Chapter 478 Platelet and Blood Vessel Disorders
Megakaryopoiesis
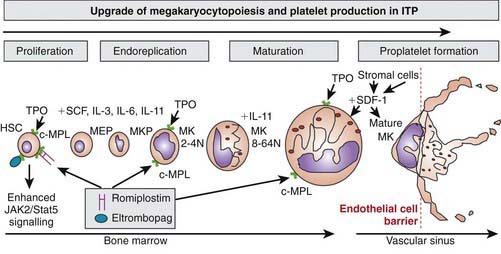
Platelets are non-nucleated cellular fragments produced by megakaryocytes within the bone marrow and other tissues. Megakaryocytes are large polyploid cells. When the megakaryocyte approaches maturity, budding of the cytoplasm occurs and large numbers of platelets are liberated. Platelets circulate with a life span of 10-14 days. Thrombopoietin (TPO) is the primary growth factor that controls platelet production (Fig. 478-1). Levels of TPO appear to correlate inversely with platelet number and megakaryocyte mass. Levels of TPO are highest in the thrombocytopenic states associated with decreased marrow megakaryopoiesis and may be variable in states of increased platelet production.
Thrombocytopenia
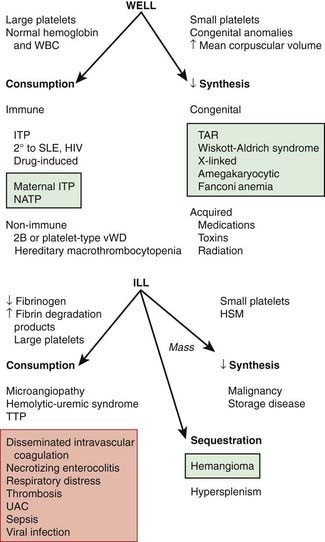
The normal platelet count is 150-450 × 109/L. Thrombocytopenia refers to a reduction in platelet count to <150 × 109/L. Causes of thrombocytopenia include: (1) decreased production on either a congenital or an acquired basis; (2) sequestration of the platelets within an enlarged spleen or other organ; and (3) increased destruction of normally synthesized platelets on either an immune or a nonimmune basis (Chapter 469; Tables 478-1 and 478-2 and Fig. 478-2).
Table 478-1 DIFFERENTIAL DIAGNOSIS OF THROMBOCYTOPENIA IN CHILDREN AND ADOLESCENTS
DESTRUCTIVE THROMBOCYTOPENIAS
Primary Platelet Consumption Syndromes
Combined Platelet and Fibrinogen Consumption Syndromes
IMPAIRED PLATELET PRODUCTION
SEQUESTRATION
HIV, human immunodeficiency virus; ITP, immune thrombocytopenic purpura; VWD, von Willebrand disease.
From Wilson DB: Acquired platelet defects. In Orkin SH, Nathan DG, Ginsburg D, et al, editors: Nathan and Oski’s hematology of infancy and childhood, ed 7, Philadelphia, 2009, WB Saunders, p 1555, Box 33-1.
Table 478-2 CLASSIFICATION OF FETAL AND NEONATAL THROMBOCYTOPENIAS*
| CONDITION | |
| Fetal | Alloimmune thrombocytopenia |
| Congenital infection (e.g., CMV, toxoplasma, rubella, HIV) | |
| Aneuploidy (e.g., trisomy 18, 13, or 21, or triploidy) | |
| Autoimmune condition (e.g., ITP, SLE) | |
| Severe Rh hemolytic disease | |
| Congenital/inherited (e.g., Wiskott-Aldrich syndrome) | |
| Early-onset neonatal (<72 hr) | Placental insufficiency (e.g., PET, IUGR, diabetes) |
| Perinatal asphyxia | |
| Perinatal infection (e.g., Escherichia coli, GBS, Haemophilus influenzae) | |
| DIC | |
| Alloimmune thrombocytopenia | |
| Autoimmune condition (e.g., ITP, SLE) | |
| Congenital infection (e.g., CMV, toxoplasma, rubella, HIV) | |
| Thrombosis (e.g., aortic, renal vein) | |
| Bone marrow replacement (e.g., congenital leukemia) | |
| Kasabach-Merritt syndrome | |
| Metabolic disease (e.g., proprionic and methylmalonic acidemia) | |
| Congenital/inherited (e.g., TAR, CAMT) | |
| Late-onset neonatal (>72 hr) | Late-onset sepsis |
| NEC | |
| Congenital infection (e.g., CMV, toxoplasma, rubella, HIV) | |
| Autoimmune | |
| Kasabach-Merritt syndrome | |
| Metabolic disease (e.g., proprionic and methylmalonic acidemia) | |
| Congenital/inherited (e.g., TAR, CAMT) |
CAMT, congenital amegakaryocytic thrombocytopenia; CMV, cytomegalovirus; DIC, disseminated intravascular coagulation; GBS, group B streptococcus; ITP, idiopathic thrombocytopenic purpura; IUGR, intrauterine growth restriction; NEC, necrotizing enterocolitis; PET, preeclampsia; SLE, systemic lupus erythematosus; TAR, thrombocytopenia with absent radii.
* The most common conditions are shown in bold.
From Roberts I, Murray NA: Neonatal thrombocytopenia: causes and management, Arch Dis Child Fetal Neonatal Ed 88:F359–F364, 2003.
Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359:1261-1270.
Newman PK, Newman DK. Platelets and the vessel wall. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1379-1399.
478.1 Idiopathic (Autoimmune) Thrombocytopenic Purpura
Pathogenesis
Why some children develop the acute presentation of an autoimmune disease is unknown. The exact antigenic target for most such antibodies in most cases of childhood acute ITP remains undetermined. although in chronic ITP most patients demonstrate antibodies against the platelet glycoprotein complexes, α11b-B3 and GPIb. After binding of the antibody to the platelet surface, circulating antibody-coated platelets are recognized by the Fc receptor on splenic macrophages, ingested, and destroyed. Most common viruses have been described in association with ITP, including Epstein-Barr virus (Chapter 246) and HIV (Chapter 268). Epstein-Barr virus-related ITP is usually of short duration and follows the course of infectious mononucleosis. HIV-associated ITP is usually chronic. In some patients ITP appears to arise in children infected with Helicobacter pylori or rarely following the measles, mumps, rubella vaccine.
Clinical Manifestations
Laboratory Findings
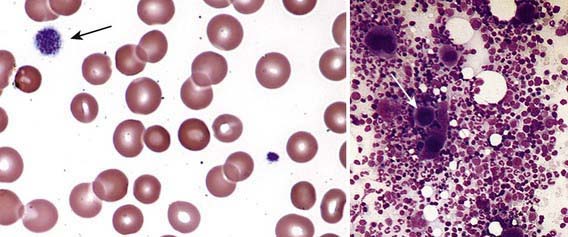
Severe thrombocytopenia (platelet count <20 × 109/L) is common, and platelet size is normal or increased, reflective of increased platelet turnover (Fig. 478-3). In acute ITP, the hemoglobin value, white blood cell (WBC) count, and differential count should be normal. Hemoglobin may be decreased if there have been profuse nosebleeds or menorrhagia. Bone marrow examination shows normal granulocytic and erythrocytic series, with characteristically normal or increased numbers of megakaryocytes. Some of the megakaryocytes may appear to be immature and are reflective of increased platelet turnover. Indications for bone marrow aspiration/biopsy include an abnormal WBC count or differential or unexplained anemia as well as findings on history and physical examination suggestive of a bone marrow failure syndrome or malignancy. Other laboratory tests should be performed as indicated by the history and physical examination. In adolescents with new-onset ITP, an antinuclear antibody test should be done to evaluate for SLE. HIV studies should be done in at-risk populations, especially sexually active teens. Platelet antibody testing is seldom useful in acute ITP. A direct antiglobulin test (Coombs) should be done if there is unexplained anemia to rule out Evans syndrome (autoimmune hemolytic anemia and thrombocytopenia) (Chapter 458) or before instituting therapy with IV anti-D.
Diagnosis/Differential Diagnosis
The well-appearing child with moderate to severe thrombocytopenia, an otherwise normal complete blood cell count (CBC), and normal findings on physical examination has a limited differential diagnosis that includes exposure to medication that induces drug-dependent antibodies, splenic sequestration due to previously unappreciated portal hypertension, and rarely, early aplastic processes, such as Fanconi anemia (Chapter 462). Other than congenital thrombocytopenia syndromes (Chapter 478.8), such as thrombocytopenia-absent radius (TAR) syndrome and MYH9-related thrombocytopenia, most marrow processes that interfere with platelet production eventually cause abnormal synthesis of red blood cells (RBCs) and WBCs and therefore manifest diverse abnormalities on the CBC. Disorders that cause increased platelet destruction on a nonimmune basis are usually serious systemic illnesses with obvious clinical findings (e.g., hemolytic-uremic syndrome [HUS], disseminated intravascular coagulation [DIC]) [see Table 477-1 and Fig. 478-2]. Isolated enlargement of the spleen suggests the potential for hypersplenism owing to either liver disease or portal vein thrombosis. Autoimmune thrombocytopenia may be an initial manifestation of SLE, HIV infection, common variable immunodeficiency, or rarely lymphoma. Wiskott-Aldrich syndrome (WAS; Chapter 120.2) must be considered in young males found to have thrombocytopenia with small platelets, particularly if there is a history of eczema and recurrent infection.
Treatment
Chronic Idiopathic Thrombocytopenic Purpura
Approximately 20% of patients who present with acute ITP have persistent thrombocytopenia for >12 mo and are said to have chronic ITP. At that time, a careful re-evaluation for associated disorders should be performed, especially for autoimmune disease, such as SLE; chronic infectious disorders, such as HIV; and nonimmune causes of chronic thrombocytopenia, such as type 2B and platelet-type von Willebrand disease, X-linked thrombocytopenia, autoimmune lymphoproliferative syndrome, common variable immunodeficiency syndrome, autosomal macrothrombocytopenia, and WAS (also X-linked). The presence of co-existing H. pylori infection should be explored and, if found, treated. Therapy should be aimed at controlling symptoms and preventing serious bleeding. In ITP, the spleen is the primary site of both antiplatelet antibody synthesis and platelet destruction. Splenectomy is successful in inducing complete remission in 64-88% of children with chronic ITP. This effect must be balanced against the lifelong risk of overwhelming postsplenectomy infection. This decision is often affected by lifestyle issues as well as the ease with which the child can be managed using medical therapy, such as IVIG, corticosteroids, IV anti-D. Rituximab, a chimeric monoclonal anti–B cell antibody, effectively induces a remission in 30-50% of children with chronic ITP. Two new effective agents that act to stimulate thrombopoiesis, romiplastin and eltrombopag (see Fig. 478-1), have been approved by the Federal Drug Administration to treat adults with chronic ITP. There are no data regarding either drug’s safety or efficacy in children.
Blanchette V, Bolton-Maggs P. Childhood immune thrombocytopenic purpura: diagnosis and management. Pediatr Clin North Am. 2008;55:393-420. ix
Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet. 2011;377:393-402.
De Waele L, Freson K, Louwette S, et al. Severe gastrointestinal bleeding and thrombocytopenia in a child with an anti-GATA1 autoantibody. Pediatr Res. 2010;67:314-319.
Edmonson MB, Riedesel EL, Williams GP, et al. Generalized petechial rashes in children during a parvovirus B19 outbreak. Pediatrics. 2010;125:e787-e792.
Fogarty PF, Segal JB. The epidemiology of immune thrombocytopenic purpura. Curr Opin Hematol. 2007;14:515-519.
France EK, Glanz J, Xu S, et al. Risk of immune thrombocytopenic purpura after measles-mumps-rubella immunization in children. Pediatrics. 2008;121:e687-e692.
George JN. Management of immune thrombocytopenia—something old, something new. N Engl J Med. 2010;363(20):1959-1960.
Glanz J, France E, Xu S, et al. A population-based, multistate cohort study of the predictors of chronic idiopathic thrombocytopenic purpura in children. Pediatrics. 2008;121:e506-e512.
Heddle NM, Cook RJ, Tinmouth A, et al. A randomized controlled trial comparing standard- and low-dose strategies for transfusion of platelets (SToP) to patients with thrombocytopenia. Blood. 2009;113:1564-1573.
Kühne T, Blanchette V, Buchanan GR, et al. Splenectomy in children with idiopathic thrombocytopenic purpura: a prospective study of 134 children from the Intercontinental Childhood ITP Study Group. Pediatr Blood Cancer. 2007;49:829-834.
Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med. 2010;363:1889-1899.
Mantadakis E, Farmaki E, Buchanan GR. Thrombocytopenic purpura after measles-mumps-rubella vaccination: a systematic review of the literature and guidance for management. J Pediatr. 2010;156:623-628.
McMillan R. The pathogenesis of chronic immune thrombocytopenic purpura. Semin Hematol. 2007;44:S3-S11.
The Medical Letter. Desirudin (Iprivask) for DVT prevention. Med Lett. 2010;52(1350):85-86.
The Medical Letter. Two new drugs for chronic ITP. Med Lett. 2009;51:10-11.
Mueller BU, Bennett CM, Feldman HA, et al. One year follow-up of children and adolescents with chronic immune thrombocytopenic purpura (ITP) treated with rituximab. Pediatr Blood Cancer. 2009;52:259-262.
Neunert CE, Buchanan GR, Imbach P, et al. Severe hemorrhage in children with newly diagnosed immune thrombocytopenic purpura. Blood. 2008;112:4003-4008.
Nurden AT. Sustaining platelet counts in chronic ITP. Lancet. 2011;377:358-360.
Panzer S, Pabinger I. Eltrombopag in chronic idiopathic thrombocytopenic purpura. Lancet. 2009;373:607-608.
Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenic (ITP): study of 40 cases. Blood. 2009;114:4777-4783.
Slichter SJ, Kaufman RM, Assmann SF, et al. Dose of prophylactic platelet transfusions and prevention of hemorrhage. N Engl J Med. 2010;362:600-612.
Thachil J, Hall GW. Is this immune thrombocytopenic purpura? Arch Dis Child. 2008;93:76-81.
478.3 Nonimmune Platelet Destruction
J. Paul Scott and Robert R. Montgomery
The syndromes of DIC (Chapter 477), HUS (Chapters 478.4 and 512), and thrombotic thrombocytopenic purpura (TTP) (Chapter 478.5) share the hematologic picture of a thrombotic microangiopathy in which there is RBC destruction and consumptive thrombocytopenia caused by platelet and fibrin deposition in the microvasculature. The microangiopathic hemolytic anemia is characterized by the presence of RBC fragments, including helmet cells, schistocytes, spherocytes, and burr cells.
478.4 Hemolytic-Uremic Syndrome
J. Paul Scott and Robert R. Montgomery
See also Chapter 512. Typical HUS is an acute disease of infancy and early childhood and usually follows an episode of acute gastroenteritis, often triggered by Escherichia coli 0157:H7. Shortly thereafter, signs and symptoms of hemolytic anemia, thrombocytopenia, and acute renal failure ensue. Sometimes neurologic symptoms are associated with these findings. E. coli 0157:H7 produces a specific toxin (verotoxin) that binds to and damages renal endothelial cells preferentially.
478.6 Kasabach-Merritt Syndrome
J. Paul Scott and Robert R. Montgomery
See also Chapter 642. The association of a giant hemangioma with localized intravascular coagulation causing thrombocytopenia and hypofibrinogenemia is called Kasabach-Merritt syndrome. In most patients, the site of the hemangioma is obvious, but retroperitoneal and intra-abdominal hemangiomas may require body imaging for detection. Inside the hemangioma there is platelet trapping and activation of coagulation, with fibrinogen consumption and generation of fibrin(ogen) degradation products. Arteriovenous malformation within the lesions can cause heart failure. Pathologically Kasabach-Merritt syndrome appears to develop more often as a result of a kaposiform hemangioendothelioma or tufted hemangioma rather than a simple hemangioma. The peripheral blood smear shows microangiopathic changes. Multiple modalities have been used to treat Kasabach-Merritt syndrome, including surgical excision (if possible), laser photocoagulation, high-dose corticosteroids, local radiation therapy, antiangiogenic agents, such as interferon-α2, and vincristine. Over time, most patients who present in infancy have regression of the hemangioma. Treatment of the associated coagulopathy may benefit from a trial of antifibrinolytic therapy with ε-aminocaproic acid (Amicar).
478.8 Congenital Thrombocytopenic Syndromes
See Table 478-2. Congenital amegakaryocytic thrombocytopenia (CAMT) usually manifests within the 1st few days to wk of life, when the child presents with petechiae and purpura caused by profound thrombocytopenia. CAMT is caused by a rare defect in hematopoiesis due to a mutation in the stem cell TPO receptor (MPL). Other than skin and mucous membrane abnormalities, findings on physical examination are normal. Examination of the bone marrow shows an absence of megakaryocytes. These patients often progress to marrow failure (aplasia) over time. Hematopoietic stem cell transplantation is curative.
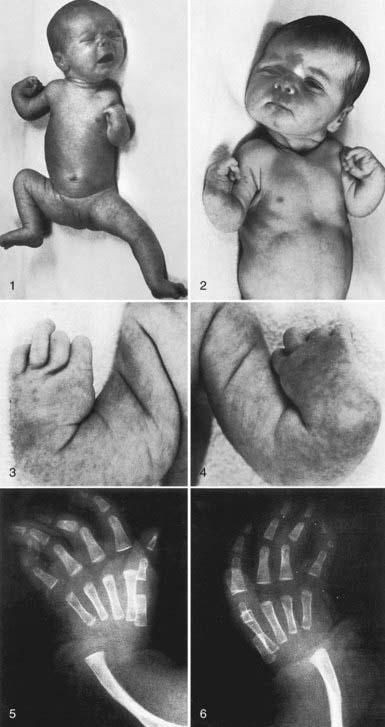
Thrombocytopenia-absent radius (TAR) syndrome consists of thrombocytopenia (absence or hypoplasia of megakaryocytes) that presents in early infancy with bilateral radial anomalies of variable severity, ranging from mild changes to marked limb shortening (Fig. 478-4). Many such individuals also have other skeletal abnormalities of the ulna, radius, and lower extremities. Thumbs are present. Intolerance to cow’s milk formula (present in 50%) may complicate management by triggering gastrointestinal bleeding, increased thrombocytopenia, eosinophilia, and a leukemoid reaction. The thrombocytopenia of TAR syndrome frequently remits over the 1st few yr of life. The molecular basis of TAR syndrome remains to be defined. A few patients have been reported to have a syndrome of amegakaryocytic thrombocytopenia with radioulnar synostosis due to a mutation in the HOXA11 gene. Different from TAR syndrome, this mutation causes marrow aplasia.
Wiskott-Aldrich syndrome (WAS) is characterized by thrombocytopenia, with tiny platelets, eczema, and recurrent infection due to immune deficiency (Chapter 120.2). WAS is inherited as an X-linked disorder, and the gene for WAS has been sequenced. The WAS protein appears to play an integral role in regulating the cytoskeletal architecture of both platelets and T lymphocytes in response to receptor-mediated cell signaling. The WAS protein is common to all cells of hematopoietic lineage. Molecular analysis of families with X-linked thrombocytopenia has shown that many affected members have a point mutation within the WAS gene, whereas individuals with the full manifestation of WAS have large gene deletions. Examination of the bone marrow in WAS shows the normal number of megakaryocytes, although they may have bizarre morphologic features. Transfused platelets have a normal life span. Splenectomy often corrects the thrombocytopenia, suggesting that the platelets formed in WAS have accelerated destruction. After splenectomy, these patients are at increased risk for overwhelming infection and require lifelong antibiotic prophylaxis against encapsulated organisms. Approximately 5-15% of patients with WAS develop lymphoreticular malignancies. Successful hematopoietic stem cell transplantation cures WAS. X-linked macrothrombocytopenia and dyserythropoiesis have been linked to mutations in the GATA-1 gene, an erythroid and megakaryocytic transcription factor.
MYH9-related thrombocytopenia: A diverse number of hereditary thrombocytopenia syndromes, given names such as Sebastian, Epstein, May-Hegglin, and Fechtner syndromes, are characterized by autosomal dominant macrothrombocytopenia, neutrophil inclusion bodies, and a variety of physical anomalies, including sensorineural deafness, renal disease, and/or eye disease. These have all been shown to be due to different mutations in the MYH9 gene (nonmuscle myosin-IIa heavy chain). The thrombocytopenia is usually mild and not progressive. Some other individuals with recessively inherited macrothrombocytopenia have abnormalities in chromosome 22q11. Mutations in the gene for glycoprotein Ibβ, an essential component of the platelet von Willebrand factor receptor, can result in Bernard-Soulier syndrome (Chapter 478.13).
478.9 Neonatal Thrombocytopenia
Thrombocytopenia in the newborn rarely is indicative of a primary disorder of megakaryopoiesis but more often is the result of either systemic illness or transfer of maternal antibodies directed against fetal platelets (see Table 478-2). Neonatal thrombocytopenia often occurs in association with congenital viral infection, especially rubella; cytomegalovirus; protozoal infection, such as toxoplasmosis; syphilis; and perinatal bacterial infection, especially those caused by gram-negative bacilli. Thrombocytopenia associated with DIC may be responsible for severe spontaneous bleeding. The constellation of marked thrombocytopenia and abnormal abdominal findings is common in necrotizing enterocolitis and other causes of necrotic bowel. Thrombocytopenia in an ill child requires a prompt search for viral and bacterial pathogens.
Two syndromes of congenital failure of platelet production often present in the newborn period. In congenital amegakaryocytic thrombocytopenia (CAMT), the newborn manifests petechiae and purpura shortly after birth. Findings on physical examination are otherwise normal. Megakaryocytes are absent from the bone marrow. This syndrome is caused by a mutation in the megakaryocyte TPO receptor that is essential for development of all hematopoietic cell lines. Pancytopenia eventually develops, and hematopoietic stem cell transplantation is curative. TAR syndrome consists of thrombocytopenia that presents in early infancy, with bilateral radial anomalies of variable severity, ranging from mild changes to marked limb shortening. Thumbs are present. In many such individuals, there are also other skeletal abnormalities of the lower extremities. Intolerance to cow’s milk formula is present in 50% of patients. TAR syndrome frequently remits over the 1st few yr of life (Chapter 478.8) (see Fig. 478-4).
Bussel JB, Sola-Visner M. Current approaches to the evaluation and management of the fetus and neonate with immune thrombocytopenia. Semin Perinatol. 2009;33:35-42.
Rivers A, Slayton WB. Congenital cytopenias and bone marrow failure syndromes. Semin Perinatol. 2009;33:20-28.
Roberts I, Murray NA. Neonatal thrombocytopenia. Semin Fetal Neonatal Med. 2008;13:256-264.
478.10 Thrombocytopenia Due to Acquired Disorders Causing Decreased Production
478.13 Congenital Abnormalities of Platelet Function
Lambert MP, Poncz M. Inherited platelet disorders. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1463.
Neunert CE, Journeycake JM. Congenital platelet disorders. Hematol Oncol Clin North Am. 2007;21:663-684. vi
Nurden P, Nurden AT. Congenital disorders associated with platelet dysfunctions. Thromb Haemost. 2008;99:253-263.
478.14 Disorders of the Blood Vessels
Henoch-Schönlein Purpura
Henoch-Schönlein purpura (HSP) is characterized by the sudden development of a purpuric rash, arthritis, abdominal pain, and renal involvement (Chapter 509). The characteristic rash, consisting of petechiae and often palpable purpura, usually involves the lower extremities and buttocks. Results of coagulation studies are normal. The pathologic lesions in the skin, intestines, and synovium are leukocytoclastic angiitis, inflammatory damage to the endothelium of the capillary and postcapillary venules mediated by WBCs and macrophages. The trigger for HSP is unknown. In the kidney, the lesion is focal glomerulonephritis with deposition of immunoglobulin A. Results of coagulation studies as well as platelet count are normal in HSP.
Ehlers-Danlos Syndrome
Ehlers-Danlos syndrome is a common disorder of collagen structure that causes easy bruising and poor wound healing (Chapter 651). Suggestive findings on physical examination include soft, velvety skin that is hyperelastic; lax joints that are easily subluxed; and unusual scarring. More than 10 variants of Ehlers-Danlos syndrome have been described. The most serious forms have been associated with sudden rupture of visceral organs. Results of coagulation screening tests are usually normal, although bleeding time may be mildly prolonged. Results of platelet aggregation studies are either normal or mildly abnormal, with deficient aggregation to collagen.
Blanchette V, Bolton-Maggs P. Childhood immune thrombocytopenic purpura: diagnosis and management. Pediatr Clin North Am. 2008;55:393-420.
Bolton-Maggs PH, Chalmers EA, Collins PW, et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol. 2006;135:603-633.
de Paepe A, Malfait F. Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br J Haematol. 2004;127:491-500.
Lambert MP, Poncz M. Inherited platelet disorders. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1463.
Nurden AT, Viallard JF, Nurden P. New-generation drugs that stimulate platelet production in chronic immune thrombocytopenic purpura. Lancet. 2009;373:1562-1568.
Wilson DW. Acquired platelet defects. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1553-1590.



