Chapter 15 Polycystic Ovary Syndrome
INTRODUCTION
In 1935, Stein and Leventhal described patients who had a constellation of amenorrhea, infertility, and enlarged, polycystic ovaries.1 They found that bilateral ovarian wedge resection resulted in regular menses and even pregnancy in some. They reasoned that the thickened tunica was preventing follicles from reaching the surface of the ovary, resulting in the classic appearance of an enlarged ovary with multiple follicles beneath the cortex. Today we know that the mere finding of a polycystic-appearing ovary is not pathognomonic of PCOS, because many females without the disorder will have similar ovarian morphology.2,3
DIAGNOSTIC CRITERIA
In 1990, a conference on PCOS sponsored by the National Institutes of Health (NIH) led to diagnostic criteria based on a majority opinion of conference participants.4 The criteria included: hyperandrogenism and/or hyperandrogenemia, chronic anovulation, and exclusion of other known disorders. In 2003, the European Society of Human Reproduction and Embryology (ESHRE)/American Society of Reproductive Medicine (ASRM)-sponsored PCOS consensus workshop in Rotterdam revised the diagnostic criteria for PCOS.5 The revised criteria state that PCOS remains a diagnosis of exclusion, but that two out of the following three criteria must be present: (1) oligo-ovulation or anovulation, (2) hyperandrogenism and/or hyperandrogenemia, and (3) polycystic ovaries.
PREVALENCE
The prevalence of PCOS is estimated to be 4% to 12% of reproductive-age women. The largest U.S. study on PCOS prevalence was published in 1998.6 Out of 277 women included in the study, 4.0% had PCOS as defined by the 1990 NIH criteria. The prevalence was 4.7% for white women and 3.4% for black women. The inclusion of polycystic ovaries in the 2003 Rotterdam criteria calls for re-evaluation of the prevalence of PCOS, because 21% to 23% of normal women have polycystic-appearing ovaries on ultrasound.2,3
CLINICAL PRESENTATION
Hyperandrogenism
Clinical manifestations of hyperandrogenemia include hirsutism, acne, and male pattern alopecia. Hirsutism is defined as the growth of coarse, pigmented hairs in androgen-dependent areas such as the face, chest, back, and lower abdomen. Approximately 80% of hirsute patients will have PCOS.7 The modified Ferriman-Gallway scoring system can be used for clinical assessment of hirsutism. This was originally used in the United Kingdom for a population of presumably white women, and scores hair growth in nine body areas from 0 (absence of terminal hairs) to 4 (extensive terminal hair growth).8 Sex hair growth differs among various ethnic and genetic groups, being decreased in Asians, even when high androgen levels are present.9
Other hyperandrogenic manifestations commonly found in PCOS patients include acne and alopecia.10,11 Acne is a result of androgen stimulation of the pilosebaceous unit with increased skin oiliness.10 Cela and colleagues investigated a multiethnic group of 89 women who presented with androgenic alopecia and found that 67% had polycystic ovaries, compared to 27% of controls.11 In addition, women with alopecia had higher androgen levels and a higher prevalence of hirsutism.
Obesity
Obesity is very common in PCOS, with the android pattern present in approximately 44% of women with PCOS.12 This central obesity is more characteristic of PCOS, because these patients have an increased waist-to-hip ratio compared to obese women without PCOS.13 Hyperinsulinemia may stimulate central adiposity, which, in turn, exacerbates underlying or latent insulin resistance.14 It has been shown that obese women with PCOS have greater insulin resistance than weight-matched controls.15
Acanthosis Nigricans
Acanthosis nigricans is a dermatologic condition of hyperkeratosis and increased skin pigmentation with raised, symmetric, darkened, velvety plaques that commonly appear on the nape of the neck. It can also be found in the axilla, groin, and other intertriginous areas of the body. This skin finding is a common manifestation of insulin resistance in patients with PCOS. Mor and coworkers found that acanthosis nigricans is more likely to be found in PCOS patients with insulin resistance compared to PCOS patients without insulin resistance (OR = 6.0, P < 0.5).16 The elevated insulin level has a mitogenic effect on basal cells of the epidermis, making acanthosis nigricans a relatively specific clinical marker of insulin resistance.17 Other pathologic conditions associated with acanthosis nigricans include insulinoma and malignant disease, especially adenocarcinoma of the stomach.
Reproductive Aberration
Miscarriage
The risk of a first-trimester spontaneous abortion (i.e., miscarriage) is reported to be significantly higher for patients with PCOS than in normal women. The spontaneous abortion rate in PCOS is reported to be 30%.18 In comparison, retrospective studies find the risk of spontaneous abortion to be 5% to 14% for normal women.19,20 Of patients with recurrent miscarriage, 36% to 82% have polycystic ovaries.18,21,22
Several explanations have been offered. For example, Homburg and colleagues demonstrated that high concentrations of LH during the follicular phase in women with polycystic ovaries have a deleterious effect on rates of conception and are associated with early pregnancy loss.23 In another study, those with elevated LH levels had a 65% miscarriage rate compared to 12% of pregnancies with normal LH levels.24 Elevated serum androgen levels, obesity, and hyperinsulinemia have also been implicated as risk factors for early pregnancy loss.25,26
Polycystic Ovarian Morphology
The Rotterdam ESHRE/ASRM-sponsored PCOS consensus workshop group included the polycystic morphology of the ovary as one of the diagnostic criteria for the syndrome.5 A polycystic ovary is defined as having 12 or more follicles in one ovary measuring 2 to 9 mm in diameter, and/or increased ovarian volume of greater than 10 mL, which is the maximum size of a normal ovary3 (Fig. 15-1).
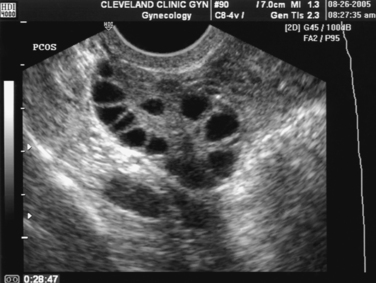
This definition should not be substituted with a subjective appearance of a polycystic ovary. The classic image is that of an enlarged ovary containing an increased number of small follicles around the periphery of the cortex (i.e., a string of pearls) with a bright echogenic stroma. The characteristic increase in stromal volume differentiates the polycystic ovary from a multifollicular ovary.3
The follicle distribution is omitted from the definition of a polycystic ovary, as well as the increase in stromal echogenicity and volume, but ovarian volume has been shown to be a good surrogate for stromal volume.27 Oral contraceptives modify ovarian morphology; thus the definition of a polycystic ovary does not apply to women taking these medications. When there is a dominant follicle (>10 mm) or a corpus luteum, the ultrasound should be repeated during the early follicular phase of the next cycle.
PATHOGENESIS
Altered Gonadotropin Secretion
One of the well-described features of PCOS is an increase in LH and relative decrease in follicle stimulating hormone (FSH).28 The relative decrease in FSH is the chief cause of anovulation, because increasing FSH will lead to folliculogenesis. The pulsatile secretion of LH from the pituitary is increased in amplitude and frequency in PCOS.29 In addition, the pituitary has a greater LH response to gonadotropin-releasing hormone (GnRH) compared with normal women.29,30 Elevations in LH could be secondary to increased GnRH pulse frequency, stimulatory factors other than GnRH, or a combination of these effects.
The pulsatile secretion of GnRH cannot be studied in humans, so it must be inferred by detecting peripheral LH patterns. A study of PCOS women by Berga and colleagues found increased pulse frequency and amplitude for LH and α-subunit, providing evidence for aberrant increases in GnRH pulse frequency (Fig. 15-2).29 Furthermore, increased GnRH pulse frequency in rats has been shown to increase LHβ gene expression.31 Elevated LH is not caused by altered pituitary sensitivity to GnRH; GnRH receptor blockade resulted in similar LH decreases in PCOS and normal women.32
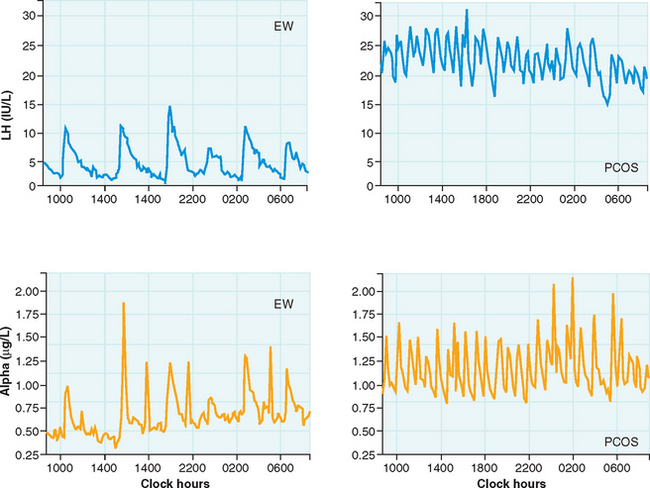
These findings suggest a derangement of the hypothalamic-pituitary axis, which appears to play a major role; many of the cardinal features of PCOS can be traced to alterations in gonadotropins. The basis for decreased FSH secretion in PCOS has not been determined, although the negative feedback effect of chronic unopposed estrogen secretion in these women has been implicated as a mechanism.33
Altered GnRH Drive
Neuroanatomic Considerations
The GnRH pulse generator refers to the synchronized pulsatile secretion of GnRH from neurons that are widely distributed in the medial basal hypothalamus. GnRH is synthesized in neuronal cell bodies that have migrated during fetal life from the olfactory placode to the hypothalamus and is secreted from neuroendocrine terminals localized in the median eminence. Knobil conducted experiments with the Rhesus monkey to establish that the GnRH system exhibits rhythmic electrical behavior in the arcuate nucleus of the medial basal hypothalamus.34 There was remarkable synchrony between pulses of GnRH in the portal blood and LH pulses in peripheral blood. This phenomenon was later studied in isolated human medial basal hypothalamus where GnRH pulses were found to occur at a frequency of 60 to 100 minutes.35 Thus, the GnRH neurons per se, with their intrinsic pulsatile secretion, appear to constitute the GnRH pulse generator.
There is compelling evidence that non-neuronal cells, such as glial and endothelial cells, regulate GnRH secretion in the median eminence. Glial processes belonging to either astrocytes or tanycytes separate GnRH nerve endings from the pericapillary space in the median eminence and, through signaling molecules such as nitric oxide, play a role in regulation of GnRH secretion into pituitary portal blood vessels.36,37 The secretion of GnRH into the portal vasculature also appears to be regulated by dynamic remodeling of GnRH neurovascular junctions. Morphologic plasticity of the median eminence during the menstrual cycle has been demonstrated, where the maximal number of GnRH neurovasculature junctions are found during the LH surge.38
In Vitro GnRH Neuroregulation
Mellon and coworkers developed a cell line of immortalized hypothalamic GnRH neurons (GT1 cells) that were determined to have an inherent ability to secrete GnRH in a pulsatile fashion.40 The development of GnRH cell lines, such as the GT1 cells, has permitted numerous studies to identify putative factors that regulate pulsatile GnRH secretion. Substances implicated in the modulation of the GnRH pulse generator include norepinephrine, dopamine, insulin-like growth factor I (IGF-I), γ-aminobutyric acid (GABA), and opioids, among others. There also appears to be an autocrine regulation of GnRH release, because GT1 cells themselves express GnRH receptors. The activation of GnRH receptors enhances action potentials, heightens synchronization of neuronal activities, and results in pulsatile release of GnRH.
In Vivo GnRH Neuroregulation in PCOS
The GnRH pulse generator in PCOS patients is intrinsically faster, and the frequency is less likely to be suppressed with continuous estrogen and progesterone treatment.40 A persistently rapid GnRH pulse generator would increase LH secretion and could suppress FSH levels low enough to prevent folliculogenesis.
Increased central adrenergic tone has been implicated as a cause of the aberrations of GnRH and gonadotropin secretion in PCOS. One possible mechanism is the increase in local blood flow and permeability of the portal vascular system, permitting the entry of increased amounts of GnRH.41 Dopamine injection into the third ventricle led to a rapid increase in GnRH and prolactin inhibitory factor in portal blood, suggesting dopamine-mediated regulation of GnRH and prolactin inhibitory factor.42 The identification of β1-adrenergic and D1-dopaminergic receptors on GT1 GnRH neurons provides a mechanism by which norepinephrine and dopamine could regulate gonadotropin release via direct synapses on GnRH neurons.43
Other studies have examined the effect of dopamine on immunoreactive LH. Dopamine exerts an inhibitory effect on LH secretion in normally cycling women44 and also has been shown to lower circulating LH in PCOS patients.45 It has been suggested that an impairment of the dopaminergic inhibitory activity on LH secretion leads to the increased frequency of LH pulses seen in PCOS women.46 In fact, the dopamine agonist bromocriptine has been shown to lower circulating LH and improve menstrual function in normoprolactinemic PCOS women.47 This finding has not been consistently demonstrated, as Lobo and coworkers did not find an increase in the ratio of bioactive to immunoreactive LH in response to dopamine alone or a dopamine-carbidopa combination, and concluded that dopamine did not play a critical role in LH secretion.48
The role of IGF-I in modulation of GnRH cells has also been investigated. IGF-I regulates growth, differentiation, survival, and reproductive function. The IGF receptor is a tyrosine kinase receptor located in the periphery and central nervous system, including the median eminence.49 Mouse studies have shown that IGF-I mRNA in the hypothalamus increases through peripubertal and adult development.50 IGF-I was found to stimulate GnRH gene expression in postnatal and peripubertal mice.50 Another study using the GT1 cell line demonstrated that IGF-I treatment caused a significant increase in GnRH nuclear primary transcript levels and cytoplasmic mRNA.51 In PCOS women, an increased ratio of IGF-I to IGF binding proteins correlated significantly with increased concentrations of circulating LH.30 These findings suggest that IGF-I can modulate GnRH neurons by inducing gene expression, resulting in more circulating LH.
Other substances found to affect GnRH and LH secretion include GABA and opioids. GABA may tonically inhibit LH secretion, as evidenced by an increase in LH secondary to a GABA-A receptor blocking drug, bicuculline.52 Naloxone is an opioid antagonist that has been shown to increase luteal phase LH, suggesting that opiates have an inhibitory effect on pituitary gonadotropins.53
Hyperandrogenemia
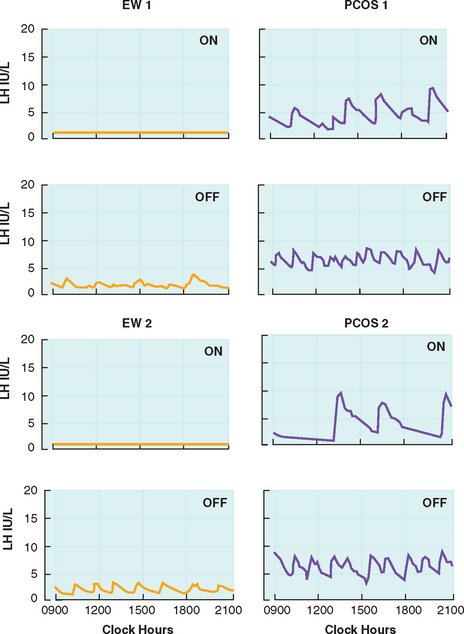
Circulating androgens are elevated in PCOS, with contributions from the ovary and adrenal glands. The elevated androgens can only be partially suppressed with combination oral contraceptive therapy. Daniels and Berga treated PCOS women with 3 weeks of combination oral contraceptives and found that androstenedione levels remained significantly higher compared to treated controls.40 Pulse frequency of LH was suppressed in both PCOS women and controls, but the frequency remained significantly higher in PCOS patients (Fig. 15-3). This suggests reduced sensitivity of the GnRH pulse generator to suppression by sex steroids. The authors also suggest that the GnRH drive in PCOS women may be intrinsically and irreversibly faster than in eumenorrheic women. GnRH output may be affected by peripheral androgens. Androgen blockade restores hypothalamic sensitivity to feedback inhibition by ovarian steroids.54 Administration of estrogen and progesterone to PCOS women and controls pretreated with the anti-androgen flutamide demonstrated similar reductions in LH pulse frequency.
Theca Cell Function
Ovarian hyperandrogenism is driven by LH acting on theca cells, and the effect is amplified by the increased sensitivity of PCOS theca cells to LH.55 Hyperandrogenism may also result from dysregulation of the androgen-producing enzyme P450c17, which has 17α-hydroxylase and 17,20-lyase activity. The dysregulation appears to be an intrinsic abnormality of P450c17, but autocrine/paracrine factors may also be involved.56
Insulin and IGF have also been shown to play a role in ovarian androgen production. Receptors for insulin, IGF-I, and IGF-II have been localized to the theca compartment of ovaries from normal and PCOS patients.57,58 When given IGF-I, IGF-II, and insulin, thecal cell cultures from euandrogenic women produced increased androgens, and the effect was potentiated when LH was added.57,59 Ovarian stroma from hyperandrogenic patients also released more androstenedione and testosterone in response to insulin, however, no synergy was found between insulin and LH.60
In contrast, in vivo studies do not find significant increases in androgen secretion in PCOS or normal women despite considerable increases in insulin levels. Dunaif and Graf studied PCOS and normal patients undergoing evaluation with the hyperinsulinemic euglycemic clamp, and found that insulin decreased androgens in PCOS women and did not increase androgens in normal women.13 This argues against a simple causal relationship between hyperinsulinemia and hyperandrogenism, but a role for insulin is strongly suggested by the observation that reduction of hyperinsulinemia is associated with decreases in serum androgens. Treatment of PCOS patients with metformin, which reduces hepatic glucose production and secondarily lowers insulin, has been shown to decrease levels of testosterone, dehydroepiandrosterone sulfate (DHEAS), and androstenedione.61
Adrenal Function
Excess adrenal androgen production is seen in PCOS women, with a 48% to 64% increase in DHEAS and 11β-hydroxyandrostenedione.9 The underlying cause of elevated adrenal androgens is yet to be elucidated, but PCOS women do not have increased corticotropin levels.62 Increased adrenal androgen production in PCOS is likely caused by either altered adrenal responsiveness to corticotropin or abnormal adrenal stimulation by factors other than corticotropin. A recent study by Moran and colleagues found that adrenal androgen excess in PCOS is associated with a greater 17α-hydroxylase activity in response to corticotropin.63 The cytochrome P450c17 (CYP17) gene regulates 17α-hydroxylase and 17,20-lyase activity. A defect in the P450c17 enzyme or in the regulation by autocrine/paracrine factors appears to be involved.56
Hyperinsulinemia may also play a role in adrenal androgen production in PCOS. PCOS patients treated with troglitazone experienced improvement in insulin resistance with a concomitant decrease in DHEAS levels, regardless of initial DHEAS levels.64 Obese PCOS women treated with pioglitazone had significant improvement in insulin sensitivity with a decrease in corticotropin-stimulated androstenedione levels.65 These data support the notion that insulin enhances corticotropin-stimulated steroidogenesis.
Anovulation
Granulosa Cell Function
FSH levels are characteristically low in PCOS women, resulting in arrested follicular development. Insufficient granulosa cell aromatase activity was the basis of earlier studies that tried to explain poor follicular development, because follicular fluid estradiol concentrations were thought to be low. To the contrary, more recent studies found that PCOS granulosa cells are hyperresponsive to FSH in vitro, and estradiol concentrations from PCOS follicles and normal follicles are no different.66 A dose–response study in PCOS women demonstrated a significantly greater capacity for estradiol production in response to recombinant human FSH compared with normal women.67 The incremental response of serum estradiol was almost two times greater and considerably accelerated compared with that found in normal women.
Van Der Meer and coworkers demonstrated that the accelerated estradiol production may simply be explained by the increased number of follicles.68 Another study of granulosa cells from polycystic ovaries discovered that there are more FSH receptors compared with cells from normal ovaries.69 Whether the increased estradiol responsiveness to FSH is due to increased number of follicles, increased granulosa cell sensitivity to FSH, or increased FSH receptor binding, women with PCOS appear to be susceptible to ovarian hyperstimulation in response to gonadotropins.
Insulin has been investigated as a modulator of both theca and granulosa cell function. As noted earlier, insulin has been shown to augment the effect of LH on thecal cell androgen secretion. The effects of insulin on granulosa cells are less well known. In vitro studies have shown that insulin augments two actions of FSH on granulosa cells: basal production of estrogen and progesterone and induction of LH responsiveness.70,71 The latter effect results in further enhanced LH-stimulated steroid production of estradiol and progesterone. Willis and Franks showed that the effects of insulin were apparent at low concentrations, underscoring the fact that insulin acts via its own receptor and suggesting that the ovary is not insulin resistant.72 The premature responsiveness of granulosa cells to LH may lead to untimely differentiation of granulosa cells, resulting in premature arrest of follicular growth. This might explain the presence of the many 5- to 10-mm follicles commonly observed in PCOS ovaries containing steroidogenically active granulosa cells that do not progress spontaneously to the preovulatory stage.
Insulin Resistance
Although 50% to 70% of PCOS patients have insulin resistance,73 it is not one of the diagnostic criteria for PCOS. The topic deservedly receives much attention because many of the clinical signs and symptoms of PCOS may be attributed to excess insulin exposure. The precise molecular basis for insulin resistance is unknown, but it appears to be a postreceptor defect.74 There is tissue specificity of insulin resistance in PCOS: muscle and adipose tissue are resistant; the ovaries, adrenals, liver, skin, and hair remain sensitive. The resistance to insulin in skeletal muscle and adipose tissue leads to a metabolic compromise of insulin function and glucose homeostasis, but there is preservation of the mitogenic and steroidogenic function in other tissues. The effect of hyperinsulinemia on the sensitive organs results in the downstream effects seen in PCOS, such as hirsutism,7 acanthosis nigricans,17 obesity,14 stimulation of androgen synthesis,57 increase in bioavailable androgens via decreased sex hormone-binding globulin (SHBG),75 and, potentially, modulation of LH secretion.60
In 1992, Hales and Barker proposed the concept that the environmental influence of undernutrition in early life increased the risk of type 2 diabetes in adulthood.76 They discovered a relationship between low birth weight and type 2 diabetes in men from England. The finding has been replicated in many different populations and ethnic groups, and the relationship has been extended to include the antecedent condition of insulin resistance.77 In the thrifty phenotype hypothesis, malnutrition serves as a fetal and infant insult that results in a state of nutritional thrift. The adaptations result in postnatal metabolic changes that prepare the individual for survival under poor nutritional conditions. One advantageous change is the development of insulin resistance in muscle and adipose tissue, with selective protection of brain growth and activity. The adaptations become detrimental when the postnatal environment changes to one of an overabundance of nutrients, resulting in obesity and diabetes.
Recognition of insulin resistance can identify those who are most likely to respond to lifestyle and pharmacologic intervention. Insulin resistance puts patients at increased risk for certain metabolic sequelae, such as diabetes, hypertension, dyslipidemia, and cardiovascular disease.78 Insulin resistance is a component of the World Health Organization (WHO) definition of the metabolic syndrome, which is a cluster of risk factors for cardiovascular disease.79 The WHO defines the metabolic syndrome as the presence of glucose intolerance or insulin resistance, with at least two of the following: hypertension, dyslipidemia, obesity, and microalbuminuria. Women with PCOS are 4.4 times more likely to have the metabolic syndrome, so it becomes prudent to screen these patients, especially in those with insulin resistance.80
LONG-TERM CONSEQUENCES OF PCOS
Metabolic Syndrome
Numerous studies have established that there is an increased risk of diabetes and cardiovascular disease in PCOS women. One third of PCOS patients have impaired glucose tolerance, and they also have a fivefold to tenfold greater risk for type 2 diabetes mellitus, regardless of ethnicity.73 By the time these patients reach menopause, they will have significantly more hypertension and diabetes compared to controls.81 Lipid abnormalities are also more prevalent in PCOS patients. There can be significant increases in total cholesterol, LDL cholesterol, and triglycerides and a decrease in HDL cholesterol compared to weight-matched controls.82 The dyslipidemia, impaired glucose intolerance, central obesity, hyperandrogenism, and hypertension seen in PCOS patients greatly increase the risk for cardiovascular disease. Based on this risk profile, women with PCOS have a sevenfold increased risk of myocardial infarction.83
Direct evidence for the increased cardiovascular risk was shown when women in their 40s with a history of PCOS were found to have subclinical atherosclerosis detected by carotid ultrasonography.84 PCOS women have also been found to have a higher prevalence of coronary artery and aortic calcification compared to age- and BMI-matched controls.80 Women with PCOS also have decreased global fibrinolytic capacity, suggesting the presence of a prothrombotic state, yet another risk for cardiovascular disease.85 Whether PCOS itself constitutes a risk factor independent of known risk factors remains to be elucidated. Despite the elevated risk of cardiovascular disease, a large retrospective study of almost 800 PCOS women in the United Kingdom did not find a markedly higher than average mortality from circulatory disease nor increased overall excess mortality.86
Cancer
Women with PCOS have unopposed estrogen and are more likely to be obese, which leads to concerns about an increased risk for endometrial hyperplasia or malignancy. One long-term follow-up study of PCOS women in the United Kingdom found a significantly elevated risk of endometrial cancer, but no increased risk of breast cancer.87 The elevated risk of endometrial cancer may be a consequence of more than just unopposed estrogen in these patients. Hormone analyses from endometrial cancer patients reveal a 3.6-fold and 2.8-fold increased risk of endometrial cancer among premenopausal and postmenopausal women, respectively, who have high levels of androstenedione.88 The elevations in LH may have a role as well, because LH/hCG receptors are overexpressed in endometrial hyperplasia and carcinoma.89 The authors concluded that the overexpression of these receptors is a feature of endometrial disease developing in younger anovulatory women, including those with PCOS. An investigation into the possible risk of postmenopausal breast cancer in PCOS women was performed using data from the Iowa Women’s Health Study. This prospective cohort study received completed questionnaires from over 41,000 women aged 55 to 69 and found no increased risk for breast cancer in women with a history of PCOS.90
LABORATORY EVALUATION
In the workup for anovulation, prolactinoma should be excluded. It is not uncommon to detect mild elevations in prolactin levels in PCOS patients, but high levels deserve magnetic resonance imaging of the pituitary to exclude prolactinoma. Thyroid-stimulating hormone (TSH) should be evaluated to exclude hyperthyroidism or hypothyroidism. Serum LH, FSH, and estradiol levels should be obtained to exclude hypothalamic amenorrhea or premature ovarian failure. A recent study suggested the combination of LH, FSH, and androstenedione levels provide the highest clinical utility in diagnostic sensitivity and specificity for PCOS.91
Diabetes Screen
The 2003 Rotterdam PCOS consensus group recommends a 2-hour oral glucose tolerance test (OGTT) for obese PCOS patients and nonobese PCOS patients with risk factors for insulin resistance, such as family history of diabetes.5 Women with PCOS are at significantly increased risk for impaired glucose tolerance and type 2 diabetes compared to age-, weight-, and ethnicity-matched controls.92 The 2-hour OGTT is the preferred method for the detection of diabetes in PCOS patients due to its increased sensitivity over fasting plasma glucose, and it can be used to detect impaired glucose tolerance, an independent risk factor for diabetes that is associated with cardiovascular disease and increased mortality. If either the fasting glucose is 126 mg/dL or more, or the 2-hour level is 200 mg/dL or more, diabetes is detected and should be confirmed with a repeat test. Impaired fasting glucose is defined as a glucose level between 100 mg/dL and 126 mg/dL. Impaired glucose tolerance is defined as a 2-hour glucose level between 140 mg/dL and 200 mg/dL. Both impaired fasting glucose and impaired glucose tolerance are independent risk factors for the future development of diabetes.
Insulin Resistance Evaluation
Defining insulin resistance is difficult because the concept is nebulous with no universally accepted diagnostic strategy. Generally speaking, insulin resistance is defined as “a subnormal biologic response to a given concentration of insulin.”93 The WHO defines insulin resistance as the lowest quartile of measures of insulin sensitivity.79 Once the diagnosis of insulin resistance is made, there are many implications given the numerous effects hyperinsulinemia may have on reproduction and metabolism.
The G0/I0 ratio was first described by Legro and coworkers as a measure of insulin sensitivity in white, obese PCOS women from Pennsylvania. The G0/I0 ratio detected insulin resistance with a sensitivity of 95% and specificity of 84% compared to the frequently sampled intravenous glucose tolerance test.94 Ducluzeau and coworkers later confirmed that the G0/I0 ratio is a predictor of insulin resiatnce in nonobese women as well.95 This study sought to identify the best markers of insulin resistance compared to the hyperinsulinemic euglycemic clamp. They measured serum SHBG, leptin, adiponectin, OGTT, and HOMA in addition to the G0/I0 ratio. The G0/I0 ratio emerged as the strongest independent parameter to appraise insulin resistance. The 2-hour OGTT can also be used to detect insulin resistance, with a 2-hour G0/I0 ratio of less than 1.0 suggestive of the diagnosis in the PCOS population.94 Correlation of insulin levels to glucose levels from the OGTT reveals how impaired glucose tolerance appears to be the result of decreased insulin sensitivity while impaired fasting glucose is a result of defective insulin secretion.96
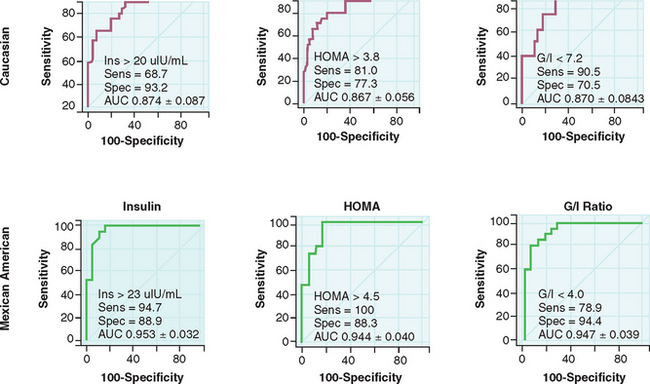
One must be cognizant that there is considerable ethnic variability when screening for diabetes and insulin resistance. No normative values have been developed that are ethnicity-specific. Kauffman and colleagues studied white and Mexican American women with PCOS and found differing values for G0/I0 ratios to detect insulin resistance in the two populations.97 A fasting insulin greater than 20 μU/mL and G0/I0 ratio less than 7.2 detected insulin resistance in white women; a fasting insulin greater than 23 μU/mL and G0/I0 ratio less than 4.0 detected insulin resistance in Mexican American women (Fig. 15-4). In the clinical setting, the 2-hour OGTT that measures both fasting and postprandial glucose and insulin levels will yield the most information about glucose intolerance and hyperinsulinemia. It is recommended that the 2-hour OGTT be used to screen for the metabolic syndrome in obese women with PCOS, as well as any nonobese women with PCOS who have risk factors for insulin resistance.98 It is also reasonable to obtain a fasting lipid profile in women suspected of having risk factors for cardiovascular disease.
TREATMENT
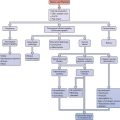
There are many considerations when deciding on therapy for PCOS (Fig. 15-5). Identification of patient concerns is necessary when prioritizing goals and formulating a treatment plan. A combination of therapies may be warranted, and the practitioner should appropriately counsel the patient on treatment expectations. Amelioration of long-term health risks should be emphasized regardless of the primary complaints of the patient.
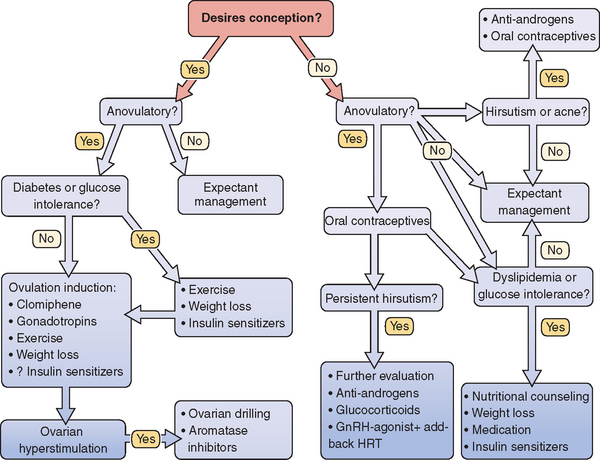
Figure 15-5 Polycystic ovary syndrome treatment algorithm.
(Data from Berga S: The obstetrician-gynecologist’s role in the practical management of polycystic ovary syndrome. Am J Obstet Gynecol 179:109S–113S, 1998.)
Weight Loss
Weight reduction should be a major component of any treatment plan for the overweight patient (BMI 26 or greater). Any sustained improvement in weight should involve diet and exercise, and consultation with a nutritionist may be helpful for those with difficulty achieving weight reduction. Obese PCOS patients who achieve weight loss will have an increase in SHBG, decrease in free testosterone, and improvement in fasting insulin levels.99
Oral Contraceptives
Korytkowski and coworkers have shown that short-term use of combination oral contraceptives in PCOS women results in a small decrease in insulin sensitivity and no change in the baseline elevation in triglyceride levels.100 However, in normal women, combination oral contraceptives were shown to produce an even more pronounced decline in insulin sensitivity, along with a significant elevation in triglyceride levels. The longer-term effects of combination oral contraceptives on insulin sensitivity and lipoprotein profiles have not been well documented. PCOS women are at greater risk for the development of diabetes and cardiovascular disease, and further investigation into the safety of long-term hormonal therapy is needed.
Antiandrogens
Antiandrogens are commonly used as an adjunct to oral contraceptive therapy for treatment of hirsutism, but they have also been found to improve ovulation and restore regular menses. It is important to remember that all antiandrogens are teratogenic and pose a risk of feminizing a male fetus, and thus should be used along with an effective form of contraception.
Spironolactone is an aldosterone antagonist and is the most commonly used adjunctive agent in the treatment of hirsutism. It competes for testosterone binding sites on the pilosebaceous unit, inhibits 5α-reductase, and inhibits androgen production by interfering with cytochrome P450.102 The potassium-sparing effect warrants judicious use in the patient on potassium supplementation or with preexisting hypertension.
Flutamide is a nonsteroidal antiandrogen that competes for the androgen receptor. Anovulatory PCOS patients treated with flutamide experienced resumption of ovulation with restoration of normal ovarian appearance with one dominant follicle.103 This study also reported a reduction in plasma levels of LH, androstenedione, and testosterone. Liver toxicity is a rare but potentially serious side effect of flutamide.
Insulin-Sensitizing Agents
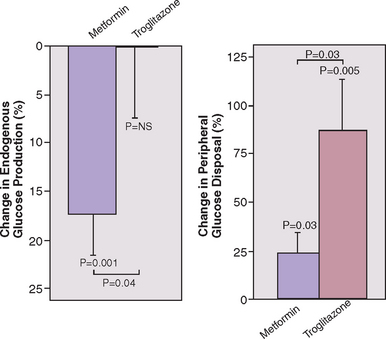
Insulin-sensitizing agents have been shown to improve endocrine and reproductive abnormalities in PCOS. Metformin is the most thoroughly investigated insulin-lowering agent used in PCOS. It is a biguanide that primarily works by suppressing hepatic gluconeogenesis and, to a lesser degree, increasing peripheral insulin sensitivity (Fig. 15-6).103 Thiazolidinediones are peroxisome proliferator activating receptor agonists that improve peripheral insulin sensitivity but do not appear to have an effect on hepatic glucose production (see Fig. 15-6).103 This class of medications includes troglitazone, pioglitazone, and rosiglitazone.
Many studies have demonstrated the positive effects of metformin on the reproductive axis of PCOS patients, with one of the most comprehensive studies recently demonstrating a dramatic improvement after 6 months of treatment. Metformin administration to nonobese hyperandrogenic PCOS patients resulted in a reduction of (1) LH pulse amplitude; (2) androstenedione levels; (3) testosterone levels; (4) ovarian volume; and (5) Ferriman-Gallway scores. Menstrual cyclicity was also improved in most patients.104 The investigators did not determine if metformin increased the likelihood of ovulation or if FSH levels rose. Similarly, troglitazone-treated PCOS patients demonstrated improved ovulation, decreased hirsutism, decreased free testosterone level, and increased SHBG.105
The improvement in ovulation and menstrual cyclicity in patients treated with insulin-sensitizing agents suggests improved fertility. Indeed, spontaneous and clomiphene-induced ovulation rates in metformin-treated PCOS women are increased.106 Spontaneous ovulation occurred in 34% of those treated with 500 mg of metformin three times daily compared to only 4% in the placebo group. Clomiphene-induced ovulation occurred in 90% of women who received metformin compared to 8% who received placebo. For those who are clomiphene-resistant, significant improvements in ovulation and pregnancy rates were reported in a randomized, double-blind, placebo-controlled trial for women pretreated with metformin.107 Troglitazone alone and the combination of troglitazone plus clomiphene is also associated with increased rates of ovulation and pregnancy in insulin-resistant women with PCOS.108
Though metformin is a category B medication, its use throughout pregnancy is becoming more attractive. In one retrospective study, Jakubowicz and colleagues found a significant reduction in the rate of early pregnancy loss for PCOS women who conceived while taking metformin and continued the agent throughout pregnancy. The rate of early pregnancy loss in the metformin group was 8.8% compared to 41.9% in controls. In the women with a prior history of miscarriage, the early loss rate was 11.1% for the metformin group compared with 58.3% in the control group.109 The efficacy of metformin for pregnancy loss is not yet clear, and safety data for this indication are lacking.
Another possible beneficial effect of metformin administration during pregnancy is the significant reduction in gestational diabetes, as seen in one prospective cohort study.110 Randomized trials are needed before use of metformin is supported to prevent gestational diabetes in PCOS women with insulin resistance.
Metformin should not be given to those with conditions associated with elevated lactate levels, such as renal or hepatic disease, because there is a risk of lactic acidosis with an associated mortality of 50%.111 Although most studies of metformin in PCOS used a dose of 500 mg three times daily, no studies have been performed to determine the optimal dosing regimen for improvement in insulin sensitivity, reduction of androgens, and resumption of ovulation. A dose–response study of type 2 diabetic patients demonstrated that the 2000 mg daily dose was optimal for improvement of glucose homeostasis,112 but the relevance of this dose to the PCOS population remains to be investigated.
Ovarian Surgery
Ovarian Drilling
A laparoscopic variant with similar results to ovarian wedge resection is called ovarian drilling (see Chapter 37). This procedure involves making multiple punctures in the ovarian cortex and destroying ovarian tissue using unipolar electrosurgery or laser. The results and complications for this approach appear to be similar or slightly less than those for ovarian wedge resection, although a prospective, randomized study has never been done. There are additional concerns about long-term effects of ovarian drilling on ovarian function.
SUMMARY
Treatment of PCOS is usually multifaceted, directed toward salient patient treatment goals while also aiming to diminish long-term risks of morbidity. When conception is desired, some forms of treatment are incongruous with others. For example, antiandrogens may be teratogenic, and HMG-CoA reductase inhibitors are contraindicated in those seeking conception. An idealized treatment algorithm is presented in Figure 15-5, which is likely to require revision as our understanding of the pathogenesis of PCOS improves.
1 Stein IF, Levinthal M. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29:181-191.
2 Farquhar CM, Birdsall M, Manning P, et al. The prevalence of polycystic ovaries on ultrasound scanning in a population of randomly selected women. Austral NZ J Obstet Gynaecol. 1994;34:67-72.
3 Polson DW, Adams J, Wadsworth J, Franks S. Polycystic ovaries—a common finding in normal women. Lancet. 1988;1:870-872.
4 Zawadski JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: Towards a rational approach. In: Dunaif A, Givens J, Haseltine F, Merriam G, editors. Polycystic Ovary Syndrome. Boston: Blackwell Scientific; 1992:377-384.
5 The Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19-25.
6 Knochenhauer ES, Key TJ, Kahsar-Miller M, et al. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeast United States: A prospective study. J Clin Endocrinol Metab. 1998;83:3078-3082.
7 O’Driscoll J, Mamtora H, Higginson J, et al. A prospective study of the prevalence of clear-cut endocrine disorders and polycystic ovaries in 350 patients presenting with hirsutism or androgenic alopecia. Clin Endocrinol. 1994;41:231-236.
8 Hatch R, Rosenfield R, Kim M, Tredway D. Hirsutism: Implications, etiology, and management. Am J Obstet Gynecol. 1981;140:815-830.
9 Carmina E, Koyama T, Chang L, et al. Does ethnicity influence the prevalence of adrenal hyperandrogenism and insulin resistance in polycystic ovary syndrome? Am J Obstet Gynecol. 1992;167:1807-1812.
10 Betti R, Bencini P, Lodi A, et al. Incidence of polycystic ovaries in patients with late-onset or persistent acne: Hormonal reports. Dermatologica. 1990;181:109-111.
11 Cela E, Robertson C, Rush K, et al. Prevalence of polycystic ovaries in women with androgenic alopecia. Eur J Endocrinol. 2003;149:439-442.
12 Carmina E, Lobo R. Polycystic ovary syndrome (PCOS): Arguably the most common endocrinopathy is associated with significant morbidity in women. J Clin Endocrinol Metab. 1999;84:1897-1899.
13 Dunaif A, Graf M. Insulin administration alters gonadal steroid metabolism independent of changes in gonadotropin secretion in insulin-resistant women with the polycystic ovary syndrome. J Clin Invest. 1989;83:23-29.
14 Arner P. Control of lipolysis and its relevance to development of obesity in man. Diabetes/Metab Rev. 1988;4:507-515.
15 Dunaif A, Segal K, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165-1174.
16 Mor E, Zograbyan A, Saadat P, et al. The insulin resistant subphenotype of polycystic ovary syndrome: Clinical parameters and pathogenesis. Am J Obstet Gynecol. 2004;190:1654-1660.
17 Barbieri R, Ryan K. Hyperandrogenism, insulin resistance and acanthosis nigricans syndrome: A common endocrinopathy with distinct pathophysiologic features. Am J Obstet Gynecol. 1983;147:90-101.
18 Sagle M, Bishop K, Ridley N. Recurrent early miscarriage and polycystic ovaries. BMJ. 1988;297:1027-1028.
19 Gray R, Wu L. Subfertility and risk of spontaneous abortion. Am J Public Health. 2000;90:1452-1454.
20 Regan L, Braude P, Trembath P. Influence of past reproductive performance on risk of spontaneous abortion. BMJ. 1989;299:541-545.
21 Liddell H, Sowden K, Farquhar CM. Recurrent miscarriage: Screening for polycystic ovaries and subsequent pregnancy outcome. Austral NZ J Obstet Gynaecol. 1997;37:402-406.
22 Clifford K, Rai R, Watson H, Regan L. An informative protocol for the investigation of recurrent miscarriage: Preliminary experience of 500 consecutive cases. Hum Reprod. 1994;9:1328-1332.
23 Homburg R, Armar N, Eshel A, et al. Influence of serum luteinising hormone concentrations on ovulation, conception, and early pregnancy loss in polycystic ovary syndrome. BMJ. 1988;297:1024-1026.
24 Regan L, Owen E, Jacobs H. Hypersecretion of luteinising hormone, infertility, and miscarriage. Lancet. 1990;336:1141-1144.
25 Fedorcsak P, Storeng R, Dale P, et al. Obesity is a risk factor for early pregnancy loss after IVF or ICSI. Acta Obstet Gynecol Scand. 2000;79:43-48.
26 Tulppala M, Stenman U, Cacciatore B, Ylikorkala O. Polycystic ovaries and levels of gonadotrophins and androgens in recurrent miscarriage: Prospective study in 50 women. Br J Obstet Gynecol. 2000;100:348-352.
27 Buckett W, Bouzayen R, Watkin K, et al. Ovarian stromal echogenicity in women with normal and polycystic ovaries. Hum Reprod. 1999;14:618-621.
28 Yen SS, Vela P, Rankin J. Inappropriate secretion of follicle-stimulating hormone and luteinizing hormone in polycystic ovary disease. J Clin Endocrinol Metab. 1970;30:435-442.
29 Berga S, Guzick D, Winters S. Increased luteinizing hormone and α-subunit secretion in women with hyperandrogenic anovulation. J Clin Endocrinol Metab. 1993;77:895-901.
30 Morales A, Laughlin GA, Butzow T, et al. Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: Common and distinct features. J Clin Endocrinol Metab. 1996;81:2854-2864.
31 Dalkin AC, Haisenleder DJ, Ortolano GA, et al. The frequency of gonadotropin-releasing hormone stimulation differentially regulates gonadotropin subunit messenger ribonucleic acid expression. Endocrinology. 1989;125:917-924.
32 Hayes F, Taylor A, Martin K, Hall J. Use of a gonadotropin-releasing hormone antagonist as a physiologic probe in polycystic ovary syndrome: Assessment of neuroendocrine and androgen dynamics. J Clin Endocrinol Metab. 1998;83:2343-2349.
33 Yen SS. The polycystic ovary syndrome. Clin Endocrinol. 1980;12:177-207.
34 Knobil E. Neuroendocrine control of the menstrual cycle. Recent Prog Hormone Res. 1980;36:53.
35 Rasmussen DD, Gambacciani M, Swartz W, et al. Pulsatile gonadotropin-releasing hormone release from the human medibasal hypothalamus in vitro: Opiate receptor-mediated suppression. Neuroendocrinol. 1989;49:150.
36 Prevot V. Glial-neuronal-endothelial interactions are involved in the control of GnRH secretion. J Neuroendocrinol. 2002;14:247-255.
37 Prevot V, Croix D, Rialas C, et al. Estradiol coupling to endothelial nitric oxide stimulates gonadotropin-releasing hormone release from rat median eminence via a membrane receptor. Endocrinology. 1999;140:652-659.
38 Prevot V, Croix D, Bouret S, et al. Definitive evidence for the existence of morphological plasticity in the external zone of the median eminence during the rat estrous cycle: Implication of neuro-glio-endothelial interactions in gonadotropin-releasing hormone release. Neuroscience. 1999;94:809-819.
39 Wetsel WC, Valenca M, Merchenthaler I, et al. Intrinsic pulsatile secretory activity of immortalized luteinizing hormone-releasing hormone-secreting neurons. Proc Natl Acad Sci. 1992;89:4149-4153.
40 Daniels T, Berga S. Resistance of gonadotropin releasing hormone drive to sex steroid-induced suppression in hyperandrogenic anovulation. J Clin Endocrinol Metab. 1997;82:4179-4183.
41 Kalro BN, Loucks TL, Berga SL. Neuromodulation in polycystic ovary syndrome. Infertil Reprod Med Clin. 2003;14:529-555.
42 Kamberi IA, Mical RS, Porter JC. Hypophysial portal vessel infusion: In vivo demonstration of LRF, FRF, and PIF in pituitary stalk plasma. Endocrinology. 1971;89:1042-1046.
43 Findell PR, Wong KH, Jackman JK, Daniels DV. β1-Adrenergic and dopamine (D1) receptors coupled to adenylyl cyclase activation in GT1 gonadotropin-releasing hormone neurosecretory cells. Endocrinology. 1993;132:682-688.
44 Leblanc H, Lachelin GC, Abu-Fadil S, Yen SS. Effects of dopamine infusion on pituitary hormone secretion in humans. J Clin Endocrinol Metab. 1976;43:668-674.
45 Quigley ME, Rakoff JS, Yen SS. Increased luteinizing hormone sensitivity to dopamine inhibition in polycystic ovary syndrome. J Clin Endocrinol Metab. 1981;52:231-234.
46 Judd SJ, Rigg LA, Yen SS. The effects of ovariectomy and estrogen treatment on the dopamine inhibition of gonadotropin and prolactin release. J Clin Endocrinol Metab. 1979;49:182-184.
47 Spruce BA, Kendall-Taylor P, Dunlop W, et al. The effect of bromocriptine in the polycystic ovary syndrome. Clin Endocrinol. 1984;20:481-488.
48 Lobo R, Shoupe D, Chang SP, Campeau J. The control of bioactive luteinizing hormone secretion in women with polycystic ovary syndrome. Am J Obstet Gynecol. 1984;148:423-428.
49 Pons S, Torres-Aleman I. Estradiol modulates insulin-like growth factor I receptors and binding proteins in neurons from the hypthalamus. J Neuroendocrinol. 1993;5:267-271.
50 Daftary S, Gore A. Developmental changes in hypothalamic insulin-like growth factor-1: Relationship to gonadotropin-releasing hormone neurons. Endocrinology. 2003;144:2034-2045.
51 Longo KM, Sun Y, Gore A. Insulin-like growth factor-1 effects on gonadotropin-releasing hormone biosynthesis in GT1-7 cells. Endocrinology. 1998;139:1125-1132.
52 Roth C, Jung H, Kim K, et al. Involvement of γ amino butyric acid (GABA) in the postnatal function of the GnRH pulse generator as determined on the basis of GnRH and GnRH receptor gene expression in the hypothalamus and the pituitary. Exper Clin Endocrinol Diabetes. 1997;105:353-358.
53 Snowden EU, Khan-Dawood FS, Dawood MY. The effect of naloxone on endogenous opioid regulation of pituitary gonadotropins and prolactin during the menstrual cycle. J Clin Endocrinol Metab. 1984;59:298-302.
54 Eagleson C, Gingrich M, Pastor C, et al. Polycystic ovarian syndrome: Evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85:4047-4052.
55 de Ziegler D, Steingold K, Cedars M, et al. Recovery of hormone secretion after chronic gonadotropin-releasing hormone agonist administration in women with polycystic ovarian disease. J Clin Endocrinol Metab. 1989;68:1111-1117.
56 Barnes R. Pathophysiology of ovarian steroid secretion in polycystic ovary syndrome. Semin Reprod Endocrinol. 1997;15:159-168.
57 Bergh C, Carlsson B, Olsson JH, et al. Regulation of androgen production in cultured human thecal cells by insulin-like growth factor I and insulin. Fertil Steril. 1993;59:323-331.
58 el-Roeiy A, Chen X, Roberts VJ, et al. Expression of the genes encoding the insulin-like growth factors (IGF-I and II), the IGF and insulin receptors, and IGF-binding proteins 1-6 and the localization of their gene products in normal and polycystic ovary syndrome ovaries. J Clin Endocrinol Metab. 1994;78:1488-1496.
59 Nahum R, Thong KJ, Hillier SG. Metabolic regulation of androgen production by human thecal cells in vitro. Hum Reprod. 1995;10:75-81.
60 Barbieri R, Makris A, Randall RW, et al. Insulin stimulates androgen accumulation in incubations of ovarian stroma obtained from women with hyperandrogenism. J Clin Endocrinol Metab. 1986;62:904-910.
61 Velazquez E, Mendoza S, Hamer T, et al. Metformin therapy in polycystic ovary syndrome reduces hyperinsulinemia, insulin resistance, hyperandrogenemia, and systolic blood pressure, while facilitating normal menses and pregnancy. Metabolism. 1994;43:647-654.
62 Chang RJ, Mandel FP, Wolfsen AR, Judd HL. Circulating levels of plasma adrenocorticotropin in polycystic ovary disease. J Clin Endocrinol Metab. 1982;54:1265-1267.
63 Moran C, Reyna R, Boots L, Azziz R. Adrenocortical hyperresponsiveness to corticotropin in polycystic ovary syndrome patients with adrenal androgen excess. Fertil Steril. 2004;81:126-131.
64 Azziz R, Ehrmann D, Legro R, et al. Troglitazone decreases adrenal androgen levels in women with polycystic ovary syndrome. Fertil Steril. 2003;79:932-937.
65 Guido M, Romualdi D, Suriano R, et al. Effect of pioglitazone treatment on the adrenal androgen response to corticotrophin in obese patients with polycystic ovary syndrome. Hum Reprod. 2004;19:534-539.
66 Mason HD, Willis DS, Beard RW, et al. Estradiol production by granulosa cells of normal and polycystic ovaries: Relationship to menstrual cycle history and concentrations of gonadotropins and sex steroids in follicular fluid. J Clin Endocrinol Metab. 1994;79:1355-1360.
67 Coffler MS, Patel K, Dahan MH, et al. Evidence for abnormal granulosa cell responsiveness to follicle-stimulating hormone in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:1742-1747.
68 Van Der Meer M, Hompes P, De Boer J, et al. Cohort size rather than follicle-stimulating hormone threshold level determines ovarian sensitivity in polycystic ovary syndrome. J Clin Endocrinol Metab. 1998;83:423-426.
69 Almahbobi G, Anderiesz C, Hutchinson P, et al. Functional integrity of granulosa cells from polycystic ovaries. Clin Endocrinol. 1996;44:571-580.
70 Willis D, Mason H, Gilling-Smith C, Franks S. Modulation of insulin of follicle-stimulating hormone and luteinizing hormone actions in human granulosa cells of normal and polycystic ovaries. J Clin Endocrinol Metab. 1996;81:302-309.
71 Greisen S, Ledet T, Ovesen P. Effects of androstenedione, insulin and luteinizing hormone on steroidogenesis in human granulosa luteal cells. Hum Reprod. 2001;16:2061-2065.
72 Willis D, Franks S. Insulin action in human granulosa cells from normal and polycystic ovaries is mediated by the insulin receptor and not the type-I insulin-like growth factor receptor. J Clin Endocrinol Metab. 1995;80:3788-3790.
73 Ovalle F, Azziz R. Insulin resistance, polycystic ovary syndrome, and type 2 diabetes mellitus. Fertil Steril. 2002;77:1095-1105.
74 Dunaif A. Insulin resistance and the polycystic ovary syndrome: Mechanism and implications for pathogenesis. Endocrine Rev. 1997;18:774-800.
75 Nestler J, Powers L, Matt D, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin lelvels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72:83-89.
76 Hales C, Barker D. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia. 1992;35:595-601.
77 Phillips D, Barker D, Hales C, et al. Thinness at birth and insulin resistance in adult life. Diabetologia. 1994;37:150-154.
78 DeFronzo R, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173-194.
79 World Health Organization (WHO). Definition, diagnosis and classification of diabetes mellitus and its complications. Report of a WHO Consultation, part 1: Diagnosis and classification of diabetes mellitus. WHO; 1999. www.who.int/diabetes/currentpublication/en.
80 Talbott E, Zborowski J, Rager J, et al. Evidence for an association between metabolic cardiovascular syndrome and coronary and aortic calcification among women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89:5454-5461.
81 Dahlgren E, Johansson S, Lindstedt G, et al. Women with polycystic ovary syndrome wedge resected in 1956 to 1965: A long-term follow-up focusing on natural history and circulating hormones. Fertil Steril. 1992;57:505-513.
82 Wild RA, Painter PC, Coulson PB, et al. Lipoprotein lipid concentrations and cardiovascular risk in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1985;61:946-951.
83 Dahlgren E, Janson PO, Johansson S, et al. Polycystic ovary syndrome and risk for myocardial infarction. Evaluated from a risk factor model based on a prospective population study of women. Acta Obstet Gynecol Scand. 1992;71:599-604.
84 Guzick S, Talbott E, Sutton-Tyrrell K, et al. Carotid atherosclerosis in women with polycystic ovary syndrome: Initial results from a case-control study. Am J Obstet Gynecol. 1996;174:1224-1229.
85 Yildiz B, Haznedaroglu I, Kirazli S, Bayraktar M. Global fibrinolytic capacity is decreased in polycystic ovary syndrome, suggesting a prothrombotic state. J Clin Endocrinol Metab. 2002;87:3871-3875.
86 Pierpoint T, McKeigue P, Isaacs A, et al. Mortality of women with polycystic ovary syndrome at long-term follow-up. J Clin Epidemiol. 1998;51:581-586.
87 Wild S, Pierpoint T, Jacobs H, McKeigue P. Long-term consequences of polycystic ovary syndrome: Results of a 31-year follow-up study. Hum Fertil. 2000;3:101-105.
88 Potischman N, Hoover R, Brinton L, et al. Case-control study of endogenous steroid hormones and endometrial cancer. J Natl Cancer Inst. 1996;88:1127-1135.
89 Konishi I, Koshiyama M, Mandai M, et al. Increased expression of LH/hCG receptors in endometrial hyperplasia and carcinoma in anovulatory women. Gynecol Oncol. 1997;65:273-280.
90 Anderson K, Sellers T, Chen P, et al. Association of Stein-Leventhal syndrome with the incidence of postmenopausal breast carcinoma in a large prospective study of women in Iowa. Cancer. 1997;79:494-499.
91 Koskinen P, Penttila T, Anttila L, et al. Optimal use of hormone determinations in the biochemical diagnosis of the polycystic ovary syndrome. Fertil Steril. 1996;65:517-522.
92 Legro R, Kunselman A, Dodson W, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: A prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84:165-169.
93 Moller DE, Flier JS. Insulin resistance—mechanisms, syndrome, and implications. NEJM. 1991;325:938-948.
94 Legro R, Finegood D, Dunaif A. A fasting glucose to insulin ratio is a useful measure of insulin sensitivity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1998;83:2694-2698.
95 Ducluzeau P, Cousin P, Malvoisin E, et al. Glucose-to-insulin ratio rather than sex hormone-binding globulin and adiponectin levels is the best predictor of insulin resistance in nonobese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:3626-3631.
96 Carnevale Schiance G, Rossi A, Sainaghi P, et al. The significance of impaired fasting glucose versus impaired glucose tolerance. Diabetes Care. 2003;26:1333-1337.
97 Kauffman RP, Baker VM, DiMarino P, et al. Polycystic ovarian syndrome and insulin resistance in white and Mexian American women: A comparison of two distinct populations. Am J Obstet Gynecol. 2002;187:1362-1369.
98 Legro R, Castracane VD, Kauffman RP. Detecting insulin resistance in polycystic ovary syndrome: Purposes and pitfalls. Obstet Gynecol Surv. 2004;59:141-154.
99 Guzick D, Wing R, Smith D, et al. Endocrine consequences of weight loss in obese, hyperandrogenic, anovulatory women. Fertil Steril. 1994;61:598-604.
100 Korytkowski M, Mokan M, Horwitz M, Berga S. Metabolic effects of oral contraceptives in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1995;80:3327-3334.
101 Cumming D, Yang J, Rebar R, Yen S. Treatment of hirsutism with spironolactone. JAMA. 1982;247:1295-1298.
102 De Leo V, Lanzetta D, D’Antona D, et al. Hormonal effects of flutamide in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1998;83:99-102.
103 Inzucchi S, Maggs D, Spollett G, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. NEJM. 1998;338:867-872.
104 Genazzani A, Battaglia C, Malavasi B, et al. Metformin administration modulates and restores luteinizing hormone spontaneous episodic secretion and ovarian function in nonobese patients with polycystic ovary syndrome. Fertil Steril. 2004;81:114-119.
105 Azziz R, Ehrmann D, Legro R, et al. Troglitazone improves ovulation and hirsutism in the polycystic ovary syndrome: A multicenter, double blind, placebo-controlled trial. J Clin Endocrinol Metab. 2001;86:1626-1632.
106 Nestler J, Daniela J, Evans W, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycystic ovary syndrome. NEJM. 1998;338:1876-1880.
107 Vandermolen D, Ratts V, Evans W, et al. Metformin increases the ovulatory rate and pregnancy rate from clomiphene citrate in patients with polycystic ovary syndrome who are resistant to clomiphene citrate alone. Fertil Steril. 2001;75:310-315.
108 Mitwally M, Kuscu N, Yalcinkaya T. High ovulatory rates with use of troglitazone in clomiphene-resistant women with polycystic ovary syndrome. Hum Reprod. 1999;14:2700-2703.
109 Jakubowicz D, Iuorno M, Jakubowicz S, et al. Effects of metformin on early pregnancy loss in the polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87:524-529.
110 Glueck C, Wang P, Kobayashi S, et al. Metformin therapy throughout pregnancy reduces the development of gestational diabetes in women with polycystic ovary syndrome. Fertil Steril. 2002;77:520-525.
111 De Leo V, La Marca A, Petraglia F. Insulin-lowering agents in the management of polycystic ovary syndrome. Endocrine Rev. 2003;24:633-667.
112 Garber A, Duncan T, Goodman A, et al. Efficacy of metformin in type II diabetes: Results of a double-blind, placebo-controlled, dose–response trial. Am J Med. 1997;103:491-497.