Poisonings
Resuscitation and Stabilization
The initial management of seriously ill poisoned patients requires assessment of airway patency, breathing difficulties, circulatory problems, and the level of consciousness. These issues, along with immediate resuscitation interventions, are usually addressed in the emergency department but may be continued in the ICU (Box 68.1).
Diagnosis
Physical Examination
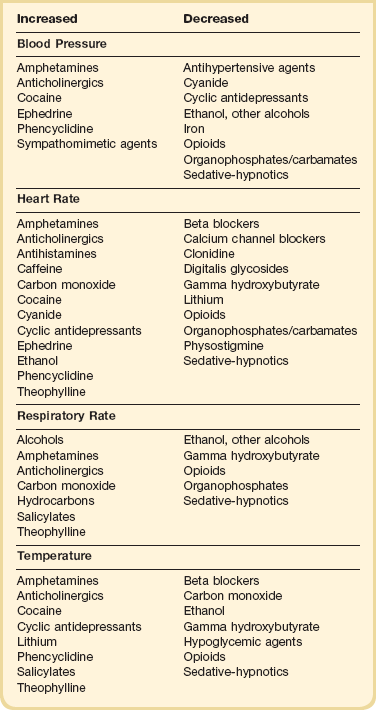
The initial physical examination should focus on vital signs and neurologic findings that may provide physiologic clues to the toxicologic cause. Many toxic substances affect the autonomic nervous system, which is responsible for changes in vital signs mediated by the sympathetic and parasympathetic pathways. Attention to these initial and subsequent clinical signs is of paramount importance in identifying patterns or changes suggesting a particular drug or category of drugs (Table 68.1). Changes in the clinical examination after a therapeutic intervention or the administration of an antidote should be noted. Continued monitoring and reevaluation are necessary because drug effects may not be present on initial evaluation.
Altered mental status is common in a toxicologic emergency. A detailed assessment of neurologic status should be made to determine if there is any alteration in level (stupor/coma or agitation) or content of consciousness (confusion/delirium).1 The evaluation should include an assessment of pupillary reactivity, ocular movements, and motor responses. Ruling out structural versus toxic or metabolic reasons for the altered state is important. Drug-induced seizures are often difficult to treat and may respond only to specific antidotal therapy. In general, benzodiazepines are more effective in terminating drug-induced seizures than other agents.
Toxidromes
A complex of signs and symptoms may be identified by physical examination and grouped into a toxic syndrome, or “toxidrome.” In many cases, recognition of this toxic pattern is more important than identifying a specific offending agent. Identifying a toxidrome enables the clinician to initiate the assessment, derive a differential diagnosis, and formulate a treatment plan. The most typical toxidromes are listed in Table 68.2.2 Importantly, the clinician should note that patients may not present with a classic toxidrome due to variable manifestations of toxins and overlapping features that exist between toxidromes.
Table 68.2
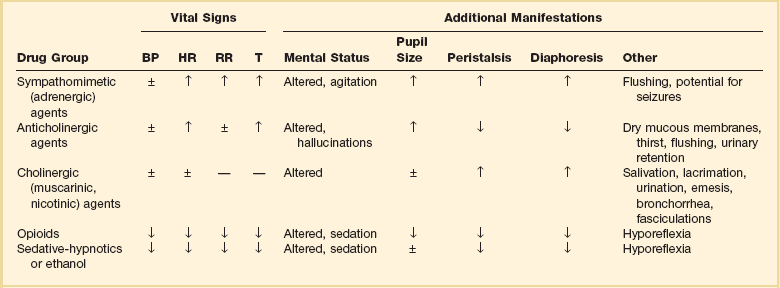
Adapted from Nelson NS, Lewin NA, Howland MA, et al: Initial evaluation of the patient: Vital signs and toxic syndromes. In Nelson NS, Lewin NA, Howland MA, et al (eds): Goldfrank’s Toxicologic Emergencies, 9th ed. New York, McGraw-Hill Medical, 2011, p 33.
Laboratory Tests
A laboratory test for a patient exposed to toxic agents should be helpful in diagnosis or monitoring.3 Select laboratory examinations may be used when appropriate to determine the three gaps of toxicology—the anion gap, the osmolar gap, and the oxygen saturation gap. An arterial blood gas (ABG) analysis will identify hypoxemia or hypoventilation, as well as acid-base abnormalities. Agents associated with a gap in oxygen saturation (>5% difference between measured and calculated saturation) include carbon monoxide (CO) and methemoglobin inducers. In these exposures, a pulse oximeter inaccurately reflects the oxygen saturation of tissues and co-oximetry is necessary to identify abnormal hemoglobins. Determination of electrolytes with blood urea nitrogen (BUN) and creatinine will detect renal abnormalities and allow calculation of the anion gap. Some common drugs associated with an anion gap acidosis are listed in Box 68.2. Hypoperfusion must also be considered as a cause of metabolic acidosis. An osmolar gap (>10 mOsm/L) may be caused by any small particle (toxin) that increases the measured osmolarity as measured by freezing point depression. Such agents include ethanol, ethylene glycol, glycerol, isopropyl alcohol, mannitol, methanol, propylene glycol, and sorbitol. An electrocardiogram (ECG) should be obtained when potential cardiac toxicity exists.
Gastrointestinal Decontamination
GI decontamination techniques in the poisoned patient with an oral ingestion have included gastric emptying procedures (ipecac-induced emesis, gastric lavage), adsorption of drugs (activated charcoal), and increasing transit through the GI tract (cathartics, whole bowel irrigation [WBI]). Use of these interventions has decreased due to uncertain evidence of benefit and recognition of adverse effects of the techniques.4 The consideration of a GI decontamination technique depends on the toxicity of the substance ingested, potential for deterioration in respiratory and mental status, severity of symptoms, dose, time since ingestion, presence of spontaneous emesis, and contraindications of the procedure.
Ipecac, which contains emetic alkaloids, stimulates gastric mucosal sensory receptors and the chemoreceptor trigger zone in the brain to produce vomiting. The amount of ingested drug removed by ipecac-induced emesis is highly variable, and no benefit of ipecac has been confirmed even when administered less than 60 minutes after ingestion. Currently, ipecac is not used in the management of adult poisoning victims.5 Complications that have been associated with ipecac administration include aspiration pneumonitis, esophageal rupture, Mallory-Weiss tear, pneumomediastinum, and protracted vomiting that can delay administration of activated charcoal.
Gastric lavage with a large bore (36- to 40-French) orogastric tube is a technique used to empty the stomach of orally ingested substances that can be associated with significant complications. After insertion of the tube, lavage is accomplished with sequential 250 mL aliquots of normal saline or water until no pill fragments are retrieved. Intubation for airway protection is required before the procedure in patients with a depressed level of consciousness or potential for sedation. No clear benefit of gastric lavage has been demonstrated, even when instituted in obtunded patients presenting within 1 hour of ingestion.6 Gastric lavage should not be employed routinely in the management of poisoned patients.7 In rare circumstances, gastric lavage may be considered with ingestion of a life-threatening amount of toxin when the procedure can be instituted within 60 minutes of ingestion or a significant amount of toxin is still likely to be present in the stomach. The clinician must consider contraindications and the potential risks before performing gastric lavage in an overdose patient. Serious complications include aspiration pneumonitis, esophageal perforation, and cardiovascular instability. Gastric lavage is contraindicated with ingestions of substances such as acid, alkali, or hydrocarbons when the risk of aspiration is increased. Patients with a risk of GI perforation or severe bleeding diathesis or who are combative should also not be subjected to gastric lavage.
Single-dose activated charcoal is one of the more frequently used interventions for GI decontamination. Activated charcoal potentially adsorbs the toxin in the GI tract and minimizes systemic absorption. The optimum dose of activated charcoal has not been established, but the usual dose for adults is 25 to 100 g (1 g/kg). Activated charcoal is not effective in adsorbing iron, lithium, cyanide, strong acids and bases, alcohols, and some hydrocarbons. Some clinical studies examining the use of activated charcoal versus no intervention found no improvement in outcomes.8,9 Volunteer studies suggest that the greatest benefit of administering activated charcoal may be within 1 hour of ingestion.10 Use of activated charcoal may be considered when a potentially toxic amount of a substance adsorbed by charcoal has been ingested within 1 hour.11,12 Later administration may be appropriate if clinical factors suggest the ingested substance has not yet been completely absorbed. Activated charcoal is contraindicated in patients with a depressed level of consciousness unless intubated, when administration increases the risk of aspiration, or the patient is known or suspected to have a GI perforation. Few complications are associated with the appropriate use of single-dose activated charcoal. Emesis has been reported but may be related to sorbitol administered with charcoal or the ingested toxin.
Cathartics have been administered in poisoning ingestions based on the hypothesis that absorption and overall bioavailability of the agent are decreased by reducing contact time in the GI tract. Sorbitol (70% solution with activated charcoal) is the most commonly used cathartic, but magnesium citrate and magnesium sulfate have also been used. No clinical studies have demonstrated beneficial effects of cathartics in poisoned patients. A cathartic alone has no role in the management of poisonings, and even the routine use of a cathartic in combination with activated charcoal cannot be recommended.13 If a cathartic is used, only a single dose should be administered. A cathartic should not be administered in patients with ileus, GI obstruction or perforation, recent GI surgery, or hemodynamic instability. Complications of cathartics include nausea, vomiting, and abdominal cramping. Multiple doses of magnesium-containing cathartics may result in significant dehydration and electrolyte abnormalities.
WBI has been proposed as a technique to prevent absorption of ingested poisons by rapidly expelling the bowel contents. WBI involves the enteral administration (usually by nasogastric tube) of large volumes (1 to 2 L/hour in adults) of polyethylene glycol electrolyte lavage solution; this is continued until the rectal effluent is clear or elimination of the toxin has been confirmed. During the procedure, the head of the bed should be elevated to 45 degrees to decrease the likelihood of vomiting and aspiration. No clinical trials have assessed the impact of WBI on patient outcomes. Currently, there are no established indications for WBI, but it may be considered for potentially toxic ingestions of sustained-release or enteric-coated drugs, iron, and illicit drug packets.14 WBI is contraindicated in the presence of ileus, GI obstruction or perforation, GI bleeding, hemodynamic instability, or intractable vomiting. In the patient with decreased level of consciousness or respiratory depression, the airway must be protected before instituting WBI.
Enhanced Elimination
Multiple-dose activated charcoal (MDAC) therapy involves the repeat oral administration of activated charcoal to prevent absorption of drug that persists in the GI tract and to enhance elimination of drugs already absorbed into the body by functioning as an adsorbent “sink” at several sites in the gut.15 First, it can interrupt enterohepatic circulation of drugs or metabolites that are actively secreted into bile. Second, it can adsorb drugs or metabolites that enter the gut by active secretion or passive diffusion and prevent reabsorption. Finally, it may prevent desorption of drugs, particularly acidic substances that bind two to three times less avidly to activated charcoal in the alkalotic milieu of the intestinal lumen than in the acidic environment of the stomach. Drugs with a prolonged elimination half-life after overdose and small volume of distribution are more likely to have elimination enhanced significantly by MDAC.
There is no convincing evidence that MDAC reduces morbidity and mortality rates in poisoned patients.16 However, MDAC may be considered if the patient has ingested a life-threatening amount of carbamazepine, dapsone, phenobarbital, quinine, or theophylline and may obviate the need for invasive extracorporeal techniques. Insufficient evidence exists to support routine use of MDAC in ingestions of other substances.
Urinary alkalinization is beneficial in increasing renal clearance of weak acids such as salicylates and phenobarbital. These weak acids are ionized at alkaline urine pH, trapped in the renal tubules, and not reabsorbed. Alkalinization can be initiated by adding 88 to 132 mEq sodium bicarbonate to 1 L of 5% dextrose in water (D5W). Urine pH should be tested every hour, and the rate of the bicarbonate infusion should be titrated to achieve a urine pH of 7.5 to 8.5. Alkalinization may be difficult to achieve if metabolic acidosis is present. Hypokalemia is a common complication and requires correction to facilitate urinary alkalinization. Increasing the urine pH with carbonic anhydrase inhibitors such as acetazolamide is not recommended because metabolic acidosis will worsen. Urine alkalinization can be considered in patients with significant salicylate ingestions who do not require hemodialysis. Phenobarbital poisonings are more effectively treated with MDAC.17
Continuous renal replacement therapies have been used less frequently for drug removal in the treatment of poisoning.18 Clearance rates achieved with these techniques are considerably lower than those achieved with hemodialysis. Such therapy may be instituted after hemodialysis or hemoperfusion to further remove the drug after it slowly redistributes from tissue to blood. This is a potential option for agents such as lithium or procainamide. Continuous renal replacement techniques may be advantageous in hemodynamically unstable patients who cannot tolerate conventional hemodialysis or hemoperfusion. Despite many case reports demonstrating significant drug clearance, there are no data demonstrating that these techniques affect outcome.
Specific Poisonings
Alcohols
Ethylene Glycol and Methanol
Practice guidelines are available for the treatment of ethylene glycol and methanol intoxication.19,20 If the patient has symptoms and is significantly acidemic, sodium bicarbonate may be administered as a temporizing measure to enhance formate and oxalate elimination by ion trapping. Fluid overload and hyperosmolarity may become significant problems as a result of bicarbonate administration. Hydration is helpful because ethylene glycol is well excreted by the kidney as long as renal function is maintained. The definitive treatment of intoxication with methanol or ethylene glycol is inhibition of the alcohol’s metabolism and hemodialysis to remove the alcohol and toxic metabolites and to correct metabolic abnormalities. Hemodialysis should be considered for the following conditions: deteriorating vital signs despite intensive supportive care, significant metabolic acidosis (pH < 7.25 to 7.3), blood level of methanol or ethylene glycol higher than 25 mg/dL, or any evidence of renal failure or electrolyte imbalances unresponsive to conventional therapy.19,20
Antidotal treatment of significant poisoning involves inhibition of alcohol dehydrogenase to prevent metabolism of the alcohols to toxic metabolites with ethanol or fomepizole. Ethanol (IV or oral) allows preferential metabolism of ethanol over methanol and ethylene glycol. Ethanol should be administered to maintain a blood level of 100 to 150 mg/dL. A loading dose should be followed by a maintenance infusion according to the established dosing requirements for nondrinkers, drinkers, and during hemodialysis (Table 68.3).21 Problems encountered during ethanol administration include CNS depression, hypoglycemia, dehydration, and fluctuating serum concentrations. A second IV line using 0.9% sodium chloride may be necessary to avoid development of hyponatremia because of the large free water content and significant hypertonicity (1713 mOsm/L) of 10% ethanol solution. Advance notice should be given to the pharmacy to allow sufficient time to locate enough ethanol for administering and preparing the solution. If IV ethanol is not available, oral ethanol can be used.
Table 68.3
Intravenous Administration of 10% Ethanol
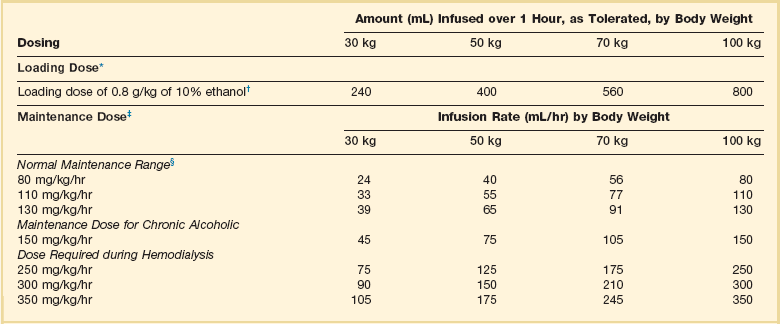
*A 10% vol/vol concentration yields approximately 100 mg/mL.
†For a 5% concentration, multiply the amount by 2.
‡Infusion to be started immediately after the loading dose. Concentrations above 10% are not recommended for intravenous administration. The dose schedule is based on the premise that the patient initially has a zero ethanol level. The aim of therapy is to maintain a serum ethanol level of 100 to 150 mg/dL, but constant monitoring of the ethanol level is required because of wide variations in endogenous metabolic capacity. Ethanol will be removed by hemodialysis.
§Rounded to the nearest milliliter.
Adapted from Howland MA: Antidotes in depth: Ethanol. In Nelson NS, Lewin NA, Howland MA, et al (eds): Goldfrank’s Toxicologic Emergencies, 9th ed. New York, McGraw-Hill Medical, 2011, p 1419.
Fomepizole, a competitive inhibitor of alcohol dehydrogenase, is approved for use in ethylene glycol and methanol overdose.22 It is easier to administer than ethanol, does not cause sedation, and is associated with fewer severe and serious adverse events.23 Fomepizole administration should be considered instead of ethanol if the patient develops altered consciousness, seizures, or a significant metabolic acidosis. Although fomepizole appears to be equally effective, there are no data to demonstrate its comparative efficacy or cost-effectiveness. Administration of ethanol or fomepizole should continue after dialysis until the serum ethylene glycol or methanol concentration is undetectable or less than 20 mg/dL or acidosis is resolved and the patient is asymptomatic. In the absence of renal dysfunction and a significant metabolic acidosis, the use of fomepizole potentially could obviate the need for hemodialysis, even though the serum ethylene glycol or methanol concentration exceeds 50 mg/dL.24 If patients with high serum concentrations of ethylene glycol are not treated with hemodialysis, then their acid-base balance should be monitored closely and hemodialysis instituted if a metabolic acidosis develops.19
Propylene Glycol
Propylene glycol is another alcohol that can cause toxicity in critically ill patients receiving high doses of IV medications containing the alcohol as a solvent. Medications that contain propylene glycol include lorazepam, diazepam, phenobarbital, pentobarbital, nitroglycerin, phenytoin, esmolol, etomidate, and sulfamethoxazole/trimethoprim. Propylene glycol toxicity is more commonly observed with lorazepam because of the use of high doses in some patients, the frequency of use for sedation in ICUs, and the high concentration of propylene glycol—approximately 830 mg/mL.25 Common manifestations of propylene glycol accumulation are anion gap metabolic acidosis and increased osmolar gap.26 Additional toxicities include renal dysfunction, hemolysis, cardiac arrhythmias, seizures, and CNS depression or agitation. Clinical studies suggest that an elevated osmolar gap correlates with propylene glycol accumulation. Accumulation can occur when doses of lorazepam exceed 0.1 mg/kg/hour and when renal or hepatic insufficiency is present. Although toxicity is more common after long periods of lorazepam infusion (>3 days), toxicity has occurred with short-term, high-dose use. The treatment of choice is to stop the lorazepam infusion and sedate with an agent that does not contain propylene glycol. Hemodialysis removes propylene glycol but is usually not required unless severe renal dysfunction develops.
Analgesics
Acetaminophen
Acetaminophen (N-acetyl-p-aminophenol [APAP]) is present in a large number of prescription and over-the-counter medications and is frequently a coingestant with other drugs. In addition, unintentional overdoses result from patients unknowingly ingesting multiple products containing acetaminophen (particularly acetaminophen-narcotic combinations). Because APAP overdose may result in significant hepatotoxicity and even death that is preventable, it is important to recognize and initiate appropriate therapy. With higher doses of APAP, a greater proportion is hepatically metabolized by the cytochrome P-450 system of mixed function oxidases (CYP450) to the toxic metabolite, N-acetyl-p-benzoquinoneimine (NAPQI), which can result in cell injury and death. Hepatic glutathione facilitates detoxification and elimination of NAPQI with therapeutic doses of APAP, but glutathione supply is overwhelmed in APAP overdoses. The clinical course of APAP toxicity has been divided into stages on the basis of the development of hepatotoxicity (Table 68.4).27
Table 68.4
Stages of Acetaminophen Toxicity
| Stage | Time Course (after Ingestion) | Characteristics |
| I | 0-24 hours | Asymptomatic or nausea, vomiting; normal LFTs |
| II | 24-72 hours (latent stage) | Right upper quadrant pain; abnormal LFTs and PT; renal dysfunction possible |
| III | 72-96 hours (hepatic stage) | Encephalopathy, jaundice, bleeding, renal dysfunction; maximal hepatic injury, synthetic dysfunction |
| IV | 4 days-2 weeks (recovery stage) | Recovery of liver function |
If possible, an estimate of the quantity and dosage form of APAP ingested and the time of ingestion should be obtained. In adults, hepatic toxicity can occur after ingestion of more than 7.5 to 10 g during 8 hours or less but has been reported with exposures of 4 g. The maximum daily dose of acetaminophen has been reduced to 3 g because of concerns for toxicity.28 The risk of toxicity may be increased in patients with low glutathione stores (malnutrition, fasting state, chronic alcoholism) or induction of CYP450 enzymes (chronic alcoholism, phenytoin or carbamazepine use). For patients with a recent single, acute ingestion, an acetaminophen level should be obtained at least 4 hours after ingestion. Liver enzymes only need to be evaluated if the APAP level indicates potential toxicity or the clinical examination suggests hepatic injury. If the time of ingestion is unknown, an APAP level should be obtained on admission. An APAP level and liver function tests should be determined in patients presenting late, patients with multiple ingestions over time, or chronic ingesters of APAP.
NAC is the antidote for APAP poisoning, but the optimal route and duration of treatment are still debated.29 NAC limits toxicity by combining with NAPQI and by serving as a precursor of glutathione, which inactivates NAPQI. For patients with a single, acute ingestion of APAP, the serum acetaminophen level assessed at least 4 hours after ingestion is compared with the Rumack-Matthew nomogram. Treatment with NAC is initiated in the United States if the value falls above the lower possible hepatotoxicity line. Only the initial APAP level is used in making the decision to initiate or continue NAC treatment. Subsequent levels are unnecessary unless extended-release preparations are ingested (see following). The Rumack-Matthew nomogram is not useful for patients with multiple ingestions of APAP over time, chronic ingesters, or those ingesting extended-release forms (see following discussion). If acetaminophen levels are not available, NAC treatment should be initiated if more than 150 mg/kg or 10 g acetaminophen is ingested. For extended-release APAP, a second level 4 hours after an initial nontoxic level should be evaluated to assess for delayed absorption. If the second value is above the lower line on the Rumack-Matthew nomogram, NAC is initiated.
NAC is most effective in preventing toxicity if administered within 8 hours of ingestion. NAC therapy can be initiated pending results of the acetaminophen level if the patient is presenting late or APAP level results will be delayed. The oral regimen for NAC includes a loading dose of 140 mg/kg followed by 17 oral maintenance doses of 70 mg/kg administered 4 hours apart (72-hour regimen). Due to the odor of the oral form, a nasogastric tube may need to be placed for administration, and antiemetic therapy may be necessary to control vomiting that occurs in up to 50% of patients. If the patient vomits the loading dose or any maintenance dose within 1 hour of administration, the dose should be repeated. IV NAC is administered as a loading dose of 150 mg/kg over 60 minutes followed by 50 mg/kg infused over 4 hours and then 100 mg/kg infused over 16 hours (21-hour regimen). Anaphylactoid reactions may occur in 14% to 18% of patients with IV NAC. Oral and IV regimens of administering NAC are similar in efficacy.30 However, the oral regimen may be more appropriate in patients who present later after ingestion (>18 hours) and when large amounts of APAP are ingested due to the higher dose of administered NAC.31,32 If the patient has a serum APAP level in the potentially toxic range, the aspartate aminotransferase (AST) or alanine aminotransferase (ALT) level should be evaluated daily. If abnormal, additional tests such as bilirubin, prothrombin time, creatinine, BUN, blood glucose, and electrolytes should also be obtained. In patients with elevated liver enzymes, NAC may be continued beyond the full course of therapy until transaminases are decreasing.
Chronic ingesters of APAP or patients with multiple ingestions over time are problematic when determining the need to administer NAC. Presentation beyond 24 hours after ingestion makes the APAP level essentially useless, and there are no established guidelines for administration of NAC in these circumstances. A marker of toxicity that may be useful is the evaluation of AST and ALT. If enzymes are elevated at the time of presentation (>50 IU/L) or the APAP level is greater than 10 µg/mL (>10 µmol/L), a course of NAC should be strongly considered.33 A course of NAC should also be administered to patients with hepatic failure caused by APAP.
Opioids
Prescription opioids obtained from physicians or illicitly now account for almost 40% of all poisoning deaths in the United States and affect all age groups.34 The agents most commonly involved in deaths include methadone, oxycodone, and hydrocodone.35 Toxicity depends on the potency of the agent, dose ingested, tolerance of the individual, and concomitant use of other drugs. These prescription opioids have overshadowed deaths due to heroin. Heroin is rapidly absorbed by all routes of administration including IV, intranasal, intramuscular, subcutaneous, and inhalation, but most fatal overdoses occur with IV administration. IV fentanyl (sometimes extracted from analgesic patches) is also associated with fatalities. Diagnosis of an opioid overdose is made by characteristic clinical findings, exposure history, qualitative urine toxicology assay, and response to naloxone. Qualitative urine assays may not detect all opioid derivatives (e.g., fentanyl).
The immediate priorities in a patient with opioid toxicity are support of ventilation, correction of hypotension, and reversal of the toxic effects with an opioid antagonist. If reversal of respiratory depression cannot be accomplished quickly, intubation may be necessary. Isotonic fluids should be administered for hypotension. Naloxone, a potent competitive opioid antagonist, is the antidote for opioid toxicity. It can be administered intravenously, intramuscularly, subcutaneously, by sublingual injection, or through an endotracheal tube. The initial dose of naloxone in a suspected opioid overdose is 0.04 to 2 mg; the lower dose should be considered in patients suspected of chronic addiction to avoid precipitating acute withdrawal symptoms. The goal of therapy is to restore adequate spontaneous respirations rather than complete arousal. Doses of naloxone up to 10 to 20 mg may be required to reverse the effects of synthetic opioids such as pentazocine, methadone, and fentanyl. The effects of naloxone last approximately 60 to 90 minutes, necessitating continued observation of the patient for resedation. Patients may require continuous infusion of naloxone to maintain adequate respirations, particularly with long-acting opioids. The dose for infusion is typically one half to two thirds of the initial amount of naloxone that reversed the respiratory depression administered on an hourly basis. Adjustments of the dose should be made to achieve clinical end points and avoid withdrawal symptoms. Nalmefene, a long-acting opioid antagonist, has also been used to treat opioid overdoses, but prolonged withdrawal symptoms may be a concern.36 Potential acetaminophen toxicity should be considered in patients ingesting opioids formulated with acetaminophen. Patients should also be observed for potential complications of opioid overdose including aspiration pneumonitis and noncardiogenic pulmonary edema. Noncardiogenic pulmonary edema is usually self-limited (24 to 36 hours) and managed with supportive care that may include intubation and mechanical ventilation.37 Other complications that may be related to injection drug use include wound botulism, endocarditis, rhabdomyolysis, and compartment syndrome.
Salicylates
Activated charcoal can be used for GI decontamination. Salicylates in large ingestions or enteric-coated forms can result in gastric concretions providing a depot for continued absorption; multiple doses of activated charcoal can be considered in this situation or when levels continue to rise despite other therapy.16,38 A salicylate ingestion is considered toxic if symptoms are present, if more than 150 mg/kg has been ingested, or if levels are higher than 35 mg/dL at 6 hours after ingestion. The Done nomogram was developed in pediatric ingestions and does not provide good clinical correlation with toxicity in adults. Acute intoxication with salicylate levels in excess of 35 mg/dL at 6 hours after ingestion should be treated with sodium bicarbonate to alkalinize the urine to a pH 7 to 8, which increases the renal clearance of salicylate metabolites through ion trapping.17 Hypokalemia will develop with correction of metabolic acidosis and must be corrected for urine alkalinization to be achieved. Alkalemia shifts the gradient of movement of salicylate from brain and tissues to blood.
Carbon Monoxide
Management of CO poisoning includes a detailed evaluation of neurologic and cardiorespiratory status. Acid-base status should be determined and an ECG examined for evidence of ischemia or arrhythmia. Oxygen is the antidote and shortens the half-life of carboxyhemoglobin by competing for binding with hemoglobin. High-concentration oxygen should be instituted as soon as possible and continued until the carboxyhemoglobin level has decreased to normal. Pulse oximetry overestimates arterial oxygenation because carboxyhemoglobin is misinterpreted as oxyhemoglobin. Analysis of arterial blood by co-oximetry is required for an accurate assessment of oxygen content. Intubation may be necessary in patients exposed to CO from fire. Hyperbaric oxygen therapy shortens the half-life of carboxyhemoglobin to 15 to 30 minutes compared with 40 to 80 minutes when patients breathe 100% oxygen. However, controversy exists over the specific indications for instituting hyperbaric oxygen therapy in CO poisoning.39–41 Coma has been used as an indication for hyperbaric oxygen therapy; other suggested indications include a period of unconsciousness, neurologic findings other than headache, carboxyhemoglobin level greater than 40%, pregnancy with carboxyhemoglobin level greater than 15%, cardiac ischemia or arrhythmia, history of ischemic heart disease with carboxyhemoglobin level greater than 20%, and symptoms that do not resolve with normobaric oxygen after 4 to 6 hours. Hyperbaric oxygen treatment may decrease postexposure cognitive deficits.42
Cardiovascular Drugs
Cardiovascular drugs are in the top five categories of substances involved in adult overdose exposures and a leading cause of fatalities from overdose.4 Although a large number of cardiovascular drugs are available, the most clinically relevant agents are beta blockers, calcium channel blockers, and digoxin. Note that the clinical manifestations and management of beta-blocker and calcium channel blocker toxicity are very similar.43
Beta Blockers
β-Adrenergic blockers differ in their lipid solubility, oral availability, first-pass effect, protein binding, metabolism, β-1 selectivity, membrane stabilization, and intrinsic sympathomimetic activity. Clinical findings with beta-blocker toxicity include bradycardia, atrioventricular (AV) conduction abnormalities (QRS prolongation, first-degree AV block), and hypotension. The hypotension is primarily due to the negative inotropic effects of these agents. With the exception of sotalol, ventricular fibrillation and other arrhythmias are not usually seen. The more lipophilic β-adrenergic antagonists such as propranolol, metoprolol, acebutolol, and timolol can cause delirium, coma, and seizures. Hypoglycemia is rare in adults. Toxicity generally occurs within 6 hours of ingestion of immediate-release preparations. Ingestion of sotalol or extended-release preparations may result in delayed toxicity, and these patients should be observed for 24 hours or longer if absorption is delayed. Propranolol is associated with the highest mortality rate, which may reflect its greater toxicity attributable to membrane-stabilizing effects.44 Bradyarrhythmias and asystole usually precede death.
The primary goal of treatment is to reverse hypotension rather than to increase heart rate. Increases in heart rate do not always result in improvements in blood pressure. Although atropine is frequently administered for bradycardia, it is usually ineffective for beta–blocker and calcium channel blocker toxicity. Isotonic fluids can be administered for hypotension, but caution is warranted due to the negative inotropic effects of beta blockers on myocardial function. Pharmacologic interventions that have been used in beta–blocker overdoses include glucagon, calcium, catecholamines, insulin euglycemia therapy, and phosphodiesterase inhibitors.43 IV lipid emulsion has recently been reported to be effective in refractory cases. Although clinical trials are lacking, glucagon is considered to be the appropriate initial intervention and is administered as 2 to 5 mg IV followed by a dose of 10 mg if necessary. If there is a positive clinical response, a continuous infusion (usually 2 to 10 mg/hour) is necessary owing to the short duration of action of glucagon. Glucagon is a chronotropic and inotropic agent that stimulates cyclic adenosine monophosphate (cAMP) by bypassing adrenergic receptors.45 Adverse effects of glucagon include nausea, vomiting, hyperglycemia, and hypocalcemia.
Calcium salts should be considered as the next intervention for reversing hypotension that does not respond to glucagon if digoxin ingestion has been excluded.43,46 Calcium chloride 10% (1 g by slow IV push) may be administered initially, and up to 3 g is recommended. Calcium chloride is preferred over calcium gluconate because it contains three times the amount of elemental calcium. It is best administered through a central venous catheter to avoid the possibility of skin necrosis from extravasation. If access is limited, 10% calcium gluconate can be administered but an increased dose is required.
High-dose regular insulin infusions and glucose administration to maintain euglycemia have been shown to be effective in beta-blocker and calcium channel blocker toxicity.46,47 The beneficial effect may be caused in part by the metabolic effects of decreasing cardiac uptake of free fatty acids and increasing carbohydrate use. Reported insulin doses have been variable, but a reasonable initial dose is 0.5 units/hour with titration based on clinical response. It is also reasonable to administer a bolus insulin dose prior to initiation of the infusion. A glucose infusion should be initiated at the same time and a glucose bolus may also be needed. Careful monitoring of glucose and potassium is required.
Catecholamine infusions are frequently administered in beta-blocker toxicity concomitantly with other interventions. No agent is known to be more effective than others and response to various agents is often inconsistent in these clinical situations. Very large doses may be required because the β-adrenergic receptors are blocked; as a result, tachyarrhythmias can occur. The combination of dobutamine and norepinephrine may allow for titration of desired effects against cardiac output and blood pressure. Alternatively, phenylephrine may be used in conjunction with dobutamine. Epinephrine has been shown to be more effective than isoproterenol.48 Infusion of any vasoactive agent should be started at usual doses and rapidly escalated to achieve a clinical response, but it should be stopped if there is a further fall in blood pressure or no beneficial effect.
Phosphodiesterase inhibitors such as milrinone and enoximone have been reported to be effective in some cases of human ingestions.43,46 These agents may be useful in patients who fail other pharmacologic interventions, although experience is limited and they may cause further hypotension through peripheral vasodilation. Invasive hemodynamic monitoring may be needed in some patients.
Recent reports suggest that a bolus administration of 20% lipid emulsion (100 mL) may be beneficial in beta–blocker and calcium channel blocker cardiovascular toxicity refractory to other interventions. An additional infusion of 0.25 mL/kg/minute has been used in some cases. The exact mechanism of effect is unknown, but it has been proposed that lipids serve as a “sink” for the toxin that reduces free drug levels and limits distribution to tissues. Another possibility is that the fatty acids of lipids improve inotropy by increasing intracellular calcium concentrations.49
Calcium Channel Blockers
Signs and symptoms of toxicity occur within 6 hours for immediate release formulations but are delayed 6 to 18 hours for sustained-release preparations.50 Gastric concretions often form, acting as a further reservoir for sustained absorption. Nausea, vomiting, and hypotension are usually accompanied by bradycardia with verapamil and diltiazem or reflex tachycardia with nifedipine. Conduction abnormalities associated with verapamil and diltiazem may include first-degree block, Wenckebach block, junctional rhythm, third-degree AV block, and AV dissociation. Hypoperfusion secondary to decreased cardiac output may lead to tissue ischemia and metabolic acidosis. The patient may present with CNS symptoms such as lethargy, confusion, and coma. Hyperglycemia can result from a decrease in insulin secretion and insulin resistance and may be a marker of severe exposure.
As with beta blockers, initial treatment of calcium channel blocker overdose should be aimed at treating hypotension and significant conduction defects. Interventions include the same pharmacologic interventions as for beta blockers with a change in order of administration. Calcium salts should be administered initially in toxicity due to calcium channel blockers.44,46 Repeat boluses of calcium may be needed every 10 minutes. If blood pressure improves with calcium, an infusion is usually needed owing to the transient effect of bolus doses of calcium. Doses up to 0.4 mL/kg/hour of 10% calcium chloride may be needed. Ionized calcium concentrations can be monitored, but high serum concentrations may be necessary for beneficial effects. Glucagon has also been reported to be beneficial in toxicity due to calcium channel blockers and it should be considered as a second intervention if there is no response to calcium.45 Glucagon is dosed as in beta-blocker overdoses. A continuous infusion of glucagon can be titrated to maintain blood pressure, cardiac output, and sinus rhythm. Insulin-glucose infusions have also been evaluated as an adjunctive treatment in severe cases and should be considered if calcium and glucagon are ineffective.47 As with beta-blocker overdose, large doses of catecholamines may be required. Administration of lipid emulsion can be considered in refractory cases.49 Transthoracic or transvenous pacing may be considered but are often ineffective. Refractory hypotension may require intra-aortic balloon pump or cardiopulmonary bypass.
Digoxin
GI decontamination in digoxin overdose consists of activated charcoal, if the timing is appropriate. Late administration of activated charcoal or MDAC may be considered due to enterohepatic metabolism of the drug.16 Steroid-binding resins such as cholestyramine and colestipol have also been used to prevent further absorption from the GI tract and reduce serum half-life in the same manner as charcoal. Forced diuresis, hemoperfusion, and hemodialysis are not effective in hastening the elimination of digoxin because of the large volume of distribution (4 to 10 L/kg). Only 1% of total body stores of digoxin is present in the serum; of that, 25% is protein bound.
The treatment for life-threatening digitalis toxicity is administration of digoxin-specific antibody fragments.51 Administration of digoxin-specific antibody fragments results in a sharp decrease in free digoxin levels, an increase in total serum digoxin, an increase in renal excretion of digoxin bound to Fab, and a decrease of serum potassium toward normal. The time to response is approximately 30 minutes (range 20 to 90 minutes). Indications for administration of digoxin-specific antibody fragments include severe ventricular arrhythmias, progressive bradyarrhythmias unresponsive to atropine, potassium concentration greater than 5 mEq/L in the setting of suspected digoxin toxicity, rapidly progressive cardiac or GI symptoms, an increasing potassium concentration, serum digoxin concentration greater than 15 ng/mL at any time or more than 10 ng/mL at steady state, ingestion of more than 10 mg of digoxin in a previously healthy adult, and to establish the diagnosis. In the event that digoxin-specific fragments are not immediately available, phenytoin (50 mg/minute up to 1000 mg) or lidocaine may be administered until control of the arrhythmia is achieved. Atropine may work for severe supraventricular bradyarrhythmias or varying degrees of AV block if administered early. Beta blockers may be used for supraventricular and ventricular tachycardias. Magnesium sulfate may be an effective temporizing measure for the treatment of ventricular arrhythmias in the absence of digoxin-specific antibodies, even in the presence of hypermagnesemia. All class Ia antiarrhythmic drugs are contraindicated. Isoproterenol should be avoided because there is an increased risk of ventricular ectopy in the presence of toxic digoxin levels. Transthoracic or transvenous pacing has limited value in this setting.52
After digoxin-specific antibodies have been administered, serum digoxin levels are no longer reliable because they represent free and bound digoxin. The digoxin-specific antibodies are effective even in anephric patients. In renal insufficiency, the Fab half-life is prolonged 10-fold with no change in volume of distribution. Fab concentrations remain detectable for 2 to 3 weeks. Although there is no dissociation of the complex in renal insufficiency, free digoxin levels rebound (redistribution from tissue sites) and Fab fragments leave the vascular space over 7 to 14 days.53 During this time symptoms may recur and a second dose may be necessary.
Cyanide
Inhalation or ingestion of cyanide is rare but can produce severe poisoning rapidly leading to death. A history of potential cyanide exposure is extremely important in suggesting the diagnosis because rapid cyanide assays are not available and clinical manifestations are nonspecific.54 Cyanide exposure may occur from incomplete combustion of products containing carbon and nitrogen in fires and from industrial processes such as electroplating, metal refining, photography, fumigation, and gold or silver extraction. Cyanogenic substances are also found in a variety of plants, although severe toxicity is rare. Iatrogenic cyanide intoxication may occur during nitroprusside administration with high doses or in the presence of hepatic dysfunction.
Hydroxocobalamin, a vitamin B12 precursor administered at an initial dose of 5 g, is commonly used in Europe for acute cyanide poisoning and has been available in the United States since 2006.55 It displaces cyanide from the cytochrome oxidase and forms cyanocobalamin, which is then excreted in the urine or metabolized by hepatic rhodanese. A second dose of 5 g can be administered for severe poisoning or lack of clinical response. Thiosulfate is administered with hydroxocobalamin. Hyperbaric oxygen has also been proposed for treating cyanide toxicity, but data supporting efficacy are not available.
Dietary and Nutritional Agents
Dietary and nutritional products are categorized as supplements and can be marketed without testing for safety or efficacy. Although some herbs and supplements may have inherent toxicity, poisoning may result from product misuse, contamination of the product, or through interaction with other medications.56–58 Patients and their families should always be questioned regarding use of nutritional supplements, herbal preparations, energy drinks, or natural remedies when considering possible toxin exposure as a cause of clinical abnormalities. Adverse effects resulting from these products should be reported to the U.S. Food and Drug Administration. Table 68.5 contains a partial list of toxicities that may result in or complicate critical illness.
Table 68.5
Toxicities of Selected Dietary and Nutritional Agents
| Agent | Toxic Effect(s) |
| Herbal teas | |
| Aconitine | Bradycardia, ventricular tachycardia and fibrillation, hypersalivation, GI disturbances, muscle weakness |
| Cardiac glycosides (digoxin-like factors) | Arrhythmias, GI disturbances, visual disturbances |
| Energy drinks (caffeine, xanthine alkaloids) | Arrhythmias, cardiac arrest |
| Weight loss products | |
| Ephedrine (ma huang) | Sympathomimetic syndrome, intracranial hemorrhage, seizures, arrhythmias, myocardial infarction, stroke, hepatic failure, rhabdomyolysis, death |
| Ephedrine-free supplements (bitter orange, synephrine, octopamine) | Myocardial ischemia, syncope, stroke, ischemic colitis |
| Ginkgo biloba | Bleeding (cerebral or extracerebral) |
| Ginseng | Hypoglycemia, potential bleeding |
| Garlic | Bleeding |
| Kava kava | Hepatic failure, potentiation of anesthetics |
Herbal teas may contain aconitine or digoxin-like substances.59 Management is usually supportive. A digoxin level should be obtained in any patient demonstrating symptoms consistent with digoxin toxicity. The level may not correlate with clinical findings because numerous cardiac glycosides will not cross-react in the digoxin immunoassay. With significant toxicity, digoxin-specific antibodies should be administered.
Products containing ephedrine and ephedrine-free products are often used for weight loss. These products can result in manifestations similar to a sympathomimetic syndrome with cardiovascular and cerebrovascular complications.60–62 Similarly, caffeinated energy drinks can precipitate atrial and ventricular arrhythmias.63
Toxicity from ingestion of herbal preparations and nutritional supplements may result from product contaminants. Products may contain heavy metals, unlisted drugs, or other ingredients.64 The California Department of Health Services, Food and Drug Branch, screened 260 Asian patent medicine products and found 32% contained undeclared pharmaceuticals or heavy metals.65 Unusual symptoms or toxidromes in patients ingesting such products may require the assistance of the local health department or toxicologist to identify a possible toxin. Additional information about specific agents can be found at www.herbmed.org or www.mskcc.org/aboutherbs.
Methemoglobin Inducers
Methemoglobin results from the oxidation of ferrous iron in hemoglobin to ferric iron. Methemoglobin is not able to bind oxygen and high concentrations lead to a functional anemia which results in cellular hypoxia by limiting oxygen delivery. The most common inducers of methemoglobin in the health care setting are topical spray anesthetics (benzocaine, tetracaine, butyl aminobenzoate) and dapsone. Procedures utilizing topical anesthetics that are associated with the development of methemoglobinemia are transesophageal echocardiography, GI endoscopy, intubation, and bronchoscopy.66,67 Predisposing factors include sepsis, anemia, and hospitalization. Dapsone may cause prolonged methemoglobinemia in patients with acquired immunodeficiency syndrome (AIDS) because of its long half-life.
Methylene blue is the antidote for methemoglobinemia and should be administered when there are significant symptoms indicating impaired oxygen delivery.68 In addition, individuals with lower methemoglobin concentrations may also warrant treatment if further oxidant stress may increase the concentrations. High-flow oxygen should be administered. The usual dose of methylene blue is 1 to 2 mg/kg IV over 5 minutes. Clinical improvement in symptoms and cyanosis is usually evident within a few minutes. A second dose can be administered if cyanosis does not resolve within 1 hour. Although there is concern for methylene blue causing hemolysis in patients with glucose-6-phosphate dehydrogenase (G-6-PD) deficiency, this information will not usually be known at the time of treatment and methylene blue should not be withheld in symptomatic patients.
Organophosphate and Carbamate Agents
Organophosphates and carbamates are cholinesterase inhibitors and are usually a component of insecticides. However, nerve agents used in chemical warfare such as sarin and VX are also organophosphate compounds. Cholinesterase inhibitors exert toxicity by blocking the activity of acetylcholinesterase resulting in acetylcholine accumulation at cholinergic receptors. When organophosphates or carbamates bind to acetylcholinesterase, they form a conjugate that is infinitely more stable than the acetylcholine-acetylcholinesterase conjugate. The carbamate-acetylcholinesterase bond spontaneously hydrolyzes in minutes to hours so that acetylcholinesterase is eventually regenerated (reversible binding). Carbamates do penetrate the CNS based on clinical symptoms and autopsy studies.69 Most carbamate poisonings spontaneously resolve within 24 to 48 hours and do not have significant morbidity or mortality risk. Phosphorylated or phosphonylated enzymes, however, degrade over days to weeks, making acetylcholinesterase essentially inactive (irreversible binding). For the physiologic enzyme activity to return, new enzyme must be generated or antidote given. After the acetylcholinesterase is phosphorylated over 24 to 48 hours, “aging” occurs, and the enzyme can no longer spontaneously hydrolyze and is permanently inactivated.70
Organophosphates may be absorbed by virtually any route including transdermal, transconjunctival, inhalation, across the GI or genitourinary mucosa, and through direct injection. Onset of systemic symptoms may occur in 5 minutes with inhalation, and most patients will develop symptoms within 12 hours of ingestion, unless exposure to fat-soluble organophosphates (e.g., fenthion, clorfenthion) has occurred or if significant metabolic activation must occur (e.g., parathion, malathion). Signs and symptoms of cholinesterase poisoning are listed in Table 68.6. Pulmonary toxicity from bronchorrhea, bronchospasm, and respiratory depression is the primary concern.71
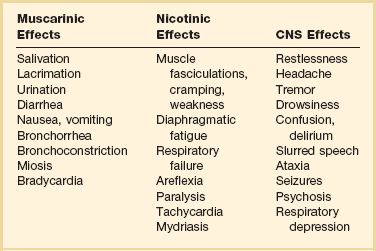
Atropine does not reverse nicotinic effects, and patients with significant respiratory muscle weakness require the use of pralidoxime. Pralidoxime is a nucleophilic oxime that regenerates acetylcholinesterase at muscarinic, nicotinic, and CNS sites. It may also prevent continued toxicity by scavenging the remaining organophosphate molecules. Treatment with pralidoxime may be most effective when started early. It may have benefit beyond the 48-hour aging limit, although the mechanisms have not been clearly elucidated. It should be continued as long as atropine is continued. The evidence for benefit of any oxime in pesticide poisoning is limited.72 Pralidoxime is usually administered as a loading dose (1 to 2 g in normal saline 500 mL administered over 30 minutes) and then as a continuous infusion at 200 to 500 mg/hour to maintain serum levels higher than 4 µg/L.73 Other dosing regimens have also been proposed.71 Pralidoxime may also be protective against the development of the intermediate syndrome and other long-term neurologic sequelae. Pralidoxime therapy is usually not needed if the toxin is known to be a carbamate.
In addition to acute toxicity, organophosphates may cause persistent effects, which may manifest while the patient is in the ICU and last several weeks to months. These include organophosphate-induced delayed neurotoxicity and delayed polyneuropathy that occur 1 to 3 weeks after exposure.74 Recovery may occur gradually or not at all. The third complication is intermediate syndrome.75 The syndrome develops 24 to 96 hours after resolution of an acute, severe cholinergic crisis, and patients develop acute respiratory paralysis, weakness in the bulbar musculature, nuchal weakness, proximal limb weakness, and depressed reflexes. Electromyography studies show decremental conduction with repetitive nerve stimulation and suggest both presynaptic and postsynaptic nerve impairment. Recovery takes 2 to 4 times longer than the development.
Psychotropic Drugs
Cyclic Antidepressants
Deaths caused by overdose with cyclic antidepressants are declining because of the increasing use of alternative antidepressants.4 The principal toxicities of cyclic antidepressants result from central and peripheral anticholinergic activity, α-adrenergic antagonism, and inhibition of norepinephrine reuptake. They also exert a membrane-depressant local anesthetic effect on the myocardium by blocking rapid sodium influx during phase 0 of the action potential. Primary toxicities include depressed level of consciousness, wide-complex arrhythmias, seizures, and hypotension. Acidosis, hypoxia, and seizures may increase the risk of wide-complex arrhythmias. Anticholinergic effects include mydriasis, fever, dry skin, delirium, agitation, tachycardia, ileus, and urinary retention. Life-threatening events usually occur within 6 hours of ingestion, most often in the first 2 hours.76 Several electrocardiographic criteria have been proposed to predict complications: QRS duration greater than 0.1 second correlates with risk of seizures, QRS duration greater than 0.16 second correlates with increased risk of arrhythmias, and the presence of an R wave in lead aVR greater than 3 mm predicts seizures and arrhythmias.77,78 However, the performance of these criteria in predicting complications including death is relatively poor.79
If poisoning with a cyclic antidepressant is suspected, electrocardiographic monitoring should be instituted and IV access obtained. Intubation may be needed in patients with CNS depression who are unable to protect their airway. If wide-complex arrhythmias (or ECG changes described previously), hypotension, or seizures are present, stabilization requires immediate alkalinization of the blood and sodium loading with sodium bicarbonate.80 Sodium bicarbonate should be administered in 50 to 100 mEq (1 to 2 mEq/kg) boluses to alkalinize the blood pH to 7.5 to 7.55. Clinical end points are normalization (narrowing) of the QRS complex, reestablishment of an adequate blood pressure, and termination of seizure activity. Alkalinization appears to decrease the free drug by increasing protein binding and shifting the concentration gradient away from tissues back into the main compartment. Sodium loading may have a greater benefit by overcoming the blockade of the myocardial sodium channels. The bolus doses of sodium bicarbonate should be immediately followed with a continuous infusion, which can be prepared by adding 150 mEq NaHCO3 (sodium bicarbonate) to 1 L of D5W. This should be titrated to the desired blood pH, QRS interval, and blood pressure. The infusion may be discontinued after 4 to 6 hours if the width of the QRS complex remains less than 100 ms without the administration of sodium bicarbonate. Hyperventilation to achieve blood alkalinization may be less effective but useful in patients who cannot tolerate the sodium and volume load or in those who develop pulmonary edema from treatment with sodium bicarbonate.80,81 Hyperventilation without the administration of sodium bicarbonate may also be considered for patients with cerebral edema, head trauma, or poorly controlled congestive heart failure. Hypertonic saline has been effective in treating cardiac toxicity refractory to initial blood alkalinization.82,83
If torsades de pointes is associated with QT prolongation, magnesium sulfate 1 to 2 g IV over 2 to 5 minutes should be administered.80 Hypotension refractory to volume expansion is best treated with a direct-acting catecholamine such as norepinephrine or phenylephrine in the setting of depleted norepinephrine stores.84 An inotropic agent such as dobutamine can be added if hypotension is the result of depressed myocardial contractility and decreased cardiac output. If hypotension remains refractory to fluids and vasopressors, the use of an intra-aortic balloon pump may be considered as a temporizing measure. Recently, IV lipid emulsion has been reported to successfully treat hemodynamic instability associated with cyclic antidepressant toxicity.49,85
After the patient with a known or suspected cyclic antidepressant overdose has been stabilized and the airway protected, activated charcoal is indicated. Gastric lavage should only be performed if the patient is seriously ill and the ingestion occurred within 1 hour of presentation. A second dose of activated charcoal may be given in several hours if it seems plausible that the drug still remains in the GI tract in the case of a massive ingestion or hypotension. MDAC to enhance elimination is not warranted given the extremely large volume of distribution (10 to 50 L/kg) and the low-protein binding of cyclic antidepressants.16 Forced diuresis, hemodialysis, and hemoperfusion are ineffective. Physostigmine should also be avoided because of the potential anticholinergic toxicity of seizures and asystole.
Lithium
Although lithium is used to treat bipolar affective disorder and other psychiatric disorders, its narrow therapeutic index predisposes to toxicity.86 After oral ingestion, lithium is absorbed within 1 to 2 hours, reaching peak blood levels in 2 to 4 hours with regular preparations or in 4 to 12 hours with sustained-release preparations. Lithium does not bind to plasma proteins and is excreted almost entirely by the kidneys. Toxicity may occur with acute, acute on chronic, or chronic ingestions. Drugs that increase lithium reabsorption (angiotensin-converting enzyme [ACE] inhibitors, thiazides, nonsteroidal anti-inflammatory drugs), sodium restriction, volume depletion, and intrinsic renal dysfunction increase the risk of toxicity. Lithium levels do not necessarily correlate with toxic symptoms. With an acute ingestion, the patient may be asymptomatic with a lithium level of 6 to 8 mmol/L. In chronic lithium ingestion, a high total-body lithium burden results in more immediate toxicity at lower serum levels.
Management decisions in treating lithium toxicity may depend on the type of ingestion (acute versus chronic) and the product ingested (regular versus sustained-release). Lithium is not adsorbed by activated charcoal, but charcoal may be administered if other drugs are ingested or suspected. A forced diuresis is not effective in enhancing lithium excretion, but isotonic saline should be administered to replete and maintain intravascular volume and promote adequate urine output.87 Diuretics can worsen lithium toxicity and should be avoided. WBI has been proposed for GI decontamination with acute or acute on chronic ingestions, ingestion of sustained-release products, or when serial lithium levels are rising. Despite lack of proven clinical benefit, this approach may be considered with appropriate precautions. Hemodialysis is effective in removing lithium, but controversy exists on the indications for treatment and duration of therapy. Proposed indications that have not been validated include renal dysfunction, severe neurologic dysfunction, inability to tolerate fluid replacement, lithium level higher than 4 mmol/L in an acute overdose, and lithium level higher than 2.5 mmol/L in chronic toxicity. The lithium level, duration of exposure, and severity of clinical symptoms should be balanced against risks of the procedure before initiating hemodialysis. Hemodialysis clears lithium only from the plasma, and a rebound increase can develop from drug redistribution. A lithium level should be assessed immediately after hemodialysis and 6 to 8 hours later. Repeat dialysis can be considered if the lithium level increases or neurologic toxicity persists at that time. Although a lithium level of 1 mmol/L is often recommended as the end point for hemodialysis, no systematic investigations have established the ideal end point for optimal outcome. Because of redistribution of lithium, improvement of neurologic toxicity lags behind the decrease in plasma level. Prolonged monitoring of lithium levels may be necessary, especially when sustained-release preparations are ingested. Continuous venovenous hemodiafiltration has also been used to remove lithium and may be associated with less rebound.88 This technique results in a slower lithium clearance compared with hemodialysis and is not recommended if hemodialysis is available and can be tolerated. Sodium polystyrene sulfonate resin has been proposed to bind and remove lithium, but it is not currently recommended and may result in hypokalemia, hypernatremia, and fluid overload. Aminophylline and low-dose dopamine infusions have also been proposed to enhance lithium excretion, but no evidence of clinical efficacy exists and they should not be used.87
Selective Serotonin Reuptake Inhibitors
Selective serotonin reuptake inhibitors (SSRIs) and related antidepressants are frequently prescribed for depression and other disorders. They have decreased lethality and fewer adverse cardiovascular effects compared with cyclic antidepressants.89 Most fatalities involving SSRIs involve coingestion of other substances. Manifestations of an acute SSRI overdose may include nausea, vomiting, dizziness, blurred vision, and rarely CNS depression. Seizures and a wide QRS occur rarely but may be more likely with citalopram and bupropion, a unicyclic antidepressant. A syndrome characteristic of SSRIs is the serotonin syndrome, which may occur after a single dose, high dose, overdose, or when combined with other serotonergic agents.90 The pathophysiology is related to excessive stimulation of central and peripheral serotonergic receptors. Clinical manifestations include altered mental status ranging from agitation to coma; autonomic dysfunction including diaphoresis, tachycardia, hyperthermia, unstable blood pressure, and diarrhea; and neuromuscular abnormalities that may range from tremors to myoclonus and rigidity.91 Severe cases may be complicated by rhabdomyolysis, renal failure, disseminated intravascular coagulation, or acute respiratory distress syndrome.
Management of an acute overdose of SSRIs is largely supportive. Gastric lavage is not warranted because of the low toxicity of these compounds, but use of activated charcoal may be considered. An ECG should be obtained to assess for the rare occurrence of a wide QRS complex or other electrocardiographic abnormality. Although clinical experience is limited, there are reports of sodium bicarbonate administration resulting in narrowing of the QRS complex. The treatment of serotonin syndrome is primarily supportive therapy after discontinuing the precipitating agents. Intubation and mechanical ventilation may be necessary for patients with significant alteration of mental status. Benzodiazepines are useful for control of agitation and external cooling for sustained hyperthermia. Rarely, neuromuscular blockers may be necessary for control of muscle rigidity or tremor. The syndrome usually resolves in 24 to 72 hours. Treatment of patients with serotonin antagonists has been proposed, but experience is limited to case reports. Cyproheptadine in varying dose regimens (12 to 32 mg/24 hour) has been most commonly recommended as a treatment option. Currently there is no role for the use of bromocriptine or dantrolene.91
Sedatives
Benzodiazepines
Although ingestions are relatively common, fatalities from benzodiazepines alone are uncommon. Benzodiazepine overdose results in a typical sedative-hypnotic toxidrome characterized by depressed level of consciousness, respiratory depression, hyporeflexia, and potentially hypotension and bradycardia. The clinical manifestations may be exacerbated by concomitant ingestion of other agents with sedating properties, such as ethanol, opioids, or antidepressants. Alprazolam is commonly seen in overdoses due to wide availability and may result in greater toxicity than other benzodiazepines.92 Flunitrazepam is a benzodiazepine not approved for use in the United States that has been associated with sexual assault. Diagnosis of benzodiazepine ingestion is primarily based on the history and clinical manifestations. Many benzodiazepines can be detected in qualitative urine toxicology assays, but a negative test does not rule out ingestion. If warranted, gas chromatography/mass spectrometry can be requested for definitive detection.
Flumazenil is a competitive benzodiazepine receptor antagonist that will reverse the sedative effects of benzodiazepines. It may be a helpful diagnostic tool in evaluating an overdose patient but should not be routinely used as a substitute for adequate airway protection.93 A dose greater than 1 mg is seldom necessary in overdose victims. The short half-life of flumazenil (0.7 to 1.3 hours) makes resedation likely because of the longer half-life of benzodiazepines. Continuous monitoring must be instituted if flumazenil is used to arouse the patient. Flumazenil use has been associated with seizures in patients with chronic benzodiazepine use and when cyclic antidepressants are present.93 It is best to avoid flumazenil in those situations and in patients with a seizure disorder or when a drug capable of causing seizures has been ingested. Slow titration of flumazenil (0.1 mg/minute) and limiting the total dose to 1 mg may minimize the risk of seizures. If seizures occur with flumazenil, benzodiazepines (often in higher doses) may be effective.
Gamma Hydroxybutyrate
Gamma hydroxybutyrate (GHB), a naturally occurring substance found in the brain and peripheral tissues, is banned in the United States except for the treatment for narcolepsy. It is one of several agents characterized as a “date rape” drug and has been promoted to build muscle, improve performance, produce euphoria, induce fat loss, and enhance sleep. Several deaths have been attributed to GHB and related agents.94 The drug is usually available as a colorless, odorless liquid with a mild, salty taste that is easy to mask in drinks. GHB is rapidly absorbed from the stomach (usually within 10 to 15 minutes) and readily crosses the blood-brain barrier. It is metabolized to carbon dioxide and water without active metabolites. Stimulatory effects occur from resulting increased dopamine levels in the brain and sedative effects by potentiation of endogenous opioids. The manifestations of GHB toxicity are dose related and include agitation, coma, seizures, respiratory depression, and vomiting.95 Other effects include amnesia, tremors, myoclonus, hypotonia, hypothermia, decreased cardiac output, and bradycardia. Coma and respiratory depression may be potentiated by the concomitant use of ethanol. GHB is not routinely detected by urine toxicology assays but can be detected in plasma or urine by gas chromatographic and mass spectrophotometric techniques. Diagnosis is usually determined by the clinical course and history of exposure elicited after the patient recovers. Gamma butyrolactone (GBL), also known as 2(3H)-furanone dihydro, and 1,4-butanediol (BD), also called tetramethylene glycol, have been abused with the same adverse effects as GHB including death. Both agents are metabolized in the body to GHB.94
No antidote for GHB, GBL, or BD exists. The primary management for ingestion of these drugs is supportive care with particular attention to airway protection and ventilation. In some cases, intubation and mechanical ventilation are required. Gastric lavage and activated charcoal are not indicated because of the small amounts involved and the rapid absorption. Naloxone and flumazenil are of no benefit. Patients with mild intoxication may be observed in the emergency department and released after symptoms resolve. A rapid recovery of consciousness from an obtunded condition is frequently observed. In patients requiring intubation and mechanical ventilation, symptoms can be expected to resolve within 2 to 96 hours unless complications such as aspiration or anoxic injury have occurred. The concomitant use of alcohol may prolong the CNS depression. Although physostigmine has been used to awaken patients with GHB intoxication, its use is not recommended.96
A withdrawal syndrome has been described in patients who frequently ingest high doses of GHB (every 1 to 3 hours).97 Mild symptoms such as anxiety, insomnia, nausea, vomiting, and tremors begin within 6 hours of the last dose and may progress to severe delirium with autonomic instability (usually mild) requiring hospitalization and sedation. The duration of symptoms requiring treatment may be as long as 2 weeks. Benzodiazepines are the initial choice for management, and high doses may be required. Propofol and barbiturates have also been used successfully.
Propofol
Propofol is a sedative-hypnotic used for general anesthesia, procedural sedation, more prolonged sedation in critically ill patients, and treatment of status epilepticus. It has also been implicated in abuse, accidental overdose, suicide, and even murder.98 The critical care clinician should be aware of a potentially fatal toxicity associated with more prolonged infusions of propofol known as propofol-related infusion syndrome (PRIS).99 The syndrome is usually associated with doses of propofol greater than 4 mg/kg/hour for longer than 48 hours’ duration. However, metabolic acidosis has been reported within 1 to 4 hours after the initiation of propofol infusion. Predisposing factors that have been associated with PRIS include young age, CNS or respiratory illness, vasopressor use, glucocorticoid use, greater severity of illness, sepsis, and impaired oxygen delivery. Mortality rates of 18% to 80% have been reported.99 Clinical features of the syndrome can include refractory bradycardia progressing to asystole, other arrhythmias, myocardial failure, lactic acidosis, rhabdomyolysis, renal failure, and hypertriglyceridemia.100 The pathophysiology is hypothesized to be related to mitochondrial utilization of free fatty acids and genetic predisposition.
Propofol infusions should be limited to less than 4 mg/kg/hour and no longer than 48 hours of infusion when possible. Prompt recognition of early signs of PRIS (elevated serum lactate, elevated creatine kinase, hypertriglyceridemia) is essential for successful intervention. Another clinical clue to possible PRIS is the unexplained need for increasing doses of pressor or inotropic agents. Management includes discontinuation of propofol and the use of alternative sedative agents. The most effective treatment for severe PRIS is cardiorespiratory support and hemodialysis or hemofiltration to decrease blood levels of metabolic acids and lipids.100
Stimulants
Amphetamines/Methamphetamines
Amphetamines, methamphetamines, and related agents cause central and peripheral release of catecholamines, which result in a sympathomimetic/adrenergic toxidrome characterized by tachycardia, hyperthermia, diaphoresis, agitation, hypertension, and mydriasis. Hallucinations (visual and tactile) and acute psychoses are frequently observed. The clinical manifestations associated with abuse of these agents may last up to 24 hours due to the longer duration of pharmacologic effects. The acute adverse consequences are similar to those seen with cocaine abuse (see following) and include myocardial ischemia and arrhythmias, pulmonary hypertension, seizures, intracranial hemorrhage, stroke, hepatotoxicity, rhabdomyolysis, necrotizing vasculitis, and death.101 Chronic use of these drugs may result in dilated cardiomyopathy.102 Poor oral hygiene and severe dental caries (“meth mouth”) can be clues to chronic methamphetamine use.103
Methamphetamine hydrochloride in a crystalline form called “ice,” “crank,” or “crystal” is one of the most popular drugs in this class. It has high purity and can be orally ingested, smoked, insufflated nasally, or injected intravenously. An amphetamine-like drug (3-4-methylenedioxymethamphetamine) is a designer drug commonly known as Ecstasy, XTC, or MDMA that acts as a stimulant and hallucinogen.104 It results in serotonin release in the brain with inhibition of serotonin reuptake and has been reported to produce serotonin syndrome. Complications are usually a result of the drug effects and excessive physical activity. Complications include hyperthermia, hyponatremia (excessive water intake or syndrome of inappropriate antidiuretic hormone secretion), rhabdomyolysis, renal failure, cardiac collapse, cerebral infarction/hemorrhage, and multiple organ failure. MDMA and other amphetamines will usually be detected on qualitative toxicology assays of urine but false positives and negatives occur.
Cocaine
Cocaine abuse is a global problem that results in significant medical complications.105 Cocaine hydrochloride is water soluble and can be injected intravenously, ingested, or snorted intranasally. Crack or rock cocaine is the alkaloid form primarily abused by inhalation. The onset, peak, and duration of physiologic effects of cocaine vary with the route of use, form of cocaine used, and concomitant use of other drugs.105 Both forms of cocaine are absorbed from all mucosal surfaces and undergo hydrolysis by plasma and liver cholinesterases. The major metabolites, benzoylecgonine and ecgonine methyl ester, are excreted in the urine and can be detected by qualitative urine assays. The metabolites of cocaine may be detectable in urine for 24 to 36 hours after use, but prolonged detection can occur in frequent users of high doses.
Cocaine inhibits the presynaptic reuptake of biogenic amines such as norepinephrine, dopamine, and serotonin throughout the body including the CNS. Characteristic clinical findings of a sympathomimetic syndrome include tachycardia, mydriasis, hypertension, hyperthermia, diaphoresis, euphoria, and agitation. The sympathetic stimulation also results in multiple potential complications (Box 68.3). Complications such as myocardial ischemia or cerebral infarction may occur several days after the last use of cocaine. Complications of transporting cocaine in body cavities may include rupture of packets with drug absorption and bowel obstruction.
No large clinical trials have evaluated therapeutic strategies for myocardial ischemia resulting from cocaine use. Aspirin should be administered if the risk of intracranial hemorrhage is low because a significant number of patients have thrombotic occlusion as the cause of ischemia. Benzodiazepines and nitroglycerin are first-line agents for relief of chest pain, but small clinical studies have yielded conflicting results on the benefit of combining the agents.106 α-Adrenergic blockers such as phentolamine and calcium channel blockers have been recommended as second-line treatment for unrelieved pain but are rarely necessary.106 Beta blockers were often considered to be contraindicated in the management of potential ischemia related to cocaine because of the potential for unopposed α-adrenergic-mediated vasoconstriction leading to elevated blood pressures. However, beta-blocker use was not found to be detrimental in two retrospective studies of patients with recent cocaine use and may be beneficial in some patients.107,108 It may be appropriate to avoid administration of beta blockers in patients manifesting acute sympathomimetic findings. In addition, routine use of IV beta blockers is no longer recommended for acute coronary syndromes. Reperfusion interventions, primarily percutaneous coronary interventions, should be considered for patients with myocardial infarction.106 Ventricular arrhythmias are not common and may be related to cocaine blockade of myocardial sodium channels leading to QRS and QT interval prolongation. Wide complex arrhythmias may respond to treatment with sodium bicarbonate.109 Class IA antiarrhythmic drugs such as procainamide should be avoided. Treatment of life-threatening arrhythmias should otherwise follow advanced life support guidelines.
Cerebral complications should be managed by standard interventions specific for the injury. Seizures are best managed with benzodiazepines. The reported incidence of underlying vascular malformations in intracranial hemorrhage has been variable, but if present these problems may require specific intervention. Severe hyperthermia is managed the same as heat stroke with either conductive or evaporative cooling.110 When there is suspicion of rhabdomyolysis, IV hydration should be instituted immediately pending assessment of renal function and CPK levels. Asymptomatic transporters of cocaine packets should be managed conservatively with activated charcoal, possible WBI, and supportive care. Surgery is reserved for patients exhibiting manifestations of cocaine poisoning or GI perforation or obstruction.111 Contamination of cocaine with other drugs or fillers can result in toxicities that are not directly related to the effects of cocaine. Levamisole, an anthelminthic agent used in veterinary medicine, has been found in cocaine supplies and linked to the development of agranulocytosis.112 Agranulocytosis resolves when drug use is discontinued. Retiform purpura and skin necrosis secondary to thrombotic vasculopathy have also been linked to cocaine contaminated with levamisole.113,114 The potential for suicidal intent should be recognized in cocaine abusers, and psychiatric consultation may be appropriate after stabilization.
Mephedrone/Methylenedioxypyrovalerone (Bath Salts)
Mephedrone and 3.4-methylenedioxpyrovalerone (MDPV) are synthetic stimulants marketed in products sold as bath salts to avoid regulation. MDPV inhibits reuptake of dopamine and norepinephrine, and mephedrone may act as a monoamine reuptake inhibitor.115 Street names include Ivory Wave, Bliss, White Lightning, Vanilla Sky, Hurricane Charlie, White Knight, and others. The products have been ingested, snorted, smoked, and injected and are often used in combination with alcohol or other drugs. These agents are not detected by qualitative toxicology screens.
Clinical manifestations are similar to those of other stimulant drugs and include agitation, tachycardia, hyperthermia, hypertension, mydriasis, chest pain, palpitations, seizures, hallucinations, delusions, and paranoia.115,116 Symptoms may be prolonged for more than 24 hours after use of mephedrone despite a short expected half-life.116 Management includes IV benzodiazepines for significant agitation and psychotic symptoms. Symptoms may recur as sedation wears off, requiring repeat dosing. A thorough clinical evaluation is required to identify other complications such as rhabdomyolysis, myocardial ischemia, and organ dysfunction. Seizures are best treated with IV benzodiazepines.
Valproic Acid
Use of valproic acid (VPA) for seizure disorders, psychiatric disorders, migraine prophylaxis, and neuropathic pain has resulted in increased toxicity reported with therapeutic doses and overdoses. The most common manifestation of toxicity in overdoses is CNS depression with higher drug levels associated with an increased incidence of coma and respiratory depression requiring intubation.117 Cerebral edema may occur 48 to 72 hours after overdoses and may be related to hyperammonemia that usually occurs in the absence of hepatotoxicity. The increase in ammonia is not well understood but may be related to carnitine deficiency, renal abnormalities, or defects in the urea cycle.117 Large VPA ingestions can result in refractory hypotension. Pancreatitis has been associated with chronic ingestion and acute overdose. Metabolic abnormalities of VPA toxicity include hypernatremia, hyperammonemia, anion gap metabolic acidosis, hypocalcemia, and acute renal failure. Hepatotoxicity is rare with VPA overdoses, but mild, asymptomatic abnormalities may occur with chronic use.
Initial and serial VPA levels should be obtained because of delayed peak serum levels in overdose. Patients may be comatose with normal serum VPA concentrations caused by unmeasured metabolites. An ammonia level should be obtained in patients with altered level of consciousness. Activated charcoal should be administered if the patient presents early after ingestion. MDAC may be beneficial because of a potential enterohepatic recirculation of drug, but routine use is not currently recommended. Although VPA is highly protein bound, protein binding is saturated at high serum concentrations resulting in more free drug being available for removal by extracorporeal techniques. Hemoperfusion, combined hemodialysis-hemoperfusion, or high flux hemodialysis may be considered in patients with severe or persistent hemodynamic instability, coma, or metabolic acidosis.118 No antidote exists for VPA toxicity, but L-carnitine administration has been proposed for patients with VPA toxicity and hyperammonemia. Chronic VPA therapy has been associated with carnitine deficiency and case reports suggest that L-carnitine administration is safe.119 However, there is no evidence that L-carnitine alters clinical outcome and more rigorous study is needed. Dosing of L-carnitine in reports has been variable but a suggested regimen is 100 mg/kg IV bolus or infusion over 15 to 30 minutes, followed by 50 mg/kg (maximum 3 g/dose) every 8 hours until the ammonia level is decreasing and the patient clinically improves.119
References
1. Hoffman, RS, Goldfrank, LR. The poisoned patient with altered consciousness: Controversies in the use of a “coma cocktail. ”. JAMA. 1995; 274:562.
2. Nelson, NS, Lewin, NA, Howland, MA, et al. Initial evaluation of the patient: Vital signs and toxic syndromes. In: Nelson NS, Lewin NA, Howland MA, et al, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York: McGraw-Hill Medical; 2011:33.
3. Wu, AHB, McKay, C, Broussard, LA, et al. National Academy of Clinical Biochemistry Laboratory Medicine practice guideline: Recommendations for the use of laboratory tests to support poisoned patients who present to the emergency department. Clin Chem. 2003; 49:357.
4. Bronstein, AC, Spyker, DA, Cantilena, LR, et al. 2010 annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 28th annual report. Clin Toxicol. 2011; 49:910.
5. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Position paper: Ipecac syrup. J Toxicol Clin Toxicol. 2004; 42:133.
6. Pond, SM, Lewis-Driver, DJ, Williams, GM, et al. Gastric emptying in acute overdose: A prospective randomized controlled trial. Med J Aust. 1995; 163:345.
7. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Position paper: Gastric lavage. J Toxicol Clin Toxicol. 2004; 42:933.
8. Merigian, KS, Blaho, KE. Single-dose oral activated charcoal in the treatment of the self-poisoned patient: A prospective, randomized, controlled trial. Am J Ther. 2002; 9:301.
9. Cooper, GM, Le Couteur, DG, Richardson, D, Buckely, NA. A randomized clinical trial of activated charcoal for the routine management of oral drug overdose. Q J Med. 2005; 98:655.
10. Jürgens, G, Hoegberg, LCG, Graudal, NA. The effect of activated charcoal on drug exposure in healthy volunteers: A meta-analysis. Clin Pharmacol Ther. 2009; 85:501.
11. American Academy of Clinical Toxicology and European Association of Poisons Centres and Clinical Toxicologists. Position paper: Single dose activated charcoal. Clin Toxicol. 2005; 43:61.
12. Isbister, GK, Kumar, VVP. Indications for single-dose activated charcoal administration in acute overdose. Curr Opin Crit Care. 2011; 17:351.
13. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Position paper: Cathartics. J Toxicol Clin Toxicol. 2004; 42:243.
14. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Position paper: Whole bowel irrigation. J Toxicol Clin Toxicol. 2004; 42:843.
15. Levy, G. Gastrointestinal clearance of drugs with activated charcoal. N Engl J Med. 1982; 307:676.
16. American Academy of Clinical Toxicology; European Association of Poison Centres and Clinical Toxicologists. Position statement and practice guidelines on the use of multidose activated charcoal in the treatment of acute poisoning. J Toxicol Clin Toxicol. 1999; 37:731.
17. Proudfoot, AT, Krenzelok, EP, Vale, JA. Position paper on urine alkalinization. J Toxicol Clin Toxicol. 2004; 42:1.
18. Goodman, JW, Goldfarb, DS. The role of continuous renal replacement therapy in the treatment of poisoning. Semin Dial. 2006; 44:803.
19. Barceloux, DG, Krenzelok, EP, Olson, K, Watson, W. American Academy of Clinical Toxicology practice guidelines on the treatment of ethylene glycol poisoning. J Toxicol Clin Toxicol. 1999; 37:537.
20. Barceloux, DG, Bong, GR, Krenzelok, EP, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol. 2002; 40:415.
21. Howland, MA. Antidotes in depth, ethanol. In: Nelson NS, Lewin NA, Howland MA, et al, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York: McGraw-Hill Medical; 2011:1419.
22. Brent, J. Fomepizole for ethylene glycol and methanol poisoning. N Engl J Med. 2009; 360:2216.
23. Lepik, KJ, Levy, AR, Sobolev, BG, et al. Adverse drug events associated with the antidotes for methanol and ethylene glycol poisoning: A comparison of ethanol and fomepizole. Ann Emerg Med. 2009; 53:439.
24. Hovda, KE, Froyshov, S, Gudmundsdottir, H, et al. Fomepizole may change indication for hemodialysis in methanol poisoning: Prospective study in seven cases. Clin Nephrol. 2005; 64:190.
25. Wilson, KC, Reardon, C, Theodore, AC, Farber, HW. Propylene glycol toxicity: A severe iatrogenic illness in ICU patients receiving IV benzodiazepines. Chest. 2005; 128(3):1674.
26. Arroliga, AC, Shehab, N, McCarthy, K, Gonzalez, JP. Relationship of continuous infusion lorazepam to serum propylene glycol concentration in critically ill adults. Crit Care Med. 2004; 32(8):1709.
27. Rumack, BH, Peterson, RG, Koch, GG, Amara, IA. Acetaminophen overdose: 662 cases with evaluation of oral acetylcysteine treatment. Arch Intern Med. 1981; 141:380.
28. Berger, E. Anxieties over acetaminophen. Ann Emerg Med. 2009; 54:13A.
29. Prescott, L. Oral or intravenous N-acetylcysteine for acetaminophen poisoning? Ann Emerg Med. 2005; 45:409.
30. Brok, J, Buckley, N, Gluud, C. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst Rev. (2):2006.
31. Yarema, MC, Johnson, DW, Berlin, RJ, et al. Comparison of the 20-hour intravenous and 72-hour oral acetylcysteine protocols for the treatment of acute acetaminophen poisonings. Ann Emerg Med. 2009; 54:606.
32. Doyon, S, Klein-Schwartz, W. Hepatotoxicity despite early administration of intravenous N-acetylcysteine for acute acetaminophen overdose. Acad Emerg Med. 2009; 16:34.
33. Daly, FFS, O’Malley, GF, Heard, K, et al. Prospective evaluation of repeated supratherapeutic acetaminophen (paracetamol) ingestion. Ann Emerg Med. 2004; 44:393.
34. Centers for Disease, Prevention, and Control. Vital signs: Overdoses of prescription opioid pain relievers—United States, 1999-2008. MMWR. 2011; 43:1487.
35. Centers for Disease, Prevention, and Control. Overdose deaths involving prescription opioids among Medicaid enrollees—Washington, 2004-2007. MMWR. 2009; 58:1171.
36. Kaplan, JL, Marx, JA, Calabro, JJ, et al. Double-blind, randomized study of nalmefene and naloxone in emergency department patients with suspected narcotic overdose. Ann Emerg Med. 1999; 34:42.
37. Sporer, KA, Dorn, E. Heroin-related noncardiogenic pulmonary edema. Chest. 2001; 120:1628.
38. Hillman, RJ, Prescott, LF. Treatment of salicylate poisoning with repeated oral charcoal. Br Med J. 1986; 291:1472.
39. Domachevsky, L, Adir, Y, Grupper, M, Keynan, Y. Hyperbaric oxygen in the treatment of carbon monoxide poisoning. Clin Toxicol. 2005; 43:181.
40. Juurlink, DN, Buckley, NA, Stanbrook, MB, et al. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. (1):2005.
41. Annane, D, Chadda, K, Gajdos, P, et al. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: Two randomized controlled trials. Intensive Care Med. 2011; 37:486.
42. Weaver, LK. Carbon monoxide poisoning. N Engl J Med. 2009; 360:1217.
43. DeWitt, CR, Waksman, JC. Pharmacology, pathophysiology and management of calcium channel blocker and β-blocker toxicity. Toxicol Rev. 2004; 23:223.
44. Reith, DM, Dawson, AH, Epid, D, et al. Relative toxicity of beta blockers in overdose. J Toxicol Clin Toxicol. 1996; 34:273.
45. White, CM. A review of potential cardiovascular uses of intravenous glucagon administration. J Clin Pharmacol. 1999; 39:442.
46. Kerns, W. Management of β-adrenergic blocker and calcium channel antagonist toxicity. Emerg Med Clin North Am. 2007; 25:309.
47. Mégarbane, B, Karyo, S, Baud, FJ. The role of insulin and glucose (hyperinsulinaemia/euglycaemia) therapy in acute calcium channel antagonist and β-blocker poisoning. Toxicol Rev. 2004; 23:215.
48. Weinstein, RS. Recognition and management of poisoning with beta-adrenergic blocking agents. Ann Emerg Med. 1984; 13:1123.
49. Cave, G, Harvey, M. Intravenous lipid emulsion as antidote beyond local anesthetic toxicity: A systematic review. Acad Emerg Med. 2009; 16:815.
50. Ramoska, EA, Spiller, HA, Myers, A. Calcium channel blocker toxicity. Ann Emerg Med. 1990; 19:649.
51. Lapostolle, F, Borron, SW, Verdier, C, et al. Digoxin-specific Fab fragments as single first-line therapy in digitalis poisoning. Crit Care Med. 2008; 36:3014.
52. Taboulet, P, Baud FJ. Bismuth, C, et al. Acute digitalis intoxication: Is pacing still appropriate? J Toxicol Clin Toxicol. 1993; 31:261.
53. Ujhelyi, MR, Robert, S. Pharmacokinetic aspects of digoxin-specific Fab therapy in the management of digitalis toxicity. Clin Pharmacokinet. 1995; 28:483.
54. Hall, AH, Rumack, BH. Clinical toxicology cyanide. Ann Emerg Med. 1986; 15:1067.
55. Borron, SW, Baud, FJ, Barriot, P, et al. Prospective study of hydroxocobalamin for acute cyanide poisoning in smoke inhalation. Ann Emerg Med. 2007; 49:794.
56. De Smet, PAGM. Herbal remedies. N Engl J Med. 2002; 347:2046.
57. Ang-Lee, MK, Moss, J, Yuan, C-S. Herbal medicines and perioperative care. JAMA. 2001; 286:208.
58. Haller, CA. Clinical approach to adverse events and interactions related to herbal and dietary supplements. Clin Toxicol. 2006; 44:605.
59. Lin, C-C, Chan, TYK, Deng, J-F. Clinical features and management of herb-induced aconitine poisoning. Ann Emerg Med. 2004; 43:574.
60. Haller, CA, Benowitz, NL. Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N Engl J Med. 2000; 343:1833.
61. Bouchard, NC, Howland, MA, Greller, HA, et al. Ischemic stroke associated with use of an ephedra-free dietary supplement containing synephrine. Mayo Clin Proc. 2005; 80:541.
62. Nykamp, DL, Fackih, MN, Compton, AL. Possible association of acute lateral-wall myocardial infarction and bitter orange supplement. Ann Pharmacother. 2004; 38:812.
63. Berger, AJ, Alford, K. Cardiac arrest in a young man following excess consumption of caffeinated “energy drinks. ”. Med J Aust. 2009; 190:41.
64. Saper, RB, Phillips, RS, Sehgal, A, et al. Lead, mercury and arsenic in US- and Indian-manufactured ayurvedic medicines sold via the internet. JAMA. 2008; 300:915.
65. Ko, RJ. Adulterants in Asian patent medicines (letter). N Engl J Med. 1998; 339:847.
66. Moore, TJ, Walsh, CS, Cohen, MR. Reported adverse event cases of methemoglobinemia associated with benzocaine products. Arch Intern Med. 2004; 164:1192.
67. Kane, GC, Hoehn, SM, Behrenbeck, TR, Mulvagh, SL. Benzocaine-induced methemoglobinemia based on the Mayo Clinic experience from 28,478 transesophageal echocardiograms. Arch Intern Med. 2007; 167:1977.
68. Wright, RO, Lewander, WJ, Woolf, AD. Methemoglobinemia: Etiology, pharmacology, and clinical management. Ann Emerg Med. 1999; 34:646.
69. Moriya, F, Hashimoto, Y. Comparative studies on tissue distribution of organophosphorus, carbamate and organochlorine pesticides in decedents intoxicated with these chemicals. J Forensic Sci. 1999; 44:1131.
70. Peter, JV, Cherian, AM. Organic insecticides. Anaesth Intensive Care. 2000; 28:11.
71. Leikin, JB, Thomas, RG, Walter, FG, et al. A review of nerve agent exposure for the critical care physician. Crit Care Med. 2002; 30:2346.
72. Buckley, NA, Eddleston, M, Li, Y, et al. Oximes for acute organophosphate pesticide poisoning. Cochrane Database of Systematic Reviews. (2):2011.
73. Eddleston, M, Szinicz, L, Eyer, P, Buckley, N. Oximes in acute organophosphorous pesticide poisoning: A systematic review of clinical trials. Q J Med. 2002; 95:275.
74. Abou-Donia, MB, Lapadula, DM. Mechanisms of organophosphorus ester-induced delayed neurotoxicity: Type I and type II. Annu Rev Pharmacol Toxicol. 1990; 30:405.
75. Abdollahi, M, Karami-Mohajeri, S. A comprehensive review on experimental and clinical in intermediate syndrome caused by organophosphate poisoning. Toxicol Appl Pharmacol. 2012; 258:309.
76. Callaham, M, Kassel, D. Epidemiology of fatal tricyclic antidepressant ingestion: Implications for management. Ann Emerg Med. 1985; 14:1.
77. Boehnert, M, Lovejoy, FH, Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985; 313:474.
78. Liebelt, EL, Francis, PD, Woolf, AD. ECG lead AVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med. 1995; 26:195.
79. Bailey, B, Buckley, NA, Amre, DK. A meta-analysis of prognostic indicators to predict seizures, arrhythmias, or death after tricyclic antidepressant overdose. J Toxicol Clin Toxicol. 2004; 42:877.
80. Bradberry, SM, Thanacoody, HKR, Watt, BE, et al. Management of the cardiovascular complications of tricyclic antidepressant poisoning: Role of sodium bicarbonate. Toxicol Rev. 2005; 24:195.
81. Pentel, PR, Benowitz, NL. Tricyclic antidepressant poisoning—Management of arrhythmias. Med Toxicol. 1986; 1:101.
82. McCabe, JL, Cobaugh, DJ, Menegazzi, JJ, et al. Experimental tricyclic antidepressant toxicity: A randomized, controlled comparison of hypertonic saline solution, sodium bicarbonate, and hyperventilation. Ann Emerg Med. 1998; 32:329.
83. McKinney, PE, Rasmussen, R. Reversal of severe tricyclic antidepressant-induced cardiotoxicity with intravenous hypertonic saline solution. Ann Emerg Med. 2003; 42:20.
84. Tran, TP, Panacek, EA, Rhee, KJ, et al. Response to dopamine vs norepinephrine in tricyclic antidepressant-induced hypotension. Acad Emerg Med. 1997; 4:864.
85. Engels, PT, Davidow, JS. Intravenous fat emulsion to reverse haemodynamic instability from intentional amitriptyline overdose. Resuscitation. 2010; 81:1037.
86. Grandjean, EM, Aubry, J-M. Lithium: Updated human knowledge using an evidence-based approach, Part III: Clinical safety. CNS Drugs. 2009; 23:397.
87. Scharman, EJ. Methods used to decrease lithium absorption or enhance elimination. J Toxicol Clin Toxicol. 1997; 35:601.
88. LeBlanc, M, Raymond, M, Bonnardeaux, A, et al. Lithium poisoning treated by high performance continuous arteriovenous and venovenous hemodiafiltration. Am J Kidney Dis. 1996; 27:365.
89. Isbister, GK, Bowe, SJ, Dawson, A, Whyte, IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol. 2004; 42:277.
90. Isbister, GK, Buckley, NA, Whyte, IM. Serotonin toxicity: A practical approach to diagnosis and treatment. Med J Aust. 2007; 187:361.
91. Boyer, EW, Shannon, M. The serotonin syndrome. N Engl J Med. 2005; 352:1112.
92. Isbister, GK, O’Regan, G, Sibbritt, D, Whyte, IM. Alprazolam is relatively more toxic than other benzodiazepines in overdose. Br J Clin Pharmacol. 2004; 58:88.
93. Seger, DL. Flumazenil-treatment or toxin. J Toxicol Clin Toxicol. 2004; 42:209–216.
94. Zvosec, DL, Smith, SW, McCutcheon, JR, et al. Adverse events, including death, associated with the use of 1,4-butanediol. N Engl J Med. 2001; 344:87.
95. Snead, OC, Gibson, KM. Gamma-hydroxybutyric acid. N Engl J Med. 2005; 352:2721.
96. Traub, SJ, Nelson, LS, Hoffman, RS. Physostigmine as a treatment for gamma-hydroxybutyrate toxicity: A review. J Toxicol Clin Toxicol. 2002; 40:781.
97. Dyer, JE, Roth, B, Hyma, BA. Gamma-hydroxybutyrate withdrawal syndrome. Ann Emerg Med. 2001; 37:147.
98. Kirby, RR, Colaw, JM, Douglas, MM. Death from propofol: Accident, suicide, or murder? Anesth Analg. 2009; 108:1182.
99. Roberts, RJ, Barletta, JF, Fong, JJ, et al. Incidence of propofol-related infusion syndrome in critically ill adults: A prospective, multicenter study. Crit Care. 2009; 13:R169.
100. Kam, PCA, Cardone, D. Propofol infusion syndrome. Anaesthesia. 2007; 62:690.
101. Lineberry, TW, Bostwick, JM. Methamphetamine abuse: A perfect storm of complications. Mayo Clin Proc. 2006; 81:77.
102. Yeo, K-K, Wijetunga, M, Ito, H, et al. The association of methamphetamine use and cardiomyopathy in young patients. Am J Med. 2007; 120:165.
103. Hamamoto, DT, Rhodus, NL. Methamphetamine abuse and dentistry. Oral Dis. 2009; 15:27.
104. de la Torre, R, Farrell, M, Roset, PN, et al. Human pharmacology of MDMA: Pharmacokinetics, metabolism, and disposition. Ther Drug Monit. 2004; 26:137.
105. Goldstein, Ram, DesLauriers, C, Burda, AM. Cocaine: History, social implications, and toxicity—A review. Dis Mon. 2009; 55:6.
106. McCord, J, Jneid, H, Hollander, JE, et al. Management of cocaine-associated chest pain and myocardial infarction. A statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008; 117:1897.
107. Dattilo, PB, Halpern, SM, Fearon, K, et al. Beta-blockers are associated with reduced risk of myocardial infarction after cocaine use. Ann Emerg Med. 2008; 51:117.
108. Rangel, C, Shu, RG, Lazar, LD, et al. β-blockers for chest pain associated with recent cocaine use. Arch Intern Med. 2010; 170:874.
109. Wood, DM, Dargan, PI, Hoffman, RS. Management of cocaine-induced cardiac arrhythmias due to cardiac ion channel dysfunction. Clin Toxicol. 2009; 47:14.
110. Bouchama, A, Knochel, JP. Heat stroke. N Engl J Med. 2002; 346:1978.
111. Traub, SJ, Hoffman, RS, Nelson, LS. Body packing—The internal concealment of illicit drugs. N Engl J Med. 2003; 349:2519.
112. Centers for Disease, Prevention, and Control. Agranulocytosis associated with cocaine use—Four states, March 2008-November 2009. MMWR. 2009; 58:1381.
113. Bradford, M, Rosenberg, B, Moreno, J. Bilateral necrosis of earlobes and cheeks: Another complication of cocaine contaminated with levamisole. Ann Intern Med. 2010; 152:758.
114. Milman, N, Smith, CD. Cutaneous vasculopathy associated with cocaine use. Arthritis Care Res. 2011; 63:1195.
115. Jerry, J, Collins, G, Streem, D. Synthetic legal intoxicating drugs: The emerging “incense” and “bath salt” phenomenon. Cleve Clin J Med. 2012; 79:258.
116. James, D, Adams, RD, Spears, R, et al. Clinical characteristics of mephedrone toxicity reported to the UK National Poisons Information Service. Emerg Med J. 2011; 38:686.
117. Sztajnkrycer, MD. Valproic acid toxicity: Overview and management. J Toxicol Clin Toxicol. 2002; 40:789.
118. Licari, E, Calzavacca, P, Warrillow, SJ, Bellomo, R. Life-threatening sodium valproate overdose: A comparison of two approaches to treatment. Crit Care Med. 2009; 37:3161.
119. Perrot, J, Murphy, NG, Zed, PJ. L-Carnitine for acute valproic acid overdose: A systematic review of published cases. Ann Pharmacother. 2010; 44:1287.