[level-membership-for-pathology-category]
Polyps of the Small Intestine
Reetesh K. Pai
Rish K. Pai
Introduction
Upper gastrointestinal (GI) endoscopy is routinely performed as part of the evaluation of patients with GI symptoms. Newer techniques such as double-balloon enteroscopy and capsule endoscopy also allow for complete visualization of the small intestine. Gastroenterologists may identify and sample a variety of polyps, nodules, excrescences, and subtle abnormalities in the mucosa during the course of the procedure that, before the endoscopic era, would have gone largely unrecognized. Many of these lesions are clinically asymptomatic and occur in the duodenum, which is amenable to upper endoscopic examination. This chapter discusses the clinicopathologic features and differential diagnoses of the most common or important lesions that may cause the clinical impression of an intestinal polyp (Box 21.1).
Inflammatory Lesions
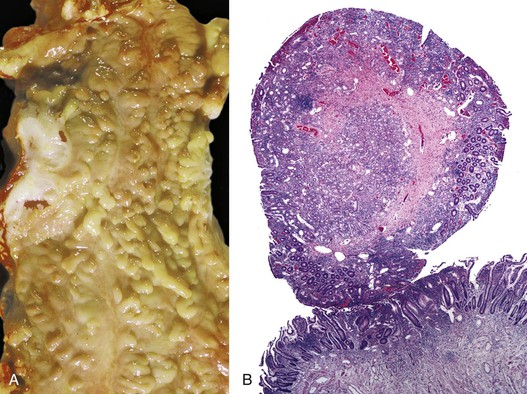
As in other areas of the GI tract, inflammatory lesions of the small intestine may produce polypoid masses. Small polyps are often incidental, whereas larger polyps may be symptomatic due to hemorrhage or luminal obstruction.1 Inflammatory pseudopolyps associated with Crohn’s disease are the most common inflammatory polyps of the small intestine; they are commonly encountered in the terminal ileum, where they demonstrate varying degrees of villous architectural distortion, pyloric gland metaplasia, and active inflammation (Fig. 21.1). They can be quite numerous and can even carpet the small bowel with finger-like projections.1
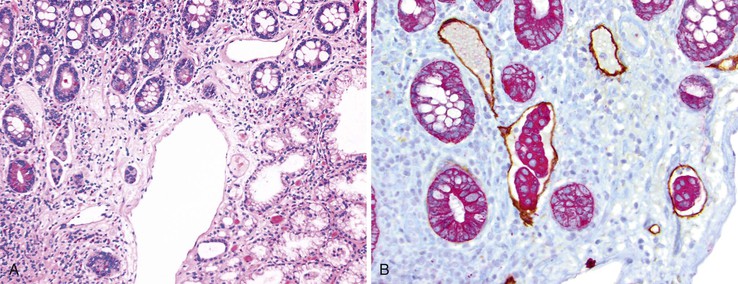
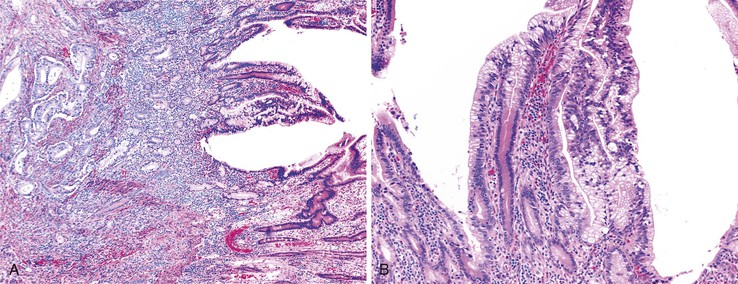
Cytomegalovirus infection can rarely manifest as isolated GI inflammatory polyps, often in immunocompromised hosts (Fig. 21.2).2,3 Xanthomas can also occur in the duodenum, although they are more rare than gastric xanthomas.4 They are characterized by collections of pale and foamy histiocytes in the lamina propria. Histiocytic differentiation can be documented by immunohistochemical staining with CD163, CD68, or both. Inflammatory fibroid polyps (described in Chapter 30) are also less common in the small bowel compared with the stomach (Fig. 21.3).5 Similar to their gastric counterparts, they often demonstrate mutations in the PDGFRA gene.6
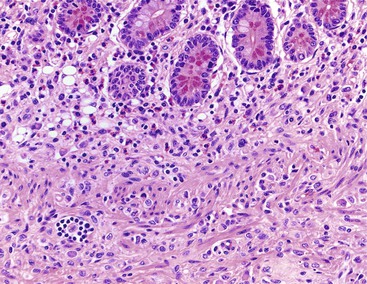
Inflammatory-type polyps of the small intestine may arise secondarily from other types of disease. Endometriosis of the bowel wall and submucosa may induce inflammatory changes including inflammatory-type polyps. Changes mimicking Crohn’s disease have also been reported.7,8 Endometriosis can also involve the mucosa, resembling dysplasia and even carcinoma (Fig. 21.4). Well-differentiated neuroendocrine tumors (NETs) may also induce an inflammatory reaction in the mucosa away from the main tumor, likely related to mucosal prolapse. These lesions contain numerous ectatic capillaries with admixed smooth muscle bundles and fibrosis.9
Hyperplasia and Heterotopia
Brunner Gland Hyperplasia/Hamartoma
Clinical and Endoscopic Features
Brunner glands are lobular collections of tubular glands within the duodenum; they are predominantly located in the duodenal submucosa. However, in the first part of the duodenum, Brunner glands commonly transgress the muscularis mucosae and extend into the lamina propria.10 Brunner glands within the lamina propria are less frequently observed in the middle to distal duodenum.
Occasionally, Brunner glands proliferate, creating small polypoid excrescences or imparting a nodular appearance to the mucosal surface (Fig. 21.5). These Brunner gland proliferative lesions are most commonly encountered in the duodenal bulb, usually as an incidental finding at endoscopy performed for other indications. However, they may be seen as one of a constellation of findings, including villous shortening and gastric foveolar mucous cell metaplasia of the villous epithelium, that are indicative of peptic injury of the duodenum (peptic duodenitis). Proliferative Brunner gland lesions have also been associated with end-stage renal disease and uremia.11,12
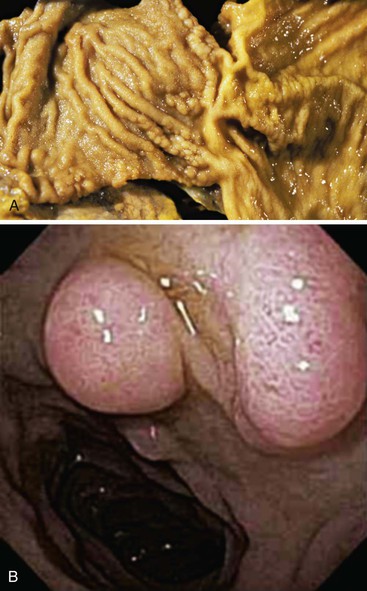
The nomenclature of Brunner gland proliferative lesions is not well established, and a number of diagnostic terms, including Brunner gland hyperplasia, Brunner gland adenoma, and Brunner gland hamartoma, have been used. The distinction between these diagnoses is arbitrary. No well-documented cases of either true glandular dysplasia or carcinoma arising in proliferative Brunner glands have been reported. Careful review of the limited literature reports of Brunner gland adenocarcinoma and dysplasia reveals that the dysplastic glandular epithelium involves the surface epithelium with likely secondary involvement of the underlying hyperplastic Brunner glands.13–17 In other cases, the adenocarcinomas appear to arise from pyloric gland adenomas and not from hyperplastic Brunner glands.16,17 Therefore, the term Brunner gland adenoma is potentially misleading, and we prefer to use the term Brunner gland hyperplasia to limit confusion among our clinicians and to highlight the non-neoplastic nature of the entity.
Some authors distinguish between Brunner gland hyperplasia and Brunner gland hamartoma based on the size of the lesion. However, the size cutoff between these two entities is arbitrary and not well established in the literature.18 The distinction is of no clinical significance, and a diagnosis of Brunner gland hyperplasia/hamartoma is usually sufficient in large polypoid lesions.
Pathologic Features
Brunner glands are composed of neutral, mucin-secreting, cuboidal to columnar cells with basally located nuclei; they are arranged in lobules containing thin, fibrous septa. Brunner glands are histologically indistinguishable from pyloric glands of the distal gastric mucosa. Indeed, biopsies of the gastroduodenal junction may demonstrate a gradual transition of these epithelial types. The predominantly submucosal location of Brunner glands helps to identify the biopsy as duodenal in origin.
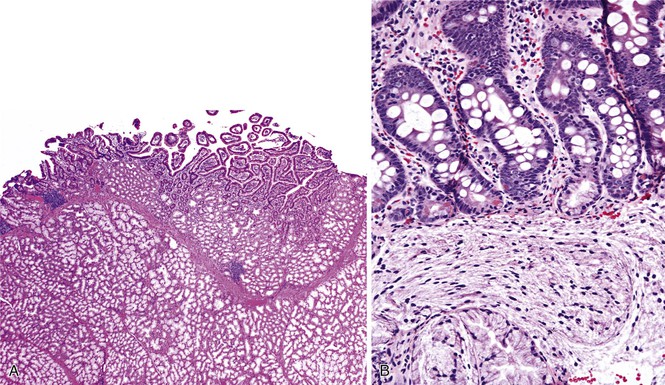
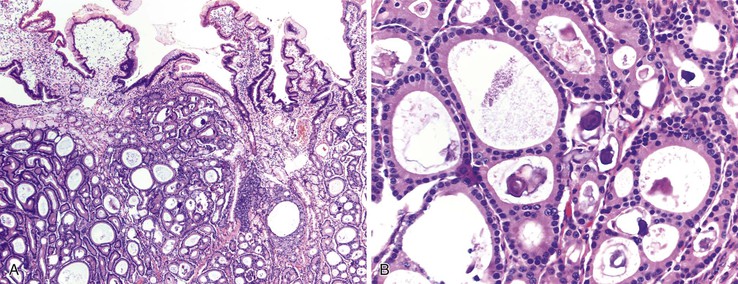
The diagnostic criteria for Brunner gland hyperplasia are subjective, because Brunner glands may be normally seen in the lamina propria of the duodenum, particularly within the duodenal bulb. In addition, proliferation of Brunner glands is commonly observed in peptic-type duodenal injury. We reserve the term Brunner gland hyperplasia for those endoscopically visible duodenal nodules that are found to contain lobules of Brunner glands within the mucosa in at least 50% of the length of a biopsy specimen (Fig. 21.6, A). Brunner gland hyperplasia may manifest as solitary or multiple nodules that are typically small (<1 cm) and characterized by lobules of glands that are increased in both size and number. The lobules extend into the mucosa and are separated by delicate fibrous septa. Cystic dilatation of Brunner glands may occur, and rare cases of Brunner gland cysts have been reported.19,20 The cells are cytologically bland with abundant neutral mucin and small, basally located nuclei with minimal to absent mitotic activity.
Large polyps (>2 cm) composed of Brunner glands may produce symptoms of obstruction, intussusceptions, melena, and anemia requiring endoscopic resection. Such polyps typically display more abundant fibromuscular stroma, likely the result of mucosal prolapse-type changes. For such large polyps, the diagnosis of Brunner gland hyperplasia/hamartoma may be appropriate, because it recognizes the arbitrary distinction between these two diagnoses.
In mucosal biopsy specimens, Brunner glands are commonly crushed, imparting a spindle cell appearance to the epithelial cells (see Fig. 21.6, B). Crushed Brunner glands may be mistaken for histiocytic collections, raising concern for Whipple disease, particularly given their intense positive reaction on periodic acid–Schiff (PAS) staining. The spindled appearance of crushed Brunner glands may also raise concern for a mesenchymal lesion. Careful histologic evaluation with comparison of adjacent partially crushed lobular collections of Brunner glands will allow for identification of this distortion artifact.
Differential Diagnosis
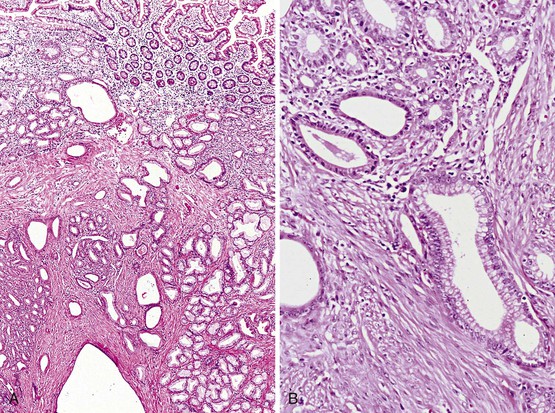
Histologic features of peptic-type injury of the duodenum include any of the following: active inflammation within the lamina propria or epithelium, Brunner gland hyperplasia (Fig. 21.7, A), gastric foveolar metaplasia of the surface epithelium (see Fig. 21.7, B), and hemorrhage and edema. Nodular duodenitis is frequently observed in patients with a history of peptic ulcer disease.21 In the setting of histologic findings indicative of peptic-type duodenal injury, the presence of Brunner gland hyperplasia should be attributed to peptic duodenitis.
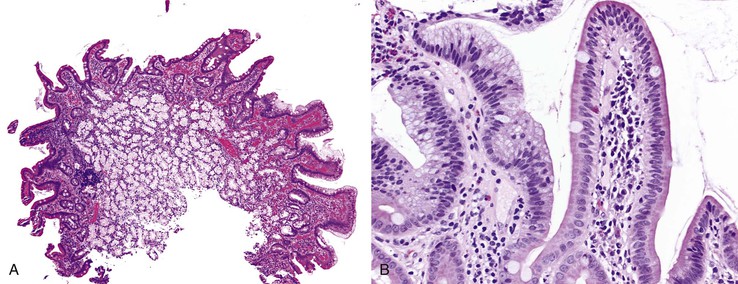
Pyloric gland adenoma is a relatively recently described adenoma subtype that can occur in the duodenum and not infrequently evolves into invasive adenocarcinoma. The distinction between pyloric gland adenoma and Brunner gland hyperplasia/hamartoma is difficult (see later discussion). Both express the mucin core peptide MUC6; however, pyloric gland adenomas usually also demonstrate labeling with MUC5AC, which is not typical of Brunner glands.22
Gastric Heterotopia
Clinical and Endoscopic Features
Gastric heterotopias in the duodenum are most commonly identified in the bulb and are usually incidental, small (<1.0 cm) nodules that may be multiple.23 Patients are typically asymptomatic, but larger polyps may manifest as masses and cause obstruction or intussusception.24,25 Peptic ulceration due to heterotopic oxyntic glands in the proximal duodenum is rare because the acid secretions are quickly diluted by the alkaline duodenal contents derived from the pancreatobiliary system. However, rare cases of massive GI bleeding have been reported, particularly in patients with extensive heterotopias.24,26 Gastric heterotopia is also associated with concurrent fundic gland polyps of the stomach, suggesting that the increased use of proton pump inhibitors (PPIs) in the general population may enhance its endoscopic detection.27
Pathologic Features
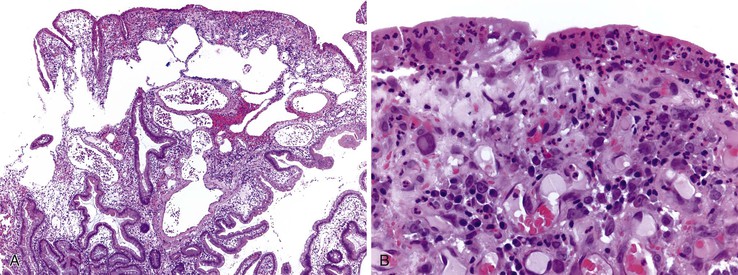
Biopsies of gastric heterotopias display well-organized oxyntic glands composed of chief and parietal cells (Fig. 21.8, A). The overlying surface often exhibits gastric foveolar-type mucinous epithelium with adjacent normal duodenal intestinal villi. Gastric heterotopia in the duodenum may harbor the same pathologic processes that affect the stomach, including Helicobacter pylori infection (see Fig. 21.8, B).27 Large gastric heterotopias may exhibit secondary mucosal prolapse-type changes including prominence of the muscularis mucosae, submucosal fibrosis, cystic dilatation of the oxyntic and mucinous glands, and surface epithelial hyperplasia.28
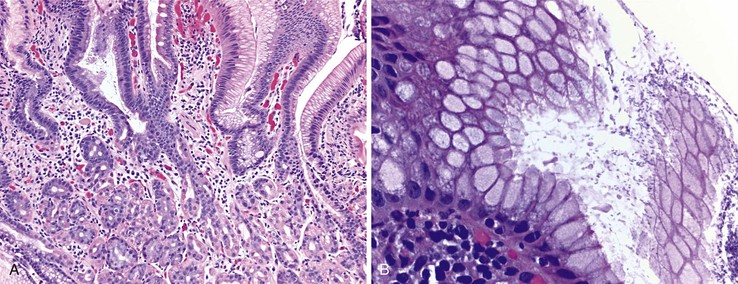
Differential Diagnosis
Gastric heterotopia differs from gastric foveolar metaplasia, which is hypothesized to be a response to inflammation caused by peptic injury associated with gastric H. pylori infection29 (Table 21.1). Histologically, peptic-type injury of the duodenum typically results in active inflammation within the lamina propria or epithelium and erosion or ulceration. The metaplastic gastric foveolar epithelium is not associated with the oxyntic glands characteristic of gastric heterotopia.
Table 21.1
Gastric Heterotopia Versus Gastric Metaplasia
| Gastric Heterotopia | Gastric Metaplasia | |
| Clinical presentation | Usually incidental | Epigastric pain |
| Endoscopic features | Nodular mucosa | Duodenitis |
| Histologic features | Oxyntic mucosa Normal duodenal mucosa |
Gastric foveolar (mucous) cells Villous blunting ± active inflammation Prominent Brunner glands |
| Clinical implications | None | Peptic injury |
Pancreatic Heterotopia
Clinical and Endoscopic Features
Pancreatic heterotopia represents the presence of pancreatic tissue outside the normal pancreas without any anatomic or vascular connection to the pancreas. It is synonymous with the terms pancreatic rest and ectopic pancreas. Pancreatic heterotopia of the small intestine is most commonly located in the proximal duodenum and can be seen either confined to the mucosa or within the muscularis propria. Pancreatic tissue may be normally identified in the region of the minor papilla and does not necessarily qualify as heterotopia.30 In most cases, pancreatic heterotopia is asymptomatic and only incidentally detected. Mural heterotopias develop most commonly in the peraampullary region and are more likely to be symptomatic due to duodenal obstruction, intussusception, and stricture or stenosis of the ampulla.31–33 Symptomatic mural pancreatic heterotopias often mimic a neoplastic process, leading to surgical resection,31 and an endoscopic biopsy diagnosis is not possible given the location of the lesion.
Pathologic Features
Pancreatic heterotopia may be composed of pancreatic acini, ducts, or islets, either alone or in combination (Fig. 21.9). Most frequently, pancreatic ducts are identified, with a variable number of acinar cells and islets and no relationship between symptoms and the cell type present.34 Mural lesions are often histologically indistinguishable from normal pancreas. Most mucosal-based lesions are unassociated with inflammation, being present in otherwise normal mucosa of the duodenum. Rarely, ductal adenocarcinoma, mucinous neoplasms, and NETs have been reported to develop in pancreatic heterotopia.34–37
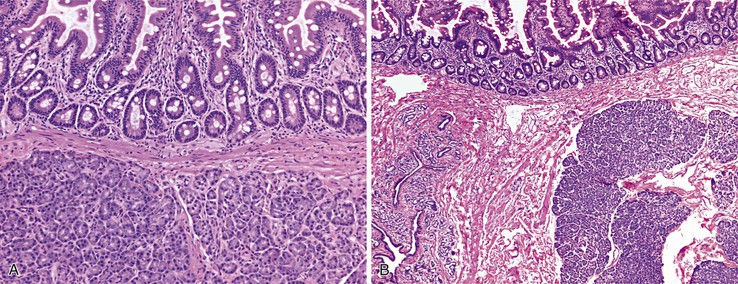
Differential Diagnosis
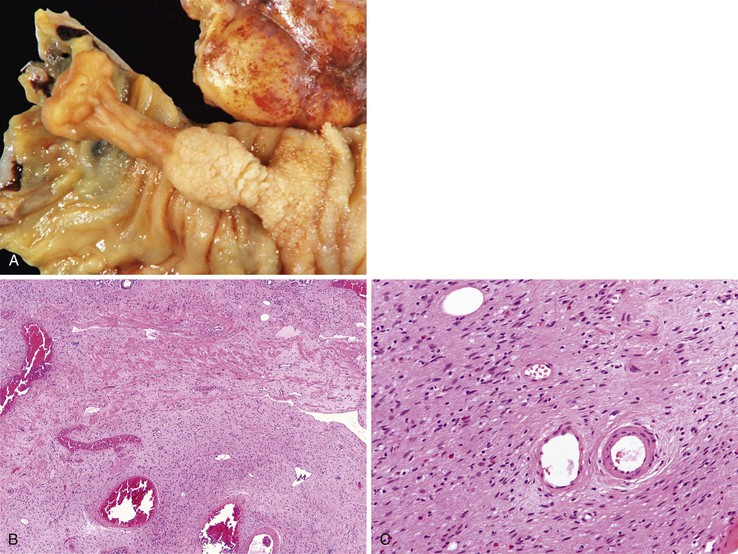
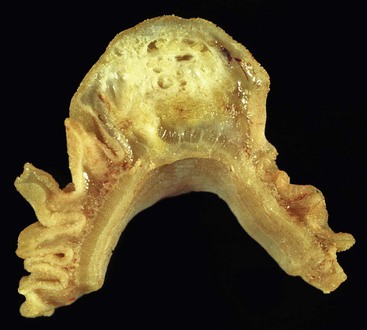
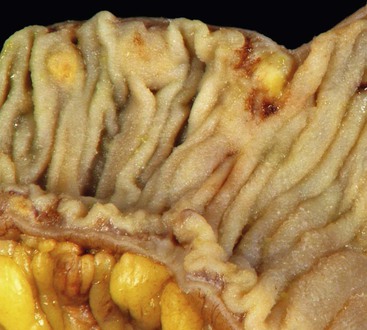
Peraampullary duodenal wall cyst (groove pancreatitis) is not often confused with mural pancreatic heterotopias. Although both may contain pancreatic ducts, acini, and islets, peraampullary duodenal wall cyst has characteristic clinicopathologic features. Most patients are young to middle-aged men who report alcohol use and typically present with pain and vomiting due to duodenal stenosis.38–40 Jaundice may also be seen late in the disease course as the result of compression of the common bile duct.38 The lesion typically involves the area of the minor papilla where pancreatic tissue may normally be identified. Fibrosis and inflammation occur in the “groove” between the superior aspect of the pancreatic head, the common bile duct, and the duodenum. In addition to groove pancreatitis, alternative names include cystic dystrophy of the duodenal wall and paraduodenal pancreatitis.30 Most cases are characterized by multiple or solitary cysts varying in size from 1 to 10 cm within the duodenal wall and pancreas (Fig. 21.10). “Solid” types have also been described and are characterized by fibrotic thickening of the duodenal wall with small (<1 cm) cysts.39 A reactive myofibroblastic spindle cell proliferation is usually present in the duodenal wall, whereas the fibrosis in the groove area is more paucicellular and hyalinized.41 Cystically dilated ducts partially lined by ductal epithelium and containing inspissated eosinophilic material are characteristic (Fig. 21.11). Squamous metaplasia, granulation tissue, calcium deposition, and giant cell reaction are also frequently encountered.39,41 It has been speculated that this disorder is a localized form of alcohol-induced pancreatitis occurring in pancreatic tissue present in the area of the minor papilla.30
Hamartomatous Polyps
By definition, hamartomatous polyps are a haphazard arrangement of normal stromal and epithelial elements. Hamartomatous polyps of the small bowel occur mainly in the setting of polyposis syndromes including Peutz-Jeghers syndrome (PJS), juvenile polyposis, and PTEN hamartoma tumor syndrome. The latter two syndromes have similar-appearing hamartomatous polyps. Correct classification of these polyps is critical to management, because many of these syndromes carry unique risks of GI and extra-GI malignancy (Table 21.2). Rarely, sporadic hamartomatous polyps resembling their syndromic counterparts occur.42 Other sporadic hamartomatous lesions have been described, including those made predominantly of mesenchymal elements such as adipose tissue, vessels, and nerves. There are numerous reports of neuromuscular and vascular hamartomas in the literature; however, these lesions are not hamartomas but are likely related to injury from nonsteroidal antiinflamatory drugs (diaphragm disease) (see Chapters 12 and 16).43,44
Table 21.2
Hamartomatous Syndromes Affecting the Small Bowel
| Syndrome | Polyp Type | Small Bowel Involvement | Histology | Dysplasia In Polyp* | Gene Mutations | Risk of Small Bowel Adenocarcinoma |
| Peutz-Jeghers syndrome | Peutz-Jeghers polyp | Frequent (65%) | Normal mucosal elements with arborizing smooth muscle | Rare; possible increased risk of dysplasia in nonpolypoid mucosa | STK11/LKB1 | 13% |
| Juvenile polyposis syndrome | Juvenile polyp | Infrequent (<20%) | Inflamed, edematous lamina propria, cystic glands, often ulcerated | Rare | SMAD4 and BMPR1A | ~10% |
| PTEN hamartoma tumor syndrome | Multiple types: juvenile polyps, ganglioneuromas, lipomas, and adenomas | Variable (33-66%) | Variable | Rare | PTEN | Unclear, likely low |
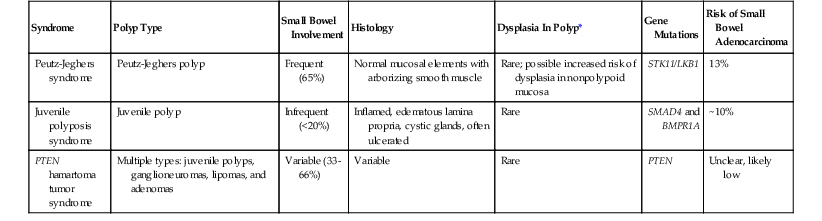
* Refers to dysplasia in polyps other than an adenoma.
PTEN, Phosphatase and tensin homolog.
Peutz-Jeghers Polyps
Clinical and Endoscopic Features
PJS is an autosomal dominant hamartomatous polyposis syndrome with a prevalence of 1 in 200,000.45,46 The majority of patients have positive family histories; however, 25% of cases develop de novo. A diagnosis of PJS should be considered if one of the following criteria is met: (1) three or more histologically confirmed Peutz-Jeghers polyps, (2) any number of Peutz-Jeghers polyps with a family history of PJS, (3) characteristic mucocutaneous pigmentation with a family history of PJS, or (4) any number of Peutz-Jeghers polyps and characteristic mucocutaneous pigmentation.45,47 More than 90% of patients with PJS develop small intestinal polyps, most commonly in the jejunum, followed by the ileum and the duodenum. PJS patients are often diagnosed at an early age (approximately 20 years), because these polyps often cause abdominal pain, obstruction due to intussusception, and bleeding.45,48 Surgical intervention often occurs in these settings. Indeed, patients are at risk for short gut syndrome due to the numerous GI surgeries undertaken to resect these polyps.49 Patients with PJS have a significantly increased risk of intestinal adenocarcinoma, particularly of the colon (cumulative risk of 40%) but also of the small bowel (13% cumulative risk).45,50–59 For this reason, patients are enrolled in an endoscopic screening protocol at a very young age.45 Affected individuals are also at increased risk for other tumors: breast (54% cumulative risk for females), endometrial, cervical (adenoma malignum), pancreatic (36% cumulative risk), ovarian (sex cord/stromal tumor with annular tubules), and testicular (large-cell calcifying Sertoli cell tumor).50,57,58,60–62 The cumulative lifetime risk for any cancer approaches 90%.
Peutz-Jeghers polyps have an irregular, multilobulated endoscopic appearance and lack the velvety texture typical of adenomas (Fig. 21.12). Because they contain prominent smooth muscle, they are tightly anchored to the bowel. Unlike juvenile polyps, they rarely autoamputate.
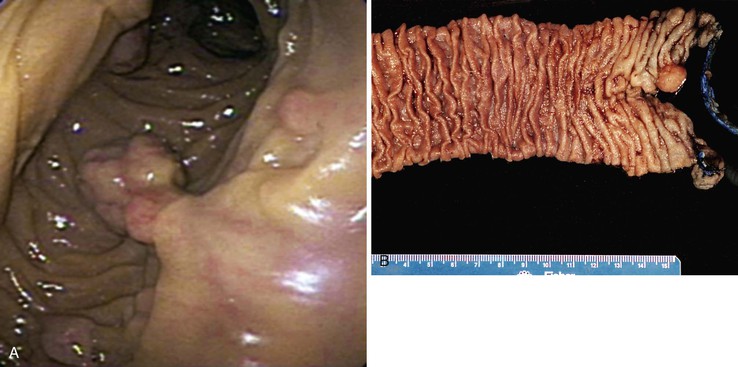
In 1998, the gene responsible for the majority of sporadic and inherited cases of PJS was identified as STK11 (also known as LKB1), a serine/threonine kinase located on chromosome 19p13.3.48,63–66 STK11 is a tumor suppressor gene that is involved in a wide spectrum of cellular functions, including cellular proliferation, cell polarity, and apoptosis.67–69 STK11 has been shown to interact with other tumor suppressors, including TP53 and PTEN.67
There have been case reports of sporadic Peutz-Jeghers polyps, but this is somewhat controversial. In a study at a tertiary care center, three patients with potential sporadic Peutz-Jeghers polyps were identified, but two of them had other features suggestive of PJS, although strict clinical criteria were not met. The authors concluded that if sporadic Peutz-Jeghers polyps do exist, they are extremely rare.42
Pathologic Features
The hamartomatous polyps of PJS are histologically distinctive and consist of disorganized mucosa with prominent smooth muscle bundles. The smooth muscle fibers of the muscularis mucosae form thick cores and permeate the polyps in an arborizing fashion (Fig. 21.13, A). The epithelium lining these polyps is similar to the normal mucosa in that there are goblet cells, absorptive enterocytes, endocrine cells, and Paneth cells (see Fig. 21.13, B). Rarely, metaplastic bone formation has been reported in these polyps.70 As much as 10% of Peutz-Jeghers polyps have foci of misplaced epithelium, in which the epithelium is located within the submucosa or the muscularis propria or both. This phenomenon is encountered more frequently in polyps larger than 3 cm in diameter (see Fig. 21.13, C).71
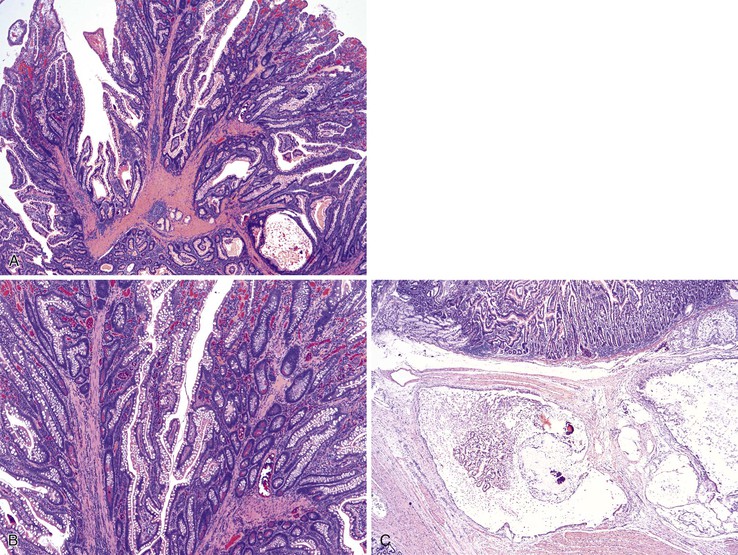
The development of Peutz-Jeghers polyps is the subject of some controversy. Given the prominent smooth muscle in these polyps and the presence of misplaced epithelium, some authors suggest that patients with PJS are prone to mucosal prolapse.72 Indeed, the differential diagnosis of Peutz-Jeghers polyps, particularly in the colon, is mucosal prolapse polyps. However, hemorrhage and hemosiderin deposits, commonly seen in prolapse polyps, are not prominent in Peutz-Jeghers polyps.
The sequence of events that results in GI dysplasia and carcinoma is also unclear in relation to Peutz-Jeghers polyps. One occasionally encounters dysplasia and even carcinoma in a Peutz-Jeghers polyp; however, in a study of 2461 Peutz-Jeghers polyps from 63 patients, only 6 polyps contained foci of dysplasia.73 These results argue that the hamartoma-adenoma-carcinoma hypothesis proposed by some authors rarely occurs.74 Peutz-Jeghers polyps have also been shown to be polyclonal, arguing against the possibility of malignant potential. Given these results, it is possible that carcinoma develops in surrounding mucosa that may have an accelerated pathway to dysplasia and carcinoma.57,72
In any case, when one encounters dysplasia in a Peutz-Jeghers polyp, the dysplasia should be classified according to a two-tiered system (i.e., low- or high-grade dysplasia). Care must be taken to not interpret misplaced epithelium as invasive adenocarcinoma.71 The misplaced epithelium should not have desmoplastic stroma or an infiltrative growth pattern. The epithelial cells should have a similar cytology to other parts of the polyps, and the glands should be invested with lamina propria.
Juvenile Polyps
Clinical and Endoscopic Features
Juvenile polyps of the small intestine are rare and occur in two polyposis syndromes: juvenile polyposis syndrome and PTEN hamartoma tumor syndrome.75 Juvenile-type polyps can also be seen in Cronkhite-Canada syndrome, an extremely rare, noninherited disorder associated with diffuse GI polyposis.76,77 Juvenile polyps tend to be friable and undergo autoamputation. Endoscopically, they appear smooth and often have evidence of hemorrhage. As a result, patients typically present with complaints related to bleeding, such as occult blood loss, hematochezia, fatigue, or anemia.
First described in 1964, juvenile polyposis syndrome is an extremely rare (1 per 100,000 births), autosomal dominant polyposis syndrome with variable penetrance. Three clinical phenotypes of juvenile polyposis syndrome have been described: infantile juvenile polyposis, juvenile polyposis coli, and generalized juvenile polyposis. Infantile juvenile polyposis syndrome, the most severe type, is characterized by early and diffuse involvement of the entire GI tract. Because these hamartomatous polyps have surface erosion, affected patients usually present with GI bleeding and anemia. Juvenile polyposis syndrome is also associated with a significantly increased risk of adenocarcinoma in involved GI sites. The colon is the site most commonly involved by disease, and the cumulative lifetime risk for colorectal cancer is 40%, with a median age at onset of 44 years.78 The lifetime risk of developing carcinomas of the stomach, small intestine, or pancreas is approximately 10% to 15%.79 Prevention of intestinal adenocarcinoma mandates intensive screening, with colonoscopy and upper GI endoscopy every 2 years, beginning at age 15.75 Severe GI bleeding may necessitate immediate surgery. Progression to dysplasia in a polyp is not necessarily an indication for surgery, provided that it can be completely removed. Given the increased risk of adenocarcinoma, genetic screening of family members of affected individuals is essential.
Approximately 25% of patients with juvenile polyposis syndrome have a negative family history, and their polyps presumably arise from a de novo mutation. Mutations in two genes have been described in patients with juvenile polyposis: SMAD4 (also known as MADH4 or DPC4) and BMPR1A (also known as ALK3).80–85 The proteins encoded by these genes are involved in the transforming growth factor-β (TGF-β) signal transduction pathway. Because the TGF-β signaling pathway mediates growth inhibitory signals, proteins in this pathway function as tumor suppressors. Mutations in SMAD4 have been shown to be more common in patients with upper GI polyps, whereas patients with BMPR1A mutations are more likely to present at a younger age than those with mutations in SMAD4.86
The term PTEN hamartoma tumor syndrome was coined to encompass all hamartoma syndromes that arise through mutations in the tumor suppressor, PTEN. This term encompasses Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome (BRRS), and Proteus syndrome, among others.75,87 Patients with these syndromes have a wide variety of GI, skin, and soft tissue tumors and an increased risk for carcinomas of the colorectum, thyroid, breast, or endometrium.88 Cowden syndrome is 50% less common than juvenile polyposis syndrome and is characterized by multiple hamartomatous tumors of endodermal, mesodermal, and ectodermal origin. The most distinctive lesions are mucocutaneous—in particular, trichlemmomas, acral keratosis, subcutaneous lipomas, palmarplantar keratoses, oral cobblestoning, and oral papillomas. Colonic polyps in Cowden syndrome are common and range from ganglioneuromas to adenomas, lipomas, hyperplastic polyps, and juvenile polyps. Polyps of the small intestine are more rare but do occur, with frequencies between 37% and 66%.89,90 BRRS is also associated with juvenile polyps of the GI tract (mostly limited to ileum and colon); however, these patients also have neurologic findings, including macrocephaly and slowed psychomotor development. Some BRRS patients also develop extraintestinal hamartomatous tumors similar to those seen in patients with Cowden syndrome.75
Pathologic Features
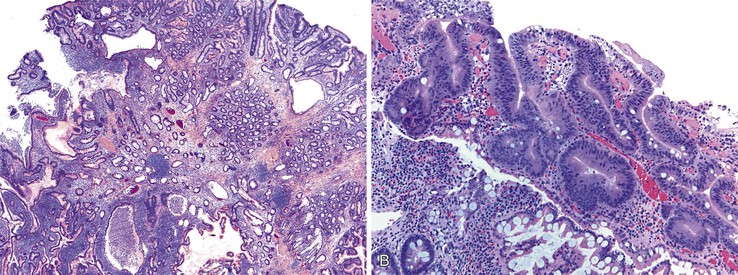
Juvenile polyps are pedunculated and are histologically characterized by abundant lamina propria, dilated mucin-filled crypts, active inflammation, and surface erosion (Fig. 21.14). The epithelium in these polyps can contain adsorptive cells, goblet cells, endocrine cells, and Paneth cells. Often, eosinophils are prominent. Unlike Peutz-Jeghers polyps, juvenile polyps lack prominent smooth muscle bundles. Juvenile polyps in the small bowel are very similar to inflammatory-type polyps. The distinction between these two polyps is mostly based on the clinical context in which they arise. In a patient with a known polyposis syndrome, the diagnosis of a juvenile polyp is appropriate. In cases where multiple inflammatory or juvenile polyps are seen, the possibility of a polyposis syndrome should considered. If a single juvenile or inflammatory polyp is encountered, one should be cautious in suggesting a polyposis syndrome.
Benign Epithelial Neoplasms
Adenomas of the Small Intestine
Clinical and Endoscopic Features
Adenomas account for approximately 25% of benign neoplasms of the small intestine and demonstrate a distinct predilection for the ampulla and the peraampullary region. Most are sporadic lesions occurring in older individuals, but they also occur in patients with adenomatous polyposis syndromes, including familial adenomatous polyposis (FAP) and MUTYH-associated polyposis (MAP). Approximately 17% of patients with MAP have duodenal polyposis, and their lifetime risk of duodenal cancer is 4%.91,92 Small bowel adenomas also occur in Lynch syndrome. Unlike adenomas in MAP and FAP, those in Lynch may not have such a strong predilection for the proximal small bowel and ampulla. As in MAP, the lifetime risk of small bowel adenocarcinoma in Lynch syndrome is 4%.93 Given that adenomas may not be amenable to upper endoscopy, capsule endoscopy has been used to identify occult adenomas and carcinomas in Lynch syndrome94; however, this is currently not recommended for routine screening.93
Although some adenomas located distal to the ampulla produce luminal obstructive symptoms or occult blood loss, most are asymptomatic and clinically silent until complicated by the development of carcinoma. In contrast, ampullary and peraampullary adenomas can produce obstructive jaundice before malignant transformation and become symptomatic earlier in their evolution.
Pathologic Features
With the exception of some ampullary neoplasms, most adenomas of the small intestine are endoscopically and histologically similar to those of the colon. Larger adenomas (>1 cm) are more likely to harbor areas of high-grade dysplasia, and approximately 50% have a villous component, consisting of long papillary projections lined by columnar, mucin-depleted epithelial cells with enlarged, hyperchromatic nuclei. Paneth cells and endocrine cells are often more numerous in adenomas of the small intestine compared with those of the colon. The criteria for grading dysplasia in adenomas of the small intestine are similar to those for colonic adenomas. Low-grade dysplasia is defined as nuclear stratification confined to the lower half of the cells in the absence of complex architectural changes (Fig. 21.15, A and B). High-grade dysplasia is characterized by the presence of marked nuclear enlargement with loss of nuclear polarity and irregular nuclear membranes. These nuclear changes are often accompanied by complex architectural abnormalities such as cribriforming and glandular crowding (see Fig. 21.15, C). The terms severe dysplasia and carcinoma in situ should no longer be used to describe these changes.
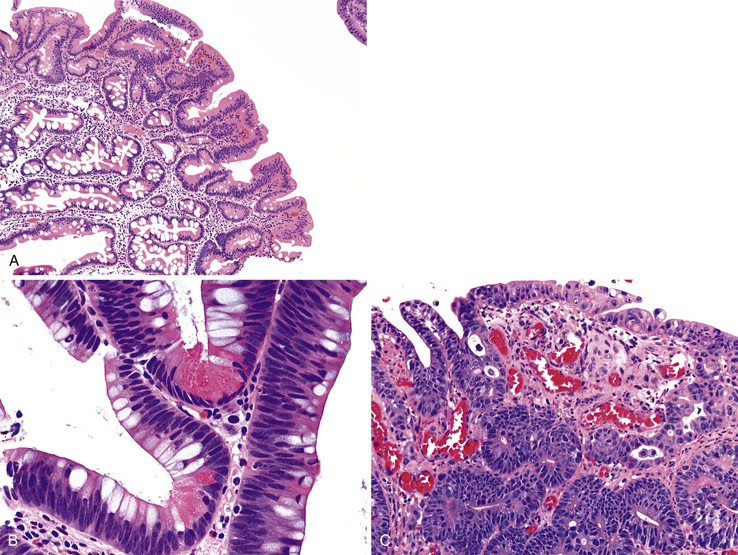
Unlike adenomas of the colon, but similar to those of the esophagus and stomach, the lamina propria in adenomas of the small intestine contains a rich lymphatic network. When dysplastic epithelial cells break through the basement membrane and infiltrate the lamina propria, the term intramucosal adenocarcinoma is appropriate. Intramucosal adenocarcinoma of the small intestine carries a risk of lymph node metastasis and is staged as pT1a carcinoma, according to the seventh edition of the Cancer Staging Manual of the American Joint Committee on Cancer.95
Ampullary and Peraampullary Adenomas
Clinical and Endoscopic Features
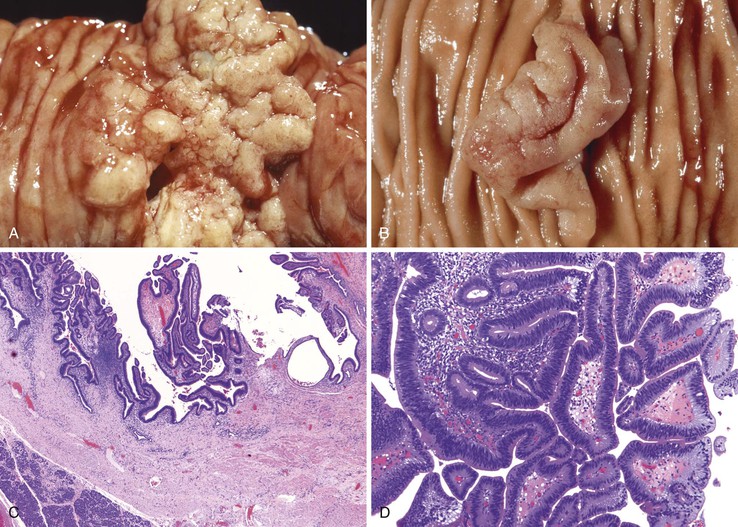
Ampullary adenomas typically occur in older adults (mean age, 64 years), and most are sporadic96 (see also Chapter 41). Small adenomas of the ampulla and peraampullary region appear as polypoid excrescences or as a subtle prominence of the ampulla, whereas larger adenomas are velvety, flat lesions (“carpet adenomas”) at high risk for the development of invasive adenocarcinoma (Fig. 21.16). Several investigators have postulated that the ampulla may be prone to neoplastic transformation because it is chronically irritated by pancreatic juices and bile salts, resulting in long-standing injury to the mucosa and culminating in epithelial cell dysplasia.97,98
Pathologic Features
Adenomas of the ampulla can be divided into intestinal and pancreaticobiliary types (although mixed lesions also occur). The intestinal-type adenomas are more common (~75%) and resemble colonic and peraampullary/duodenal adenomas (Fig. 21.16, C and D). Pancreaticobiliary-type adenomas—or neoplasms, as preferred by the current World Health Organization (WHO) classification99—often have prominent papillary architecture and resemble intraductal papillary neoplasms of the pancreatic duct. The papillary projections are lined by a monolayer of cuboidal epithelial cells with round nuclei and eosinophilic cytoplasm (see Fig. 21.17, A and B). These lesions are often high-grade with complex architecture including extensive arborization of the papillae, cribriforming, and marked nuclear atypia. Some authors have used immunohistochemistry to differentiate these histologic subtypes. Intestinal-type adenomas express apomucin MUC2 and the transcription factor CDX2. Pancreaticobiliary-type neoplasms express MUC1, MUC5AC, and MUC6.100 However, there is considerable overlap in the expression of these proteins in these lesions, limiting their clinical utility.
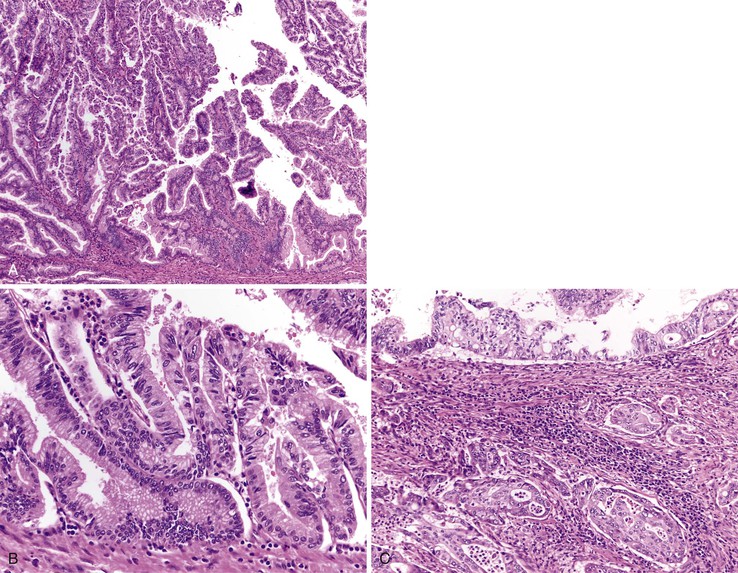
Both intestinal and pancreaticobiliary mass-forming lesions of the ampulla are often associated with adjacent invasive adenocarcinoma (~80%). Although most adenocarcinomas of the ampulla arise from adenomas, nonpolypoid (or flat) dysplastic lesions of the ampulla also occur. These dysplastic lesions are almost always associated with an adjacent invasive adenocarcinoma. Some of these lesions have a micropapillary architecture lined by dysplastic cuboidal to columnar cells.
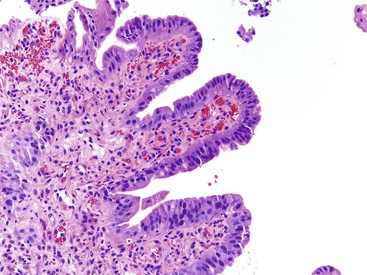
The evaluation of biopsy or resection specimens of ampullary and peraampullary adenomas may be difficult for several reasons. Ampullary ducts and glands are in close proximity to bundles of smooth muscle, and when dysplastic epithelium extends into these structures, a mistaken diagnosis of invasive adenocarcinoma may be rendered (Fig. 21.18). The lobular architecture and lack of desmoplasia can be helpful in distinguishing this finding from invasive adenocarcinoma. As with their colonic counterparts, misplacement of the dysplastic epithelium into the submucosa may occur as a consequence of mechanical injury or prolapse in these adenomas. The presence of lamina propria around these dysplastic glands and the presence of hemosiderin deposits and acellular mucin pools are helpful features to differentiate epithelial misplacement from invasive adenocarcinoma. Finally, because many patients with ampullary lesions have obstructive jaundice, biliary stents are often placed. Stents cause inflammatory epithelial injury and erosions of the ampullary epithelium with concomitant reactive and regenerative epithelial changes, as well as inflammation of the underlying stroma (Fig. 21.19). This constellation of findings may mimic dysplasia or even invasive cancer. Therefore, it is imperative to know the patient’s status regarding stenting of the common bile duct to evaluate the specimen properly and avoid the pitfall of overdiagnosing neoplasia. The presence of active inflammation and foveolar metaplasia should make one cautious in interpreting the epithelial changes as dysplastic.
Immunohistochemical and Molecular Features
Sporadic peraampullary and ampullary adenomas harbor mutations in the APC gene (discussed in the next section) in approximately two thirds of cases, although loss of heterozygosity at chromosome 5q is infrequent.88 Almost half demonstrate KRAS mutations, but TP53 mutations are usually lacking, particularly in the absence of high-grade dysplasia.88–91 Mutations in BRAF are only rarely seen in ampullary and peraampullary adenomas. Therefore, the molecular features of sporadic ampullary adenomas are similar to those of colonic adenomas and small intestinal adenomas associated with FAP.
Familial Adenomatous Polyposis
Clinical and Endoscopic Features
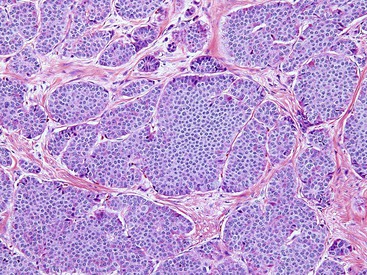
FAP is an autosomal dominant inherited disease occurring in 1 to 2 of 100,000 persons.87 Fortunately, FAP is relatively easy to diagnose clinically, because most affected patients have strong family histories and commonly have 100 to 1000 colonic polyps (classic FAP is defined as the presence of >100 colonic adenomas). An attenuated form of FAP has been described that manifests with significantly fewer colonic polyps, often more than 15 but always less than 100. Small intestinal adenomas occur in approximately 90% of FAP patients (Fig. 21.20). Typically, they are more prominent in the duodenum and ampulla.101 The incidence of small bowel or ampullary carcinoma was estimated to be 4.5% among those patients enrolled in a screening program.102,103

In 1987, the gene responsible for FAP was localized to chromosome 5q21, and in 1991 it was identified as the adenomatous polyposis coli gene (APC).104 The APC gene contains 15 transcribed exons encoding a 312-kDa tumor suppressor. Mutations in APC constitute the initial step in the development of colorectal carcinoma in FAP patients. APC is involved in a wide variety of cellular functions, including cell adhesion, migration, chromosome segregation, and signal transduction. One of the most important roles of APC is to regulate the activity of β-catenin, a protein involved in the Wnt signaling pathway and cellular adhesion. In conjunction with other proteins, APC is essential in inducing the phosphorylation and subsequent degradation of β-catenin. Loss of APC function through mutation leads to accumulation of β-catenin, which subsequently translocates to the nucleus, leading to transcription of many proto-oncogenes such as MYC. Because APC also regulates the microtubule spindle apparatus, mutations predispose to aneuploidy and further neoplastic transformation.105 More than 700 mutations in APC have been identified in FAP patients. Interestingly, the location of the mutation has striking effects on the phenotype of FAP. Particularly those occurring in the central portion of the APC gene (codons 279-1309) correlate with increased numbers of duodenal polyps as well as the size of the adenomas.106
Risk of Malignancy
In the early 1990s, Spigelman developed a classification scheme based on the number and size of adenomas, their architectural features, and the degree of dysplasia, in an attempt at risk stratification of FAP patients (Table 21.3).98 Originally, the dysplasia was classified as mild, moderate, or severe; however, more recent studies have modified the original Spigelman classification to reflect the division of dysplasia into high-grade and low-grade.107 The risk of progression to duodenal or ampullary adenocarcinoma is likely strongly correlated to the Spigelman stage at initial endoscopy. In a study of 114 patients with FAP, 6 duodenal or ampullary carcinomas were identified. In five cases, the original Spigelman stage was III (one case) or IV (four cases). Only 17% of patients progressed to a higher Spigelman stage during the surveillance program.108 A more recent study demonstrated a much higher rate of progression in Spigelman stage (40%); however, no patient developed adenocarcinoma.107 Given that the Spigelman stage depends on villousity and degree of dysplasia, it is possible that interobserver variability among pathologists could account for these differences. Currently, endoscopic mucosal resection or prophylactic pancreaticoduodenectomy is considered for patients with advanced Spigelman stage.
Table 21.3
Modified Spigelman Score*
| Factor | 1 Point | 2 Points | 3 Points |
| Polyp number | 1-4 | 5-20 | >20 |
| Polyp size, mm | 1-4 | 5-10 | >10 |
| Architecture | Tubular | Tubulovillous | Villous |
| Dysplasia | Low-grade | — | High-grade |

* Stage grouping: stage 0, no polyps; stage I, 1-4 points; stage II, 5-6 points; stage III, 7-8 points; stage IV, 9-12 points.
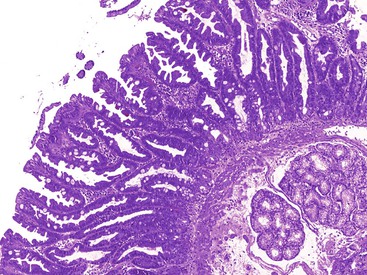
Pyloric Gland Adenoma
Pyloric gland adenomas are composed of tightly packed glands lined by cuboidal epithelium with an even monolayer of round nuclei with abundant mucinous cytoplasm reminiscent of pyloric-type glands (Fig. 21.21, A).109,110 These polyps are likely more common in the stomach, but 46% of pyloric gland adenomas occurred in the duodenum in one study.109 Given their resemblance to Brunner glands, the distinction between pyloric gland adenoma and Brunner gland hamartoma/hyperplasia can be difficult. However, pyloric gland adenomas often do not have overlying intestinalized epithelium when they occur in the duodenum. Furthermore, there is no lobular configuration in these lesions, in contrast to Brunner gland hamartoma/hyperplasia.
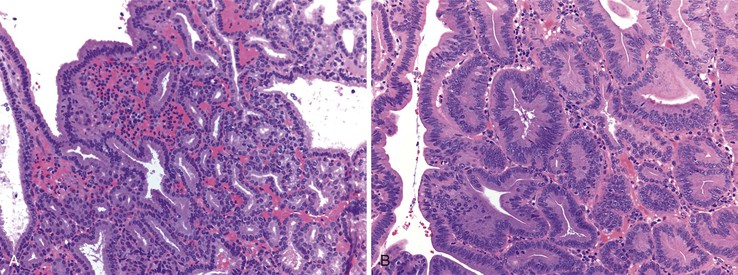
By definition, these lesions are neoplastic. However, despite being called adenomas, some are fairly bland without areas of conventional dysplasia. Therefore, nondysplastic pyloric gland adenomas do occur. However, often there are areas of dysplasia (classified as either low-grade or high-grade) (see Fig. 21.21, B). Between 10% and 30% of pyloric gland adenomas are associated with invasive adenocarcinoma.
Hyperplastic Polyp
Recent reports have described small intestinal polyps with morphologic features resembling colonic microvesicular hyperplastic polyps with prominent epithelial serrations and abundant cytoplasm with small mucin droplets (Fig. 21.22).111,112 In a series of nine polyps, five were found in the second portion of the duodenum. Molecular analysis performed on six of these polyps found a V600E BRAF mutation in two polyps and KRAS mutations in another two polyps.112 These polyps also expressed gastric mucins similar to serrated colorectal polyps. Although these findings are suggestive, additional studies need to be performed to further characterize these lesions.
Neuroendocrine Tumors
A detailed discussion of NETs of the GI tract is provided in Chapter 29. Although alternative terms, such as neuroendocrine neoplasm and endocrine neoplasm, are also acceptable and strongly advocated by some authors, for the sake of clarity and in keeping with the WHO classification, we use the term neuroendocrine tumor (NET).113 The GI tract is a common location for NETs, and they occur predominantly in the small intestine.114 In the small intestine, most NETs are encountered in the ileum, whereas only 2% to 3% occur in the duodenum.115 NETs of the duodenum and proximal jejunum (i.e., foregut NETs) are frequently associated with characteristic clinical and pathologic features distinct from NETs of the distal jejunum and ileum (midgut NETs). Duodenal NETs can be associated with clinical syndromes due to hormone hypersecretion detected by serologic evaluation for elevated circulating hormones. Immunohistochemical staining for peptide hormones can be performed to confirm the clinical impression of a syndromic NET. However, routine immunohistochemical staining for peptide hormones is not suggested for clinically nonfunctional NETs and can be potentially confusing. If peptide hormone immunohistochemistry is performed and demonstrates that the majority of the tumor cells exhibit hormone production, then it is acceptable but not necessary to supplement the diagnosis of NET to reflect the corresponding subtype (e.g., gastrin-producing NET).113 However, specific functional terms such as “gastrinoma” should not be used to describe NETs in the absence of clinical and serologic findings indicative of a hormonal syndrome. Although the functional status of a NET is defined by the clinical and serologic findings, it is useful to divide NETs into categories based on their location in the small intestine and the corresponding cell subtype, because this allows for discussion of site-related differences among NETs (Table 21.4).
Table 21.4
Clinicopathologic Features of NETs according to Site and Hormone Production
| Tumor Type | Anatomic Site Predilection | Associations | Clinical Course |
| Gastrin-producing NET | Duodenum, pancreas, and lymph nodes (primary tumor may be microscopic) | Functional: MEN1 (25%) | Functional: frequent regional and distant metastasis |
| Nonfunctional: Helicobacter infection, long-term PPI? | Nonfunctional: indolent | ||
| Somatostatin-producing NET | Ampulla/peraampullary area and pancreas | NF type 1, adrenal pheochromocytomas | Typically indolent, but larger tumors may metastasize |
| Gangliocytic paraganglioma | Ampulla/peraampullary area | NF type 1 | Typically indolent, but larger tumors may metastasize |
| Serotonin-producing NET | Distal jejunum and ileum | True carcinoid tumor due to serotonin production | Malignant with frequent regional and distant metastasis |
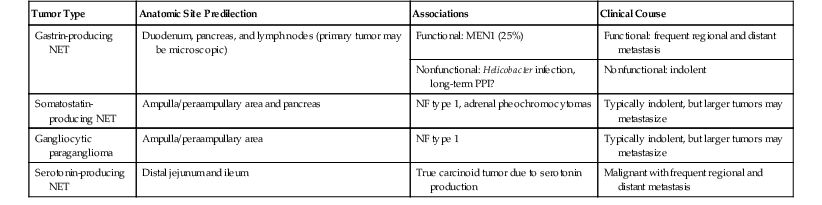
NET, Neuroendocrine tumor; MEN1, multiple endocrine neoplasia type 1; NF, neurofibromatosis; PPI, proton pump inhibitors.
NETs of the small intestine are further classified by using the criteria applicable to all GI and pancreatic NETs published by the WHO in 2010.113 Grading is performed primarily by assessment of mitotic activity and the Ki67 proliferation index. A NET is defined as a well-differentiated neuroendocrine neoplasm expressing general markers of neuroendocrine differentiation (such as chromogranin A and synaptophysin) with a low number of mitoses (<20 per 10 high-power fields [HPF]) and a low Ki67 proliferation index (≤20%); this encompasses G1 NETs (<2 mitoses per 10 HPF and ≤2% Ki67 index) and G2 NETs (2 to 20 mitoses per 10 HPF, or 3% to 20% Ki67 index, or both) (Table 21.5).
Table 21.5
World Health Organization (WHO) 2010 Classification of Neuroendocrine Tumors
| WHO Grade | Mitotic Count | Ki67 Index | Terminology |
| G1 | <2 per 10 HPF | ≤2% | Neuroendocrine tumor |
| G2 | 2-20 per 10 HPF | 3-20% | Neuroendocrine tumor |
| G3 | >20 per 10 HPF | >20% | Neuroendocrine carcinoma |

HPF, High-power field.
NETs of the Duodenum, Ampulla, and Proximal Jejunum
Gastrin-Producing NETs
Clinical Features
Gastrin-producing NETs represent the largest group NETs of the duodenum and proximal jejunum, accounting for approximately two thirds of NETs in the upper GI tract.116,117 Gastrin-producing NETs can be either nonfunctioning or associated with Zollinger-Ellison syndrome (ZES), a syndrome of hypergastrinemia, gastric hypersecretion, and refractory peptic ulcer disease.118 The functional status of the NET is defined by clinical and serologic findings and not by the immunohistochemical expression of peptide hormones. Gastrin-producing NETs develop in the pancreaticoduodenal region (gastrinoma triangle); the primary tumor is found with near-equal frequency in the pancreas and in the duodenum. Rarely, a gastrin-producing NET is identified in a lymph node in the gastrinoma triangle without an apparent duodenal or pancreatic primary tumor, suggesting the possibility of a primary lymph node gastrinoma.119 However, it is likely that so-called primary lymph node gastrinomas represent metastases from microscopic primary tumors that are easily overlooked on routine examination.120–124
The association of ZES is found in approximately 40% to 50% of duodenal gastrin-producing NETs.116,117,125 Most ZES NETs are sporadic, and approximately 25% are associated with multiple endocrine neoplasia type 1 (MEN1). Sporadic ZES NETs most commonly occur in the duodenum (60% to 75%) but can also be found in the pancreas.122,126 In contrast, most MEN1-associated ZES NETs occur in the duodenum rather than the pancreas, and they can frequently be multifocal within the duodenum.126
Nonfunctional gastrin-producing NETs typically occur in the duodenal bulb and are localized in the lamina propria and submucosa.127 Most nonfunctioning gastrin-producing NETs are clinically indolent, small (<1cm), and asymptomatic; they are often discovered during endoscopy for other reasons or during surgical resections for unrelated causes. Nonfunctioning gastrin-producing NETs of the duodenum have been associated with H. pylori infection of the stomach or long-term PPI therapy.127 It is postulated that H. pylori infection and PPI therapy result in G-cell hyperplasia within the duodenum, leading to formation of sporadic nonfunctional gastrin-producing NETs.127
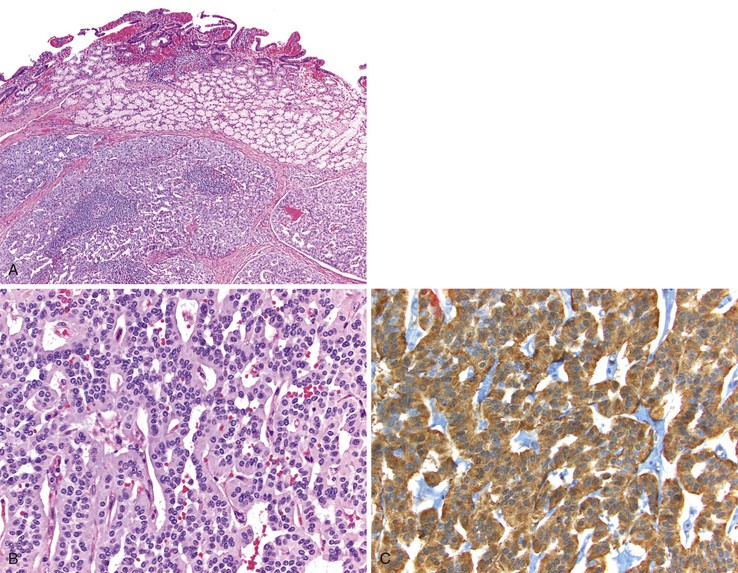
Pathologic Features
Gastrin-producing NETs resemble NETs elsewhere in the gastroenteropancreatic system and are composed of uniform cells with lightly eosinophilic cytoplasm arranged in anastomosing cords, trabeculae, and tubules (Fig. 21.23, A and B). Immunohistochemistry often reveals staining with synaptophysin, chromogranin A, and gastrin (see Fig. 21.23, C). Other peptides may be detected in a subset of tumor cells.116,117 Gastrin-producing NETs infrequently label with CDX2128 but may express PAX8 by immunohistochemistry.129,130 The nontumorous duodenum can demonstrate gastrin cell proliferation in MEN1-associated gastrinomas and in sporadic nonfunctional gastrin-producing NETs associated with H. pylori gastritis and long-term PPI therapy.127,131 However, sporadic ZES NETs typically do not exhibit proliferative gastrin cell changes in the nontumorous mucosa.131 Both sporadic and MEN1-associated ZES NETs may be multifocal. However, the pathogenesis varies in these two conditions. Multifocal sporadic ZES NETs from the same patient probably represent metastases, because they frequently demonstrate the same somatic MEN1 gene mutation and similar patterns of X-chromosome inactivation in tumors at different sites within the same patient.132 Multifocal ZES NETs in patients with MEN1 syndrome have varied genetic abnormalities and probably represent independent primary tumors.133
Natural History
Incidentally discovered, nonfunctioning gastrin-producing NETs of the duodenal bulb appear to be indolent tumors without significant risk of aggressive behavior based on early evidence.127 In contrast, ZES NETs may behave in a clinically malignant fashion. Patients with functional gastrin-producing NETs should be evaluated for MEN1 syndrome, because patients with this syndrome and their family members are at risk for synchronous or metachronous syndrome-related tumors and would benefit from close surveillance and genetic counseling.
The prognosis of sporadic and MEN1-associated duodenal gastrin-producing NETs is better than that of pancreatic gastrin-producing NETs.126 Patients who have sporadic ZES NETs with distant metastatic disease typically undergo surgical exploration and resection of the primary tumor and regional lymph nodes.118,122,124,134 Between 50% and 60% of sporadic duodenal ZES NETs are associated with regional lymph node metastases.124,134 The management of patients with MEN1 is more complicated, because surgery is rarely curative.135 Nevertheless, recent data suggest that patients with MEN1 syndrome benefit from surgical removal of the NET, compared with medical management alone.124,136
Somatostatin-Producing NETs
Clinical Features
Somatostatin-producing NETs have a predilection to affect the ampulla and peraampullary region (Fig. 21.24).137–139 Somatostatin inhibits secretion of a number of endocrine and exocrine products and diminishes peristaltic contractions of the gallbladder and stomach.140 The somatostatinoma syndrome—consisting of diabetes mellitus (due to inhibition of insulin secretion), steatorrhea (due to inhibition of pancreatic enzyme secretion), hypohydria or achlorydia, and cholelithiasis (due to suppression of cholecytokinin-pancreozymin release and gallbladder contraction)—is rarely observed in duodenal somatostatin-producing NETs.141–144 In contrast, somatostatin-producing NETs of the pancreas may produce the somatostatinoma syndrome.138,145
Because of the preferred ampullary and peraampullary location, somatostatin-producing NETs of the duodenum typically manifest with symptoms and signs related to bile duct obstruction, abdominal pain, or cholelithiasis.146,147 Somatostatin-producing ampullary/peraampullary NETs have been associated with von Recklinghausen disease (neurofibromatosis type 1, or NF1), although the frequency with which patients with NF1 develop these tumors is not clear.144,148–159 Some patients with ampullary somatostatin-producing NETs also have a pheochromocytoma involving one or both adrenal glands.150
Pathologic Features
Somatostatin-producing ampullary/peraampullary NETs have characteristic morphologic features that help distinguish these tumors from other NETs of the duodenum. Approximately 60% of somatostatin-producing NETs display a characteristic mixed pseudoglandular and trabecular architectural arrangement with polarized cells arranged in pseudoglandular structures; the central lumina frequently contain diastase-resistant proteinaceous secretions (Fig. 21.25, A).133,150 Psammoma bodies are seen in approximately 60% of sporadic somatostatin-producing ampullary/peraampullary NETs and in almost all NF1-associated somatostatin-producing NETs (see Fig. 21.25, B).133,136 In contrast, pancreatic somatostatin-producing NETs less frequently exhibit a pseudoglandular architecture (~20%) or psammoma bodies (~40%).133 Somatostatin-producing NETs are strongly positive for somatostatin, chromogranin A, and synaptophysin. A subset of the tumor cells may also express other peptide hormones, including gastrin, calcitonin, and serotonin.141,160
Natural History
Both sporadic and NF1-associated somatostatin-producing duodenal NETs may metastasize to regional lymph nodes and to the liver.133 The risk of metastasis appears to be related to tumor size. Tumors smaller than 2.0 cm have a low metastatic potential, whereas larger lesions are at increased risk of metastatic disease.152 Even in the setting of metastatic disease, patients with somatostatin-producing duodenal NETs typically have a protracted clinical course.
Gangliocytic Paraganglioma
Clinical Features
Gangliocytic paragangliomas are rare tumors that have a predilection for the ampulla/peraampullary area. Gangliocytic paragangliomas occur slightly more frequently in men and usually occur in middle age (mean age, 54 years) but have been reported to occur in younger patients.161–163 These lesions manifest as polypoid submucosal nodules ranging in size from 2 to 4 cm. Many patients present with GI bleeding due to mucosal erosion, but ampullary/peraampullary tumors can cause obstructive jaundice.162,164,165 Similar to somatostatin-producing NETs, gangliocytic paragangliomas are associated with NF1.164,166,167
Pathologic Features
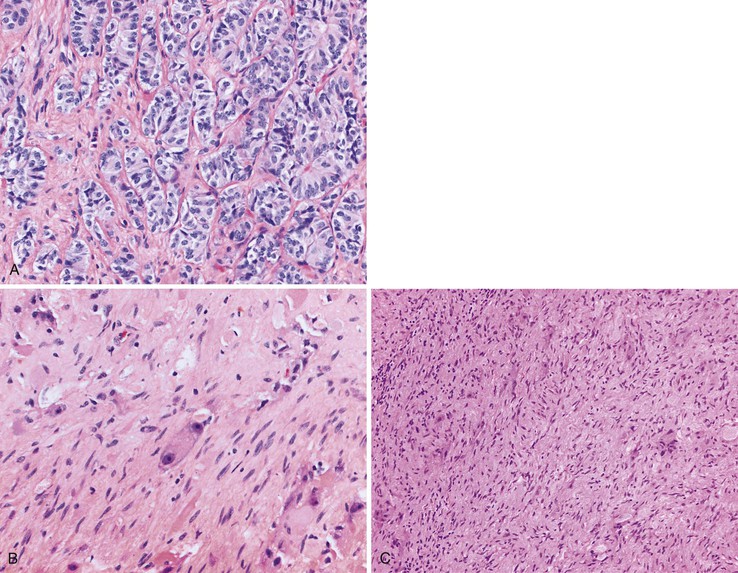
Gangliocytic paragangliomas are composed of a variable mixture of (1) nests of uniform polygonal neuroendocrine cells that occasionally form tubules, (2) mature ganglion cells, and (3) spindle cells with schwannian differentiation arranged in broad fascicles or associated with the polygonal cells and ganglion cells (Fig. 21.26).163,168,169 Psammoma bodies may be identified in the neuroendocrine cell population.141 The neuroendocrine cells typically stain for both chromogranin A and synaptophysin but can also label for the peptide hormones somatostatin, pancreatic polypeptide, vasoactive intestinal peptide (VIP), and gastrin. The spindle cells show tapered ends, contain faintly eosinophilic cytoplasm, and stain intensely for S100 and neurofilament. The ganglion cells may be dispersed singly, or they may form small clusters and stain for synaptophysin.
Natural History
Gangliocytic paragangliomas usually follow a benign course. However, there are reports of gangliocytic paragangliomas with metastasis to regional lymph nodes, particularly large tumors (>2 cm).170–174 Most frequently, the metastatic tumor in lymph nodes consists of the neuroendocrine component of the tumor, although cases of metastasis containing all three components have been described.172 To our knowledge, none of the reported cases of metastatic gangliocytic paraganglioma has resulted in patient death. Therefore, a conservative management approach seems appropriate.
NETs of the Distal Jejunum and Ileum
Clinical Features
In contrast to duodenal NETs, NETs of the distal jejunum and ileum are derived from serotonin-producing enterochromaffin (EC) cells and may secrete serotonin. Therefore, jejunoileal NETs are true carcinoid tumors. These tumors also elicit peritumoral or mesenteric fibrosis that may produce symptoms of recurrent small bowel obstruction, bowel ischemia and infarction, and protracted abdominal pain.175 The mechanism by which jejunoileal NETs stimulate fibrosis remains poorly understood but may be related to the secretion of serotonin or possibly growth factors by these tumors.175 A significant percentage (approximately one third) of jejunoileal NETs are associated with second synchronous or metachronous malignancies, most frequently adenocarcinomas of the GI, genitourinary, and gynecologic tracts.176–179 Gene expression profiling and comparative genomic hybridization analysis of jejunoileal NETs has revealed frequent loss of chromosome 18q with frequent gain of chromosomes 4 and 7 seen in metastases.180–182 Other genetic analyses have suggested that gain of chromosome 14 predicts poor outcome in ileal NETs.183
Pathologic Features
Jejunoileal NETs have a variable gross appearance, ranging from the presence of polypoid tan-yellow submucosal nodules to annular masses or strictures secondary to marked fibrosis characteristic of these lesions (Fig. 21.27). Approximately 25% of tumors manifest as multiple mucosa-based polypoid lesions, with younger patients more frequently presenting with multiple tumors.184 X-chromosome inactivation analysis of multifocal carcinoid tumors of the small intestine has demonstrated identical X-chromosome inactivation patterns, indicating that multiple tumors represent mucosal metastases in some cases.185
Jejunoileal NETs are typically composed of rounded nests of uniform tumor cells with lightly eosinophilic cytoplasm (Fig. 21.28). Anastomosing cords, trabeculae, and cribriform and pseudoglandular structures may also be observed. The tumor cells are associated with densely fibrotic stroma that may compress the tumor cells into a single-file and cord-like arrangement. Most tumors demonstrate perineural and lymphovascular invasion. Jejunoileal NETs are typically strongly positive for synaptophysin and chromogranin and usually display diffuse, strong reactivity for CDX2.128,186–190 Jejunoileal NETs are negative for PAX8, which may help to localize the primary site of a NET in the setting of metastatic disease to the liver with an unknown primary.129,130 However, the antibody commonly used for PAX8 may actually recognize another related PAX protein.191
Natural History
Because of their relatively small size and submucosal location, jejunoileal NETs are rarely diagnosed until the development of metastatic disease to lymph nodes or distant sites, particularly the liver. In contrast to the better 5-year survival rates for NETs of the duodenum, patients with jejunoileal NETs have a relatively poor 5-year survival rate (55%).192 Factors associated with aggressive clinical behavior include size (>1cm), regional lymph node metastases, and metastases to the liver.193–195 Some groups have found that female sex, age younger than 50 years, multiple mucosal nodules, and the presence of carcinoid syndrome are also associated with a poorer clinical outcome.184,193
Neuroendocrine Carcinoma of the Small Intestine and Ampulla
Clinical Features
Neuroendocrine carcinomas are defined by the presence of either a high mitotic rate (>20 per 10 HPF) or a high Ki67 index (>20%).113 Neuroendocrine carcinoma can be further subdivided into small cell and large cell types, comparable to neuroendocrine carcinomas seen in the lung. Within the small intestine, neuroendocrine carcinomas are relatively rare but appear to have a predilection for the ampulla/peraampullary area.196,197 Neuroendocrine carcinomas appear to occur more frequently in males and in older patients (mean age, 68 years).196,198
Pathologic Features
The distinction between small cell and large cell neuroendocrine carcinoma is based on a variety of cytoarchitectural features. Small cell neuroendocrine carcinoma tumor cells are typically small (less than 3 times the size of a small lymphocyte) and are arranged in large sheets with areas of necrosis. The nuclei display finely granular chromatin, inconspicuous nucleoli, and prominent nuclear molding.116,117,197 Large cell neuroendocrine carcinomas usually demonstrate an architectural arrangement similar to that of well-differentiated NETs, often displaying an organoid or trabecular architecture. The tumor cells have larger, more vesicular nuclei, prominent nucleoli, and moderate amounts of cytoplasm.116,117,197 Both small cell and large cell neuroendocrine carcinomas can be associated with adenomas of the overlying surface epithelium. In addition, adenocarcinoma has been reported to be associated with neuroendocrine carcinomas, particularly of the ampulla.196,197,199 If the tumor contains a significant component (>30%) of these tumor types, the term mixed adenoneuroendocrine carcinoma (MANEC) should be used.116,117 Rare cases of composite small cell neuroendocrine carcinoma and squamous cell carcinoma of the ampulla have also been reported.200
Mesenchymal Lesions
A complete discussion of the clinical and pathologic features of mesenchymal tumors is presented in Chapter 30. Most previously reported cases of leiomyomas, schwannomas, and neurofibromas of the small intestine likely represent gastrointestinal stromal tumors (GISTs), because true smooth muscle and nerve sheath tumors are extremely rare in the duodenum, being more likely to occur elsewhere in the GI tract. Lymphatic lesions are commonly encountered; they frequently form endoscopically visible polypoid lesions in the small intestine.
Lymphatic Lesions
Dilated lymphatics are not infrequently encountered during endoscopic evaluation of the small intestine, where they appear as yellow or white plaques or polyps. Abnormalities of the small bowel lymphatics may represent a primary disorder of the small intestine, or they may be associated with numerous secondary processes. However, in one analysis, approximately 13% of patients undergoing endoscopic evaluation of the small bowel harbored incidental yellow or white plaques containing dilated lymphatics that were not associated with any other primary or secondary disorder.201
Primary Lymphangiectasia
Primary intestinal lymphangiectasia is a rare condition affecting children and young adults that is characterized by severe edema, thickening of the small bowel wall, protein-losing enteropathy, ascites, and pleural effusion.202 Secondary generalized edema may occur as a result of hypoproteinemia. The pathogenesis of this disorder is unclear. Computed tomographic imaging is most often used in the diagnosis; it reveals diffuse, nodular thickening of the bowel wall with ascites and hypodense streaks in the small bowel due to markedly dilated lymphatics.203,204 Mucosal biopsies are not specific, revealing only multiple dilated lymphatic spaces, but they are often necessary to confirm the presence of dilated lymphatics. Mesenteric lymphatics are also dilated and thickened; however, surgical resections are not performed for this disorder. The diagnosis requires knowledge of the clinical and radiographic findings.
Secondary Lymphangiectasia
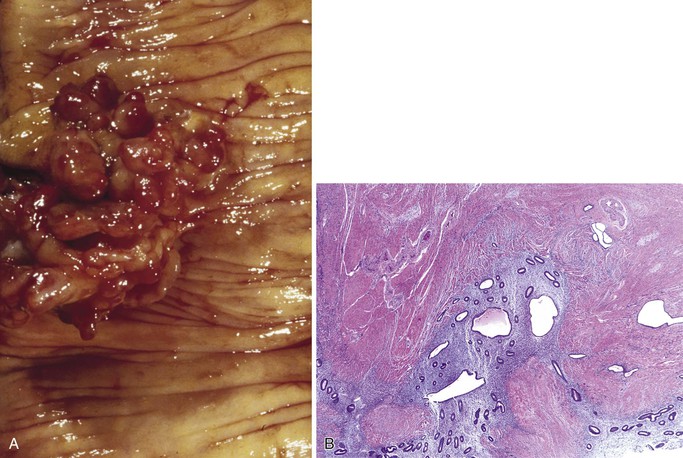
A number of disorders can give rise to dilated lymphatics in the small intestine. Acute inflammatory disorders including infections frequently display dilated lymphatics. In particular, dilated lymphatics are a common histologic finding in Whipple disease, and their presence should prompt a careful evaluation for the characteristic PAS-positive macrophages in the lamina propria. Dilated lymphatics may also be the result of extraluminal compression due to malignancy elsewhere in the GI tract or regional lymph nodes, malrotation, fibrosis, or lymph node obstruction or infiltration by inflammation, tumor (Fig. 21.29), or infection.
Lymphoid Lesions
Benign Lymphoid Hyperplasia
The lamina propria of the small intestine normally contains a mild chronic inflammatory infiltrate composed predominantly of plasma cells and mature lymphocytes with occasional eosinophils, histiocytes, and mast cells. Lymphoid aggregates, many with germinal centers, are also seen in the small intestine and are found in increasing concentration distally, forming nodular aggregates of lymphocytes in the terminal ileum (Peyer patches). The specialized epithelium overlying Peyer patches is distinct from the surrounding mucosa in that it contains fewer goblet cells and is associated with more abundant intraepithelial lymphocytes as well as occasional neutrophils; these findings should not be interpreted as representing enteritis. Reactive lymphoid hyperplasia in the distal ileum may occur in association with a wide variety of inflammatory disorders, including infections (e.g., Salmonella, Yersinia, Mycobacterium), as well as idiopathic inflammatory bowel disease.
Nodular lymphoid hyperplasia, also known as follicular lymphoid hyperplasia, is characterized by hyperplastic germinal centers with well-defined lymphocytic mantles located in the mucosa and submucosa (Fig. 21.30).205 Endoscopically, the nodules may resemble small polyps, prompting biopsy. In children, nodular lymphoid hyperplasia is usually an incidental finding and may be seen in the rectum, colon, terminal ileum, and duodenum. It typically follows a benign course with spontaneous regression.206 In adults, nodular lymphoid hyperplasia in the duodenum is typically a reactive process caused by a variety of conditions including peptic duodenitis, celiac disease, autoimmune enteritis, and Crohn’s disease. A recent report has also linked diffuse duodenal nodular lymphoid hyperplasia with concurrent H. pylori–associated gastritis.207
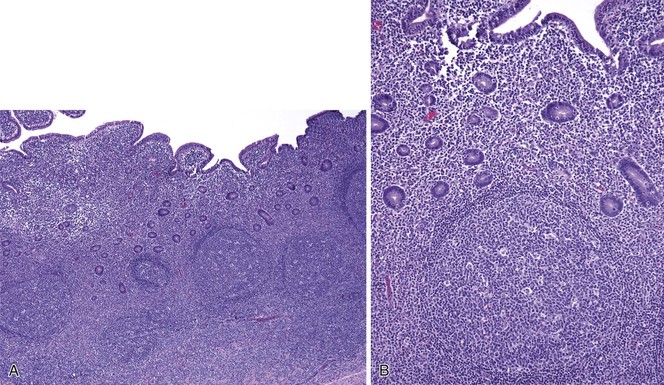
Diffuse nodular lymphoid hyperplasia of the small intestine, distal to the ampulla, in adults is rare and usually implies the presence of an underlying immune disorder. In adults, nodular lymphoid hyperplasia is associated most often with common variable immunodeficiency (CVID) and is occasionally seen in patients with selective immunoglobulin A (IgA) deficiency or HIV/AIDS.206,208,209 Approximately 50% of adults with CVID have nodular lymphoid hyperplasia, which can occur diffusely throughout the small intestine.210 A wide spectrum of histologic changes can be observed in CVID. In addition to reactive nodular lymphoid hyperplasia, prominent mononuclear inflammation in the lamina propria in association with villous blunting and increased intraepithelial lymphocytes may be seen.210,211 A marked decrease, or complete absence, of plasma cells is present in up to two thirds of patients.210 Concurrent infection with Giardia lamblia is seen in many patients with either IgA deficiency or CVID.206,210,211 Nodular lymphoid hyperplasia may also been seen with Giardia infection in the absence of an identifiable humoral immunodeficiency.212,213
Finally, nodular lymphoid hyperplasia has been reported to be a possible risk factor, or premalignant condition, for the development of lymphoma in the small intestine.214 Literature reports describe the presence of nodular lymphoid hyperplasia in the mucosa adjacent to primary small intestinal malignant lymphomas.209,211,214–216 In such cases, there is often a gradual transition between the hyperplastic and neoplastic lymphoid tissue.214,215 Although the data linking nodular lymphoid hyperplasia and the development of malignant lymphoma are limited, the presence of persistent and diffuse nodular lymphoid hyperplasia should prompt careful evaluation for the possibility of malignant lymphoma.
Malignant Lymphoma
A detailed discussion of hematopoietic malignancies that may affect the small intestine is presented in Chapter 31. With the exception of enteropathy-associated T-cell lymphoma, most lymphomas of the small intestine are B-cell lymphomas, and specific subtypes demonstrate a predilection to affect different regions of the small intestine. Enteropathy-associated T-cell lymphomas usually develop in the jejunum or distal duodenum, reflecting the disease distribution of celiac disease. Most lymphomas of the ileum are diffuse large B-cell lymphomas. Primary follicular lymphoma of the GI tract has a predilection for the duodenum and is typically low-grade with a favorable prognosis.217–219
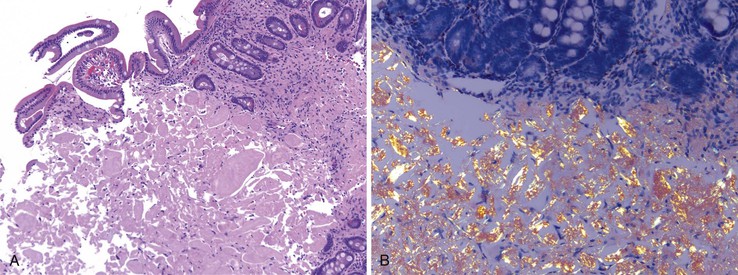
Malignant lymphoid neoplasms can also result in amyloid deposition in any portion of the GI tract including the small bowel (primary amyloidosis). Secondary amyloidosis and inherited amyloidotic disorders can also involve the GI tract.220,221 The small bowel is most commonly affected. Whereas amyloid deposition within the gut is usually around submucosal vessels, occasionally amyloid deposition can manifest as nodules or polyps (Fig. 21.31, A and B).
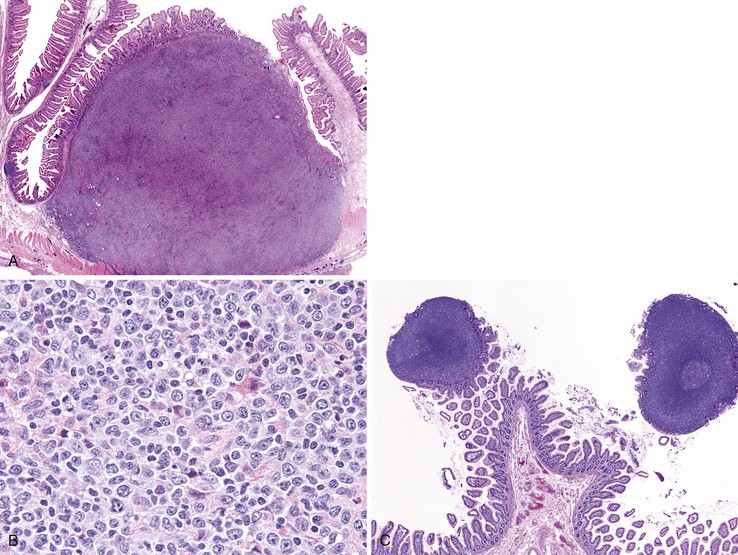
Multiple Lymphomatous Polyposis
Lymphomatous polyposis is an unusual form of GI involvement by lymphoma that is characterized by innumerable polypoid excrescences (Fig. 21.32). It can affect any portion of the GI tract but frequently produces large polyps in the ileocecal region. Lymphomatous polyposis usually occurs as a manifestation of mantle cell lymphoma, although other lymphoma types, including follicular lymphoma, diffuse large B-cell lymphoma, marginal zone lymphoma, and T-cell lymphoma, can give rise to lymphomatous polyposis.222–225
Metastases
Adenocarcinoma is the most common primary malignancy of the small intestine and is typically identified in the duodenum in the area around the ampulla of Vater, with decreasing incidence in the jejunum and ileum.226 However, virtually any type of malignancy may secondarily involve the small intestine and simulate a primary malignancy, particularly underlying pancreatic adenocarcinomas that invade into the small bowel (Fig. 21.33). When entertaining a diagnosis of primary small intestinal carcinoma, the possibility of metastasis must always be considered, because metastatic disease is more common than primary small intestinal adenocarcinoma. Secondary involvement of the small intestine may occur via direct extension from another organ, peritoneal dissemination, or hematogenous or lymphovascular spread. Identifying a metastasis to the small intestine is often straightforward when there is a known history of a primary neoplasm elsewhere. If the initial primary diagnosis is remote or unknown, distinguishing a metastasis from a primary small intestinal adenocarcinoma can be difficult.227
Most metastatic carcinomas to the small intestine originate elsewhere in the GI tract, particularly the colorectum, or in the gynecologic tract.228 These lesions usually involve the small intestine as a result of peritoneal dissemination and typically demonstrate extensive, often multifocal, involvement of the subserosal connective tissue and muscularis propria. Primary carcinomas of the lung, genitourinary tract, and head and neck can also secondarily involve the small intestine as a result of hematogenous dissemination and may produce solitary lesions limited to the mucosa and submucosa, imitating a primary intestinal carcinoma.229–231 Breast carcinoma, particularly infiltrating lobular carcinoma, can also involve the small intestine and typically infiltrates the lamina propria and submucosa without destroying the crypt epithelium (Fig. 21.34).232
Features favoring a diagnosis of metastatic carcinoma include multifocal involvement, extensive mural and serosal disease with minimal mucosal involvement, and extensive lymphovascular invasion. Small intestinal adenocarcinomas are of an intestinal type, and the presence of signet ring cell differentiation or a diffuse-type growth pattern is uncommon in primary carcinomas of the small intestine, especially in areas distal to the ampulla or in tumors unrelated to Crohn’s disease.233 Frequently, metastatic adenocarcinoma to the small intestine can demonstrate maturation of the neoplastic epithelium as it grows from the serosa toward the bowel lumen. Metastatic carcinoma can also reach the mucosal surface of the small intestine and exhibit an in situ growth pattern along intact basement membranes, simulating a “precursor” dysplastic lesion (adenoma). In one analysis, mucosal colonization was observed in 60% of metastatic GI tract adenocarcinomas to the small intestine that involved the mucosa and in 26% of adenocarcinomas from non–GI tract primary sites.228 Therefore, the presence of an apparent in situ dysplastic lesion does not provide evidence for a primary small intestinal carcinoma.
Immunohistochemical stains are particularly helpful when considering metastasis from non–GI tract primary sites, such as the lung or breast. However, immunohistochemistry is not helpful in distinguishing primary small intestinal adenocarcinoma from metastatic GI tract adenocarcinomas. Primary small intestinal adenocarcinomas often demonstrate a CK7-positive/CK20-negative or CK7-positive/CK20-positive immunophenotype. Some authors have suggested that this CK7 and CK20 expression profile may help to distinguish primary small intestinal adenocarcinomas from secondary colorectal adenocarcinomas, which typically are CK7-negative and CK20-positive.234 However, colorectal adenocarcinomas, particularly those with high levels of microsatellite instability, can coexpress CK7 and CK20 or display a CK7-positive/CK20-negative expression profile, limiting the utility of CK7 and CK20 immunohistochemistry in this setting.235
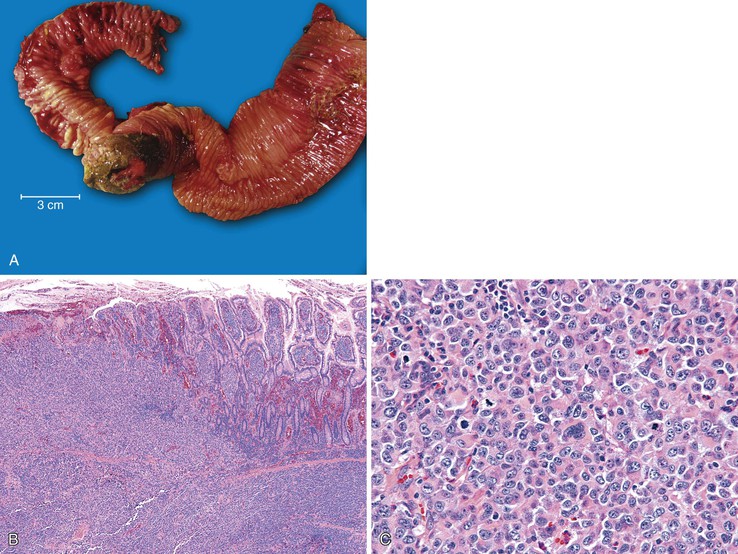
Malignant melanoma shows a predilection for the GI tract and usually affects the small intestine (Fig. 21.35).236,237 Metastatic melanomas to the GI tract can be either pigmented or amelanotic and may mimic a variety of primary neoplasms, including carcinomas, sarcomas or GISTs, and large cell lymphomas. Immunohistochemical stains (S100 protein, HMB-45, A103 [MART-1], tyrosinase, microphthalmia transcription factor, and SOX10) have been shown to demonstrate high sensitivity and variable specificity for melanoma.238,239 Importantly, when one is evaluating high-grade spindle cell lesions for which GIST is in the differential diagnosis, one or more melanoma stains should be included in the panel, because as much as 50% of malignant melanomas can express CD117.240–242
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Polyps of the Small Intestine
Reetesh K. Pai
Rish K. Pai
Introduction
Upper gastrointestinal (GI) endoscopy is routinely performed as part of the evaluation of patients with GI symptoms. Newer techniques such as double-balloon enteroscopy and capsule endoscopy also allow for complete visualization of the small intestine. Gastroenterologists may identify and sample a variety of polyps, nodules, excrescences, and subtle abnormalities in the mucosa during the course of the procedure that, before the endoscopic era, would have gone largely unrecognized. Many of these lesions are clinically asymptomatic and occur in the duodenum, which is amenable to upper endoscopic examination. This chapter discusses the clinicopathologic features and differential diagnoses of the most common or important lesions that may cause the clinical impression of an intestinal polyp (Box 21.1).
Inflammatory Lesions
As in other areas of the GI tract, inflammatory lesions of the small intestine may produce polypoid masses. Small polyps are often incidental, whereas larger polyps may be symptomatic due to hemorrhage or luminal obstruction.1 Inflammatory pseudopolyps associated with Crohn’s disease are the most common inflammatory polyps of the small intestine; they are commonly encountered in the terminal ileum, where they demonstrate varying degrees of villous architectural distortion, pyloric gland metaplasia, and active inflammation (Fig. 21.1). They can be quite numerous and can even carpet the small bowel with finger-like projections.1
Cytomegalovirus infection can rarely manifest as isolated GI inflammatory polyps, often in immunocompromised hosts (Fig. 21.2).2,3 Xanthomas can also occur in the duodenum, although they are more rare than gastric xanthomas.4 They are characterized by collections of pale and foamy histiocytes in the lamina propria. Histiocytic differentiation can be documented by immunohistochemical staining with CD163, CD68, or both. Inflammatory fibroid polyps (described in Chapter 30) are also less common in the small bowel compared with the stomach (Fig. 21.3).5 Similar to their gastric counterparts, they often demonstrate mutations in the PDGFRA gene.6
Inflammatory-type polyps of the small intestine may arise secondarily from other types of disease. Endometriosis of the bowel wall and submucosa may induce inflammatory changes including inflammatory-type polyps. Changes mimicking Crohn’s disease have also been reported.7,8 Endometriosis can also involve the mucosa, resembling dysplasia and even carcinoma (Fig. 21.4). Well-differentiated neuroendocrine tumors (NETs) may also induce an inflammatory reaction in the mucosa away from the main tumor, likely related to mucosal prolapse. These lesions contain numerous ectatic capillaries with admixed smooth muscle bundles and fibrosis.9
Hyperplasia and Heterotopia
Brunner Gland Hyperplasia/Hamartoma
Clinical and Endoscopic Features
Brunner glands are lobular collections of tubular glands within the duodenum; they are predominantly located in the duodenal submucosa. However, in the first part of the duodenum, Brunner glands commonly transgress the muscularis mucosae and extend into the lamina propria.10 Brunner glands within the lamina propria are less frequently observed in the middle to distal duodenum.
Occasionally, Brunner glands proliferate, creating small polypoid excrescences or imparting a nodular appearance to the mucosal surface (Fig. 21.5). These Brunner gland proliferative lesions are most commonly encountered in the duodenal bulb, usually as an incidental finding at endoscopy performed for other indications. However, they may be seen as one of a constellation of findings, including villous shortening and gastric foveolar mucous cell metaplasia of the villous epithelium, that are indicative of peptic injury of the duodenum (peptic duodenitis). Proliferative Brunner gland lesions have also been associated with end-stage renal disease and uremia.11,12
The nomenclature of Brunner gland proliferative lesions is not well established, and a number of diagnostic terms, including Brunner gland hyperplasia, Brunner gland adenoma, and Brunner gland hamartoma, have been used. The distinction between these diagnoses is arbitrary. No well-documented cases of either true glandular dysplasia or carcinoma arising in proliferative Brunner glands have been reported. Careful review of the limited literature reports of Brunner gland adenocarcinoma and dysplasia reveals that the dysplastic glandular epithelium involves the surface epithelium with likely secondary involvement of the underlying hyperplastic Brunner glands.13–17 In other cases, the adenocarcinomas appear to arise from pyloric gland adenomas and not from hyperplastic Brunner glands.16,17 Therefore, the term Brunner gland adenoma is potentially misleading, and we prefer to use the term Brunner gland hyperplasia to limit confusion among our clinicians and to highlight the non-neoplastic nature of the entity.
Some authors distinguish between Brunner gland hyperplasia and Brunner gland hamartoma based on the size of the lesion. However, the size cutoff between these two entities is arbitrary and not well established in the literature.18 The distinction is of no clinical significance, and a diagnosis of Brunner gland hyperplasia/hamartoma is usually sufficient in large polypoid lesions.
Pathologic Features
Brunner glands are composed of neutral, mucin-secreting, cuboidal to columnar cells with basally located nuclei; they are arranged in lobules containing thin, fibrous septa. Brunner glands are histologically indistinguishable from pyloric glands of the distal gastric mucosa. Indeed, biopsies of the gastroduodenal junction may demonstrate a gradual transition of these epithelial types. The predominantly submucosal location of Brunner glands helps to identify the biopsy as duodenal in origin.
The diagnostic criteria for Brunner gland hyperplasia are subjective, because Brunner glands may be normally seen in the lamina propria of the duodenum, particularly within the duodenal bulb. In addition, proliferation of Brunner glands is commonly observed in peptic-type duodenal injury. We reserve the term Brunner gland hyperplasia for those endoscopically visible duodenal nodules that are found to contain lobules of Brunner glands within the mucosa in at least 50% of the length of a biopsy specimen (Fig. 21.6, A). Brunner gland hyperplasia may manifest as solitary or multiple nodules that are typically small (<1 cm) and characterized by lobules of glands that are increased in both size and number. The lobules extend into the mucosa and are separated by delicate fibrous septa. Cystic dilatation of Brunner glands may occur, and rare cases of Brunner gland cysts have been reported.19,20 The cells are cytologically bland with abundant neutral mucin and small, basally located nuclei with minimal to absent mitotic activity.
Large polyps (>2 cm) composed of Brunner glands may produce symptoms of obstruction, intussusceptions, melena, and anemia requiring endoscopic resection. Such polyps typically display more abundant fibromuscular stroma, likely the result of mucosal prolapse-type changes. For such large polyps, the diagnosis of Brunner gland hyperplasia/hamartoma may be appropriate, because it recognizes the arbitrary distinction between these two diagnoses.
In mucosal biopsy specimens, Brunner glands are commonly crushed, imparting a spindle cell appearance to the epithelial cells (see Fig. 21.6, B). Crushed Brunner glands may be mistaken for histiocytic collections, raising concern for Whipple disease, particularly given their intense positive reaction on periodic acid–Schiff (PAS) staining. The spindled appearance of crushed Brunner glands may also raise concern for a mesenchymal lesion. Careful histologic evaluation with comparison of adjacent partially crushed lobular collections of Brunner glands will allow for identification of this distortion artifact.
Differential Diagnosis
Histologic features of peptic-type injury of the duodenum include any of the following: active inflammation within the lamina propria or epithelium, Brunner gland hyperplasia (Fig. 21.7, A), gastric foveolar metaplasia of the surface epithelium (see Fig. 21.7, B), and hemorrhage and edema. Nodular duodenitis is frequently observed in patients with a history of peptic ulcer disease.21 In the setting of histologic findings indicative of peptic-type duodenal injury, the presence of Brunner gland hyperplasia should be attributed to peptic duodenitis.
Pyloric gland adenoma is a relatively recently described adenoma subtype that can occur in the duodenum and not infrequently evolves into invasive adenocarcinoma. The distinction between pyloric gland adenoma and Brunner gland hyperplasia/hamartoma is difficult (see later discussion). Both express the mucin core peptide MUC6; however, pyloric gland adenomas usually also demonstrate labeling with MUC5AC, which is not typical of Brunner glands.22
Gastric Heterotopia
Clinical and Endoscopic Features
Gastric heterotopias in the duodenum are most commonly identified in the bulb and are usually incidental, small (<1.0 cm) nodules that may be multiple.23 Patients are typically asymptomatic, but larger polyps may manifest as masses and cause obstruction or intussusception.24,25 Peptic ulceration due to heterotopic oxyntic glands in the proximal duodenum is rare because the acid secretions are quickly diluted by the alkaline duodenal contents derived from the pancreatobiliary system. However, rare cases of massive GI bleeding have been reported, particularly in patients with extensive heterotopias.24,26 Gastric heterotopia is also associated with concurrent fundic gland polyps of the stomach, suggesting that the increased use of proton pump inhibitors (PPIs) in the general population may enhance its endoscopic detection.27
Pathologic Features
Biopsies of gastric heterotopias display well-organized oxyntic glands composed of chief and parietal cells (Fig. 21.8, A). The overlying surface often exhibits gastric foveolar-type mucinous epithelium with adjacent normal duodenal intestinal villi. Gastric heterotopia in the duodenum may harbor the same pathologic processes that affect the stomach, including Helicobacter pylori infection (see Fig. 21.8, B).27 Large gastric heterotopias may exhibit secondary mucosal prolapse-type changes including prominence of the muscularis mucosae, submucosal fibrosis, cystic dilatation of the oxyntic and mucinous glands, and surface epithelial hyperplasia.28
Differential Diagnosis
Gastric heterotopia differs from gastric foveolar metaplasia, which is hypothesized to be a response to inflammation caused by peptic injury associated with gastric H. pylori infection29 (Table 21.1). Histologically, peptic-type injury of the duodenum typically results in active inflammation within the lamina propria or epithelium and erosion or ulceration. The metaplastic gastric foveolar epithelium is not associated with the oxyntic glands characteristic of gastric heterotopia.
Table 21.1
Gastric Heterotopia Versus Gastric Metaplasia
| Gastric Heterotopia | Gastric Metaplasia | |
| Clinical presentation | Usually incidental | Epigastric pain |
| Endoscopic features | Nodular mucosa | Duodenitis |
| Histologic features | Oxyntic mucosa Normal duodenal mucosa |
Gastric foveolar (mucous) cells Villous blunting ± active inflammation Prominent Brunner glands |
| Clinical implications | None | Peptic injury |
Pancreatic Heterotopia
Clinical and Endoscopic Features
Pancreatic heterotopia represents the presence of pancreatic tissue outside the normal pancreas without any anatomic or vascular connection to the pancreas. It is synonymous with the terms pancreatic rest and ectopic pancreas. Pancreatic heterotopia of the small intestine is most commonly located in the proximal duodenum and can be seen either confined to the mucosa or within the muscularis propria. Pancreatic tissue may be normally identified in the region of the minor papilla and does not necessarily qualify as heterotopia.30 In most cases, pancreatic heterotopia is asymptomatic and only incidentally detected. Mural heterotopias develop most commonly in the peraampullary region and are more likely to be symptomatic due to duodenal obstruction, intussusception, and stricture or stenosis of the ampulla.31–33 Symptomatic mural pancreatic heterotopias often mimic a neoplastic process, leading to surgical resection,31 and an endoscopic biopsy diagnosis is not possible given the location of the lesion.
Pathologic Features
Pancreatic heterotopia may be composed of pancreatic acini, ducts, or islets, either alone or in combination (Fig. 21.9). Most frequently, pancreatic ducts are identified, with a variable number of acinar cells and islets and no relationship between symptoms and the cell type present.34 Mural lesions are often histologically indistinguishable from normal pancreas. Most mucosal-based lesions are unassociated with inflammation, being present in otherwise normal mucosa of the duodenum. Rarely, ductal adenocarcinoma, mucinous neoplasms, and NETs have been reported to develop in pancreatic heterotopia.34–37
Differential Diagnosis
Peraampullary duodenal wall cyst (groove pancreatitis) is not often confused with mural pancreatic heterotopias. Although both may contain pancreatic ducts, acini, and islets, peraampullary duodenal wall cyst has characteristic clinicopathologic features. Most patients are young to middle-aged men who report alcohol use and typically present with pain and vomiting due to duodenal stenosis.38–40 Jaundice may also be seen late in the disease course as the result of compression of the common bile duct.38 The lesion typically involves the area of the minor papilla where pancreatic tissue may normally be identified. Fibrosis and inflammation occur in the “groove” between the superior aspect of the pancreatic head, the common bile duct, and the duodenum. In addition to groove pancreatitis, alternative names include cystic dystrophy of the duodenal wall and paraduodenal pancreatitis.30 Most cases are characterized by multiple or solitary cysts varying in size from 1 to 10 cm within the duodenal wall and pancreas (Fig. 21.10). “Solid” types have also been described and are characterized by fibrotic thickening of the duodenal wall with small (<1 cm) cysts.39 A reactive myofibroblastic spindle cell proliferation is usually present in the duodenal wall, whereas the fibrosis in the groove area is more paucicellular and hyalinized.41 Cystically dilated ducts partially lined by ductal epithelium and containing inspissated eosinophilic material are characteristic (Fig. 21.11). Squamous metaplasia, granulation tissue, calcium deposition, and giant cell reaction are also frequently encountered.39,41 It has been speculated that this disorder is a localized form of alcohol-induced pancreatitis occurring in pancreatic tissue present in the area of the minor papilla.30
Hamartomatous Polyps
By definition, hamartomatous polyps are a haphazard arrangement of normal stromal and epithelial elements. Hamartomatous polyps of the small bowel occur mainly in the setting of polyposis syndromes including Peutz-Jeghers syndrome (PJS), juvenile polyposis, and PTEN hamartoma tumor syndrome. The latter two syndromes have similar-appearing hamartomatous polyps. Correct classification of these polyps is critical to management, because many of these syndromes carry unique risks of GI and extra-GI malignancy (Table 21.2). Rarely, sporadic hamartomatous polyps resembling their syndromic counterparts occur.42 Other sporadic hamartomatous lesions have been described, including those made predominantly of mesenchymal elements such as adipose tissue, vessels, and nerves. There are numerous reports of neuromuscular and vascular hamartomas in the literature; however, these lesions are not hamartomas but are likely related to injury from nonsteroidal antiinflamatory drugs (diaphragm disease) (see Chapters 12 and 16).43,44
Table 21.2
Hamartomatous Syndromes Affecting the Small Bowel
| Syndrome | Polyp Type | Small Bowel Involvement | Histology | Dysplasia In Polyp* | Gene Mutations | Risk of Small Bowel Adenocarcinoma |
| Peutz-Jeghers syndrome | Peutz-Jeghers polyp | Frequent (65%) | Normal mucosal elements with arborizing smooth muscle | Rare; possible increased risk of dysplasia in nonpolypoid mucosa | STK11/LKB1 | 13% |
| Juvenile polyposis syndrome | Juvenile polyp | Infrequent (<20%) | Inflamed, edematous lamina propria, cystic glands, often ulcerated | Rare | SMAD4 and BMPR1A | ~10% |
| PTEN hamartoma tumor syndrome | Multiple types: juvenile polyps, ganglioneuromas, lipomas, and adenomas | Variable (33-66%) | Variable | Rare | PTEN | Unclear, likely low |
* Refers to dysplasia in polyps other than an adenoma.
PTEN, Phosphatase and tensin homolog.
Peutz-Jeghers Polyps
Clinical and Endoscopic Features
PJS is an autosomal dominant hamartomatous polyposis syndrome with a prevalence of 1 in 200,000.45,46 The majority of patients have positive family histories; however, 25% of cases develop de novo. A diagnosis of PJS should be considered if one of the following criteria is met: (1) three or more histologically confirmed Peutz-Jeghers polyps, (2) any number of Peutz-Jeghers polyps with a family history of PJS, (3) characteristic mucocutaneous pigmentation with a family history of PJS, or (4) any number of Peutz-Jeghers polyps and characteristic mucocutaneous pigmentation.45,47 More than 90% of patients with PJS develop small intestinal polyps, most commonly in the jejunum, followed by the ileum and the duodenum. PJS patients are often diagnosed at an early age (approximately 20 years), because these polyps often cause abdominal pain, obstruction due to intussusception, and bleeding.45,48 Surgical intervention often occurs in these settings. Indeed, patients are at risk for short gut syndrome due to the numerous GI surgeries undertaken to resect these polyps.49 Patients with PJS have a significantly increased risk of intestinal adenocarcinoma, particularly of the colon (cumulative risk of 40%) but also of the small bowel (13% cumulative risk).45,50–59 For this reason, patients are enrolled in an endoscopic screening protocol at a very young age.45 Affected individuals are also at increased risk for other tumors: breast (54% cumulative risk for females), endometrial, cervical (adenoma malignum), pancreatic (36% cumulative risk), ovarian (sex cord/stromal tumor with annular tubules), and testicular (large-cell calcifying Sertoli cell tumor).50,57,58,60–62 The cumulative lifetime risk for any cancer approaches 90%.
Peutz-Jeghers polyps have an irregular, multilobulated endoscopic appearance and lack the velvety texture typical of adenomas (Fig. 21.12). Because they contain prominent smooth muscle, they are tightly anchored to the bowel. Unlike juvenile polyps, they rarely autoamputate.
In 1998, the gene responsible for the majority of sporadic and inherited cases of PJS was identified as STK11 (also known as LKB1), a serine/threonine kinase located on chromosome 19p13.3.48,63–66 STK11 is a tumor suppressor gene that is involved in a wide spectrum of cellular functions, including cellular proliferation, cell polarity, and apoptosis.67–69 STK11 has been shown to interact with other tumor suppressors, including TP53 and PTEN.67
There have been case reports of sporadic Peutz-Jeghers polyps, but this is somewhat controversial. In a study at a tertiary care center, three patients with potential sporadic Peutz-Jeghers polyps were identified, but two of them had other features suggestive of PJS, although strict clinical criteria were not met. The authors concluded that if sporadic Peutz-Jeghers polyps do exist, they are extremely rare.42
Pathologic Features
The hamartomatous polyps of PJS are histologically distinctive and consist of disorganized mucosa with prominent smooth muscle bundles. The smooth muscle fibers of the muscularis mucosae form thick cores and permeate the polyps in an arborizing fashion (Fig. 21.13, A). The epithelium lining these polyps is similar to the normal mucosa in that there are goblet cells, absorptive enterocytes, endocrine cells, and Paneth cells (see Fig. 21.13, B). Rarely, metaplastic bone formation has been reported in these polyps.70 As much as 10% of Peutz-Jeghers polyps have foci of misplaced epithelium, in which the epithelium is located within the submucosa or the muscularis propria or both. This phenomenon is encountered more frequently in polyps larger than 3 cm in diameter (see Fig. 21.13, C).71
The development of Peutz-Jeghers polyps is the subject of some controversy. Given the prominent smooth muscle in these polyps and the presence of misplaced epithelium, some authors suggest that patients with PJS are prone to mucosal prolapse.72 Indeed, the differential diagnosis of Peutz-Jeghers polyps, particularly in the colon, is mucosal prolapse polyps. However, hemorrhage and hemosiderin deposits, commonly seen in prolapse polyps, are not prominent in Peutz-Jeghers polyps.
The sequence of events that results in GI dysplasia and carcinoma is also unclear in relation to Peutz-Jeghers polyps. One occasionally encounters dysplasia and even carcinoma in a Peutz-Jeghers polyp; however, in a study of 2461 Peutz-Jeghers polyps from 63 patients, only 6 polyps contained foci of dysplasia.73 These results argue that the hamartoma-adenoma-carcinoma hypothesis proposed by some authors rarely occurs.74 Peutz-Jeghers polyps have also been shown to be polyclonal, arguing against the possibility of malignant potential. Given these results, it is possible that carcinoma develops in surrounding mucosa that may have an accelerated pathway to dysplasia and carcinoma.57,72
In any case, when one encounters dysplasia in a Peutz-Jeghers polyp, the dysplasia should be classified according to a two-tiered system (i.e., low- or high-grade dysplasia). Care must be taken to not interpret misplaced epithelium as invasive adenocarcinoma.71 The misplaced epithelium should not have desmoplastic stroma or an infiltrative growth pattern. The epithelial cells should have a similar cytology to other parts of the polyps, and the glands should be invested with lamina propria.
Juvenile Polyps
Clinical and Endoscopic Features
Juvenile polyps of the small intestine are rare and occur in two polyposis syndromes: juvenile polyposis syndrome and PTEN hamartoma tumor syndrome.75 Juvenile-type polyps can also be seen in Cronkhite-Canada syndrome, an extremely rare, noninherited disorder associated with diffuse GI polyposis.76,77 Juvenile polyps tend to be friable and undergo autoamputation. Endoscopically, they appear smooth and often have evidence of hemorrhage. As a result, patients typically present with complaints related to bleeding, such as occult blood loss, hematochezia, fatigue, or anemia.
First described in 1964, juvenile polyposis syndrome is an extremely rare (1 per 100,000 births), autosomal dominant polyposis syndrome with variable penetrance. Three clinical phenotypes of juvenile polyposis syndrome have been described: infantile juvenile polyposis, juvenile polyposis coli, and generalized juvenile polyposis. Infantile juvenile polyposis syndrome, the most severe type, is characterized by early and diffuse involvement of the entire GI tract. Because these hamartomatous polyps have surface erosion, affected patients usually present with GI bleeding and anemia. Juvenile polyposis syndrome is also associated with a significantly increased risk of adenocarcinoma in involved GI sites. The colon is the site most commonly involved by disease, and the cumulative lifetime risk for colorectal cancer is 40%, with a median age at onset of 44 years.78 The lifetime risk of developing carcinomas of the stomach, small intestine, or pancreas is approximately 10% to 15%.79 Prevention of intestinal adenocarcinoma mandates intensive screening, with colonoscopy and upper GI endoscopy every 2 years, beginning at age 15.75 Severe GI bleeding may necessitate immediate surgery. Progression to dysplasia in a polyp is not necessarily an indication for surgery, provided that it can be completely removed. Given the increased risk of adenocarcinoma, genetic screening of family members of affected individuals is essential.
Approximately 25% of patients with juvenile polyposis syndrome have a negative family history, and their polyps presumably arise from a de novo mutation. Mutations in two genes have been described in patients with juvenile polyposis: SMAD4 (also known as MADH4 or DPC4) and BMPR1A (also known as ALK3).80–85 The proteins encoded by these genes are involved in the transforming growth factor-β (TGF-β) signal transduction pathway. Because the TGF-β signaling pathway mediates growth inhibitory signals, proteins in this pathway function as tumor suppressors. Mutations in SMAD4 have been shown to be more common in patients with upper GI polyps, whereas patients with BMPR1A mutations are more likely to present at a younger age than those with mutations in SMAD4.86
The term PTEN hamartoma tumor syndrome was coined to encompass all hamartoma syndromes that arise through mutations in the tumor suppressor, PTEN. This term encompasses Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome (BRRS), and Proteus syndrome, among others.75,87 Patients with these syndromes have a wide variety of GI, skin, and soft tissue tumors and an increased risk for carcinomas of the colorectum, thyroid, breast, or endometrium.88 Cowden syndrome is 50% less common than juvenile polyposis syndrome and is characterized by multiple hamartomatous tumors of endodermal, mesodermal, and ectodermal origin. The most distinctive lesions are mucocutaneous—in particular, trichlemmomas, acral keratosis, subcutaneous lipomas, palmarplantar keratoses, oral cobblestoning, and oral papillomas. Colonic polyps in Cowden syndrome are common and range from ganglioneuromas to adenomas, lipomas, hyperplastic polyps, and juvenile polyps. Polyps of the small intestine are more rare but do occur, with frequencies between 37% and 66%.89,90 BRRS is also associated with juvenile polyps of the GI tract (mostly limited to ileum and colon); however, these patients also have neurologic findings, including macrocephaly and slowed psychomotor development. Some BRRS patients also develop extraintestinal hamartomatous tumors similar to those seen in patients with Cowden syndrome.75
Pathologic Features
Juvenile polyps are pedunculated and are histologically characterized by abundant lamina propria, dilated mucin-filled crypts, active inflammation, and surface erosion (Fig. 21.14). The epithelium in these polyps can contain adsorptive cells, goblet cells, endocrine cells, and Paneth cells. Often, eosinophils are prominent. Unlike Peutz-Jeghers polyps, juvenile polyps lack prominent smooth muscle bundles. Juvenile polyps in the small bowel are very similar to inflammatory-type polyps. The distinction between these two polyps is mostly based on the clinical context in which they arise. In a patient with a known polyposis syndrome, the diagnosis of a juvenile polyp is appropriate. In cases where multiple inflammatory or juvenile polyps are seen, the possibility of a polyposis syndrome should considered. If a single juvenile or inflammatory polyp is encountered, one should be cautious in suggesting a polyposis syndrome.
Benign Epithelial Neoplasms
Adenomas of the Small Intestine
Clinical and Endoscopic Features
Adenomas account for approximately 25% of benign neoplasms of the small intestine and demonstrate a distinct predilection for the ampulla and the peraampullary region. Most are sporadic lesions occurring in older individuals, but they also occur in patients with adenomatous polyposis syndromes, including familial adenomatous polyposis (FAP) and MUTYH-associated polyposis (MAP). Approximately 17% of patients with MAP have duodenal polyposis, and their lifetime risk of duodenal cancer is 4%.91,92 Small bowel adenomas also occur in Lynch syndrome. Unlike adenomas in MAP and FAP, those in Lynch may not have such a strong predilection for the proximal small bowel and ampulla. As in MAP, the lifetime risk of small bowel adenocarcinoma in Lynch syndrome is 4%.93 Given that adenomas may not be amenable to upper endoscopy, capsule endoscopy has been used to identify occult adenomas and carcinomas in Lynch syndrome94; however, this is currently not recommended for routine screening.93
Although some adenomas located distal to the ampulla produce luminal obstructive symptoms or occult blood loss, most are asymptomatic and clinically silent until complicated by the development of carcinoma. In contrast, ampullary and peraampullary adenomas can produce obstructive jaundice before malignant transformation and become symptomatic earlier in their evolution.
Pathologic Features
With the exception of some ampullary neoplasms, most adenomas of the small intestine are endoscopically and histologically similar to those of the colon. Larger adenomas (>1 cm) are more likely to harbor areas of high-grade dysplasia, and approximately 50% have a villous component, consisting of long papillary projections lined by columnar, mucin-depleted epithelial cells with enlarged, hyperchromatic nuclei. Paneth cells and endocrine cells are often more numerous in adenomas of the small intestine compared with those of the colon. The criteria for grading dysplasia in adenomas of the small intestine are similar to those for colonic adenomas. Low-grade dysplasia is defined as nuclear stratification confined to the lower half of the cells in the absence of complex architectural changes (Fig. 21.15, A and B). High-grade dysplasia is characterized by the presence of marked nuclear enlargement with loss of nuclear polarity and irregular nuclear membranes. These nuclear changes are often accompanied by complex architectural abnormalities such as cribriforming and glandular crowding (see Fig. 21.15, C). The terms severe dysplasia and carcinoma in situ should no longer be used to describe these changes.
Unlike adenomas of the colon, but similar to those of the esophagus and stomach, the lamina propria in adenomas of the small intestine contains a rich lymphatic network. When dysplastic epithelial cells break through the basement membrane and infiltrate the lamina propria, the term intramucosal adenocarcinoma is appropriate. Intramucosal adenocarcinoma of the small intestine carries a risk of lymph node metastasis and is staged as pT1a carcinoma, according to the seventh edition of the Cancer Staging Manual of the American Joint Committee on Cancer.95
Ampullary and Peraampullary Adenomas
Clinical and Endoscopic Features
Ampullary adenomas typically occur in older adults (mean age, 64 years), and most are sporadic96 (see also Chapter 41). Small adenomas of the ampulla and peraampullary region appear as polypoid excrescences or as a subtle prominence of the ampulla, whereas larger adenomas are velvety, flat lesions (“carpet adenomas”) at high risk for the development of invasive adenocarcinoma (Fig. 21.16). Several investigators have postulated that the ampulla may be prone to neoplastic transformation because it is chronically irritated by pancreatic juices and bile salts, resulting in long-standing injury to the mucosa and culminating in epithelial cell dysplasia.97,98
Pathologic Features
Adenomas of the ampulla can be divided into intestinal and pancreaticobiliary types (although mixed lesions also occur). The intestinal-type adenomas are more common (~75%) and resemble colonic and peraampullary/duodenal adenomas (Fig. 21.16, C and D). Pancreaticobiliary-type adenomas—or neoplasms, as preferred by the current World Health Organization (WHO) classification99—often have prominent papillary architecture and resemble intraductal papillary neoplasms of the pancreatic duct. The papillary projections are lined by a monolayer of cuboidal epithelial cells with round nuclei and eosinophilic cytoplasm (see Fig. 21.17, A and B). These lesions are often high-grade with complex architecture including extensive arborization of the papillae, cribriforming, and marked nuclear atypia. Some authors have used immunohistochemistry to differentiate these histologic subtypes. Intestinal-type adenomas express apomucin MUC2 and the transcription factor CDX2. Pancreaticobiliary-type neoplasms express MUC1, MUC5AC, and MUC6.100 However, there is considerable overlap in the expression of these proteins in these lesions, limiting their clinical utility.
Both intestinal and pancreaticobiliary mass-forming lesions of the ampulla are often associated with adjacent invasive adenocarcinoma (~80%). Although most adenocarcinomas of the ampulla arise from adenomas, nonpolypoid (or flat) dysplastic lesions of the ampulla also occur. These dysplastic lesions are almost always associated with an adjacent invasive adenocarcinoma. Some of these lesions have a micropapillary architecture lined by dysplastic cuboidal to columnar cells.
The evaluation of biopsy or resection specimens of ampullary and peraampullary adenomas may be difficult for several reasons. Ampullary ducts and glands are in close proximity to bundles of smooth muscle, and when dysplastic epithelium extends into these structures, a mistaken diagnosis of invasive adenocarcinoma may be rendered (Fig. 21.18). The lobular architecture and lack of desmoplasia can be helpful in distinguishing this finding from invasive adenocarcinoma. As with their colonic counterparts, misplacement of the dysplastic epithelium into the submucosa may occur as a consequence of mechanical injury or prolapse in these adenomas. The presence of lamina propria around these dysplastic glands and the presence of hemosiderin deposits and acellular mucin pools are helpful features to differentiate epithelial misplacement from invasive adenocarcinoma. Finally, because many patients with ampullary lesions have obstructive jaundice, biliary stents are often placed. Stents cause inflammatory epithelial injury and erosions of the ampullary epithelium with concomitant reactive and regenerative epithelial changes, as well as inflammation of the underlying stroma (Fig. 21.19). This constellation of findings may mimic dysplasia or even invasive cancer. Therefore, it is imperative to know the patient’s status regarding stenting of the common bile duct to evaluate the specimen properly and avoid the pitfall of overdiagnosing neoplasia. The presence of active inflammation and foveolar metaplasia should make one cautious in interpreting the epithelial changes as dysplastic.
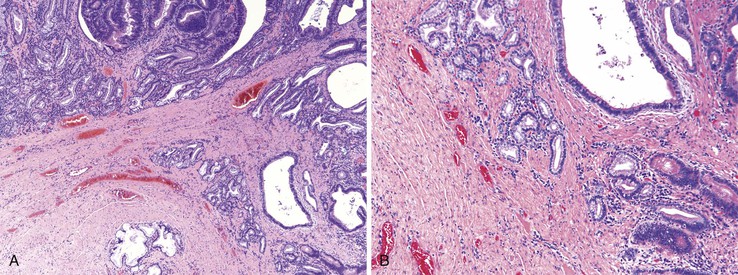
[/not-level-membership-for-pathology-category]
