatic vein manifests predominantly as biliary disease, mainly biliary strictures and cholangitis, and high-output cardiac failure. A fistula between the hepatic artery and portal vein results in portal hypertension and biliary strictures, whereas a fistula between the portal vein and hepatic vein, which is rare, results in hepatic encephalopathy.143 Nodular regenerative hyperplasia, which develops in some patients with HHT, may worsen portal hypertension.144 Although symptomatic liver disease in HHT is rare, involvement of the liver is found in a majority of patients.145
CLINICAL ASSESSMENT OF PATIENTS WITH PORTAL HYPERTENSION-RELATED BLEEDING
Portal hypertension should be suspected in all patients with gastrointestinal bleeding and peripheral stigmata of liver disease—namely, jaundice, spider angiomata, palmar erythema, Dupuytren’s contractures, parotid enlargement, testicular atrophy, loss of secondary sexual characteristics, ascites, and encephalopathy. Splenomegaly is an important clue to the presence of portal hypertension, and the presence of ascites makes the presence of esophageal varices even more likely. Caput medusae, suggestive of an intrahepatic cause of portal hypertension, is present around the umbilicus; the flow of blood is away from the umbilicus. In Budd-Chiari syndrome, by contrast, veins are dilated in the flanks and back, and blood flows in a cephalic direction.85 A bruit may be heard in the left or the right upper quadrant in a patient with a splanchnic arteriovenous fistula. A venous hum may be heard in the epigastrium of a patient with portal hypertension and represents collateral flow in the falciform ligament.
TREATMENT OF PORTAL HYPERTENSION-RELATED BLEEDING
PHARMACOLOGIC THERAPY
The pharmacologic agents used in the treatment of portal hypertension are divided into two groups: those that decrease splanchnic blood flow and those that decrease intrahepatic vascular resistance (Table 90-3). The agents that decrease splanchnic blood flow acutely are vasopressin and its analogs and somatostatin and its analogs. β-adrenergic blocking agents (beta blockers) also decrease portal blood flow but are used only to prevent variceal bleeding and rebleeding. Agents that target intrahepatic vascular resistance include α-adrenergic blocking agents, angiotensin receptor blocking agents, and nitrates, but only nitrates are now considered for clinical use. Diuretics, by decreasing plasma volume, may reduce portal pressure but are not recommended as sole agents for the treatment of portal hypertension. Metoclopramide and other gastric prokinetic agents may decrease intravariceal pressure by contracting the lower esophageal sphincter, but these agents have not been evaluated in clinical trials and are not recommended.
Table 90-3 Drugs Used in the Treatment of Portal Hypertension
Somatostatin and Its Analogs
Somatostatin is a 14-amino-acid peptide. Five somatostatin receptors—SRTR 1 to SRTR 5—are recognized, but the actual distribution of the receptors in humans is not clear. Following intravenous injection, somatostatin has a half-life in the circulation of one to three minutes; therefore, longer-acting analogs of somatostatin have been synthesized. The best known of these analogs are octreotide, lanreotide, and vapreotide.146 Somatostatin decreases portal pressure and collateral blood flow by inhibiting release of glucagon.147 The optimal dose and duration of use of somatostatin have not been adequately studied. Following a single 250-µg bolus injection of somatostatin, portal and azygous blood flow decrease, but the effect lasts only a few minutes.148 Use of higher doses is associated with a more impressive decrease in HVPG. Somatostatin also decreases portal hypertension by decreasing postprandial splanchnic blood flow.149 Following a variceal bleed, blood in the gastrointestinal tract acts like a meal, leading to an increase in portal flow and elevation in the portal pressure; this elevation in pressure is ameliorated by the use of somatostatin.
Following intravenous administration, octreotide has a half-life in the circulation of 80 to 120 minutes. Its effect on reducing portal pressure is not prolonged, however. Moreover, continuous infusion of octreotide does not decrease portal pressure despite decreasing the postprandial increase in portal pressure.62,150
Available evidence is insufficient to prove the superiority of somatostatin and its analogs to placebo in the control of acute variceal bleeding.134 Some randomized controlled trials, however, support the view that somatostatin or octreotide may be equivalent in efficacy to sclerotherapy or terlipressin for controlling acute variceal bleeding. Also, early administration of vapreotide may be associated with improved control of bleeding but without a significant reduction in mortality rate.151 In clinical practice, treatment with somatostatin or octreotide is combined with endoscopic management of variceal bleeding (see later).
β-Adrenergic Blocking Agents
Nonselective β-adrenergic blocking agents have been used extensively since the landmark study of Lebrec and colleagues demonstrated the efficacy of these agents in preventing variceal rebleeding.152 Nonselective beta blockers such as propranolol or nadolol are preferred. Blockade of β1-adrenergic receptors in the heart decreases cardiac output. Blockade of β2-adrenergic receptors, which cause vasodilatation in the mesenteric circulation, allows unopposed action of α1-adrenergic receptors and results in decreased portal flow. The combination of decreased cardiac output and decreased portal flow leads to a decrease in portal pressure. Nadolol has advantages over propranolol in that it is excreted predominantly by the kidney, has low lipid solubility, and is associated with a lower risk of central nervous system side effects such as depression. The effectiveness of beta blockers is assessed most accurately by monitoring the HVPG; this approach is not widely used in clinical practice. The usual method of monitoring the efficacy of beta blockers is to observe a decrease in the heart rate, which is a measure of β1-adrenergic receptor blockade. Despite adequate β1-adrenergic receptor blockade, however, some patients might benefit from a further increase in the dose of beta blocker, to increase the degree of β2-adrenergic blockade. Raising the dose, however, results in more side effects and the likelihood that treatment will need to be withdrawn.153
Drugs That Decrease Intrahepatic Vascular Resistance
The ideal agent for treatment of portal hypertension would be a drug that decreases intrahepatic vascular resistance. Unfortunately, such a drug is not currently available. A desirable drug would be one that selectively decreases intrahepatic vascular resistance without worsening systemic vasodilatation. Agents that may decrease intrahepatic resistance include α1-adrenergic blockers such as prazosin,154 but long-term administration of prazosin causes worsening of the systemic hyperdynamic circulation associated with portal hypertension and consequent sodium retention and ascites.154 The addition of propranolol to prazosin may ameliorate the adverse affects of prazosin on the systemic circulation. Losartan, an angiotensin II receptor type I antagonist, causes a reduction in portal pressure without significant effects on the systemic circulation.155 In randomized, controlled trials of losartan or another angiotensin II receptor antagonist, irbesartan, however, portal pressure was not reduced significantly. In fact, renal function has worsened in patients given losartan or irbesartan.156,157 ET-receptor blockers and liver-selective NO donors are promising investigational agents for therapies that target intrahepatic vascular resistance.16 Studies suggest that simvastatin may decrease intrahepatic resistance and maintain hepatic blood flow while decreasing portal pressure. This effect probably results from simvastatin-mediated NO release.158
ENDOSCOPIC THERAPY
Sclerotherapy
Endoscopic sclerotherapy has largely been supplanted by endoscopic band ligation, except when poor visualization precludes effective band ligation of bleeding varices. Available evidence does not support emergency sclerotherapy as first-line treatment of variceal bleeding (Table 90-4).159 The technique involves injection of a sclerosant into (intravariceal) or adjacent to (paravariceal) a varix. Some paravariceal injection usually takes place during attempted intravariceal therapy. The sclerosants used include sodium tetradecyl sulfate, sodium morrhuate, ethanolamine oleate, and absolute alcohol; the choice of a sclerosant is based on availability, rather than on superior efficacy of one agent over another.
Variceal Ligation
Endoscopic variceal ligation is the preferred endoscopic modality for control of acute esophageal variceal bleeding and prevention of rebleeding; however, the utility of band ligation in the treatment of gastric varices is limited. Variceal ligation is simpler to perform than injection sclerotherapy. The procedure involves suctioning of the varix into the channel of an endoscope and deploying a band around the varix. The band strangulates the varix, thereby causing thrombosis. Multi-band devices can be used to apply several bands without requiring withdrawal and reinsertion of the endoscope. Varices at the gastroesophageal junction are banded initially, and then more proximal varices are banded in a spiral manner at intervals of approximately 2 cm; the endoscope is then withdrawn. Varices in the mid- or proximal esophagus do not need to be banded. Endoscopic variceal ligation is associated with fewer complications than sclerotherapy and requires fewer sessions to achieve variceal obliteration. Moreover, esophageal variceal ligation during an acute bleed is not associated with a sustained elevation in HVPG, as occurs with sclerotherapy.160
Endoscopic variceal ligation can cause local complications, including esophageal ulcers (Fig. 90-9), strictures, and dysmotility, albeit less frequently than does sclerotherapy. Banding-induced ulcers can be large and potentially serious if gastric fundal varices are banded. Now that overtubes are no longer used to facilitate repeated insertion of the endoscope during a banding session, the mechanical complications seen in the past (mucosal tears and esophageal perforations) are uncommon. A proton pump inhibitor is usually recommended after variceal ligation even though data to support the use of a proton pump inhibitor are limited.
TRANSJUGULAR INTRAHEPATIC PORTOSYSTEMIC SHUNT
A transjugular intrahepatic portosystemic shunt (TIPS)—also referred to as a transjugular intrahepatic portosystemic stent shunt (TIPSS)—reduces elevated portal pressure by creating a communication between the hepatic vein and an intrahepatic branch of the portal vein. A percutaneous transjugular approach is used to insert the shunt. A TIPS functions as a side-to-side portacaval shunt and has been used to treat complications of portal hypertension, mainly variceal bleeding and refractory ascites, as well as Budd-Chiari syndrome, hepatic hydrothorax, and hepatorenal syndrome (see Chapters 83, 91, and 92). A TIPS can be placed by an interventional radiologist, with a mortality rate of less than 1% to 2%. TIPS placement usually is carried out with the patient under sedation. A platelet count greater than 60,000/mm3 and an acceptable prothrombin time as reflected by an international normalized ratio (INR) less than 1.4 usually are recommended but are not essential in an emergency. Broad-spectrum antibiotic coverage is recommended when TIPS placement is carried out in a patient with primary sclerosing cholangitis and as an emergency procedure.
For this procedure, the hepatic vein is cannulated through a transjugular approach, and using a Rosch needle, the portal vein is cannulated. A guidewire is then passed to connect the hepatic vein and a branch of the portal vein. Following dilation of the tract, a stent is placed and dilated as required to reduce the portacaval pressure gradient (the pressure difference between the portal vein and the inferior vena cava at the confluence of the hepatic vein) to below 12 mm Hg (Fig. 90-10). Whether a lesser reduction of portacaval pressure gradient, to only 15 mm Hg or so (instead of 12 mm Hg), could be associated with reduced bleeding or a lower frequency of hepatic encephalopathy requires further study.
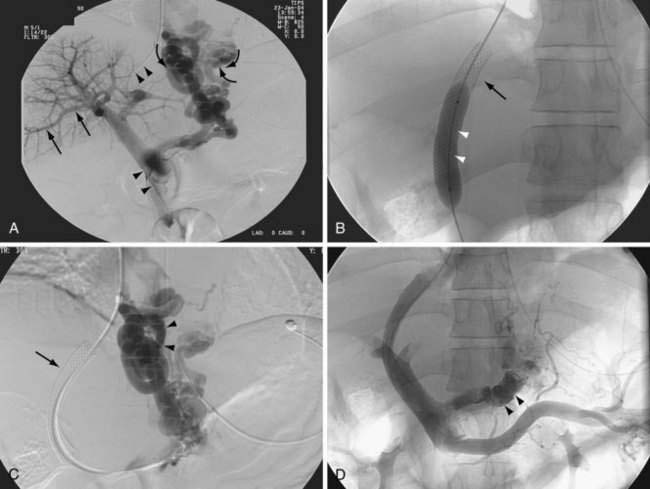
In the past, the stents most commonly used for a TIPS were the Wallstent and the Palmaz stent. Nowadays, a coated stent is used (Viatorr, Gore, Flagstaff, Arizona). This stent has an uncoated portion that anchors the stent to the portal vein and a polytetrafluoroethylene-coated portion that lines the tract in the liver parenchyma and the draining hepatic vein. The frequency of shunt stenosis is reduced when coated stents are used instead of uncoated stents.161
A TIPS can be placed successfully by an experienced operator in greater than 95% of cases. Complications following the procedure are classified as procedure related, early (occurring before 30 days), or late (after 30 days) (Table 90-5). The prevention and treatment of procedure-related, early, and late post-TIPS complications are outlined in Table 90-6.
Table 90-5 Complications of Transjugular Intrahepatic Portosystemic Shunt Placement
| TIMING OF COMPLICATION | COMPLICATION |
|---|---|
| Procedure-related (life-threatening) | Cardiopulmonary failure |
| Carotid artery puncture | |
| Intraperitoneal hemorrhage | |
| Sepsis | |
| Early post-procedure (1-30 days) | Cardiac arrhythmias |
| Fever | |
| Hematoma at puncture site | |
| Hemolytic anemia | |
| Hepatic encephalopathy | |
| Pain | |
| Progressive hepatic failure | |
| Pulmonary artery hypertension | |
| Shunt thrombosis | |
| Stent migration | |
| Reactions to contrast media | |
| Late post-procedure (>30 days) | Hepatic encephalopathy |
| Liver failure | |
| Portal vein thrombosis | |
| Progressive hepatic failure | |
| Shunt stenosis |
Modified from Kamath PS, McKusick M. Transjugular portosystemic shunt (TIPS). Bailliere Clin Gastroenterol 1997; 11:327-49.
Table 90-6 Prevention and Treatment of Transjugular Intrahepatic Portosystemic Shunt-Related Complications
| COMPLICATION | PREVENTION | TREATMENT |
|---|---|---|
| Inadvertent injury to carotid artery during jugular vein access | Perform with ultrasound guidance to facilitate venous access | Manual compression of carotid puncture site to prevent hematoma |
| Hepatic capsular laceration during portal vein access | Avoid atrophic lobes and limit needle passes to 3-4 cm of excursion | Usually requires no treatment |
| For severe hemorrhage, transfuse with blood products until stable; obtain abdominal CT and surgical consultation | ||
| Extrahepatic puncture of portal venous system | Delineate bifurcation of portal vein on preprocedure CT | Leave catheter in place for portogram; use as a guide for intrahepatic portal vein puncture |
| Work quickly to establish a functioning shunt, then remove the errant catheter | ||
| Intrahepatic arterial or biliary puncture | Work centrally within the liver | Usually no treatment is required; remove the catheter and continue |
| If a fistula develops, embolize the arterial feeder with steel coils | ||
| Sepsis after shunt placement | Give prophylactic antibiotics | Broad-spectrum antibiotic coverage |
| Adhere to strict sterile technique | ||
| Early shunt thrombosis | Avoid sharp angles when placing the stent Ends should not abut against the intima of the vein |
Shunt venogram and clot lysis using tPA delivered by pulse-spray techniqueExtend the shunt to ensure stent coverage of the intrahepatic tract and to ensure adequate length in hepatic and portal veins |
| Uncontrollable encephalopathy after shunt placement | Use narrow shunts in high-risk patients | Reduce the diameter of the shunt with additional concentrically placed stents |
| Embolize the shunt with steel coils | ||
| Shunt stenosis | Use wider or covered stents | Dilation or atherectomy of the shunt |
| Avoid bile duct injury | Place an additional stent if necessary | |
| Post-shunt liver failure | Avoid procedure in patients with a MELD score ≥24 | Consider early liver transplantation |
CT, computed tomography; MELD, Model for End-stage Liver Disease; tPA, tissue plasminogen activator.
Modified from Kamath PS, McKusick M. Transjugular portosystemic shunt (TIPS). Bailliere Clin Gastroenterol 1997; 11:327-49.
Portal Hypertension-Related Bleeding
TIPS has been effective in the management of uncontrolled esophageal variceal bleeding in patients with decompensated cirrhosis of the liver.162 Hemorrhage is controlled in more than 90% of patients, but the mortality rate in such patients is high—greater than 60% within 90 days. A similar outcome is observed in patients who undergo TIPS placement for refractory gastric variceal bleeding.163
In a meta-analysis of twelve randomized controlled trials that compared TIPS with endoscopic therapy, the rate of rebleeding was lower with TIPS, but the frequency of encephalopathy was higher, and no effect on survival was observed.164 Therefore, TIPS cannot be recommended as a first choice of treatment for preventing variceal rebleeding; rather, it is reserved for patients who have failed endoscopic or pharmacologic therapy.
Selection of Patients
The presence of a TIPS may worsen liver function by depriving the liver of portal venous blood, thereby increasing the risk of hepatic encephalopathy, with decreased survival in some patients. Therefore, the procedure should be used selectively. Emergency TIPS is clearly associated with a high mortality rate.165,166 In patients in whom TIPS placement has been carried out to prevent variceal rebleeding, 30-day mortality rates may be as high as 44%. Factors associated with a poor prognosis include a serum alanine aminotransferase (ALT) level greater than 100 U/L, serum bilirubin level greater than 3 mg/dL, and pre-TIPS hepatic encephalopathy unrelated to bleeding.166 Patients with a high CTP score (Table 90-7) also have reduced survival. The Child classification has some limitations, however; for example, it does not discriminate survival well among patients within each Child class. Furthermore, some parameters that make up the CTP score, such as ascites and encephalopathy, are assessed by subjective interpretation. The need for a more accurate method to assess survival in patients undergoing TIPS has led to creation of a new tool to predict survival, the Model for End-stage Liver Disease (MELD) (see http://www.mayoclinic.org/gi-rst/mayomodel6.html and Chapter 73).165 With data from four centers within the United States, this mathematical model originally was composed of the serum creatinine level, INR as a measure of the prothrombin time, serum bilirubin level, and etiology of liver disease. Subsequently, the MELD formula was modified to include only the first three parameters (creatinine, INR, and bilirubin).167 The MELD has been widely validated for predicting survival in patients with cirrhosis, including patients who have undergone TIPS placement, and is more accurate for this purpose than the Child classification.
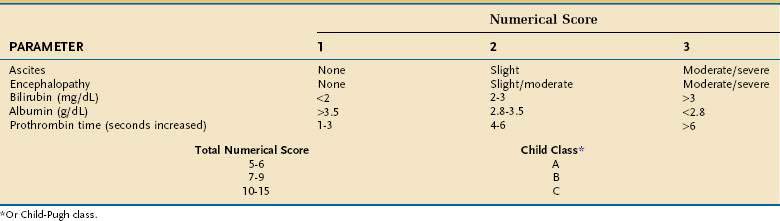
Patients with a MELD score of 14 or less have an excellent survival rate after TIPS placement; therefore, TIPS may be carried out routinely in such patients when indicated. Patients with a MELD score higher than 24 have reduced survival following TIPS placement, with a mortality rate approaching 30% at three months. Because these patients are at high priority for liver transplantation, TIPS should be avoided unless needed to control acute variceal bleeding. In the intermediate group with MELD scores ranging from 15 to 24, TIPS placement can be carried out depending on the patient’s preference and the physician’s judgment and taking into consideration the likelihood of liver transplantation. This approach has been validated independently.168
SURGICAL THERAPY
Liver transplantation should be considered in all patients with variceal bleeding who meet minimal listing criteria for liver transplantation (currently, a CTP score of 7 or greater). Selection and prioritization of patients for liver transplantation are discussed in Chapter 95.
Non-shunt Procedures
Esophageal Transection
Esophageal transection, in which the esophagus is stapled and transected, is highly effective in controlling variceal bleeding and is associated with a lower risk of encephalopathy than that for portosystemic shunts. Esophageal transection was considered in the past when two sessions of endoscopic therapy had failed to control variceal bleeding within a 24-hour period.79 Mortality rates are not improved over those observed with endoscopic sclerotherapy, however. With the advent of TIPS, esophageal staple transection is now seldom used.
Devascularization Procedures
Devascularization procedures typically have been used to prevent recurrent variceal bleeding in patients with extensive splenic and portal vein thrombosis when a suitable vein is not available for creation of a portosystemic shunt.169 In the original operation described by Sugiura and Futagawa, both a thoracotomy and a laparotomy were carried out.170 Subsequently, the operation has been carried out through an abdominal approach and combined with a splenectomy. The procedure consists of total devascularization of the greater curvature of the stomach combined with devascularization of the upper two thirds of the lesser curvature of the stomach and circumferential devascularization of the lower 7.5 cm of the esophagus. The rate of recurrent bleeding following this procedure is variable but may be as high as 40%, depending on the population being treated and duration of follow-up.
Portosystemic Shunts
Selective Shunts
The most widely used selective shunt is the distal splenorenal shunt, originally described by Warren and colleagues.171 With this shunt, only varices at the gastroesophageal junction and spleen are decompressed, and portal hypertension is maintained in the superior mesenteric vein and portal vein; therefore, variceal bleeding is controlled, but ascites persists. The shunt procedure involves a portal-azygous disconnection and subsequent anastomosis between the splenic vein and left renal vein in an end-to-side fashion (Fig. 90-11). The entire length of the pancreas must be mobilized, and the left adrenal vein must be ligated. The distal splenorenal shunt has been associated with control of variceal bleeding in approximately 90% of patients and a lower rate of hepatic encephalopathy than that reported for total shunts.172
Partial Portosystemic Shunts
A partial portosystemic shunt is carried out using a synthetic interposition graft between the portal vein and the inferior vena cava. When the shunt diameter is 8 mm, portal pressure is reduced below 12 mm Hg, and antegrade flow to the liver is maintained in most patients.173 Rates of preventing variceal rebleeding and encephalopathy following the shunt are similar to those seen with a distal splenorenal shunt. As in patients who have had a distal splenorenal shunt, ascites may occur in approximately 20% of patients who have had a partial portosystemic shunt because hepatic sinusoidal pressure is not reduced.174,175
Portacaval Shunts
End-to-side and side-to-side portacaval shunts have been described, but nowadays only the side-to-side portacaval shunt is in use.176 Any portacaval shunt that is greater than 12 mm in diameter is likely to result in a total shunting of portal blood. A shunt with a diameter <12 mm is created with an interposition graft, or alternatively a direct vein-to-vein anastomosis may be constructed. Variceal bleeding, as well as ascites, is well controlled because the hepatic sinusoids are decompressed. Variceal rebleeding following a total shunt is seen in less than 10% of patients, but hepatic encephalopathy occurs in 30% to 40% of patients.99 Liver transplantation in patients who have had a portacaval shunt is associated with increased operative morbidity and intraoperative transfusion requirements. The outcome of liver transplantation is not otherwise significantly different, however, from that for patients who have not had a portacaval shunt. Nonetheless, surgical portacaval shunts should be avoided in patients who are potential candidates for liver transplantation.
MANAGEMENT OF SPECIFIC LESIONS
ESOPHAGEAL VARICES
Natural History
Esophageal varices are present in approximately 40% of patients with cirrhosis and in as many as 60% of patients with cirrhosis and ascites.99 In cirrhotic patients who do not have esophageal varices at initial endoscopy, new varices will develop at a rate of approximately 5% per year. In patients with small varices at initial endoscopy, progression to large varices occurs at a rate of 10% to 15% per year and is related predominantly to the degree of liver dysfunction.177 On the other hand, improvement in liver function in patients with alcoholic liver disease who abstain from alcohol is associated with a decreased risk, and sometimes even disappearance, of varices.178
Up to 25% of patients with newly diagnosed varices will bleed within two years.177 The best clinical predictor of bleeding appears to be variceal size. The risk of bleeding in patients with varices less than 5 mm in diameter is 7% by two years, and the risk in patients with varices greater than 5 mm in diameter is 30% by two years.177 Even more important, however, is the HVPG because the risk of bleeding is virtually absent when the HVPG is below 12 mm Hg.87 Nevertheless, measurement of HVPG is not routinely performed in clinical practice to assess bleeding risk.
The prognosis for variceal bleeding in patients with cirrhosis has improved since the 1980s. Initial treatment is associated with cessation of bleeding in approximately 90% of patients.177,179 Approximately one half of patients with a variceal bleed stop bleeding spontaneously because hypovolemia leads to splanchnic vasoconstriction, which results in a decrease in portal pressure. Excessive transfusions may, in fact, increase the chance of rebleeding.180 Active bleeding at endoscopy, a lower initial hematocrit value, higher serum aminotransferase levels, higher Child class, bacterial infection, an HVPG greater than 20 mm Hg, and portal vein thrombosis are associated with failure to control bleeding at five days.179,181–183 Of patients who have stopped bleeding, approximately one third will rebleed within the next six weeks. Of all rebleeding episodes, approximately 40% will take place within five days of the initial bleed.184 Predictors of rebleeding include active bleeding at emergency endoscopy, bleeding from gastric varices, hypoalbuminemia, renal insufficiency, and an HVPG greater than 20 mm Hg.177 The risk of death with acute variceal bleeding is 5% to 8% at one week and about 20% at six weeks.177 Patients who rebleed early, have a MELD score >18, require >4 units of packed red blood cell transfusions,185 and in whom renal failure develops have the highest risk of death. Alcohol as the cause of cirrhosis, a higher serum bilirubin level, a lower serum albumin level, hepatic encephalopathy, and hepatocellular carcinoma are additional factors associated with an increased six-week mortality rate.
Prevention of Bleeding
The utility of pre-primary prophylaxis, that is, the efficacy of beta blockers to prevent the formation of varices, has not been demonstrated.79,186 Patients with Class C cirrhosis who have small varices may be considered for treatment with a beta blocker. All patients with large varices (diameter greater than 5 mm) should be considered for prophylactic therapy (“primary prophylaxis”) to prevent variceal bleeding. The presence of additional endoscopic signs such as red wales does not influence the decision regarding prophylactic therapy. Twelve trials have addressed the use of a nonselective beta blocker for primary prophylaxis of variceal bleeding and have demonstrated a decrease in the risk of variceal bleeding from 25% in patients in the control group to 15% in patients taking a beta blocker. The absolute risk reduction is thus approximately 10%, and the number needed to treat to prevent one variceal bleed is approximately 10 patients. The mortality rate is reduced from 28.4% in control patients to 23.9% in patients taking a beta blocker; the absolute risk reduction is 4.5%. The number of patients needed to be treated to prevent one death is approximately 22. In patients who do not bleed during therapy and who do not experience side effects, treatment should be continued indefinitely because withdrawal of a beta blocker can result in an increased risk of bleeding.187,188
The side effects of beta-blocker treatment are probably overemphasized because only approximately 15% of patients need to discontinue the drug.189 A baseline heart rate and blood pressure recording will help determine whether a patient is a candidate for pharmacologic treatment with a beta blocker. A resting heart rate of less than 55 to 60 beats per minute or a systolic blood pressure less than 90 mm Hg indicates that the patient is likely to be intolerant of beta blockers. In other patients, the HVPG ideally should be measured at baseline (Fig. 90-12). A long-acting preparation of propranolol or nadolol may be started; the usual starting dose of long-acting propranolol is 60 mg once daily and that of nadolol is 20 mg once daily. Because the risk of bleeding is greatest at night, the beta blocker should probably be administered in the evening.108 The dose of propranolol or nadolol can be increased gradually every three to five days until the target heart rate of 25% below baseline or 55 to 60 beats per minute or the maximum tolerated dose is reached, provided that the systolic blood pressure remains above 90 mm Hg. The daily dose of long-acting propranolol or nadolol required to reach the target heart rate ranges from 40 to 160 mg. Patients with a decrease in systolic blood pressure below 90 mm Hg are most likely to experience side effects.
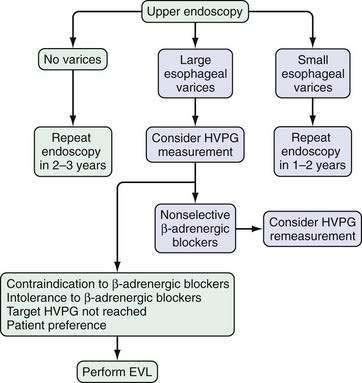
In patients on pharmacologic therapy, follow-up endoscopy is unnecessary unless gastrointestinal bleeding occurs. When the target heart rate is reached, however, a repeat HVPG measurement may be carried out as close to one month as possible. In patients in whom the HVPG has decreased to less than 12 mm Hg, the risk of bleeding is virtually eliminated. Patients in whom the HVPG decreases by at least 20% have a risk of variceal bleeding of less than 10%. Unfortunately, only 30% to 40% of patients respond to a beta blocker; those with better liver function show the best response.189 In patients who are intolerant of or who have contraindications to beta blockers, isosorbide mononitrate has been tried but is no better than placebo in preventing variceal bleeding.190 In these patients, endoscopic prophylaxis should be pursued. Unfortunately, patients who do not achieve a decrease in the HVPG to less than 12 mm Hg, or of greater than 20%, on a beta blocker may not respond well to endoscopic variceal ligation either.191
Endoscopic Prevention
Prophylactic sclerotherapy for the prevention of variceal bleeding has been studied extensively but cannot be recommended.192 The preferred method of endoscopic treatment is variceal band ligation. Variceal band ligation has been compared with no treatment in five trials. A meta-analysis of these studies demonstrated that, compared with no treatment, endoscopic variceal ligation decreases the risk of first bleeding and the mortality rate.193 Meta-analysis of the nine trials that compared endoscopic variceal ligation with a beta blocker demonstrated a lower bleeding risk with endoscopic variceal ligation, with no difference in mortality rates.194 A subsequent study has suggested that nonbleeding-related mortality may actually be reduced by beta blockers.195 Side effects with beta blockers are more frequent than with variceal ligation, but complications of variceal ligation can be potentially life threatening. Therefore, current evidence does not support endoscopic variceal ligation as the preferred method of primary prophylaxis against variceal bleeding. The risks and benefits of the alternative therapies should be discussed and treatment individualized. Beta blockers are cheaper and more convenient to use and may potentially reduce the risk of bleeding from gastric varices and portal hypertensive gastropathy. Band ligation is the only option for patients with high-risk varices who have contraindications to beta blockers or who have not responded to or are intolerant of beta blockers. Combined use of propranolol and endoscopic variceal ligation to prevent primary prophylaxis is not currently recommended.
Control of Acute Bleeding
Acute esophageal variceal bleeding constitutes a life-threatening emergency and requires management by a well-trained team of hepatologists, endoscopists, intensive care personnel, radiologists, and surgeons. Treatment is aimed at resuscitating the patient, controlling the bleeding, and preventing complications (see Chapter 19). Two large-bore intravenous access lines should be inserted immediately. Red blood cells should be transfused with the goal of maintaining the hematocrit value around 25%. Normal saline may be infused intravenously until packed red blood cells are available for transfusion. In patients with active bleeding, the airway needs to be protected, and endotracheal intubation is advised. Antibiotics should be administered to all patients to prevent bacteremia and spontaneous bacterial peritonitis. Norfloxacin, 400 mg orally twice daily for seven days, is the preferred choice.196 When oral intake is not possible, intravenous ciprofloxacin, 400 mg every 12 hours; levofloxacin, 500 mg every 24 hours; or ceftriaxone, 1 g every 24 hours for 7 days is recommended. The addition of recombinant factor VIIa to standard therapy has not been shown to improve control of bleeding.197
A combination of endoscopic therapy and pharmacologic therapy of variceal bleeding may be superior to pharmacologic treatment alone. Pharmacologic agents should be started as early as possible; in some centers, they are started while the patient is being transferred by ambulance to the hospital. Somatostatin, octreotide, terlipressin, and vasopressin plus nitroglycerin are the options for pharmacologic therapy. The specific agent chosen depends on availability and physician preference. In the United States, octreotide is the agent used most commonly. Terlipressin is the first choice in many other countries because it is the only drug that has been associated with improved survival.198 Pharmacologic treatment should be continued for up to five days to prevent early rebleeding.
At upper endoscopy, the actively bleeding varix is ligated (Fig. 90-13). Ligation initially should be at or immediately below the bleeding site. Other large varices also should be banded during the same session. If active bleeding is not seen, ligation should be carried out beginning with varices at the gastroesophageal junction and proceeding proximally at intervals of 2 cm in a spiral fashion. If bleeding obscures the varices, then multiple bands are placed at the gastroesophageal junction circumferentially until bleeding can be controlled, but the long-term risks of esophageal stricture are increased in such patients. Bleeding can be controlled in up to 90% of patients with a combination of pharmacologic and endoscopic treatment.
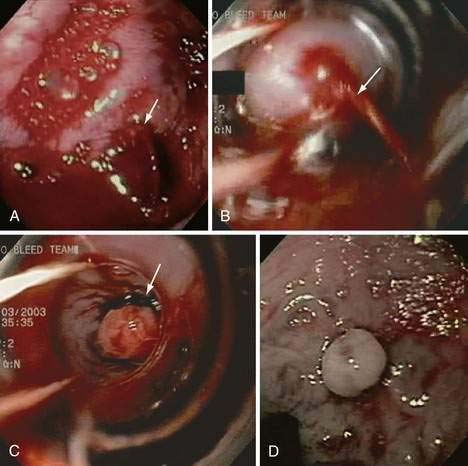
Bleeding cannot be controlled in approximately 10% of patients, as defined by any of the following three factors: (1) transfusion of four units of red blood cells or more to maintain the hematocrit value between 25% and 30%; (2) inability to increase the systolic blood pressure by 20 mm Hg or to greater than 70 mm Hg; or (3) persistence of a heart rate greater than 100 beats per minute.199 Rebleeding is defined as recurrence of bleeding after initial control for 24 hours during which the vital signs and hemoglobin level are stable. When two sessions of endoscopic treatment within a 24-hour period have failed to control variceal bleeding, salvage therapies such as TIPS should be carried out (Fig. 90-14), although the mortality rate in this group of patients is high. Surgical portosystemic shunts, although extremely effective in controlling variceal bleeding, have largely been abandoned because of high mortality rates. Balloon tamponade sometimes is used to stabilize the patient until definitive treatment can be carried out.
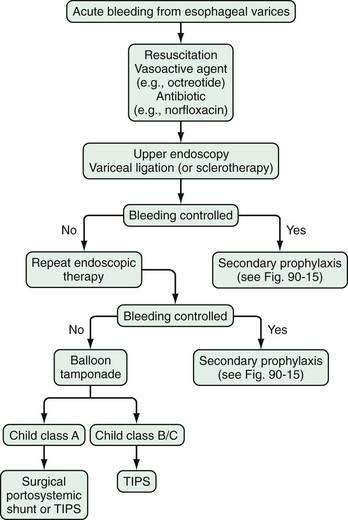
Prevention of Rebleeding
Endoscopic variceal ligation alone may be performed to prevent variceal rebleeding in patients who have poor liver function (Fig. 90-15). In practice, the first endoscopic session is carried out 7 to 14 days after the initial variceal ligation to control bleeding. Endoscopic therapy is then repeated at three- to four-week intervals; this approach has been suggested because bands might still be in place if endoscopy is repeated earlier. If the HVPG is monitored, a reduction in HVPG to less than 12 mm Hg or by greater than 20% should obviate the need for variceal ligation. For patients who bleed during pharmacologic treatment, variceal ligation should be carried out. Conversely, for patients who have undergone variceal ligation alone and experience recurrent bleeding, a beta blocker should be started. Patients who have a variceal rebleed despite receiving optimal pharmacologic and endoscopic treatment require a portosystemic shunt. Even in patients with Child class A cirrhosis, a TIPS may be as effective as a distal splenorenal shunt, and the choice of therapy should depend on local expertise.200
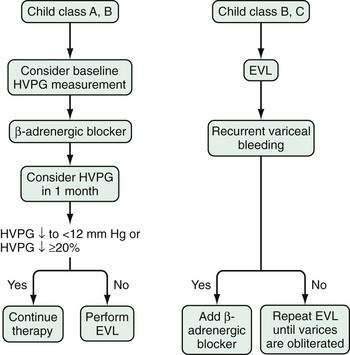
GASTRIC VARICES
The most widely used classification of gastric varices is the Sarin classification.201 According to this classification, type 1 gastroesophageal varices (GOV1) extend 2 to 5 cm below the gastroesophageal junction and are in continuity with esophageal varices; type 2 gastroesophageal varices (GOV2) are in the cardia and fundus of the stomach and in continuity with esophageal varices; varices that occur in the fundus of the stomach in the absence of esophageal varices are called isolated gastric varices type 1 (IGV1), whereas varices that occur in the gastric body, antrum, or pylorus are called isolated gastric varices type 2 (IGV2).
Natural History
Gastric varices typically occur in association with more advanced portal hypertension. Bleeding is thought to be more common in patients with GOV2 and IGV1 than in those with other types of gastric varices; in other words, bleeding is more common from fundal varices than from varices at the gastroesophageal junction. Whereas intraesophageal pressure is negative, intra-abdominal pressure is positive, and the transmural pressure gradient across gastric varices is smaller than that across esophageal varices. Gastric varices, however, tend to be larger in diameter than esophageal varices. Gastric varices are supported by gastric mucosa, whereas esophageal varices tend to be unsupported in the lower third of the esophagus. Therefore, gastric varices are likely to bleed only when they are large, as demonstrated in a study in which larger gastric varices (greater than 5 to 10 mm in diameter) were more likely to bleed than smaller ones.202 Although gastric varices have been thought to bleed less frequently than esophageal varices, the bleeding rates probably are comparable if patients are matched for the severity of cirrhosis (CTP score).201 In contrast with esophageal varices, bleeding from gastric varices has been described with an HVPG less than 12 mm Hg.203,204 Gastric varices in continuity with esophageal varices may regress following treatment of the esophageal varices. When gastric varices persist despite obliteration of esophageal varices, the prognosis is poorer, probably because of the severity of liver disease.
Control of Acute Bleeding
The approach to treating esophageal variceal hemorrhage also applies to acute gastric variceal hemorrhage and includes volume resuscitation, avoidance of overtransfusion, and antibiotic prophylaxis with norfloxacin, 400 mg twice daily for seven days. Upper endoscopy is carried out after patients have been volume resuscitated and stabilized and often following endotracheal intubation to protect the airway. The endoscopic diagnosis of gastric variceal bleeding may be difficult because of pooling of blood in the fundus. A diagnosis of gastric variceal hemorrhage should be considered if bleeding is noted from a gastric varix (Fig. 90-16); blood is found to appear at the gastroesophageal junction or the gastric fundus; blood is found in the stomach and gastric varices with a “white nipple sign” (indicating a fibrin-platelet plug) are seen in the absence of other causes of bleeding; and gastric varices are noted in the absence of other lesions in the esophagus and stomach.205
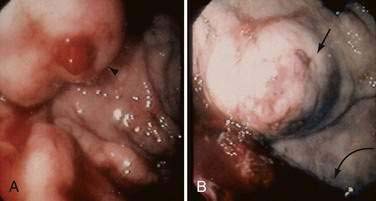
Because controlled studies evaluating pharmacologic therapy for gastric variceal bleeding are lacking, the agents used are based on extension of the data relating to esophageal varices. Medical management with vasoactive agents should be started as early as possible, preferably at least 30 minutes before endoscopic therapy is carried out. The preferred endoscopic therapy for fundal gastric variceal bleeding is injection of polymers of cyanoacrylate, usually N-butyl-2-cyanoacrylate,206,207 but these tissue adhesives are not currently available in the United States. Obliteration of the varices occurs when the injected cyanoacrylate adhesive hardens on contact with blood. The mucosa overlying the varix eventually sloughs, and the hardened polymer is extruded. Fortunately, the resulting ulcers occur late, and the risk of bleeding is lower than that associated with sclerotherapy-related ulcers. Cyanoacrylate injection has been found to be superior to both variceal band ligation and sclerotherapy using alcohol.207 Complications of cyanoacrylate injection include bacteremia and variceal ulceration. Pulmonary and cerebral emboli have been reported on occasion, usually in patients with spontaneous large portosystemic or intrapulmonary shunts. The endoscope may be damaged by the glue, but the risk is minimized if silicone gel is used and suction is avoided for 15 to 20 seconds following injection.208
For injection of GOV2 or IGV1, a retroflexed endoscopic approach is recommended. Sclerosants such as sodium tetradecyl sulfate, ethanolamine oleate, and sodium morrhuate are not particularly effective for control of gastric variceal bleeding.209 When sclerotherapy is carried out for gastric varices, the volume of sclerosant required is larger than that used for esophageal varices, and fever and retrosternal pain are more common. It is much easier to obliterate GOV1 than GOV2 or IGV1. IGV1 are the most difficult gastric varices to obliterate and, when present, should prompt early consideration of definitive treatment such as portosystemic shunting if cyanoacrylate is not available.
Although some investigators recommend ligation of gastric varices up to 20 mm in diameter,210 this recommendation is not supported by our experience. Band ligation of varices greater than 10 mm in diameter usually is unsafe. Ligation is safest if the varices are in the cardia of the stomach. Because gastric fundal varices are covered by mucosa, drawing the entire varix into the ligation device is often not possible. Application of bands results in creation of a large ulcer on the varix, sometimes with disastrous results (see Fig. 90-9).
If endoscopic and pharmacologic therapies fail to control gastric variceal bleeding, then a Linton-Nachlas tube, which has a 600-mL balloon, may be passed as a temporizing measure. The commonly used Minnesota tube and Sengstaken-Blakemore tube, with only 250-mL gastric balloons, are not as effective as the Linton-Nachlas tube for controlling bleeding from gastric fundal varices.211,212 Nevertheless, most patients in whom endoscopic and pharmacologic treatment fails to control gastric variceal bleeding will require a TIPS, which can control bleeding in greater than 90% of patients—a rate of efficacy equivalent to that for TIPS in controlling esophageal variceal bleeding (Fig. 90-17).163,213
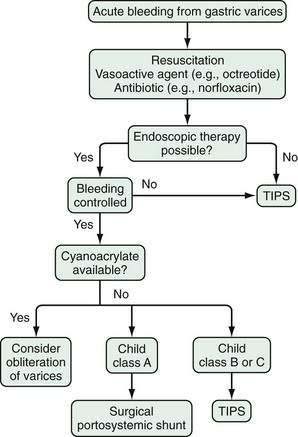
Prevention of Rebleeding
Endoscopic Therapy
In the absence of large, well-conducted trials, recommendations regarding pharmacologic or endoscopic therapy to prevent gastric variceal rebleeding are derived from retrospective studies. Cyanoacrylate glue has been used to prevent gastric variceal rebleeding, with favorable results.214 In a small study, the 2-octyl-cyanoacrylate polymer (Dermabond) has been used to prevent gastric variceal rebleeding, with excellent results.215 Patients require an average of two or three sessions for obturation of gastric varices with cyanoacrylate polymers. Detachable snares have been used to ligate gastric varices that may be too large for band ligation, but the experience with this procedure is limited.216
Transvenous Obliteration of Gastric Varices
Balloon-occluded retrograde transvenous obliteration of gastric fundal varices is carried out in patients with demonstrable splenorenal shunts. Such shunts can be demonstrated on multidetector CT in a large number of patients with bleeding gastric varices. In these patients, a catheter is passed into the left renal vein, usually through a transfemoral approach, and into the varices, which drain into the renal vein. Following balloon occlusion of the varices, a sclerosant is injected retrograde into the varix under fluoroscopic guidance. An extension of this technique is to occlude the varix through a transfemoral approach, while, at the same time, the other end of the varix is approached through a percutaneous transhepatic portal venous route. This technique allows occlusion of the varices at both ends, after which sclerosant can be injected.217 The use of these techniques, which require considerable radiologic skill, is currently limited to some centers in the Far East.
Portosystemic Shunts
Limited data are available regarding use of surgical portosystemic shunts for the treatment of gastric varices in patients with cirrhosis. Two studies performed in patients with good liver function, most of whom had extrahepatic portal vein thrombosis, demonstrated excellent results, with a low long-term risk of bleeding and encephalopathy, after creation of a surgical shunt.218,219
TIPS also is effective in preventing gastric variceal rebleeding. Because TIPS for this indication does not always result in a decrease in the size of gastric varices,220 the target HVPG is uncertain in these patients.204
ECTOPIC VARICES
Varices that occur at a site other than the gastroesophageal junction are termed ectopic varices and account for less than 5% of all varix-related bleeding episodes. Ectopic varices most commonly manifest with melena or hematemesis. They also may manifest with hemobilia, hematuria, hemoperitoneum, or retroperitoneal bleeding. Ectopic varices occur with both extrahepatic portal vein obstruction and cirrhosis. The duodenum is a common site of ectopic varices, and varices typically are associated with portal vein obstruction, but in the West, the usual cause of duodenal varices is cirrhosis. The common occurrence of duodenal varices in patients with portal vein obstruction probably relates to the formation of collateral vessels around the thrombosed portal vein, which connect pancreaticoduodenal veins to retroduodenal veins, which drain into the inferior vena cava.221 In some of those patients with extrahepatic portal vein obstruction, varices form around the gallbladder and bile duct.
The other common site of ectopic varices is peristomal, in patients with inflammatory bowel disease and primary sclerosing cholangitis who have undergone a proctocolectomy with creation of an ileostomy.222 Varices develop at the level of the mucocutaneous border of the stoma and are termed stomal varices. They are recognized by a bluish halo surrounding the stoma and by a dusky appearance and friable consistency of the stomal tissue; no obvious variceal lesions are seen.
Anorectal varices are reported in 10% to 40% of cirrhotic patients who undergo colonoscopy and must be distinguished from hemorrhoids (Fig. 90-18). Rectal varices are dilated superior and middle hemorrhoidal veins, whereas hemorrhoids are dilated vascular channels above the dentate line. Rectal varices collapse with digital pressure, but hemorrhoids do not.
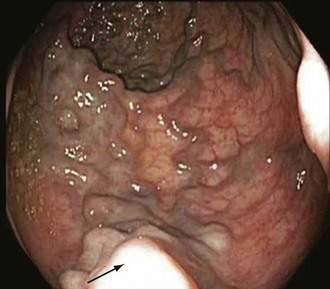
Management
In patients suspected of having ectopic variceal bleeding, vasoactive drugs may be administered initially to control the bleeding. If the bleeding ectopic varix is visualized at endoscopy, as typically is the case with duodenal or colonic varices, then endoscopic therapy can be carried out.223 Endoscopic glue injection or band ligation is the preferred approach for bleeding duodenal varices. Colonic varices tend to be larger in diameter and may require application of hemostatic clips. Patients with bleeding stomal varices can be trained to compress the site locally if bleeding is obvious. Because bleeding from stomal varices is visible and detected early, the mortality rate for bleeding stomal varices is low.224
At present, no recommendations support primary prophylaxis to prevent bleeding from ectopic varices. To prevent rebleeding from ectopic varices, pharmacologic treatment with a beta blocker is usually tried, although no studies are available to support this approach. If the portal vein is patent, then transhepatic embolization of stomal varices can be carried out (Fig. 90-19). Embolization of varices using a transhepatic approach can control bleeding in most patients with stomal varices. The rate of rebleeding is high, however, because the portal hypertension persists. In patients in whom embolization fails to prevent rebleeding, TIPS placement may be considered. A surgical portosystemic shunt is recommended in patients with Child class A cirrhosis and in patients with portal hypertension from extrahepatic portal vein thrombosis in whom a vein suitable for a shunt is available. Placement of a nonselective portosystemic shunt, such as a portacaval shunt, mesocaval shunt, or central splenorenal shunt, should be carried out in a patient with stomal varices.
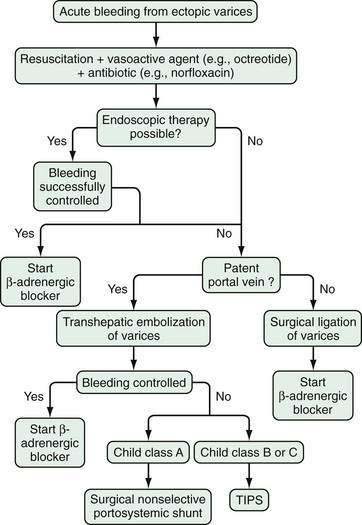
PORTAL HYPERTENSIVE GASTROPATHY AND GASTRIC VASCULAR ECTASIA
Mucosal changes in the stomach in patients with portal hypertension include portal hypertensive gastropathy (PHG) and gastric vascular ectasia. In all likelihood, these lesions are distinct, as demonstrated by histologic features and differences in the response to a TIPS. An appearance analogous to PHG in the colon is termed portal hypertensive colopathy (see Chapter 36).
The diagnosis of PHG is based on the presence of a characteristic mosaic-like pattern of the gastric mucosa on endoscopic examination. This pattern is characterized by small polygonal areas with a depressed border. Superimposed on this mosaic-like pattern may be red point lesions that usually are greater than 2 mm in diameter. PHG is considered mild when only a mosaic-like pattern is present and severe when superimposed discrete red spots are also seen (Fig. 90-20).225 The cause and pathogenesis of PHG are poorly understood. Development of PHG correlates with the duration of cirrhosis but not necessarily the degree of liver dysfunction. The frequency of PHG following endoscopic treatment of esophageal varices is increased—possibly a result of longer duration of portal hypertension in these patients.
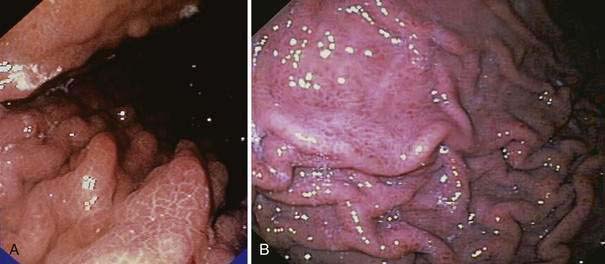
In gastric vascular ectasia, aggregates of ectatic vessels can be seen on endoscopic examination as red spots without a mosaic background.226 When the aggregates are confined to the antrum of the stomach, the term gastric antral vascular ectasia (GAVE) is used (see Chapters 19 and 36). If aggregates in the antrum are linear, the term watermelon stomach is used to describe the lesion (Fig. 90-21). When the red spots are distributed diffusely, in both the distal and the proximal stomach, the term diffuse gastric vascular ectasia is preferred.227
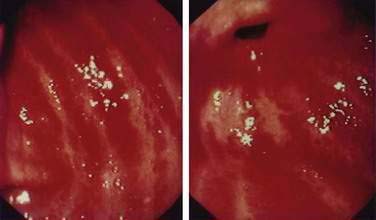
Distinguishing PHG from GAVE is sometimes difficult. A background mosaic pattern and proximal distribution favor PHG (Table 90-8). GAVE is less common, occurs in the absence of a background mosaic pattern, and typically is antral in location, although lesions may be present in the proximal stomach. Mucosal biopsies are recommended when the endoscopic diagnosis is uncertain. GAVE appears histologically as dilated mucosal capillaries with focal areas of fibrin thrombi or ectasia in combination with proliferation of spindle cells.228 Similar ectatic lesions may be seen in the small bowel and may cause acute or chronic gastrointestinal blood loss.
Table 90-8 Comparison of Portal Hypertensive Gastropathy (PHG) and Gastric Antral Vascular Ectasia (GAVE)
| FEATURE | PHG | GAVE |
|---|---|---|
| Distribution | Proximal stomach | Distal stomach |
| Mosaic pattern | Present | Absent |
| Red color signs | Present | Present |
| Findings on gastric mucosal biopsy | ||
| Thrombi | − | +++ |
| Spindle cell proliferation | + | ++ |
| Fibrohyalinosis | − | +++ |
| Treatment | β-adrenergic blockers | ?Antrectomy |
| TIPS | ?Liver transplantation |
TIPS, transjugular intrahepatic portosystemic shunt.
Management
PHG accounts for approximately one fourth of all cases of bleeding (acute and chronic) in patients with portal hypertension, but for less than 10% of all acute bleeding episodes. The more common presentation is one of chronic, slow bleeding and anemia. Pharmacologic therapy to prevent bleeding (primary prophylaxis) in patients with severe portal hypertensive gastropathy is not currently recommended. Small studies have suggested that octreotide may be useful for controlling acute bleeding.229 Beta blockers are recommended for preventing chronic blood loss in patients who have bled from severe PHG.230,231 When patients are transfusion-dependent despite beta blockade and iron supplementation, a TIPS may be inserted (Fig. 90-22). A TIPS decreases transfusion requirements and results in reversal of the mucosal lesions on endoscopic examination.227
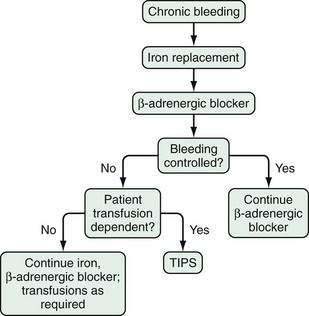
Management of GAVE is more problematic. Initial treatment involves repletion of iron and red blood cell transfusions to treat symptomatic anemia. If lesions are localized, the platelet count is greater than approximately 45,000/mm3, and the INR is less than 1.4, thermoablative therapy, as with argon plasma coagulation, may be helpful (Fig. 90-23). The usual settings for argon plasma coagulation are an energy level of 60 to 90 watts and a gas flow rate of 1 to 2 L per minute. If the coagulation parameters are suboptimal, thermal coagulation is associated with an increase in mucosal bleeding in many patients. When the vascular ectasias are diffuse and extensive in the stomach, cryotherapy using liquid nitrogen or CO2 may be used.232 If endoscopic therapy fails, therapy with an oral estrogen-progesterone combination may be useful in reducing transfusion requirements.233 The usual dose is estradiol, 35 µg, plus norethindrone, 1 mg, daily. Because the medication is taken daily, no risk of breakthrough vaginal bleeding exists. Rarely, painful gynecomastia may limit use of this combination in men. In the patient with preserved hepatic synthetic function who continues to bleed despite thermoablative therapy and an estrogen-progesterone combination, a surgical antral resection may be carried out. TIPS does not reduce the bleeding risk in patients with GAVE and is associated with a substantial risk of hepatic encephalopathy.227 Therefore, TIPS placement is not recommended as therapy for GAVE. Nevertheless, GAVE is reversed with liver transplantation, even in the presence of portal hypertension, suggesting that GAVE is related to liver failure, rather than to portal hypertension.50,234
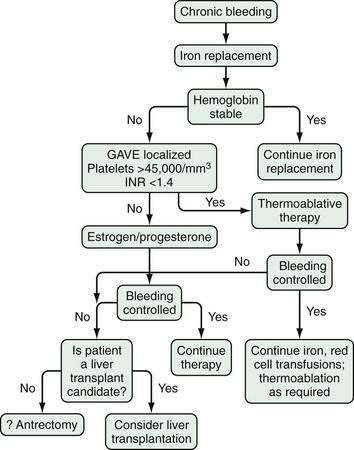
Bernard B, Grange JD, Khac EN, et al. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: A meta-analysis. Hepatology. 1999;29:1655-61. (Ref 196.)
Bosch J, Thabut D, Albillos A, et al. Recombinant factor VIIa for variceal bleeding in patients with advanced cirrhosis: A randomized, controlled trial. Hepatology. 2008;47:1604-14. (Ref 197.)
Cales P, Masliah C, Bernard B, et al. French Club for the study of portal hypertension. Early administration of vapreotide for variceal bleeding in patients with cirrhosis. N Engl J Med. 2001;344:23-8. (Ref 151.)
de Franchis R, Eisen GM, Laine L, et al. Esophageal capsule endoscopy for screening and surveillance of esophageal varices in patients with portal hypertension. Hepatology. 2008;47:1595-603. (Ref 96.)
Failli P, DeFranco R, Caligiuri A, et al. Nitrovasodilators inhibit platelet-derived growth factor-induced proliferation and migration of activated human hepatic stellate cells. Gastroenterology. 2000;119:479-92. (Ref 16.)
Fernandez M, Vizzutti F, Garcia-Pagan J, et al. Anti-VEGF receptor-2 monoclonal antibody prevents portal-systemic collateral vessel formation in portal hypertensive mice. Gastroenterology. 2004;126:886-94. (Ref 55.)
Garcia-Tsao G, Bosch J, Groszmann RJ. Portal hypertension and variceal bleeding—unresolved issues. Summary of an American Association for the Study of Liver Diseases and European Association for the Study of the Liver single-topic conference. Hepatology. 2008;47:1764-72. (Ref 83.)
Henderson JM, Boyer TD, Kutner MH, et al. Distal splenorenal shunt versus transjugular intrahepatic portal systematic shunt for variceal bleeding: A randomized trial. Gastroenterology. 2006;130:1643-51. (Ref 200.)
Iwakiri Y, Grisham M, Shah V. Vascular biology and pathobiology of the liver: Report of a single-topic symposium. Hepatology. 2008;47:1754-63. (Ref 48.)
Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33:464-70. (Ref 167.)
Lebrec D, Poynard T, Hillon P, et al. Propranolol for prevention of recurrent gastrointestinal bleeding in patients with cirrhosis: A controlled study. N Engl J Med. 1981;305:1371-4. (Ref 152.)
Perri RE, Chiorean MV, Fidler JL, et al. A prospective evaluation of computerized tomographic (CT) scanning as a screening modality for esophageal varices. Hepatology. 2008;47:1587-94. (Ref 97.)
Pinzani M, Milani S, De Franco R, et al. Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology. 1996;110:534-48. (Ref 21.)
Semela D, Das A, Langer D, et al. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135:671-9. (Ref 15.)
Talwalkar JA, Yin M, Fidler JL, et al. Magnetic resonance imaging of hepatic fibrosis: Emerging clinical applications. Hepatology. 2008;47:332-42. (Ref 109.)
1. Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med. 2001;345:1683-8.
2. Douglas BE, Baggenstoss AH, Hollingshead WN. The anatomy of the portal vein and its tributaries. Surg Gynecol Obst. 1950;91:562-76.
3. Sarin S, Groszmann R, Mosca P, et al. Propranolol ameliorates the development of portal-systemic shunting in a chronic murine schistosomiasis model of portal hypertension. J Clin Invest. 1991;87:1032-6.
4. Angelico M, Carli L, Piat C, et al. Effects of isosorbide-5-mononitrate compared with propranolol on first bleeding and long-term survival in cirrhosis. Gastroenterology. 1997;113:1632-9.
5. Rodriguez-Perez F, Groszmann R. Pharmacologic treatment of portal hypertension. Gastroenterol Clin North Am. 1992;21:15-40.
6. Bhathal P, Grossman H. Reduction of the increased portal vascular resistance of the isolated perfused cirrhotic rat liver by vasodilators. J Hepatol. 1985;1:325-7.
7. Gupta T, Toruner M, Chung M, et al. Endothelial dysfunction and decreased production of nitric oxide in the intrahepatic microcirculation of cirrhotic rats. Hepatology. 1998;28:926-31.
8. Shah V, Garcia-Cardena G, Sessa W, et al. The hepatic circulation in health and disease: Report of a single-topic symposium. Hepatology. 1998;27:279-88.
9. Rockey DC, Chung JJ. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: Endothelial dysfunction in portal hypertension. Gastroenterology. 1998;114:344-51.
10. Shah V, Toruner M, Haddad F, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental liver cirrhosis. Gastroenterology. 1999;117:1222-8.
11. Sarela A, Mihaimeed F, Batten J, et al. Hepatic and splanchnic nitric oxide activity in patients with cirrhosis. Gut. 1999;44:749-53.
12. Liu S, Premont RT, Kontos CD, et al. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med. 2005;11:952-8.
13. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655-69.
14. Lee JS, Semela D, Iredale J, et al. Sinusoidal remodeling and angiogenesis: A new function for the liver-specific pericyte? Hepatology. 2007;45:817-25.
15. Semela D, Das A, Langer D, et al. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135:671-9.
16. Failli P, DeFranco R, Caligiuri A, et al. Nitrovasodilators inhibit platelet-derived growth factor-induced proliferation and migration of activated human hepatic stellate cells. Gastroenterology. 2000;119:479-92.
17. Bellis L, Berzigotti A, Abraldes J, et al. Low doses of isosorbide mononitrate attenuate the postprandial increase in portal pressure in patients with cirrhosis. Hepatology. 2003;37:378-84.
18. Loureiro-Silva M, Cadelina G, Iwakiri Y, et al. A liver-specific nitric oxide donor improves the intra-hepatic vascular response to both portal blood flow increase and methoxamine in cirrhotic rats. J Hepatol. 2003;39:940-6.
19. Dudenhoefer A, Loureiro-Silva M, Cadelina G, et al. Bioactivation of nitroglycerin and vasomotor response to nitric oxide are impaired in cirrhotic rat livers. Hepatology. 2002;36:381-5.
20. Rockey DC, Weisiger RA. Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: Implications for regulation of portal pressure and resistance. Hepatology. 1996;24:233-40.
21. Pinzani M, Milani S, De Franco R, et al. Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology. 1996;110:534-48.
22. Kamath P, Tyce G, Miller V, et al. Endothelin-1 modulates intrahepatic resistance in a rat model of noncirrhotic portal hypertension. Hepatology. 1999;30:401-7.
23. Rockey D, Fouassier L, Chung J, et al. Cellular localization of endothelin-1 and increased production in liver injury in the rat: Potential for autocrine and paracrine effects on stellate cells. Hepatology. 1998;27:472-80.
24. Alam I, Bassd N, Bacchetti P, et al. Hepatic tissue endothelin-1 levels in chronic liver disease correlate with disease severity and ascites. Am J Gastroenterol. 2000;95:199-203.
25. Douggrell SA. The therapeutic potential of endothelin-1 receptor antagonists and endothelin-converting enzyme inhibitors on the cardiovascular system. Expert Opin Investig Drugs. 2002;11:1537-52.
26. Reynaert H, Vaeyens F, Qin H, et al. Somatostatin suppresses endothelin-1 induced rat hepatic stellate cell contraction via somatostatin receptor subtype 1. Gastroenterology. 2001;121:915-30.
27. Graupera M, Garcia-Pagan J, Titos E, et al. 5-lipoxygenase inhibition reduces intrahepatic vascular resistance of cirrhotic rat livers: A possible role of cysteinyl-leukotrienes. Gastroenterology. 2002;122:387-93.
28. Yokoyama M, Xu H, Kresge N, et al. Role of thromboxane A2 in early BDL-induced portal hypertension. Am J Physiol. 2003;284:G453-60.
29. Blendis L, Wong F. Does losartan work after all? Am J Gastroenterol. 2003;98:1222-4.
30. Sikuler E, Groszmann RJ. Hemodynamic studies in long- and short-term portal hypertensive rats: The relation to systemic glucagon levels. Hepatology. 1986;6:414-18.
31. Atucha N, Shah V, Garcia-Cardena G, et al. Role of endothelium in the abnormal response of mesenteric vessels in rats with portal hypertension and liver cirrhosis. Gastroenterology. 1996;111:1627-32.
32. Sieber CC, Groszmann RJ. Nitric oxide mediates hyporeactivity to vasopressors in mesenteric vessels of portal hypertensive rats. Gastroenterology. 1992;103:235-9.
33. Sieber CC, Groszmann RJ. In vitro hyporeactivity to methoxamine in portal hypertensive rats: reversal by nitric oxide blockade. Am J Physiol. 1992;262:G996-G1001.
34. Sieber C, Lopez-Talavera JC, Groszmann RJ. Role of nitric oxide in the in vitro splanchnic vascular hyporeactivity in ascitic cirrhotic rats. Gastroenterology. 1993;104:1750-4.
35. Niederberger M, Gines P, Martin P, et al. Comparison of vascular nitric oxide production and systemic hemodynamics in cirrhosis versus prehepatic portal hypertension in rats. Hepatology. 1996;24:947-51.
36. Cahill P, Foster C, Redmond E, et al. Enhanced nitric oxide synthase activity in portal hypertensive rabbits. Hepatology. 1995;22:598-606.
37. Cahill P, Redmond E, Hodges R, et al. Increased endothelial nitric oxide synthase activity in the hyperemic vessels of portal hypertensive rats. J Hepatol. 1996;25:370-8.
38. Garcia-Pagan J, Fernandez M, Bernadich C, et al. Effects of continued NO inhibition on portal hypertensive syndrome after portal vein stenosis in rat. Am J Physiol. 1994;267:G984-90.
39. Sogni P, Sabry S, Moreau R, et al. Hyporeactivity of mesenteric resistance arteries in portal hypertensive rats. J Hepatol. 1996;24:487-90.
40. Martin P, Xu D, Niederberger M, et al. Upregulation of endothelial constitutive NOS: A major role in the increased NO production in cirrhotic rats. Am J Physiol. 1996;270:F494-9.
41. Wiest R, Das S, Cadelina G, et al. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J Clin Invest. 1999;104:1223-33.
42. Shah V, Wiest R, Garcia-Cardena G, et al. Hsp90 regulation of endothelial nitric oxide synthase contributes to vascular control in portal hypertension. Am J Physiol. 1999;277:G463-8.
43. Morales-Ruiz M, Jimenez W, Perez-Sala D, et al. Increased nitric oxide synthase expression in arterial vessels of cirrhotic rats with ascites. Hepatology. 1996;24:1481-6.
44. Tazi K, Barriere E, Moreau R, et al. Role of shear stress in aortic eNOS up-regulation in rats with biliary cirrhosis. Gastroenterology. 2002;122:1869-77.
45. Pateron D, Tazi K, Sogni P, et al. Role of aortic nitric oxide synthase 3 (eNOS) in the systemic vasodilation of portal hypertension. Gastroenterology. 2000;119:196-200.
46. Iwakiri Y, Tsai M, McCabe T, et al. Phosphorylation of eNOS initiates excessive NO production in early phases of portal hypertension. Am J Physiol. 2002;282:H2084-90.
47. Rasaratnam B, Kaye D, Jennings G, et al. The effect of selective intestinal decontamination on the hyperdynamic circulatory state in cirrhosis. A randomized trial. Ann Intern Med. 2003;139:186-93.
48. Iwakiri Y, Grisham M, Shah V. Vascular biology and pathobiology of the liver: Report of a single-topic symposium. Hepatology. 2008;47:1754-63.
49. La Villa G, Barletta G, Pantaleo P, et al. Hemodynamic, renal, and endocrine effects of acute inhibition of nitric oxide synthase in compensated cirrhosis. Hepatology. 2001;34:19-27.
50. Spahr L, Villeneuve JP, Dufresne MP, et al. Gastric antral ectasia in cirrhotic patients: Absence of relation with portal hypertension. Gut. 1999;44:739-42.
51. Ros J, Claria J, To-Figueras J, et al. Endogenous cannabinoids: A new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat. Gastroenterology. 2002;122:85-93.
52. Batkai S, Jarai Z, Wagner J, et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med. 2001;7:827-32.
53. Wagner J, Varga K, Ellis E, et al. Activation of peripheral CB sub 1 cannabinoid receptors in haemorrhagic shock. Nature. 1997;390:518-21.
54. Fernandez M, Lambrecht R, Bonkovsky H. Increased heme oxygenase activity in splanchnic organs from portal hypertensive rats: Role in modulating mesenteric vascular reactivity. J Hepatol. 2001;34:936-9.
55. Fernandez M, Vizzutti F, Garcia-Pagan J, et al. Anti-VEGF receptor-2 monoclonal antibody prevents portal-systemic collateral vessel formation in portal hypertensive mice. Gastroenterology. 2004;126:886-94.
56. Fallon M, Abrams G, Luo B, et al. The role of eNOS in the pathogenesis of a rat model of hepatopulmonary syndrome. Gastroenterology. 1997;113:606-14.
57. Hou M, Cahill P, Zhang S, et al. Enhanced cyclooxygenase-1 expression within the superior mesenteric artery of portal hypertensive rats: Role in the hyperdynamic circulation. Hepatology. 1998;27:20-7.
58. Heinemann A, Stauber RE. Vasodilator responses to nitric oxide are enhanced in mesenteric arteries of portal hypertensive rats. Eur J Clin Invest. 1996;26:824-6.
59. Heinemann A, Wachter C, Holzer P, et al. Nitric oxide-dependent and -independent vascular hyporeactivity in mesenteric arteries of portal hypertensive rats. Br J Pharmacol. 1997;121:1031-7.
60. Tazi K, Moreau R, Cailmail S, et al. Altered growth and lack of responsiveness to angiotensin II in aortic vascular smooth muscle cells from cirrhotic rats. Gastroenterology. 1997;112:2065-72.
61. Tazi K, Moreau R, Heller J, et al. Changes in protein kinase C isoforms in association with vascular hyporeactivity in cirrhotic rat aortas. Gastroenterology. 2000;119:201-10.
62. Escorsell A, Bandi JC, Andreu V, et al. Desensitization to the effects of intravenous octreotide in cirrhotic patients with portal hypertension. Gastroenterology. 2001;120:161-9.
63. Gupta T, Chen L, Groszmann R. Pathophysiology of portal hypertension. Baillieres Clin Gastroenterol. 1997;11:203-19.
64. Vianna A, Hayes PC, Moscoso G. Normal venous circulation of the gastroesophageal junction. A route to understanding varices. Gastroenterology. 1987;93:876-89.
65. Watanabe K, Kimura K, Matsutani S, et al. Portal hemodynamics in patients with gastric varices. A study in 230 patients with esophageal and/or gastric varices using portal vein catheterization. Gastroenterology. 1988;95:434-40.
66. Dilawari JB, Chawla YK. Spontaneous (natural) splenoadrenorenal shunts in extrahepatic portal venous obstruction: A series of 20 cases. Gut. 1987;28:198-2000.
67. Sumanovski L, Battegay E, Stumm M, et al. Increased angiogenesis in portal hypertensive rats: Role of nitric oxide. Hepatology. 1999;29:1044-9.
68. Fernandez-Varo G, Ros J, Morales-Ruiz M, et al. Nitric oxide synthase 3-dependent vascular remodeling and circulatory dysfunction in cirrhosis. Am J Pathol. 2003;162:1985-93.
69. Sieber C, Sumanovski L, Stumm M, et al. In vivo angiogenesis in normal and portal hypertensive rats: Role of basic fibroblast growth factor and nitric oxide. J Hepatol. 2001;34:644-50.
70. Lee FY, Colombato LA, Albillos A, et al. Administration of N omega-nitro-L-arginine ameliorates portal-systemic shunting in portal-hypertensive rats. Gastroenterology. 1993;105:1464-70.
71. Mosca P, Lee F-Y, Kaumann A, et al. Pharmacology of portal-systemic collaterals in portal hypertensive rats: Role of endothelium. Am J Physiol. 1992;263:G544-50.
72. Chan C, Lee F, Wang S, et al. Effects of vasopressin on portal-systemic collaterals in portal hypertensive rats: Role of nitric oxide and prostaglandin. Hepatology. 1999;30:630-5.
73. Huang H, Lee F, Chan C, et al. Effects of somatostatin and octreotide on portal-systemic collaterals in portal hypertensive rats. J Hepatol. 2002;36:163-8.
74. Huang H, Wang S, Chan C, et al. Chronic inhibition of nitric oxide increases the collateral vascular responsiveness to vasopressin in portal hypertensive rats. J Hepatol. 2004;40:234-8.
75. Sakurabayashi S, Koh K, Chen L, et al. Octreotide ameliorates the increase in collateral blood flow during postprandial hyperemia in portal hypertensive rats. J Hepatol. 2002;36:507-12.
76. Chan C, Wang S, Lee F, et al. Endothelin-1 induces vasoconstriction on portal-systemic collaterals of portal hypertensive rats. Hepatology. 2001;33:816-20.
77. Escorsell A, Gines A, Llach J, et al. Increased intra-abdominal pressure increases pressure, volume, and wall tension in esophageal varices. Hepatology. 2002;36:936-40.
78. Myers JD, Taylor WJ. An estimation of portal venous pressure by occlusive catheterization of a hepatic venule. J Clin Invest. 1951;30:662-3.
79. Groszmann RJ, Garcia-Tsao G, Bosch J, et al. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med. 2005;353:2254-61.
80. Huet PM, Vincent C, Deslaurier J, et al. Portal hypertension and primary biliary cirrhosis: Effect of long-term ursodeoxycholic acid treatment. Gastroenterology. 2008;135:1552-60.
81. Groszmann RJ. Hepatic venous pressure gradient: Anything worth doing should be done right. Hepatology. 2004;39:280-2.
82. Ripoll C, Groszmann R, Garcia-Tsao G, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133:481-8.
83. Garcia-Tsao G, Bosch J, Groszmann RJ. Portal hypertension and variceal bleeding—unresolved issues. Summary of an American Association for the Study of Liver Diseases and European Association for the Study of the Liver single-topic conference. Hepatology. 2008;47:1764-72.
84. Thalheimer U, Mela M, Patch D, et al. Targeting portal pressure measurements: A critical reappraisal. Hepatology. 2004;39:286-90.
85. Menon KV, Shah VH, Kamath PS. The Budd-Chiari syndrome. N Engl J Med. 2004;350:578-85.
86. Nevens F, Bustami R, Schley SI, et al. Variceal pressure is a factor predicting the risk of a first variceal bleed. A prospective cohort study in cirrhotic patients. Hepatology. 1998;27:15-19.
87. Escorsell A, Bordas JM, Castaneda B, et al. Predictive value of the variceal pressure response to continued pharmacological therapy in patients with cirrhosis and portal hypertension. Hepatology. 2000;31:1061-7.
88. Rigau J, Bosch J, Bordas JM, et al. Endoscopic measurement of variceal pressure in cirrhosis: Correlation of portal pressure and variceal hemorrhage. Gastroenterology. 1989;96:873-88.
89. Ruiz del Arbul L, Martin de Argila C, Vasquez M. Endoscopic measurement of variceal pressure during hemorrhage from esophageal varices. Hepatology. 1992;16:147.
90. Gertsch P, Fischer G, Kleber G. Manometry as it relates to varices: Comparison of an endoscopic balloon technique with needle puncture. Gastroenterology. 1993;105:1159-66.
91. Brensing KA, Neubrand M, Textor J. Endoscopic manometry of esophageal varices: Evaluation of balloon technique compared with direct portal pressure measurement. J Hepatology. 1998;29:94-102.
92. Scheurlen C, Roleff A, Neubrand M, et al. Noninvasive endoscopic determination of intravascular pressure in patients with portal hypertension: Clinical experience with a new balloon technique. Endoscopy. 1998;30:326-32.
93. Grace ND, Groszmann RJ, Garcia-Tsao G, et al. Portal hypertension and variceal bleeding: An AASLD Single Topic Symposium. Hepatology. 1998;28:868-80.
94. de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol. 2005;43:167-76.
95. Qamar AA, Grace ND, Groszmann RJ, et al. Platelet count is not a predictor of the presence or development of gastroesophageal varices in cirrhosis. Hepatology. 2008;47:153-9.
96. de Franchis R, Eisen GM, Laine L, et al. Esophageal capsule endoscopy for screening and surveillance of esophageal varices in patients with portal hypertension. Hepatology. 2008;47:1595-603.
97. Perri RE, Chiorean MV, Fidler JL, et al. A prospective evaluation of computerized tomographic (CT) scanning as a screening modality for esophageal varices. Hepatology. 2008;47:1587-94.
98. Beppu K, Inoquachi K, Koyanagi N, et al. Prediction of variceal hemorrhage by esophageal endoscopy. Gastrointest Endosc. 1981;27:213-18.
99. Bosch J, Abraldes JG, Groszmann R. Current management of portal hypertension. J Hepatology. 2003;38:S54-68.
100. The North Italian Endoscopic Club (NIEC) for the Study and Treatment of Esophageal Varices. Prediction of the first variceal hemorrhage in patients with cirrhosis of the liver and esophageal varices. A prospective multicenter study. N Engl J Med. 1988;319:983-9.
101. Cottone M, D’Amico G, Maringhini A, et al. Predictive value of ultrasonography in the screening of non-ascitic cirrhotic patients with large varices. J Ultrasound Med. 1986;5:189-92.
102. Schepis F, Camma C, Niceforo D, et al. Which patients with cirrhosis should undergo endoscopic screening for esophageal varices detection? Hepatology. 2001;33:333-8.
103. Van Gansbekeb AEF, Delcour C, Engelholm L, et al. Sonographic features of portal vein thrombosis. Am J Roentgenol. 1985;144:749-52.
104. Sabbas S, Ferraioli G, Buanamico P, et al. A randomized study of propranolol on postprandial hyerpemia in cirrhotic patients. Gastroenterology. 1992;102:1009-16.
105. Lim JK, Groszmann RJ. Transient elastography for diagnosis of portal hypertension in liver cirrhosis: Is there still a role for hepatic venous pressure gradient measurement? Hepatology. 2007;45:1087-90.
106. Willmann JK, Weishaupt D, Bohm T, et al. Detection of submucosal gastric fundal varices with multi-detector row CT angiography. Gut. 2003;52:886-92.
107. Matsuo M, Kanematsu M, Kim T, et al. Esophageal varices: Diagnosis with gadolinium-enhanced MR imaging of the liver for patients with chronic liver damage. Am J Roentgenol. 2003;180:461-6.
108. Sugan OS, Yamamoto K, Sasao K, et al. Daily variation of azygous and portal blood flow and the effect of propranolol administration once an evening in cirrhotics. J Hepatol. 2001;34:26-31.
109. Talwalkar JA, Yin M, Fidler JL, et al. Magnetic resonance imaging of hepatic fibrosis: Emerging clinical applications. Hepatology. 2008;47:332-42.
110. Konishi Y, Nakamura T, Kida H, et al. Catheter ultrasound probe EUS evaluation of gastric cardia and perigastric vascular structures to predict esophageal variceal recurrence. Gastrointest Endosc. 2002;55:197-203.
111. Escorsell A, Bordas JM, Feu F, et al. Endoscopic assessment of variceal volume and wall tension in cirrhotic patients: Effects of pharmacological therapy. Gastroenterology. 1997;113:1640-6.
112. Leung VK, Sung JJ, Ahuja AT, et al. Large paraesophageal varices on endosonography predict recurrence of esophageal varices and re-bleeding. Gastroenterology. 1997;112:1811-6.
113. Brugge WR. EUS is an important new tool for accessing the portal vein. Gastrointest Endosc. 2008;67:343-4.
114. Pomier-Layrargues GP, Kusielewic ZD, Willems B, et al. Presinusoidal portal hypertension in non-alcoholic cirrhosis. Hepatology. 1985;5:415-18.
115. Czaja AJ, Wolf AM, Summerskill WH. Development and early prognosis of esophageal varices in severe chronic active liver disease (CALD) treated with prednisone. Gastroenterology. 1979;77:629-33.
116. Fracanzani AL, Fargion S, Romano R, et al. Portal hypertension and iron depletion in patients with genetic hemochromatosis. Hepatology. 1995;22:1127-31.
117. Gores GJ, Wiesner RH, Dickson ER, et al. Prospective evaluation of esophageal varices in primary biliary cirrhosis: Development, natural history, and influence on survival. Gastroenterology. 1989;96:1552-9.
118. Afroudakis A, Caplowitz N. Liver histopathology in chronic common bile duct stenosis due to chronic alcoholic pancreatitis. Hepatology. 1981;1:65-72.
119. Ross AJP, Bartley PB, Sleigh AC, et al. Current concepts: Schistosomiasis. N Engl J Med. 2002;346:1212-20.
120. Kamal SM, Turner B, He Q, et al. Progression of fibrosis in hepatitis C with and without schistosomiasis: Correlation with serum markers of fibrosis. Hepatology. 2006;43:771-9.
121. Valla DC. Thrombosis and anticoagulation in liver disease. Hepatology. 2008;47:1384-93.
122. Okuda K, Kono K, Ohnishi K, et al. Clinical study of 86 cases with idiopathic portal hypertension in comparison with cirrhosis with splenomegaly. Gastroenterology. 1984;86:600-10.
123. Dilawari JB, Chawla YK. Pseudosclerosing cholangitis in extrahepatic portal venous obstruction. Gut. 1992;33:272-6.
124. Condat B, Pessione F, Hillar RES. Current outcome of portal vein thrombosis in adults: Risks and benefits of anticoagulant therapy. Gastroenterology. 2001;120:490-7.
125. D’Cruz AJ, Kamath PS, Ramachandra C, et al. Nonconventional portosystemic shunts in children with extrahepatic portal vein obstruction. Acta Paediatr Jpn. 1995;37:17-20.
126. Ludwig J, Hashimoto E, Obata H, et al. Idiopathic portal hypertension. Hepatology. 1993;17:1157-62.
127. Kamath PS, Carpenter HA, Lloyd RV, et al. Hepatic localization of endothelin-1 in patients with idiopathic portal hypertension in cirrhosis of the liver. Liver Transpl. 2000;6:596-602.
128. Sama SK, Bhargava S, Nath NG, et al. Non-cirrhotic portal fibrosis. Am J Med. 1971;51:160-9.
129. Boyer JL, Sen Gupta KP, Biswas SK, et al. Idiopathic portal hypertension. Comparison with portal hypertension of cirrhosis and extrahepatic vein obstruction. Ann Intern Med. 1967;66:41-68.
130. Sarin SK, Selhi KK, Nanda R. Measurement and condition of wedged hepatic, intrahepatic, intrasplenic and intravariceal pressures in patients with cirrhosis of the liver and non-cirrhotic portal fibrosis. Gut. 1987;28:260-6.
131. Nevens F, Staessen D, Sciot R, et al. Clinical aspects of incomplete septal cirrhosis in comparison with macronodular cirrhosis. Gastroenterology. 1994;106:459-63.
132. Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: A report of 64 cases among 2500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11:787-97.
133. Gane E, Portmann B, Saxena R, et al. Nodular regenerative hyperplasia of the liver graft after liver transplantation. Hepatology. 1994;20:88-94.
134. Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: A study in 17 explanted livers. Hepatology. 2003;37:510-19.
135. Devarbhavi H, Abraham S, Kamath PS. Significance of nodular regenerative hyperplasia occurring de novo following liver transplantation. Liver Transpl. 2007;13:1552-6.
136. Sherlock S, Feldman CA, Moran B, et al. Partial nodular transformation of the liver with portal hypertension. Am J Med. 1966;40:195-203.
137. Kerr DNS, Okonkwo S, Choa RE. Congenital hepatic fibrosis: The long-term prognosis. Gut. 1978;19:514-20.
138. Qian Q, Lia R, King BF, et al. Clinical profile of autosomal-dominant polycystic liver disease. Hepatology. 2003;37:164-71.
139. Johns CJ, Michele TM. The clinical management of sarcoidosis: A 50-year experience at the Johns Hopkins Hospital. Medicine. 1999;78:65-111.
140. Grundfest A, Cooperman A, Ferguson R. Portal hypertension associated with systemic mastocystosis and splenomegaly. Gastroenterology. 1980;78:370-4.
141. Hyun B, Singer E, Sharriett R. Esophageal varices and metastatic carcinoma of the liver: A report of three cases and a review of the literature. Arch Path. 1976;77:292-5.
142. Pasha SF, Gloviczki P, Stanson AW, et al. Splanchnic artery aneurysms. Mayo Clin Proc. 2007;82:472-9.
143. Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000;343:931-6.
144. Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol. 2007;46:499-507.
145. Ianora AA, Memo M, Sabba C, et al. Hereditary hemorrhagic telangiectasia: Multi-detector row helical CT assessment of hepatic involvement. Radiology. 2004;230:250-9.
146. Albraldes JG, Bosch J. Somatostatin and analogs in the portal hypertension. Hepatology. 2002;35:1305-12.
147. Bosch J, Kravetz D, Rodes J. Effects of somatostatin on hepatic and systemic hemodynamics in patients with cirrhosis of the liver: Comparison with vasopressin. Gastroenterology. 1981;80:518-25.
148. Cirera I, Feu F, Luca A, et al. Effects of bolus injections and continuous infusions of somatostatin and placebo in patients with cirrhosis: A double-blind hemodynamic investigation. Hepatology. 1995;22:106-11.
149. Villanueva C, Ortiz J, Sabat M, et al. Somatostatin alone or combined with emergency sclerotherapy in the treatment of acute esophageal variceal bleeding: A prospective randomized trial. Hepatology. 1999;30:384-9.
150. Buanamico P, Sabba C, Garcia-Tsao G, et al. Octreotide blunts postprandial splanchnic hyperemia in cirrhotic patients: A double-blind randomized echo Doppler study. Hepatology. 1995;21:134-9.
151. Cales P, Masliah C, Bernard B, et al. French Club for the study of portal hypertension. Early administration of vapreotide for variceal bleeding in patients with cirrhosis. N Engl J Med. 2001;344:23-8.
152. Lebrec D, Poynard T, Hillon P, et al. Propranolol for prevention of recurrent gastrointestinal bleeding in patients with cirrhosis: A controlled study. N Engl J Med. 1981;305:1371-4.
153. Lui HF, Stanley AJ, Forrest EH, et al. Primary prophylaxis of variceal hemorrhage: A randomized controlled trial comparing band ligation, propranolol, and isosorbide mononitrate. Gastroenterology. 2002;123:735-44.
154. Albillos A, Lledo JL, Rossi I, et al. Continuous prazosin administrastion in cirrhotic patients: Effects on portal hemodynamics and on liver and renal function. Gastroenterology. 1995;109:1257-65.
155. Schneider AW, Friedrich J, Klein CP. Effect of losartan, an angiotensin II receptor antagonist, on portal pressure in cirrhosis. Hepatology. 1999;29:334-9.
156. Schepke M, Werner E, Biecker E, et al. Hemodynamic effects of the angiotensin II receptor antagonist irbesartan in patients with cirrhosis and portal hypertension. Gastroenterology. 2001;121:389-95.
157. Gonzalez-Abraldes J, Albillos A, Banares R, et al. Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology. 2001;121:382-8.
158. Zafra C, Abraldes JG, Turnes J, et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology. 2004;126:749-55.
159. D’Amico G, Pietras G, Tarantino I, Pagliaro L. Emergency sclerotherapy versus vasoactive drugs for variceal bleeding in cirrhosis: A Cochrane meta-analysis. Gastroenterology. 2003;124:1277-91.
160. Avgerinos A, Armonis A, Stefanidis G, et al. Sustained rise of portal pressure after sclerotherapy, but not band ligation, in acute variceal bleeding in cirrhosis. Hepatology. 2004;39:1623-30.
161. Bureau C, Garcia-Pagan JC, Otal P, et al. Improved clinical outcome using polytetrafluoroethylene-coated stents for TIPS. Results of a randomized study. Gastroenterology. 2004;126:469-73.
162. Azoulay D, Castaing D, Majno P, et al. Salvage transjugular intrahepatic portosystemic shunt for uncontrolled variceal bleeding in patients with decompensated cirrhosis. J Hepatology. 2001;35:590-7.
163. Barange K, Peron JM, Imani K, et al. Transjugular intrahepatic portosystemic shunt in the treatment of refractory bleeding from ruptured gastric varices. Hepatology. 1999;30:1139-43.
164. Papatheodoridis GV, Goulis J, Leandro G, et al. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal re-bleeding: A meta-analysis. Hepatology. 1999;30:612-22.
165. Malinchoc M, Kamath PS, Gordon FD, et al. A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology. 2000;31:864-71.
166. Chalasani N, Clark WS, Martin LG, et al. Determinants of mortality in patients with advanced cirrhosis after transjugular intrahepatic portosystemic shunting. Gastroenterology. 2000;118:138-44.
167. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33:464-70.
168. Ferral H, Gamboa P, Postoak DW, et al. Survival after elective transjugular intrahepatic portosystemic shunt creation: Prediction with model for end-stage liver disease score. Radiology. 2004;231:231-6.
169. Henderson JM. Therapies for refractory variceal hemorrhage. Clin Liver Dis. 2001;5:709-25.
170. Sugiura M, Futagawa S. Esophageal transection with para-esophagogastric devascularization (Sugiura procedure) in the treatment of esophageal varices. World J Surg. 1984;8:673-9.
171. Warren WD, Zeppa R, Foman JS. Selective trans-splenic decompression of gastroesophageal varices by distal splenorenal shunts. Ann Surg. 1967;166:437-46.
172. Rikkers LF, Jin G, Langnas AN. Shunt surgery during the era of liver transplantation. Ann Surg. 1997;226:51-7.
173. Sarfeh IJ, Rypins E. A systemic appraisal of portacaval H-graft diameters. Clinical and hemodynamic perspectives. Ann Surg. 1986;204:356-62.
174. Rosemurgy AS, Goode SE, Swiebel BR. A prospective trial of TIPS versus small diameter prosthetic H graft portocaval shunt in the treatment of bleeding varices. Ann Surg. 1996;224:378-86.
175. Orloff MJ, Orloff MS, Orloff SL. Three decades of experience with emergency portocaval shunt for acute bleeding esophageal varices in 400 unselected patients with cirrhosis of the liver. J Am Coll Surg. 1995;180:257-72.
176. Stipa S, Balducci G, Ziparo V. Total shunting in elective management of variceal bleeding. World J Surg. 1994;18:200-4.
177. de Franchis R, Primignani M. Natural history of portal hypertension in patients with cirrhosis. Clin Liver Dis. 2001;5:645-63.
178. Vorobioff J, Groszmann R, Picabea E, et al. Prognostic value of hepatic venous pressure gradient measurements in alcoholic cirrhosis: A 10-year prospective study. Gastroenterology. 1996;111:701-9.
179. D’Amico G, de Franchis R. Upper digestive bleeding in cirrhosis. Post therapeutic outcome and prognostic indicators. Hepatology. 2003;38:599-612.
180. Castaneda B, Morales J, Lionette R, et al. Effects of blood volume restitution following a portal hypertensive-related bleeding in anesthetized cirrhotic rats. Hepatology. 2001;33:821-5.
181. McCormick PA, O’Keefe C. Improving prognosis following a first variceal haemorrhage over four decades. Gut. 2001;49:682-5.
182. Ben Ari Z, Cardin F, McCormick AP, et al. A predictive model for failure to control bleeding during acute variceal haemorrhage. J Hepatology. 1999;31:443-50.
183. Goulis J, Armonis A, Patch D, et al. Bacterial infection is independently associated with failure to control bleeding in cirrhotic patients with gastrointestinal hemorrhage. Hepatology. 1998;27:1207-12.
184. Graham D, Smith J. The course of patients after variceal hemorrhage. Gastroenterology. 1981;80:800-6.
185. Bambha K, Kim WR, Pedersen R, et al. Predictors of early re-bleeding and mortality after acute variceal haemorrhage in patients with cirrhosis. Gut. 2008;57:814-20.
186. Groszmann R, Glickman M, Blei AT, et al. Wedged and free hepatic venous pressure measured with a balloon catheter. Gastroenterology. 1979;76:253-8.
187. Abraczinskas DR, Ookubo R, Grace ND, et al. Propranolol for the prevention of first esophageal variceal hemorrhage: A lifetime commitment? Hepatology. 2001;34:1096-102.
188. Garcia-Pagan JC, Villanueve C, Vila MC, et al. Isosorbide mononitrate in the prevention of first variceal bleed in patients who cannot receive beta-blockers. Gastroenterology. 2001;121:908-14.
189. Escorsell A, Ferayomi L, Bosch J, et al. The portal pressure response to beta-blockade is greater in cirrhotic patients without varices than in those with varices. Gastroenterology. 1997;112:2012-16.
190. Garcia-Pagan JC, Villanueve C, Vila MC, et al. Isosorbide-5-mononitrate in the prevention of the first variceal bleed in patients who cannot receive beta-blockers. Gastroenterology. 2001;121:908-14.
191. Bureau C, Peron JM, Alric L, et al. “A la carte” treatment of portal hypertension: Adapting medical therapy to hemodynamic response for the prevention of bleeding. Hepatology. 2002;36:1361-6.
192. de Franchis R. Updating consensus in portal hypertension: Report of the Baveno III consensus workshops on definitions, methodology and therapeutic strategies in portal hypertension. J Hepatology. 2000;33:846-52.
193. Imperiale TF, Chalasani N. A meta-analysis of endoscopic variceal ligation for primary prophylaxis of esophageal variceal bleeding. Hepatology. 2001;33:802-7.
194. Tripathi D, Graham C, Hayes PC. Variceal band ligation versus beta-blockers for primary prevention of variceal bleeding: A meta-analysis. Eur J Gastroenterol Hepatol. 2007;19:835-45.
195. Lo GH, Chen WC, Lin CK, et al. Improved survival in patients receiving medical therapy as compared with banding ligation for the prevention of esophageal variceal rebleeding. Hepatology. 2008;48:580-7.
196. Bernard B, Grange JD, Khac EN, et al. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: A meta-analysis. Hepatology. 1999;29:1655-61.
197. Bosch J, Thabut D, Albillos A, et al. Recombinant factor VIIa for variceal bleeding in patients with advanced cirrhosis: A randomized, controlled trial. Hepatology. 2008;47:1604-14.
198. Levacher S, Letoumelin P, Pateron D, et al. Early administration of terlipression plus glyceryl trinitrate to control active upper gastrointestinal bleeding in cirrhotic patients. Lancet. 1995;346:865-8.
199. de Franchis R. Portal hyperension II. Proceedings of the Second Baveno International Consensus Workshop on Definitions, Methodology, and Therapeutic Strategies. Oxford: Blackwell Science; 1996.
200. Henderson JM, Boyer TD, Kutner MH, et al. Distal splenorenal shunt versus transjugular intrahepatic portal systematic shunt for variceal bleeding: A randomized trial. Gastroenterology. 2006;130:1643-51.
201. Sarin SK, Lahoti D, Saxena SP, et al. Relevance, classification and natural history of gastric varices: A long-term follow-up study in 568 portal hypertension patients. Hepatology. 1992;16:1343-9.
202. Kim T, Shijo H, Kokawa H, et al. Risk factors for hemorrhage from gastric fundal varices. Hepatology. 1997;25:307-12.
203. Tripathi D, Therapondos G, Jackson E, et al. The role of the transjugular intrahepatic portosystemic stent shunt (TIPSS) in the management of bleeding gastric varices: Clinical and haemodynamic correlations. Gut. 2002;51:270-4.
204. Rinella ME, Shah D, Vogelzang RL, et al. Fundal variceal bleeding after correction of portal hypertension in patients with cirrhosis. Gastrointest Endosc. 2003;58:122-7.
205. Siringo S, McCormick PA, Mistry P. Prognostic significance of the white nipple sign in variceal bleeding. Gastrointest Endosc. 1991;37:51-5.
206. Lo G, Lai K, Cheng J, et al. A prospective, randomized trial of butyl cyanoacrylate injection versus band ligation in the management of bleeding gastric varices. Hepatology. 2001;33:1060-4.
207. Sarin SK, Jain AK, Jain M, et al. A randomized controlled trial of cyanoacrylate versus alcohol injection in patients with isolated fundic varices. Am J Gastroenterol. 2002;97:1010-15.
208. Binmoeller KF. Glue for gastric varices: Some sticky issues. Gastrointest Endosc. 2000;52:298-301.
209. Trudeau W, Prindville T. Endoscopic injection sclerosis in bleeding gastric varices. Gastrointest Endosc. 1986;32:264-8.
210. Ryan BM, Stockbrugger RW, Ryan JM. A pathophysiologic, gastroenterologic, and radiologic approach to the management of gastric varices. Gastroenterology. 2004;126:1175-89.
211. Panés J, Teres J, Bosch J, et al. Efficacy of balloon tamponade in treatment of bleeding gastric and esophageal varices. Results in 151 consecutive episodes. Dig Dis Sci. 1988;33:454-9.
212. Teres J, Cecilia A, Bordas JM, et al. Esophageal tamponade for bleeding varices. Controlled trial between the Sengstaken-Blakemore tube and the Linton-Nachlas tube. Gastroenterology. 1978;75:566-9.
213. Chau T, Patch D, Chan YW, et al. “Salvage” transjugular intrahepatic portosystemic shunts: Gastric fundal compared with esophageal variceal bleeding. Gastroenterology. 1998;114:981-7.
214. Mahadeva S, Bellamy MC, Kessel D, et al. Cost-effectiveness of N-butyl-2-cyanoacrylate (histoacryl) glue injections versus transjugular intrahepatic portosystemic shunt in the management of acute gastric variceal bleeding. Am J Gastroenterol. 2003;98:2688-93.
215. Greenwald B, Caldwell SH, Hespensheide E, et al. N-2-butyl-cyanoacrylate for bleeding gastric varices: A United States pilot study and cost analysis. Am J Gastroenterol. 2003;98:1982-8.
216. Yoshida T, Harada T, Shigemitsu T, et al. Endoscopic management of gastric varices using a detachable snare and simultaneous endoscopic sclerotherapy and o-ring ligation. J Gastroenterol Hepatol. 1999;14:730-5.
217. Shiba M, Higuchi K, Nakamura K, et al. Efficacy and safety of balloon-occluded endoscopic injection for sclerotherapy as a prophylactic treatment for high-risk gastric varices: A prospective, randomized, comparative clinical trial. Gastrointest Endosc. 2002;56:522-8.
218. Orloff MJ, Orloff MS, Girard B, et al. Bleeding esophagogastric varices from extrahepatic portal hypertension: 40 years’ experience with portal-systemic shunt. J Am Coll Surg. 2002;194:717-28.
219. Thomas PG, D’Cruz AJ. Distal splenorenal shunting for bleeding gastric varices. Br J Surg. 1994;81:241-4.
220. Sanyal AJ, Freedman AM, Luketic VA, et al. The natural history of portal hypertension after transjugular intrahepatic portosystemic shunts. Gastroenterology. 1997;112:889-98.
221. Itzchak Y, Glickman MG. Duodenal varices in extrahepatic portal obstruction. Radiology. 1977;124:619-24.
222. Weisner RH, LaRusso NF, Dozois RR. Peristomal varices after proctocolectomy in patients with primary sclerosing cholangitis. Gastroenterology. 1986;90:316-22.
223. Norton ID, Andrews J, Kamath PS. Management of ectopic varices. Hepatology. 1998;28:1154-8.
224. Ackerman NB, Graeber GM, Fey J. Enterostomal varices secondary to portal hypertension: Progression of disease in conservatively managed cases. Arch Surg. 1980;115:1454-5.
225. Primignani M, Materia M, Preatoni P, et al. Natural history of portal hypertensive gastropathy in patients with liver cirrhosis. Gastroenterology. 2000;119:181-7.
226. Jabbari M, Cherry R, Lough JO. Gastric antral vascular ectasia: The watermelon stomach. Gastroenterology. 1984;87:1165-70.
227. Kamath PS, Lacerda M, Ahlquist D, et al. Gastric mucosal responses to intrahepatic portosystemic shunting in patients with cirrhosis. Gastroenterology. 2000;118:905-11.
228. Payen JL, Cales P, Voigt JJ. Severe portal hypertensive gastropathy and antral vascular ectasia are distinct entities in patients with cirrhosis. Gastroenterology. 1995;108:138-44.
229. Zhou Y, Qiao WJ, Hu H, et al. Control of bleeding in portal hypertensive gastropathy. Comparison of the efficacy of octreotide, vasopressin, and omeprazole in the control of acute bleeding in patients with portal hypertensive gastropathy: A controlled study. J Gastroenterol Hepatol. 2002;17:973-9.
230. Perez-Ayuso R, Pique J, Bosch J, et al. Propranolol in prevention of recurrent bleeding from severe portal hypertensive gastropathy in cirrhosis. Lancet. 1991;337:1431-4.
231. Panes J, Bordas JM, Pique J, et al. Effects of propranolol on gastric mucosal perfusion in cirrhotic patients with portal hypertensive gastropathy. Hepatology. 1993;17:213-18.
232. Cho S, Zanati S, Yong E, et al. Endoscopic cryotherapy for the management of gastric antral vascular ectasia. Gastrointest Endosc. 2008;68:895-902.
233. Tran A, Villeneuve J-P, BIlodeau M, et al. Treatment of chronic bleeding from gastric antral vascular ectasia (GAVE) with estrogen-progesterone in cirrhotic patients: An open pilot study. Am J Gastroenterol. 1999;94:2909-11.
234. Vincent C, Pomier-Layrargues GP, Dagenais M, et al. Cure of gastric antral vascular ectasia by liver transplantation despite persistent portal hypertension: A clue for pathogenesis. Liver Transpl. 2002;8:717-20.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 90 Portal Hypertension and Gastrointestinal Bleeding
Variceal hemorrhage, hepatic encephalopathy, and ascites—the major complications of cirrhosis of the liver—result from portal hypertension, defined as an increase in hepatic sinusoidal pressure to 6 mm Hg or greater. Portosystemic collaterals decompress the hypertensive hepatic sinusoids and give rise to varices at the gastroesophageal junction and elsewhere. These portosystemic collaterals also may allow ammonia derived from the intestine to reach the brain, thereby resulting in hepatic encephalopathy through a pathologic process of several intermediary steps involving the peripheral benzodiazepine-type receptors, neurosteroids, and γ-aminobutyric acid (GABA) receptors (see Chapter 92). Additionally, portal hypertension is associated with renal retention of sodium and water and the formation of ascites (see Chapter 91). Indeed, portal hypertension and its complications remain important clinical problems despite advances in treatment and improved understanding of both the molecular basis and pathophysiology of portal hypertension.
NORMAL PORTAL CIRCULATION
The portal venous system carries capillary blood from the esophagus, stomach, small and large intestine, pancreas, gallbladder, and spleen to the liver. The portal vein is formed by the confluence of the splenic vein and the superior mesenteric vein behind the neck of the pancreas.1 The inferior mesenteric vein usually drains into the splenic vein. The left gastric vein, also called the left coronary vein, usually drains into the portal vein at the confluence of the splenic vein and superior mesenteric vein (Fig. 90-1). The portal vein is approximately 7.5 cm in length and runs dorsal to the hepatic artery and bile duct into the hilum of the liver. The uppermost 5 cm of the portal vein does not receive any tributaries.2 In the hilum of the liver, the portal vein divides into the left and right portal vein branches, which supply the left and right sides of the liver, respectively. The umbilical vein drains into the left portal vein. The cystic vein from the gallbladder drains into the right portal vein, whereas the portal venules drain into hepatic sinusoids that, in turn, are drained by the hepatic veins into the inferior vena cava. The left and middle hepatic veins usually join and drain into the inferior vena cava separately but adjacent to the confluence of the right hepatic vein with the inferior vena cava. The caudate lobe drains separately into the inferior vena cava (see Chapter 71).
The sinusoids are highly permeable and thus facilitate the transport of macromolecules to the parenchymal hepatocytes that reside on the extraluminal side of the endothelial cells. The hepatic sinusoids are highly permeable because they lack a proper basement membrane and because the endothelial cells that line the sinusoids contain fenestrae. Other unique aspects of the hepatic sinusoids are the space of Disse, a virtual space located extraluminal to the endothelial cell and adjacent to the hepatocyte, and its cellular constituents, the hepatic stellate cell (HSC) and the Kupffer cell (Fig. 90-2; see also Chapters 71 and 72). These two cell types probably play an important role, in concert with the endothelial cell, in regulating sinusoidal hemodynamics and homeostasis and may contribute to the sinusoidal derangements that occur in portal hypertension. Under basal conditions, HSCs maintain a quiescent phenotype and accumulate vitamin A. On activation, however, as occurs in cirrhosis and portal hypertension, these cells are postulated to develop contractile abilities that permit them to function as sinusoidal pericytes. Kupffer cells contribute to vascular homeostasis by generating cytokines with potent cellular and vasoregulatory actions, including tumor necrosis factor. Endothelial cells and smooth muscle cells in nonsinusoidal hepatic vessels such as the portal venule and the terminal hepatic venule are important in hepatic vasoregulation, particularly in the normal liver, where HSCs are quiescent, unactivated, and presumably less contractile.
HEMODYNAMIC PRINCIPLES OF PORTAL HYPERTENSION
in which the pressure gradient in the portal circulation (ΔP) is a function of portal flow (F) and resistance to flow (R). Increases in portal resistance or portal flow can contribute to increased pressure. Portal hypertension almost always results from increases in both portal resistance and portal flow (Fig. 90-3). One exception is that of an arteriovenous fistula, which in the initial stages causes portal hypertension largely through an increase in portal flow in the absence of an increase in resistance. The mechanism of the increase in portal resistance depends on the site and cause of portal hypertension; in the Western world, the most common cause is liver cirrhosis (see later). Because of the increase in hepatic resistance and the decrease in hepatic compliance, small changes in flow that do not increase pressure in the normal liver can have a prominent stimulatory effect on portal pressure in the cirrhotic liver. The increase in portal venous inflow is part of a generalized systemic derangement termed the hyperdynamic circulatory state. Collateral vessels that dilate and new vascular sprouts that form connect the high-pressure portal venous system with lower-pressure systemic veins. Unfortunately, this process of angiogenesis and collateralization is insufficient for normalizing portal pressure and actually causes complications of portal hypertension, such as esophageal varices.3 Approaches to block this angiogenic process are a compelling target for drug development.
The changes in portal flow and resistance also can be viewed as originating from mechanical and vascular factors. Mechanical factors include the fibrosis and nodularity of the cirrhotic liver with distortion of the vascular architecture and the remodeling that is recognized to occur in the systemic and splanchnic vasculature in response to the chronic increases in flow and shear stress that characterize the hyperdynamic circulatory state. Vascular factors include intrahepatic vasoconstriction, which contributes to increased intrahepatic resistance, and the splanchnic and systemic vasodilatation that accompanies the hyperdynamic circulatory state. The vascular factors that contribute to portal hypertension are particularly important because they are reversible and dynamic and therefore compelling targets for experimental therapies (Fig. 90-4). Conversely, effective therapies for the fixed, mechanical component of portal hypertension caused by scar, regenerative nodules, and vascular remodeling are currently lacking. Indeed, most available therapies for portal hypertension focus on correction of hemodynamic alterations in the portal circulation. Approaches include use of nonselective β-adrenergic blocking agents, octreotide, and vasopressin to reduce the hyperdynamic circulation, portal venous inflow, and splanchnic vasodilatation.4,5 Alternative agents reduce the increased intrahepatic resistance and include angiotensin receptor blockers and mononitrates.
INCREASED INTRAHEPATIC RESISTANCE
In cirrhosis, increased portal resistance occurs in great part as a result of mechanical factors that reduce vessel diameter. In addition to regenerative nodules and fibrotic bands, these mechanical factors include capillarization of the sinusoids and swelling of cells, including hepatocytes and Kupffer cells. As discussed earlier, however, reduced hepatic vessel diameter resulting in increased portal resistance, even when caused by cirrhosis, is not a purely mechanical phenomenon.6 Hemodynamic changes in the hepatic circulation also contribute to increased intrahepatic resistance.7,8 These changes are characterized by hepatic vasoconstriction and impaired responses to vasodilatory stimuli. The increase in intrahepatic resistance is determined largely by changes in vessel radius, with small reductions in vessel radius causing prominent increases in resistance. Blood viscosity and vessel length also can influence resistance, albeit to a much smaller extent. The factors that regulate resistance can be viewed in the context of the law of Poiseuille:
in which R is resistance, ηL is the product of blood viscosity and vessel length, and r is vessel radius.
Although vasoactive changes were estimated initially to account for 10% to 30% of the increase in portal resistance in cirrhosis, subsequent studies have suggested that these figures actually may underestimate the contribution of hepatic vasoconstriction to the increased resistance observed in the cirrhotic liver. In noncirrhotic causes of portal hypertension, the increase in resistance may occur at sites upstream (prehepatic) or downstream (posthepatic) of the liver, as in portal vein thrombosis and hepatic vein thrombosis, respectively (Fig. 90-5). Furthermore, the site of increased intrahepatic resistance can be further delineated as the sinusoids (sinusoidal), upstream from the sinusoids within the portal venules (presinusoidal), or downstream from the sinusoids in the hepatic venules (postsinusoidal), as in alcoholic cirrhosis, schistosomiasis, and sinusoidal obstruction syndrome, respectively. Pressure is increased only in the portal circulation behind the site of increased resistance, and in isolated portal vein thrombosis, hepatic function frequently remains largely preserved despite prominent portal hypertension.
Most evidence suggests that a decrease in the production of the vasodilator NO and an increase in the production of the vasoconstrictor ET-1 jointly contribute to the increase in hepatic vascular resistance. In experimental models of cirrhosis, the bioavailability of hepatic NO is diminished because of a reduction in the production of NO by endothelial cells.7,9,10 A similar paradigm is observed in the human cirrhotic liver.11 Most studies indicate that the reduction in NO production occurs not through a reduction in hepatic eNOS protein levels9,10 but through defects in the steps necessary to activate existing eNOS protein. For example, increases in the production of the eNOS-inhibiting protein caveolin-1 have been observed in experimental models of cirrhosis10 and in human cirrhosis. Another pathway that contributes to deficient generation of NO by eNOS is a reduction in the level of AKT (protein kinase B) phosphorylation of eNOS and upregulation of the eNOS inhibiting protein, GRK (G protein-coupled receptor kinase), in the cirrhotic liver.12 Irrespective of the mechanism of deficiency, the lack of availability of NO is thought to allow HSCs, which are activated and highly contractile in liver cirrhosis, to constrict the sinusoids that they envelop, thereby increasing portal pressure. The role of the HSCs in this process remains controversial, however, because evidence is mixed regarding whether the site of the increase in intrahepatic resistance in cirrhosis is the sinusoids, where stellate cells reside, or the pre- or postsinusoidal venules (or both), which are devoid of stellate cells and in which endothelial cells signal smooth muscle cells. Furthermore, increasing evidence points toward diverse origins of these myofibroblastic cells within the cirrhotic sinusoids, with portal myofibroblasts as well as HSC postulated to play important roles.13 In this regard, therapies that target myofibroblast migration may be a compelling therapeutic target by limiting the density of these contractile cells within the hepatic sinusoids.14 Finally, the contribution of HSCs to hepatic angiogenesis may also be an important target for treating fibrosis and portal hypertension.15
In clinical practice, NO can be delivered by NO donor agents such as mononitrates. NO donor agents exert their beneficial effects in part by relaxing the actively contractile stellate cells.16,17 The systemic actions of these agents, however, tend to cause side effects and exacerbate the hyperdynamic circulatory state. In studies utilizing a liver-specific NO donor compound, the increased intrahepatic vascular resistance could be corrected by the generation of additional NO and consequent relaxation of HSCs.18 In cirrhosis, however, deficient endothelial cell NO generation may be accompanied by impaired stellate cell relaxation in response to NO,19 perhaps because of diminished response of the NO second messenger cyclic guanosine monophosphate (cGMP) in activated cells.16 In this situation, a prominent beneficial effect of NO donors is less predictable.
Excessive ET-1 also contributes to increased intrahepatic vasoconstriction in portal hypertension through vasoconstrictive effects in the liver, presumably by enhancing HSC contractility.20,21 In experimental models, ET-1 protein and receptor expression are increased, most notably in HSCs and endothelial cells.20,22,23 In humans with portal hypertension, plasma and liver ET-1 levels also are increased.24 The reason for activation of the ET-1 system in portal hypertension is not known, but this effect may be secondary to transforming growth factor-β (TGF-β), a key fibrogenic growth factor.23 Clinical trials of ET antagonists in patients with portal hypertension are in progress; however, the variable effects of ET modulation in experimental models of portal hypertension, as well as the possible hepatotoxicity of these compounds, have limited enthusiasm for studies in humans.25 Other therapies for portal hypertension may provide benefit through the ET pathway. For example, somatostatin, which reduces portal pressure by constricting the splanchnic circulation, also may act by inhibiting ET-1–dependent HSC contraction.26
Other vasoactive mediators, including cysteinyl leukotrienes, thromboxane, angiotensin, and hydrogen sulfide, also have been implicated in the development of increased intrahepatic resistance in cirrhosis.27,28 Some of these mediators, particularly angiotensin, which causes contraction of HSCs, have been studied in humans. Attempts to reduce portal pressure using pharmacologic agents that inhibit angiotensin activation of HSC contraction have met with mixed results thus far.29
HYPERDYNAMIC CIRCULATION
In addition to the increases in portal resistance discussed earlier, a major factor in the development and perpetuation of portal hypertension is an increase in portal venous flow, or the hyperdynamic circulation. The term portal venous inflow indicates the total blood that drains into the portal circulation, not the blood flow in the portal vein itself, which may actually be diminished in portal hypertension because of portosystemic collateral shunts. The hyperdynamic circulation is characterized by peripheral and splanchnic vasodilatation, reduced mean arterial pressure, and increased cardiac output. Vasodilatation, particularly in the splanchnic bed, permits an increase in inflow of systemic blood into the portal circulation.30
Splanchnic vasodilatation is caused in large part by relaxation of splanchnic arterioles and ensuing splanchnic hyperemia. Studies of experimental portal hypertension have demonstrated that splanchnic vascular endothelial cells are primarily responsible for mediating splanchnic vasodilatation and enhanced portal venous inflow through excess generation of NO.31–39 This excess generation of NO and ensuing vasodilatation, hyperdynamic circulation, and hyperemia in the splanchnic and systemic circulation contrasts with the hepatic circulation, in which NO deficiency contributes to increased intrahepatic resistance.
The mechanism of excess NO production from the endothelial cells of the systemic and splanchnic arterial circulation is an area of active investigation. Some of the increase in NO production probably occurs from shear stress–dependent and shear stress–independent increases in the expression of eNOS, which can be corrected in part by beta blockers.37,40–45 Activation of existing eNOS by cytokines or mechanical factors also seems to contribute to excess systemic and splanchnic NO generation through pathways that include eNOS phosphorylation and protein interactions.42–46 The physiologic stimuli that mediate this process are not well understood but may include ET-1, which is increased in the serum of patients with portal hypertension, and the cytokine tumor necrosis factor-α (TNF-α) because inhibitors of TNF improve portal pressure and the splanchnic circulatory disturbances in both human and experimental portal hypertension. TNF-α may be derived from intestinal endotoxin, and intestinal decontamination appears to correct the hyperdynamic circulation in humans, suggesting a link with intestinal inflammation.47 Vascular endothelial growth factor (VEGF) has also been implicated in this process by excessively activating eNOS.48 In humans with portal hypertension, therapeutic inhibition of NOS has met with mixed clinical results.
In one study, inhibition of NOS corrected altered systemic hemodynamics,49 but other studies have not demonstrated significant portal pressure–reducing effects of systemic NOS inhibition.50 Other mediators that may contribute to systemic and splanchnic vasodilatation include anandamide, an endogenous vasodilatory cannabinoid,51–53 heme oxygenase,17,54–56 and cyclooxygenase.57 Compelling evidence also supports a primary defect in smooth muscle cells in portal hypertension, perhaps because of defects in potassium channels.58–62 In fact, many pharmacologic therapies for portal hypertension target the splanchnic arteriolar smooth muscle cells, rather than endothelial cells, to reduce splanchnic vasodilatation. For example, octreotide, a synthetic analog of somatostatin, causes marked but transient reductions in portal pressure by contracting splanchnic smooth muscle cells, thereby limiting portal venous inflow, especially after meals. Nonselective beta blockers and vasopressin also reduce portal pressure by constricting splanchnic arterioles and thereby reducing portal venous inflow. Because intrahepatic resistance persists, therapies targeted toward the increase in portal venous inflow usually do not normalize portal pressure entirely but often blunt the prominent increases in portal venous inflow that occur in response to a meal. Combination therapy with an agent that reduces increased intrahepatic resistance, such as a nitrate, and an agent that reduces portal venous inflow, such as a beta blocker, are more effective in reducing portal pressure than is either agent alone.
COLLATERAL CIRCULATION AND VARICES
The portal vein–systemic collateral circulation develops and expands in response to elevation of the portal pressure.63 Blood flow in the low volumes that normally perfuse these collaterals and flow toward the portal circulation is reversed in portal hypertension because the increased portal pressure exceeds systemic venous pressure. Therefore, flow is reversed in these collateral vessels, and blood flows out of the portal circulation toward the systemic venous circulation.
Four distinct zones of venous drainage at the gastroesophageal junction are particularly relevant to the formation of esophageal varices.64 The gastric zone, which extends for 2 to 3 cm below the gastroesophageal junction, comprises veins that are longitudinal and located in the submucosa and lamina propria. They come together at the upper end of the cardia of the stomach and drain into short gastric and left gastric veins. The palisade zone extends 2 to 3 cm proximal to the gastric zone into the lower esophagus. Veins in this zone run longitudinally and in parallel in four groups corresponding to the esophageal mucosal folds. These veins anastomose with veins in the lamina propria. The perforating veins in the palisade zone do not communicate with extrinsic (periesophageal) veins in the distal esophagus. The palisade zone is the dominant watershed area between the portal and systemic circulations. More proximal to the palisade zone in the esophagus is the perforating zone, where there is a network of veins. These veins are less likely to be longitudinal and are termed perforating veins because they connect the veins in the esophageal submucosa and the external veins. The truncal zone, the longest zone, is approximately 10 cm in length, located proximally to the perforating zone in the esophagus, and usually characterized by four longitudinal veins in the lamina propria.
The fundus of the stomach drains through short gastric veins into the splenic vein. In the presence of portal hypertension, varices may therefore form in the fundus of the stomach. Splenic vein thrombosis usually results in isolated gastric fundal varices. Because of the proximity of the splenic vein to the renal vein, spontaneous splenorenal shunts may develop and are more common in patients with gastric varices than in those with esophageal varices.65,66
The predominant collateral flow pattern in intrahepatic portal hypertension is through the right and left coronary veins, with only a small portion of flow through the short gastric veins. Therefore, most patients with intrahepatic causes of portal hypertension have esophageal varices or gastric varices in continuity with esophageal varices. Unfortunately, portal hypertension caused by cirrhosis generally persists and progresses despite the development of even an extensive collateral circulation. Progression of portal hypertension results from (1) the prominent obstructive resistance in the liver; (2) resistance within the collaterals themselves; and (3) continued increase in portal vein inflow. The collateral circulatory bed develops through a combination of angiogenesis, the development of new blood vessels, and dilatation and increased flow through preexisting collaterals.3,67 Experimental evidence suggests that VEGF, a key NO stimulatory growth factor, may contribute to both the angiogenic and collateral vessel responses.55,68 Inhibition of VEGF or NO may attenuate the collateral vessel propagation by inhibiting angiogenic responses in experimental models of portal hypertension and collateralization.67–72 Some pharmacologic agents used in the management of portal hypertension, such as beta blockers and octreotide, may act in part by constricting collateral vessels.73–76 Approaches to inhibiting VEGF and angiogenesis are worth studying therapeutically.48
The development of gastroesophageal varices requires a portal pressure gradient of at least 10 mm Hg. Furthermore, a portal pressure gradient of at least 12 mm Hg is thought to be required for varices to bleed; other local factors that increase variceal wall tension also are needed77 because all patients with a portal pressure gradient of greater than 12 mm Hg do not necessarily bleed. Factors that influence variceal wall tension can be viewed in the context of the law of Laplace:
where T is variceal wall tension, P is the transmural pressure gradient between the variceal lumen and esophageal lumen, r is the variceal radius, and w is the variceal wall thickness. When the variceal wall thins and the varix increases in diameter and pressure, the tolerated wall tension is exceeded and the varix will rupture. These physiologic observations are manifested clinically by the observation that patients with larger varices (r) in sites of limited soft tissue support (w), with elevated portal pressure (P), tend to be at greatest risk for variceal rupture from variceal wall tension (T) that becomes excessive. One notable site in which soft tissue support is limited is at the gastroesophageal junction. The lack of tissue support and high vessel density may contribute to the greater frequency of bleeding from varices at the gastroesophageal junction. The law of Laplace also has implications for the relevance of pharmacologic therapies aimed at reducing portal pressure. Reductions in portal pressure will reduce the variceal transmural pressure gradient, thereby reducing the risk that variceal wall tension will become excessive and varices will rupture. Clinically, a reduction in the hepatic venous pressure gradient to less than 12 mm Hg almost negates the risk of variceal hemorrhage. The changes in portal pressure and local variceal factors, however, are dynamic and influenced by a number of physiologic (an increase in intra-abdominal pressure, meal-induced increases in portal pressure), diurnal (circadian changes in portal pressure), and pathophysiologic (acute alcohol use) factors, and portal pressure and esophageal variceal pressure may vary at different times.
MEASUREMENT OF PORTAL PRESSURE
HEPATIC VEIN PRESSURE GRADIENT
The HVPG is the difference between the wedged hepatic venous pressure (WHVP) and free hepatic vein pressure (FHVP). The HVPG has been used to assess portal hypertension since its first description in 1951,78 and has been validated as the best predictor for the development of complications of portal hypertension.
Measurement of the HVPG requires passage of a catheter into the hepatic vein under radiologic guidance until the catheter can be passed no further, that is, until the catheter has been “wedged” in the hepatic vein. The catheter can be passed into the hepatic vein through the femoral vein or using a transjugular venous approach. The purpose of wedging the catheter is to form a column of fluid that is continuous between the hepatic sinusoids and the catheter. Therefore, the measured pressure of fluid within the catheter reflects hepatic sinusoidal pressure. One of the drawbacks of using a catheter that is wedged in the hepatic vein is that the WHVP measured in a more fibrotic area of liver may be higher than the pressure measured in a less fibrotic area because of regional variation in the degree of fibrosis. Using a balloon-occluding catheter in the right hepatic vein to create a stagnant column of fluid in continuity with the hepatic sinusoids eliminates this variation in measurement of WHVP because the balloon catheter measures the WHVP averaged over a wide segment of the liver.79 HVPG is not effective for detecting presinusoidal causes of portal hypertension. For example, in portal hypertension secondary to portal vein thrombosis, the HVPG is normal. Moreover, the HVPG may underestimate sinusoidal pressure in primary biliary cirrhosis and other presinusoidal causes of portal hypertension.80 Therefore, HVPG is accurate for detecting only sinusoidal and postsinusoidal causes of portal hypertension.
The HVPG represents the gradient between the pressure in the portal vein and the intra-abdominal inferior vena caval pressure. An elevation in intra-abdominal pressure increases both WHVP and FHVP equally, so that the HVPG is unchanged. The advantage of the HVPG is that variations in the “zero” reference point have no impact on the HVPG.81 The HVPG is measured at least three times to demonstrate that the values are reproducible. Total occlusion of the hepatic vein by the inflated balloon to confirm that the balloon is in a wedged position is demonstrated by injecting contrast into the hepatic vein. A sinusoidal pattern should be seen, with no collateral circulation to other hepatic veins. The contrast washes out promptly with deflation of the balloon. Correct positioning of the balloon also is demonstrated by a sharp increase in the recorded pressure on inflation of the balloon. The pressure then becomes steady until the balloon is deflated, when the pressure drops sharply. In experienced hands, measurement of the HVPG is highly reproducible, accurate, and safe.
Measurement of the HVPG has been proposed for the following indications: (1) to monitor portal pressure in patients taking drugs used to prevent variceal bleeding; (2) as a prognostic marker82; (3) as an end-point in trials using pharmacologic agents for the treatment of portal hypertension83; (4) to assess the risk of hepatic resection in patients with cirrhosis; and (5) to delineate the cause of portal hypertension (i.e., presinusoidal, sinusoidal, or postsinusoidal (Table 90-1), usually in combination with venography, right-sided heart pressure measurements, and transjugular liver biopsy. Although the indication for HVPG measurement with the most potential for widespread use is monitoring the efficacy of therapies to reduce portal pressure, HVPG monitoring is not done routinely in clinical practice because no controlled trials have yet demonstrated its usefulness.84
PORTAL VEIN PRESSURE
Direct measurement of the pressure in the portal vein is a rarely used method that can be carried out through a percutaneous transhepatic route, transvenous approach, or, rarely, intraoperatively (although anesthesia can affect portal pressure). The transhepatic route requires portal vein puncture performed under ultrasound guidance. A catheter is then threaded over a guidewire into the main portal vein. With increasing use of the transjugular intrahepatic portosystemic shunt (TIPS) (see later), radiologists have gained expertise in puncturing the portal vein and measuring portal vein pressure by a transjugular route. Direct portal pressure measurements are carried out when HVPG cannot be measured, as in patients with occluded hepatic veins caused by the Budd-Chiari syndrome, in whom a surgical portosystemic shunt is being contemplated,85 or in patients with intrahepatic, presinusoidal causes of portal hypertension, such as idiopathic portal hypertension, in which the HVPG may be normal.
ENDOSCOPIC VARICEAL PRESSURE
Varices rupture and bleed when the expanding force of intravariceal pressure exceeds variceal wall tension. Measurement of the difference between intravariceal pressure and pressure within the esophageal lumen (the transmural pressure gradient across the varices) is potentially a more important indicator of bleeding risk than measurement of HVPG,86,87 especially in patients with portal vein thrombosis and other causes of portal hypertension associated with a normal HVPG.
A miniature pneumatic pressure sensitivity gauge attached to the tip of an endoscope (Varipres Solid Components, Barcelona, Spain) allows noninvasive measurement of variceal pressure. Patients with previous variceal bleeding have been demonstrated to have higher variceal pressures than those in patients without previous bleeding.88 A variceal pressure greater than 18 mm Hg during a bleeding episode is associated with failure to control bleeding and predicts early rebleeding.89 Moreover, patients on pharmacologic therapy who show a decrease in variceal pressure of greater than 20% from baseline have a low probability of bleeding, as compared with patients who do not demonstrate a greater than 20% decrease in variceal pressure, in whom the risk of variceal bleeding is 46%.88 Variceal pressure measurements determined with use of Varipres are considered satisfactory when they meet the following criteria: (1) a stable intraesophageal pressure; (2) absence of artifacts caused by esophageal peristalsis; and (3) correct placement of the gauge over the varix, as shown by fine fluctuations in the pressure tracing that correspond to the cardiac cycle and respirations. Therefore, measurement of variceal pressure requires both a skilled endoscopist and a cooperative patient, and, even in expert hands, accurate variceal pressure measurements cannot be obtained in 25% of patients.
Manometry using an endoscopic balloon to measure variceal pressure is subject to observer bias because it relies on visual appearance to determine whether the varices have collapsed.90–92 With this technique, a balloon is inserted into the esophagus and inflated until the varices are noted on endoscopy to collapse. The pressure in the balloon required to collapse the varices represents the variceal pressure. In general, techniques of measuring variceal pressure are still considered experimental and not suitable for routine clinical use.
DETECTION OF VARICES
UPPER GASTROINTESTINAL ENDOSCOPY
Upper gastrointestinal endoscopy is the most commonly used method to detect varices. The current consensus is that all patients with cirrhosis of the liver should be screened for esophageal varices by endoscopy. In patients in whom no varices are detected on initial endoscopy, endoscopy to look for varices should be repeated in 2 to 3 years. If small varices are detected on the initial endoscopy, endoscopy should be repeated in 1 to 2 years.93,94 None of the various noninvasive methods of determining which patients benefit most from endoscopic screening are accurate enough to recommend for routine use in clinical practice.95 The role of noninvasive markers in predicting the risk of large esophageal varices requires study in large multicenter trials. Preliminary data suggest that wireless video capsule endoscopy (see Chapter 19)96 and computed tomography (CT) imaging are alternative screening modalities in patients who are not candidates for upper endoscopy. Moreover, CT screening may be more cost-effective than endoscopy.97
Endoscopic grading of esophageal varices is subjective. Various criteria have been used to try to standardize the reporting of esophageal varices. The best known of these criteria are those compiled by the Japanese Research Society for Portal Hypertension. The descriptors include red color signs, color of the varix, form (size) of the varix, and location of the varix.98 Red color signs include red “wale” markings, which are longitudinal whip-like marks on the varix; cherry-red spots, which usually are 2 to 3 mm or less in diameter; hematocystic spots, which are blood-filled blisters 4 mm or greater in diameter; and diffuse redness. The color of the varix can be white or blue. The form of the varix at endoscopy is described most commonly. Esophageal varices may be small and straight (grade I); tortuous and occupying less than one third of the esophageal lumen (grade II); or large and occupying more than one third of the esophageal lumen (grade III). Varices can be in the lower third, middle third, or upper third of the esophagus. Of all of the aforementioned descriptors, the size of the varices in the lower third of the esophagus is the most important. The size of the varices in the lower third of the esophagus is determined during withdrawal of the endoscope (Fig. 90-6). As much air as possible should be aspirated from the stomach while the esophageal lumen is fully inflated. Small varices, that is, those occupying less than one third of the lumen, are less than 5 mm in diameter, whereas large varices are greater than 5 mm in diameter.98,99 As a point of reference, any varix larger in diameter than an open pinch biopsy forceps is likely to be greater than 5 mm in diameter. Patients with large esophageal varices, Child (or Child-Pugh) class C cirrhosis (see later), and red color signs on varices have the highest risk of variceal bleeding within 1 year.100 The increase in bleeding risk attributable to the presence of red color signs, however, is not independent of the risk associated with large variceal size. Therefore, prophylactic treatment to prevent variceal bleeding is recommended in all patients with large esophageal varices irrespective of the presence or absence of red color signs (see later).
ULTRASONOGRAPHY
Ultrasound examination of the liver with Doppler study of the vessels has been used widely to assess patients with portal hypertension. Features suggestive of portal hypertension on ultrasonography include splenomegaly, portosystemic collateral vessels, and reversal of the direction of flow in the portal vein (hepatofugal flow). Some studies have demonstrated that a portal vein diameter greater than 13 mm and the absence of respiratory variations in the splenic and mesenteric veins are sensitive but nonspecific markers of portal hypertension.101,102 These criteria are not used routinely in clinical practice in most centers. Ultrasound examination can detect thrombosis of the portal vein, which appears as nonvisualization or cavernous transformation (a cavernoma) of the portal vein; the latter finding indicates an extensive collateral network in place of the portal vein.103 Splenic vein thrombosis also can be demonstrated. Portal blood flow can be measured by Doppler ultrasonography, which is the easiest research method for detecting postprandial increases in splanchnic blood flow.104 Although Doppler ultrasonography is clinically useful in the initial evaluation of portal hypertension, the technique is not widely used to provide quantitative assessments of the degree of portal hypertension. Transient elastography may be useful in detecting portal hypertension but is not sufficiently sensitive to recommend as a modality to monitor decreases in portal pressure in patients on pharmacotherapy (see Chapter 73).105
COMPUTED TOMOGRAPHY
Computed tomography (CT) is useful for demonstrating many features of portal hypertension, including abnormal configuration of the liver, ascites, splenomegaly, and collateral vessels (Fig. 90-7). Detection of varices may be an emerging indication for CT. Diagnosis of fundal varices by multidetector row CT (MDCT) is at least as accurate as endoscopic ultrasonography (see later). CT is especially helpful in distinguishing submucosal from perigastric fundal varices106 and is considered a less invasive alternative to conventional angiographic portography in assessing portosystemic collaterals. At present, however, CT is not a recommended screening method for detecting large esophageal varices, but it may be a cost-effective method of screening for varices and preferred to endoscopy by patients.97
MAGNETIC RESONANCE IMAGING
Gadolinium-enhanced magnetic resonance imaging (MRI) is becoming recognized as a potentially useful method of detecting esophageal varices.107 In addition, MRI can be used to measure portal and azygous blood flow, which is increased in patients with portal hypertension.108 MRI provides excellent detail of the vascular structures of the liver and can detect portal venous thrombosis and spleen stiffness in patients with portal hypertension, but the role of MRI in the assessment of portal hypertension requires further study. Unlike transient elastography using ultrasound, MRI can accurately assess the stiffness of even fatty livers.109
ENDOSCOPIC ULTRASONOGRAPHY
Endoscopic ultrasound examination (endosonography) using radial or linear array echo-endoscopes or endoscopic ultrasound mini-probes passed through the working channel of a diagnostic endoscope has been applied as an investigational tool in the evaluation of patients with varices. Endoscopic ultrasonography has been used to study several aspects of esophageal varices, including the cross-sectional area of varices to identify patients at increased risk of bleeding77; size of and flow in the left gastric vein, azygous vein, and paraesophageal collaterals; changes after endoscopic therapy; and recurrence of esophageal varices following variceal ligation (see later).110 Endosonography can be combined with endoscopic measurement of transmural variceal pressure to allow estimation of variceal wall tension, which is a predictor of variceal bleeding (see earlier).111–113
CAUSES OF PORTAL HYPERTENSION
The usual classification of causes of portal hypertension is based on the site of increased resistance to portal blood flow—namely, prehepatic, intrahepatic, and posthepatic—and is outlined in Figure 90-5. Intrahepatic sites of increased resistance can be presinusoidal, sinusoidal, or postsinusoidal. Many causes of portal hypertension are associated with an increase in resistance at more than one site. For example, alcoholic cirrhosis may be associated with increased resistance at the presinusoidal, sinusoidal, and postsinusoidal levels. Therefore, classification based on the site of resistance may not be possible for all diseases that cause portal hypertension. A more useful classification is clinically based and considers common and less common causes of portal hypertension (Table 90-2).
COMMON CAUSES
Cirrhosis
Complications related to portal hypertension are the usual clinical manifestations of cirrhosis of the liver. Although all causes of cirrhosis are associated with portal hypertension, some features are disease specific. In alcoholic liver disease, elevation of the portal pressure is accurately reflected by the HVPG; moreover, portal hypertension may occur in the absence of cirrhosis but is more marked when cirrhosis is present. Perivenular lesions implicated in the pathogenesis in noncirrhotic alcoholic liver injury account for the presinusoidal component of portal hypertension in these patients.114 Autoimmune hepatitis also may be associated with portal hypertension in the absence of cirrhosis115; however, the risk of variceal bleeding is low in patients with autoimmune hepatitis. In patients with hemochromatosis, portal hypertension may be seen even before cirrhosis; the severity of portal hypertension increases with increasing fibrosis. Patients with hemochromatosis may bleed from varices despite an HVPG less than 12 mm Hg, indicating a presinusoidal component of portal hypertension. Phlebotomy therapy in patients with hemochromatosis may result in a decrease in portal hypertension.116 In patients with primary biliary cirrhosis, portal hypertension also may occur before cirrhosis has developed. The risk of variceal bleeding increases with an increase in the histologic stage of the disease.117 In earlier stages of primary biliary cirrhosis, portal hypertension is predominantly presinusoidal, but as the disease progresses, a sinusoidal component develops. Therefore, the HVPG may underestimate portal pressure in patients with primary biliary cirrhosis.80 Portal hypertension occurs in patients with primary sclerosing cholangitis and in those with biliary strictures. A long duration of biliary obstruction usually is required, although portal hypertension has been known to develop in a few months in patients with chronic bile duct obstruction caused by chronic alcoholic pancreatitis.118 Portal hypertension in patients with biliary obstruction regresses following relief of the biliary obstruction.
Schistosomiasis
Schistosomiasis may be the most common cause of portal hypertension worldwide (see Chapter 82). Bleeding from esophageal varices is a major cause of death in patients with hepatosplenic schistosomiasis. Portal hypertension results from presinusoidal obstruction caused by deposition of eggs of Schistosoma mansoni and Schistosoma japonicum in the presinusoidal portal venules. The host reaction results in granulomatous inflammation, which causes presinusoidal and periportal fibrosis.119 The fibrosis that results is sometimes called “clay pipestem” or simply “pipestem” fibrosis and usually is associated with sustained heavy infection. The periportal collagen deposition leads to progressive obstruction of portal blood flow, portal hypertension, and variceal bleeding, along with splenomegaly and hypersplenism. Lobular architecture usually is preserved. Coinfection with hepatitis B or C virus in patients with hepatic schistosomiasis can result in more rapid progression of fibrosis, hepatic failure, and an increased risk of hepatocellular carcinoma.120
Extrahepatic Portal Vein Thrombosis
Extrahepatic portal vein thrombosis is a prehepatic, presinusoidal cause of portal hypertension and a common cause of portal hypertension in children (see Chapter 83). The most common causes of portal vein thrombosis include hematologic disorders such as polycythemia vera or other myeloproliferative disorders. Other causes include a prothrombotic state, such as antithrombin, protein C, or protein S deficiency; antiphospholipid syndrome (or antiphospholipid antibody syndrome); paroxysmal nocturnal hemoglobinuria; oral contraceptive use; a neoplasm, usually intra-abdominal; an inflammatory disease, such as pancreatitis, inflammatory bowel disease, or diverticulitis; abdominal trauma; and postoperative states, especially postsplenectomy. Cirrhosis is a cause of portal vein thrombosis.121 Older studies suggested that portal vein thrombosis occurs in approximately 6% of patients with cirrhosis and in up to 25% of those with cirrhosis and hepatocellular carcinoma.122 With improved imaging, portal vein thrombosis is now known to be a more common complication of cirrhosis, and the association with hepatocellular carcinoma may not be as strong as previously thought. Isolated splenic vein thrombosis caused by a pancreatic neoplasm or pancreatitis usually is not associated with a thrombophilia. Umbilical vein sepsis may be an etiologic factor in children with portal vein thrombosis, but even in these cases, an associated prothrombotic state may predispose the patient to portal vein thrombosis.
Acute and subacute portal vein thrombosis usually does not manifest with variceal bleeding.1 Chronic portal vein thrombosis is suggested by nonvisualization of the portal or splenic vein and an extensive collateral circulation. Patients may present with nonspecific symptoms or with variceal bleeding and hypersplenism. Bleeding usually is from gastroesophageal varices but may be from duodenal varices and, rarely, other ectopic sites. Gallbladder varices (Fig. 90-8) also have been described in patients with portal vein thrombosis.123
The treatment of portal vein thrombosis is symptomatic, with the aim of controlling variceal bleeding or preventing recurrent variceal bleeding. Patients in whom esophageal varices are not large, and a thrombophilia is detected, are best managed with anticoagulation because in these patients, the benefits of anticoagulation outweigh the risks.124 Local or systemic thrombolytic therapy is seldom required and is generally reserved for patients in whom a portal vein thrombus extends into the superior mesenteric vein, with danger of impending intestinal ischemia. Endoscopic therapy is used to control acute variceal bleeding and to prevent recurrent bleeding. Use of pharmacologic agents such as beta blockers to prevent variceal bleeding is probably also effective in patients with portal vein thrombosis, but this approach has not been well studied. Patients with portal vein thrombosis have lower mortality and morbidity rates from variceal bleeding than those reported in patients with cirrhosis and variceal bleeding, owing to the lack of coagulopathy and synthetic liver dysfunction. Surgical portosystemic shunt procedures are carried out in patients in whom bleeding cannot be controlled by conservative measures. If a suitable vein is not available for anastomosis, a large collateral vein may be anastomosed to a systemic vein.125 Placement of a TIPS is possible in some patients with chronic portal vein thrombosis.
Idiopathic Portal Hypertension
Idiopathic portal hypertension is uncommon in Western countries but is common in parts of Asia such as India and Japan. This disorder is diagnosed when the portal pressure is elevated in the absence of significant histologic changes in the liver or extrahepatic portal vein obstruction.126 A liver biopsy specimen from affected patients may be entirely normal,122 although increased concentrations of ET-1 have been noted in the periportal hepatocytes, portal venules, and hepatic sinusoids of patients with idiopathic portal hypertension.127 Various terms used (rather loosely) to describe idiopathic portal hypertension include hepatoportal sclerosis, noncirrhotic portal fibrosis, and Banti’s syndrome.128,129 Use of the term idiopathic portal hypertension probably is best restricted to portal hypertension in patients in whom no hepatic lesion is found on light microscopy. The term hepatoportal sclerosis suggests obliterative portal venopathy with subendothelial thickening of the intrahepatic portal veins; thrombosis and recanalization of these veins may follow. Fibrosis of the portal tracts is prominent later in the course.
The cause of idiopathic portal hypertension is unclear in a majority of patients, although chronic arsenic intoxication, exposure to vinyl chloride, and hypervitaminosis A have been implicated (see Chapter 87). These etiologic factors are present in only a minority of patients. The dominant clinical features of the condition are variceal bleeding and hypersplenism related to a markedly enlarged spleen. Liver biochemical test levels are usually normal, although the serum alkaline phosphatase level may be mildly elevated. Ascites is uncommon. The HVPG in this disorder usually is normal because the site of increased resistance is presinusoidal.130 Surgical portosystemic shunts are well tolerated in these patients, although hepatic encephalopathy may occur on long-term follow-up evaluation.122 Liver transplantation is rarely required in these patients.
Idiopathic portal hypertension may be confused with incomplete septal cirrhosis, which probably is an unrelated condition characterized by incomplete septa and liver nodularity.131 Patients with incomplete septal cirrhosis are clinically similar to patients with cirrhosis and may progress to end-stage liver disease and require liver transplantation.
LESS COMMON CAUSES
Nodular Regenerative Hyperplasia
Nodular regenerative hyperplasia is a histopathologic diagnosis characterized by atrophy of zone 3 hepatocytes and hypertrophy of zone 1 hepatocytes without significant fibrosis (see Chapters 35 and 94).132 This disorder has been recognized increasingly as a cause of portal hypertension and may even occur after liver transplantation.133 Similar histologic changes may be seen in well-established Budd-Chiari syndrome.134 The nodular hyperplasia may not be apparent on histologic examination unless a reticulin stain is carried out to demonstrate the micronodules. These regenerative nodules are believed to result from an imbalance between hyperperfused areas of the liver, with resulting regenerative nodules, and poorly perfused areas, with resulting atrophy. Nodular regenerative hyperplasia is associated with a variety of conditions, predominantly hematologic and rheumatologic in nature. Liver biochemical abnormalities include mild elevation of the serum aminotransferase levels. Portal hypertension manifesting as variceal bleeding is the predominant clinical presentation. Ascites also may develop in these patients, suggesting that an increase in sinusoidal pressure occurs.135 Hepatocellular carcinoma does not occur, but liver transplantation may be required in some patients.
Partial Nodular Transformation of the Liver
Partial nodular transformation of the liver is an uncommon lesion that is characterized by large nodules in the perihilar region.136 These nodules may be visible on imaging studies of the liver. The rest of the liver may be normal or may show changes of nodular regenerative hyperplasia. Liver biochemical test levels usually are normal. Like nodular regenerative hyperplasia, partial nodular transformation of the liver is believed to be related to an imbalance in portal perfusion of the liver, but the abnormality is restricted to the hilar branches, whereas in nodular regenerative hyperplasia the abnormality is more diffuse. Variceal bleeding is the predominant presentation in partial nodular transformation of the liver, although patients with large nodules may experience abdominal pain. Hepatocellular carcinoma may rarely develop in these regenerating nodules. Treatment with a surgical portosystemic shunt is associated with good long-term results.
Fibropolycystic Liver Disease
Fibropolycystic liver disease is a term which encompasses Caroli’s disease, Caroli’s complex (Caroli’s disease with congenital hepatic fibrosis), congenital hepatic fibrosis, and polycystic liver disease. Congenital hepatic fibrosis usually occurs in association with Caroli’s disease of the liver, polycystic disease of the kidney, and medullary sponge kidney (see Chapter 62). The major manifestation of congenital hepatic fibrosis is variceal bleeding.137
[/not-level-membership-for-gastroenterology-and-hepatology-category]
