24 Pituitary Tumors
Epidemiology
Pituitary adenomas are benign neoplasms and comprise the majority of tumors arising in the sella turcica. With approximately 1 in 10,000 people diagnosed annually, these neoplasms are common in the general population.1 According to updated Central Brain Tumor Registry of the United States (CBTRUS) data, pituitary adenomas account for approximately 9% to 11% of all primary intracranial tumor diagnoses.2,3 However, the epidemiology of pituitary adenomas is complicated by the high frequency of small, asymptomatic tumors. Thus, tumor registry data likely underestimate the true prevalence of pituitary tumors. Better estimates of the true prevalence of pituitary adenomas are derived from studies of autopsy specimens or incidental findings on imaging scans. In a recent meta-analysis, the prevalence of pituitary adenomas was determined to be 16.7%, (14.4% from autopsy studies and 22.5% from radiography studies).4 In addition, a recent cross-sectional case-finding survey conducted in a province of Belgium concluded that the prevalence of pituitary adenomas might be even 3 to 5 times higher than previous estimates.5
Anatomy
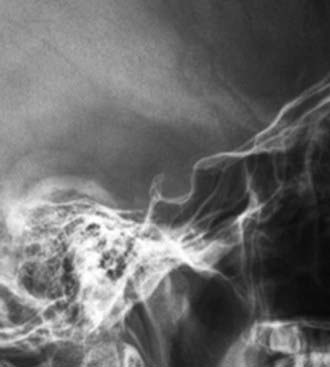
The pituitary gland is situated within the hypophyseal fossa, a fibro-osseous compartment near the center of the cranial base. This fossa is demarcated laterally and superiorly by reflections of dura and elsewhere by the sella turcica (Fig. 24-1), a depression in the body of the sphenoid bone. The diaphragma sellae is an extension of the dura that separates the pituitary from the neural structures located superiorly including the optic chiasm. A central perforation of the diaphragma sellae allows for passage of the infundibulum. The folds of dura mater that form the lateral walls of the hypophyseal fossa contain the cavernous sinuses, which consist of a series of compartmentalized venous channels separated by fibrous trabeculae. The oculomotor nerve, trochlear nerve, and first two divisions of the trigeminal nerve are embedded in the lateral wall of the cavernous sinus, lying between the endothelial lining and the dura mater, whereas the abducens nerve is contained within the sinus itself. The cavernous sinus also envelops a portion of the internal carotid artery and the sympathetic nerve plexus encircling it.
Clinical Presentation
Secretory pituitary tumors present with clinical signs and symptoms attributed to disorders associated with the relevant hypersecreted hormone (Table 24-1). The most common hypersecretory tumor type is prolactinoma. Prolactinomas cause amenorrhea and galactorrhea in women and impotence and infertility in men. The second most common functioning-pituitary tumor is a GH-secreting adenoma which results in acromegaly in adults and in gigantism in children. Cushing’s disease results from the excessive secretion of cortisol as a result of an ACTH-secreting tumor. In the setting of an ACTH-secreting tumor, surgical bilateral adrenalectomy may lead to Nelson’s syndrome, a disorder characterized by aggressive growth of the primary corticotroph adenoma. TSH-secreting adenomas, characterized by thyrotropin-induced hyperthyroidism, are rare. Nonfunctioning pituitary adenomas, arising in the majority of cases from the gonadotropin-producing cells, are not associated with a classical hypersecretory syndrome.
Table 24-1 Clinical Syndromes Associated With Hormonally Functional Pituitary Adenomas
| Hormone Produced | Clinical Syndrome |
|---|---|
| Prolactin (prl) |
The modified Hardy classification is sometimes used to describe the tumor size, local growth pattern and extension.6,7 Table 24-2 summarizes the grading of sellar floor destruction and staging of suprasellar or parasellar extension of pituitary adenomas on the basis of radiographic and operative findings.
Table 24-2 Grading of Pituitary Adenomas
| Grading of Sellar Floor Destruction | ||
| Intact sellar floor | I | Sella normal or focally expanded, tumor <10 mm |
| II | Sella enlarged, tumor ≥10 mm | |
| Sellar floor not intact | III | Localized perforation of the sellar floor |
| IV | Diffuse destruction of the sellar floor | |
| V | Spread via cerebrospinal fluid or blood | |
| Staging of Degree of Suprasellar/Parasellar Extension | ||
| 0 | Confined within sella | |
| Suprasellar extension | A | Occupies suprasellar cistern |
| B | Obliteration of the recesses of the third ventricle | |
| C | Gross displacement of the third ventricle | |
| Parasellar extension | D | Intradural extension into anterior, middle, or posterior cranial fossa |
| E | Extension into or beneath cavernous sinus | |
Types of Pituitary Adenomas
Secretory Adenomas
Prolactinoma
Prolactinomas are the most common type of pituitary adenoma, and account for approximately 40% to 45% of all pituitary tumors.8 In young adults, prolactinomas are more common in women, though this gender discrepancy is less apparent in middle-aged subjects. This gender discrepancy may reflect earlier detection in women, as hyperprolactinemia may cause oligo/amenorrhea, galactorrhea, infertility, and androgenization with hirsutism and acne. By contrast, diagnosis is often delayed in men, as the primary symptoms include reduced sexual function and libido.9 The finding of a higher prevalence of macroprolactinomas (>1 cm) and accompanying local mass effects in men may reflect this delay in diagnosis, though a gender effect on prolactinoma growth may be present as prolactinomas in men have higher proliferative indices (e.g., Ki-67) than in women.10,11 In all subjects, indications for therapy include the presence of hypogonadism, bothersome galactorrhea, infertility, headache, and local mass effects including visual field loss. The primary mode of therapy is medical with use of dopamine agonists, which rapidly reduce prolactin secretion and pituitary adenomas size in more than 90% of subjects. Administration of the dopamine agonist bromocriptine, either daily or in split daily doses, results in normalization of serum prolactin or return of ovulatory function in 80% to 90% of subjects.12 Another dopamine agonist, cabergoline, is an ergot-derived dopamine agonist that is administered orally once or twice a week and, in macroprolactinomas, can normalize serum prolactin in 75% and reduce tumor size by about 33% in two thirds of cases.13 Cabergoline may be more efficacious and better tolerated than bromocriptine.14 A potential concern with cabergoline is the finding that use of high doses of cabergoline for Parkinson’s disease may be associated with an increased risk of valvular heart disease,15,16 although the relevance of this finding to prolactinoma management is unclear. Surgery and radiation therapy are indicated in situations where medical therapy fails or the medication is poorly tolerated, or there is a significant cystic component (which does not respond fully to dopamine agonist therapy).17 Because administration of a dopamine agonist at the time of radiosurgery may limit long-term efficacy, it has been suggested that dopamine agonists should be withheld at the time of radiation treatment.18
Acromegaly
Acromegaly is a chronic, debilitating disease characterized by hypersecretion of GH and elevated levels of insulin-like growth factor-1 (IGF-1). This is an uncommon disorder, with prevalence for example in the Newcastle area of Great Britain of approximately 53 individuals per million and an incidence of 3 to 4 new cases per million over 11 years,19 although a recent study from Belgium suggests that the prevalence may be at least five times higher.5 More than 95% of cases are due to a pituitary, somatotroph adenoma, but, in rare cases, acromegaly may be due to neoplasms ectopically producing either GH or GH-releasing hormone.20 Because of the slowly progressive, insidious nature of acromegaly, the diagnosis may be delayed for up to 10 years.21,22 This delay in diagnosis may exaggerate the complications due to the tumor and GH hypersecretion, and is consistent with the finding that the majority of tumors are macroadenomas at detection. Diagnosis and disease control are imperative, as acromegaly is associated with enhanced, premature mortality, and this risk may be negated with biochemical control, including normalization of serum IGF-1 and attainment of safe GH levels.23,24 Acromegaly is also associated with comorbidities including hypertrophic cardiomyopathy, sleep apnea syndrome, arthropathy, colon polyps, carpal tunnel syndrome, along with headaches: biochemical control can significantly improve most of these outcomes as well. Transsphenoidal surgery is the treatment of choice for most patients because it leads to a rapid fall in serum GH levels, is necessary for tumor debulking if there are local mass effects, and, in contrast to medical therapy, may lead to biochemical cure (vs long-term medical “control”).
Approximately 70% to 80% of patients with microadenomas and <50% of patients with macroadenomas attain biochemical normalization after surgery.25,26 Medical therapy is largely utilized as adjuvant therapy for patients who have failed surgery. Somatostatin analogs (octreotide and lanreotide) are available in monthly depot preparations, and control GH and normalize serum IGF-1 levels in approximately 50% to 70% of cases.27 Dopamine agonists, including cabergoline and bromocriptine (both oral agents), are in general less effective than somatostatin analogs.28 The GH receptor antagonist, pegvisomant, is highly effective in reducing serum IGF-1 levels, and is often utilized as a secondary agent for subjects with incomplete or lack of response to somatostatin analogs.29 Radiation therapy is considered as an adjuvant option for patients who have failed surgery and/or are unresponsive to or poorly tolerant of medical therapy. One study has suggested that concomitant administration of a somatostatin analog at the time of radiotherapy may be radioprotective,30 though this finding was not reproduced in another study involving radiosurgery for acromegaly.31 Nevertheless, it is common practice to withhold somatostatin analogs at the time or radiation treatment, if possible.
Cushing’s Disease
Cushing’s disease results from overproduction of glucocorticoids because of excessive ACTH secretion by a corticotroph cell tumor of the pituitary gland. Cushing’s disease is uncommon, with an incidence between 0.7 and 2.4 cases per million per year.32 Consequences of Cushing’s disease include obesity, diabetes mellitus, hypertension, muscle wasting, osteoporosis, depression, coagulopathy, and cognitive deficits, and the 5-year cardiovascular mortality for untreated disease is 50%.32 The primary therapy is endonasal, transsphenoidal resection, and remission rates in patients with a microadenoma undergoing selective adenomectomy by an expert pituitary surgeon are in the range of 65% to 90%.33,34 The recurrence rate in these patients may approach 20% at 10 years,33,35 and may reflect dura mater invasion. In patients with persistent disease despite surgery, options include radiation therapy, medical adrenalectomy, and surgical adrenalectomy. Radiation therapy is reserved in an adjuvant role, in subjects with persistent, recurrent Cushing’s disease. Because of the lack of available medical therapeutics that target the pituitary adenoma, medical therapy, including use of ketoconazole, metyrapone, aminoglutethamide, and mitotane, is directed at reducing adrenal gland production of cortisol, and is largely utilized as a temporizing agent while awaiting effects of radiotherapy or plans for further surgery. Bilateral surgical adrenalectomy is a definitive treatment that provides immediate control of hypercortisolism, though the resultant adrenal insufficiency will require lifelong glucocorticoid and mineralocorticoid replacement therapy. A potential complication of bilateral adrenalectomy is Nelson’s syndrome, characterized by elevated serum ACTH levels, hyperpigmentation, and progressively enlarging, often invasive, pituitary corticotroph tumors.36,37 The ACTH-producing adenomas in Nelson’s syndrome are aggressive, and surgical resection may be difficult. Directed radiotherapy would be indicated in this situation.
Nonfunctioning Pituitary Adenomas
Because patients with nonfunctioning pituitary tumors show no evidence of hypersecretory syndromes such as Cushing’s disease or acromegaly, these tumors are often detected incidentally, or in the workup of visual field loss, headache, modest hyperprolactinemia (from compression of the hypophyseal stalk), or hypopituitarism. Though these adenomas are called nonfunctioning because of the lack of an associated hypersecretory syndrome, these tumors are comprised of different subtypes. The detection of gonadotropin hormone and steroidogenic factor-1 production in a subset of tumors allows the classification of such tumors as gonadotrophic in origin.38,39 Null cell and oncocytomas are terms relating to tumors that lack glycoprotein hormone production, though these terms are largely historical. Clinically nonfunctioning adenomas with invasive growth, elevated mitotic index, Ki-67 labeling index >3% and extensive nuclear reactivity for p53 are considered atypical, although the impact of this diagnosis on management strategies is unclear.38 The primary mode of therapy of nonfunctioning adenomas is surgery. However, because these tumors are often detected incidentally and there is a delay in diagnosis, these adenomas are often macroadenomas with extrasellar extension at diagnosis, and surgery is utilized for decompression of adjacent structures and is usually subtotal in extent.40 Medical therapies, including somatostatin analogs, are of limited use for these tumors.41 Radiation therapy is utilized in an adjuvant role for residual tumors following surgery, particularly in adenomas that involve the cavernous sinus.
Surgery
The goals of surgery for pituitary tumors are to (1) remove as much abnormal hormonally active tissue as possible; (2) eliminate tumor mass effect on the optic pathways; (3) preserve normal pituitary function; and (4) minimize the potential for recurrence. The most common surgical approach to these tumors is a transsphenoidal procedure to debulk the lesion and decompress parasellar and suprasellar structures (Fig. 24-2). Historical approaches to transsphenoidal surgery including sublabial and transnasal, transseptal surgery, although still useful, have been progressively supplanted by the techniques of direct transnasal transsphenoidal microsurgery using either a microscope or endoscope.42 Transsphenoidal surgery yields low morbidity and mortality rates and leads to improvement in visual symptoms in 87% to 90% of cases. For suprasellar tumors that are difficult to resect transsphenoidally, a variety of transcranial approaches (pterional, subfrontal, anterior interhemispheric, and transcallosal) allow adequate visualization and decompression of the optic nerves and chiasm. Surgical decompression remains the treatment of choice for symptomatic pituitary tumors. A common indication for surgery of pituitary adenomas is a macroadenoma that produces progressive visual loss from mass effect. In some cases, hemorrhage or necrosis into an existing pituitary tumor can cause precipitous visual loss associated with headache, cranial neuropathies, and sometimes acute adrenal insufficiency, a condition termed pituitary apoplexy. For prolactinomas, a pharmacologic approach is ordinarily attempted before surgery. Prolactinomas shrink dramatically with medical management (dopamine agonist therapy, usually bromocriptine or cabergoline); however, in rare cases some tumors (particularly cystic lesions) may be refractory to medical treatment, do not shrink, and maintain persistently high levels of prolactin. Alternatively, some patients may not tolerate effective doses of medical therapy. These tumors may require surgical removal.
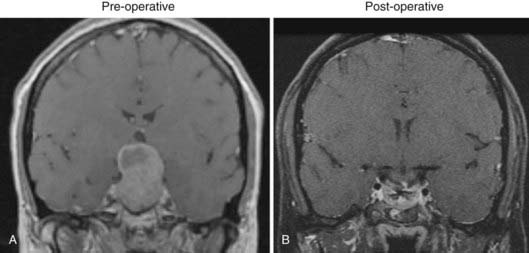
For all pituitary adenoma subtypes, postoperative anterior pituitary insufficiency has been reported to range from 1% to 27%.43 Immediate postoperative polyuria and delayed hyponatremia must be considered in the early postsurgical follow up. Following transsphenoidal surgery, transient central diabetes insipidus (DI) has been reported in 10% to 60% of cases. However, permanent DI is uncommon, and has been reported in 0.5% to 15% of surgically treated patients. Studies using endoscopic approaches report generally report lower rates of this complication. In a recent quality of life study after endoscopic pituitary surgery, transient DI occurred in 5.5% and there were no cases of permanent DI.43 In this study, worsening of preoperative visual function was present in 1% to 4% of patients and postoperative CSF leak and meningitis occurred in 0.5% to 3.9% of cases. The development of the endoscopic transsphenoidal approach to the pituitary region, which has similar indications to conventional transsphenoidal microsurgery, offers potential advantages over traditional surgical approaches because of its minimal invasiveness and panoramic visualization.
According to published series, long-term tumor control after transsphenoidal surgery alone ranges between 50% and 80%.44 Recurrences can develop over time and as many as 16% of patients who have pituitary adenoma may experience recurrent tumor growth within 10 years after surgical intervention. The incidence of recurrence has been correlated with dural invasion.45 Only 6% of patients experiencing recurrence requiring repeat surgery. Recurrent or residual tumors may require additional medical or radiation therapy.
Radiotherapy
Principles, Indications, and Clinical Results of Radiation Therapy for Pituitary Adenoma
Radiation is generally applied in the setting of residual or recurrent tumor following surgery with the goals of preventing tumor growth and normalizing elevated hormone levels. Studies evaluating radiation therapy report excellent tumor growth control rates of more than 95% at 10 years and more than 90% at 20 years (Table 24-3).46–48 Because these are considered benign tumors and patients generally have long life expectancies, care must be taken to minimize the exposure of radiation to normal tissues and to avoid radiation complications.
Studies have shown that there is a dose response for tumor control. For example, Grigsby49 et al. determined that tumor control was only 28.6% when tumors were treated with <30 Gray (Gy). Ninety-four percent tumor control was achieved with 50 to 54 Gy; 85% with 40 to 49.99 Gy; and 75% with 30 to 39.99 Gy. Investigators at University of Florida determined that late recurrences can be largely avoided and that durable tumor control, more than 90% is achievable with radiation doses of ≥45 Gy in 25 fractions.50 No significant dose response has been established for conventionally fractionated radiation doses between 45 Gy and 60 Gy. Therefore, the current accepted treatment dose schedule is 45 Gy in 1.8 Gy fractions for most pituitary adenomas, with 50.4 Gy in 1.8 Gy fractions being reserved for TSH and ACTH producing tumors.
For hormonally-active tumors, tumor control is also defined by control of hypersecretion. The normalization of hormone levels appears to increase over the length of follow-up. In Cushing’s disease, radiotherapy yields remission rates of approximately 50% to 95%, with most patients achieving normalization of plasma and urine cortisol within 2 years of treatment.51–53 The interpretation of results is more complicated for GH tumors because there is variability in the definitions of endocrine remission. For acromegaly, the time to 50% reduction of GH levels after radiotherapy has been reported to be approximately 2 years, and 75% by 5 years.54 Using GH <5 mU/L and normal IGF-1 levels as more strict criteria for endocrine cure, Biermasz et al. showed that both criteria were met in 68% of patients over the mean follow-up of 123 months.55 Normalization of prolactin levels in prolactinoma occurs in approximately 45% to 90% of cases at 5 to 10 years.49,56,57
Radiotherapy Techniques and Treatment Planning
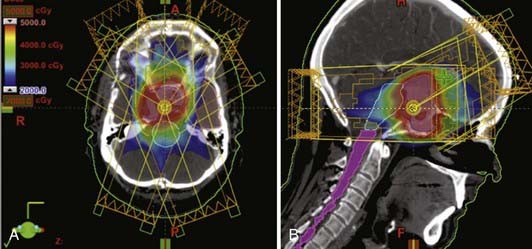
In preparation for radiotherapy, patients are typically immobilized with a rigid tilted head holder. The head and neck flexed and the chin tucked such that the head is held at roughly a 45-degree angle. This position is held in place by a thermoplastic facemask. Conventional radiotherapy treatment techniques for pituitary tumors include (1) the bicoronal wedged arc technique where bilateral 110-degree arc rotations are used with a 30-degree wedge (the wedge is reversed for the contralateral arc), and (2) the 3-field technique that uses a 3-field fixed beam arrangement of one anterior oblique and two lateral fields. Three-dimensional treatment planning that uses 4 to 6 coplanar and noncoplanar fields reduces the volume of brain tissue in the high dose region. At many institutions, conventional radiotherapy techniques using simple immobilization and limited beam angles have been largely supplanted by modern techniques including IMRT, fractionated stereotactic radiotherapy, and stereotactic radiosurgery. Improvements in immobilization with more precise thermoplastic masks, fixed or relocatable frames allow reduction of the planning margin from 1.5 to 2 cm down to 0.5 to 1.0 cm. Figure 24-3 shows an example of a 7-field coplanar and noncoplanar IMRT radiation isodose plan.
Complications of Radiotherapy
The risk of visual loss after pituitary radiotherapy is estimated to be less than 1% using modern techniques and conventional fraction sizes. Higher risks of visual loss, over 2%, have been correlated with higher fraction sizes. Although the development of secondary radiation-induced tumors is relatively infrequent, a study of 334 patients treated by postoperative radiotherapy with more than 3760 person-years of follow-up determined that the relative risk of developing a secondary tumor in these patients was 9.38 compared to the normal population.58 In this study, the cumulative risk of a second brain tumor at 10 years was 1.3% and 1.9% at 20 years. Brain necrosis is exceedingly rare at doses of 45 to 50 Gy in <2 Gy. Becker et al. reported a long-term overall risk for brain necrosis 0.2% in 1,388 patients receiving pituitary irradiation.59
Stereotactic Radiosurgery for Pituitary Adenoma
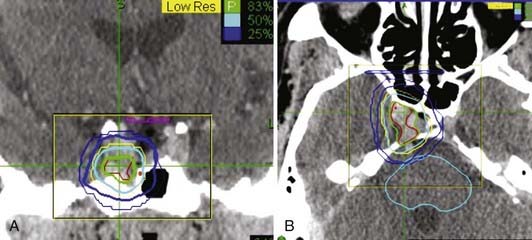
A variety of stereotactic radiosurgery techniques using gamma (γ) radiation, x-rays, or heavy charged particles have been used to treat pituitary adenomas. The goals of these techniques are similar: the highly precise delivery of conformal radiation to the target pituitary tumor while avoiding exposure to adjacent critical normal structures. In the sellar region, the optic apparatus, the native hypothalamic-pituitary structures, and the adjacent normal cerebrum are of most importance. In order to optimize tumor killing, stereotactic radiosurgery is typically delivered in up to 5 high dose fractions; although conventional fractionation has been used to deliver fractionated stereotactic radiotherapy (FSRT). Fig. 24-4 shows an example of a single fraction radiosurgery isodose plan for a small residual pituitary adenoma involving the right aspect of the sella and right cavernous sinus.
The prevailing radiobiologic factors that determine the effectiveness of radiosurgery are not well understood. However, the radiosensitivity of secretory pituitary adenomas may be influenced by the administration of antisecretory medications. Landolt et al. were the first to describe a phenomenon a radioprotective effect of antisecretory medications on growth hormone secreting pituitary adenomas and prolactinomas which resulted in lower rates of hormonal control.30,62 These authors hypothesize that radioprotection resulted from a change in the tumor’s metabolic rate induced by the medications. Pollock reported similar findings in a study of 43 patients with hormone-producing pituitary adenomas undergoing radiosurgery.63 Forty-seven percent of patients achieved normalization of hormone secretion at a median time of 14 months. The absence of antisecretory medications at the time of radiosurgery as well as maximum radiation dose >40 Gy were the only factors correlated with cure on multivariate analysis.63 Of note, these studies were all retrospective and did not involve randomization to use of somatostatin analogs at the time of radiation therapy, and it is possible that the somatostatin analog groups reflected patients with more aggressive tumors. At least two groups were unable to replicate these findings with radiosurgery.31,45 Nevertheless, it has become widely recommended that antisecretory medications be discontinued for 1 to 2 months before administering radiosurgery.62,64
Stereotactic radiosurgery series have reported growth control rates of more than 95% on average, although few series have median follow-up times beyond 4 years. The endocrinologic control rates have been highly variable, due in part to the variability of definitions of criteria of control. Biochemical control of plasma and urine cortisol levels in Cushing’s disease is reported to be 17% to 83%. Table 24-4 lists selected radiotherapy series of Cushing’s disease with at least five patients and at least 2 years of follow-up.
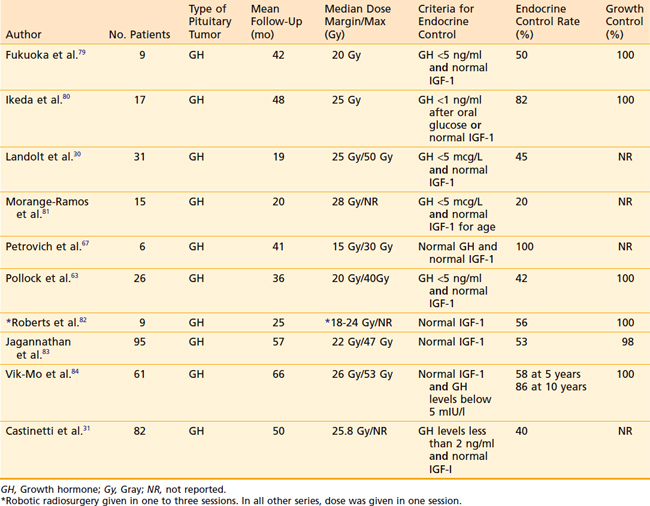
For acromegaly, it is generally appreciated that normalization of IGF-1 levels is an accurate criterion for cure, though there has been variability in criteria defining biochemical “cure,” such as with use of GH levels. Thus, it is not surprising that reported cure rates vary from 0% to 100%; studies defining the extremes of this range are quite small and may not be representative.65–67 Using serum IGF-1 criteria for biochemical response and with relatively limited follow-up, studies involving gamma knife radiosurgery have shown that only a minority of subjects achieve cure without requiring concomitant medical therapy: 17% of 82 subjects followed for a mean of 4 years in one study,31 and 17% of 53 patients followed for a mean of 5.5 years.84 Table 24-5 lists studies with at least 2-year follow-up that used the criterion of normal IGF-1 as a component of the definition of endocrine cure. The rate of endocrine cure from these sources is 20% to 100%. Radiosurgical cure rates for prolactinoma range from 0% to 84%. Although the rates of normalization of prolactin levels appears low, clinical improvements are observed in a larger number of patients.68
1 Kovacs K, Horvath E, Tumors of the pituitary gland, Washington, DC, Fascicle 21; 1986.
2 CBTRUS: Supplement Report: Primary Brain Tumors in the United States, 2004. Published by the Central Brain Tumor Registry of the United States, Hinsdale, IL.
3 Surawicz TS, McCarthy BJ, Kupelian V, et al. Descriptive epidemiology of primary brain and CNS tumors: results from the Central Brain Tumor Registry of the United States, 1990–1994. Neuro Oncol. 1999;1:14-25.
4 Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613-619.
5 Daly AF, Rixhon M, Adam C, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006;91:4769-4775.
6 Vezina JL, Hardy J, Yamashita M. [Microadenomas and hypersecreting pituitary adenomas]. Arq Neuropsiquiatr. 1975;33:119-127.
7 Wilson CB. Surgical managment of endocrine-active pituitary adenomas. In: Walker MD, editor. Oncology of the Nervous System. Boston: Martinus Nijhoff; 1983:117.
8 Mindermann T, Wilson CB. Age-related and gender-related occurrence of pituitary adenomas. Clin Endocrinol (Oxf). 1994;41:359-364.
9 Pinzone JJ, Katznelson L, Danila DC, et al. Primary medical therapy of micro- and macroprolactinomas in men. J Clin Endocrinol Metab. 2000;85:3053-3057.
10 Delgrange E, Trouillas J, Maiter D, et al. Sex-related difference in the growth of prolactinomas: a clinical and proliferation marker study. J Clin Endocrinol Metab. 1997;82:2102-2107.
11 Katznelson L, Klibanski A. Prolactinomas. Cancer Treat Res. 1997;89:41-55.
12 Molitch ME. Pathologic hyperprolactinemia. Endocrinol Metab Clin North Am. 1992;21:877-901.
13 Biermasz NR, van Dulken H, Roelfsema F. Direct postoperative and follow-up results of transsphenoidal surgery in 19 acromegalic patients pretreated with octreotide compared to those in untreated matched controls. J Clin Endocrinol Metab. 1999;84:3551-3555.
14 Webster J, Piscitelli G, Polli A, et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group. N Engl J Med. 1994;331:904-909.
15 Schade R, Andersohn F, Suissa S, et al. Dopamine agonists and the risk of cardiac-valve regurgitation. N Engl J Med. 2007;356:29-38.
16 Zanettini R, Antonini A, Gatto G, et al. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med. 2007;356:39-46.
17 Katznelson L, Klibanski A. Prolactin and its disorders. In: Becker KL, editor. Principles and Practice of Endocrinology and Metabolism. ed 2. Philadelphia: J.B. Lippincott; 1995:140-147.
18 Pouratian N, Sheehan J, Jagannathan J, et al. Gamma knife radiosurgery for medically and surgically refractory prolactinomas. Neurosurgery. 2006;59:255-266.
19 Alexander L, Appleton D, Hall R, et al. Epidemiology of acromegaly in the Newcastle region. Clin Endocrinol (Oxf). 1980;12:71-79.
20 Colao A, Ferone D, Marzullo P, et al. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev. 2004;25:102-152.
21 Katznelson L. Diagnosis and treatment of acromegaly. Growth Horm IGF Res. 2005.
22 Melmed S. Acromegaly. N Engl J Med. 1990;322:966-977.
23 Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89:667-674.
24 Swearingen B, Barker FG2nd, Katznelson L, et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998;83:3419-3426.
25 Fahlbusch R, Honegger J, Buchfelder M. Surgical management of acromegaly. Endocrinol Metab Clin North Am. 1992;21:669-692.
26 Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical “cure”. Eur J Endocrinol. 2005;152:379-387.
27 Freda PU, Katznelson L, van der Lely AJ, et al. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005.
28 Abs R, Verhelst J, Maiter D, et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998;83:374-378.
29 van der Lely AJ, Hutson RK, Trainer PJ, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet. 2001;358:1754-1759.
30 Landolt AM, Haller D, Lomax N, et al. Octreotide may act as a radioprotective agent in acromegaly. J Clin Endocrinol Metab. 2000;85:1287-1289.
31 Castinetti F, Taieb D, Kuhn JM, et al. Outcome of gamma knife radiosurgery in 82 patients with acromegaly: correlation with initial hypersecretion. J Clin Endocrinol Metab. 2005;90:4483-4488.
32 Lindholm J, Juul S, Jorgensen JO, et al. Incidence and late prognosis of cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86:117-123.
33 Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93:2454-2462.
34 De Tommasi C, Vance ML, Okonkwo DO, et al. Surgical management of adrenocorticotropic hormone-secreting macroadenomas: outcome and challenges in patients with Cushing’s disease or Nelson’s syndrome. J Neurosurg. 2005;103:825-830.
35 Patil CG, Prevedello DM, Lad SP, et al. Late recurrences of Cushing’s disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab. 2008;93:358-362.
36 Assie G, Bahurel H, Bertherat J, et al. The Nelson’s syndrome … revisited. Pituitary. 2004;7:209-215.
37 Assie G, Bahurel H, Coste J, et al. Corticotroph tumor progression after adrenalectomy in Cushing’s Disease: a reappraisal of Nelson’s Syndrome. J Clin Endocrinol Metab. 2007;92:172-179.
38 Al-Shraim M, Asa SL. The 2004 World Health Organization classification of pituitary tumors: what is new? Acta Neuropathol. 2006;111:1-7.
39 Jameson JL, Klibanski A, Black PM, et al. Glycoprotein hormone genes are expressed in clinically nonfunctioning pituitary adenomas. J Clin Invest. 1987;80:1472-1478.
40 Shomali ME, Katznelson L. Medical therapy for gonadotroph and thyrotroph tumors. Endocrinol Metab Clin North Am. 1999;28:223-240. viii
41 Shomali ME, Katznelson L. Medical therapy of gonadotropin-producing and nonfunctioning pituitary adenomas. Pituitary. 2002;5:89-98.
42 Chandler WF, Barkan AL. Treatment of pituitary tumors: a surgical perspective. Endocrinol Metab Clin North Am. 2008;37:51-66. viii
43 Karabatsou K, O’Kelly C, Ganna A, et al. Outcomes and quality of life assessment in patients undergoing endoscopic surgery for pituitary adenomas. Br J Neurosurg. 2008:1-6.
44 Laws ER, Sheehan JP, Sheehan JM, et al. Stereotactic radiosurgery for pituitary adenomas: a review of the literature. J Neurooncol. 2004;69:257-272.
45 Attanasio R, Epaminonda P, Motti E, et al. Gamma-knife radiosurgery in acromegaly: a 4-year follow-up study. J Clin Endocrinol Metab. 2003;88:3105-3112.
46 Gittoes NJ, Bates AS, Tse W, et al. Radiotherapy for non-function pituitary tumours. Clin Endocrinol (Oxf). 1998;48:331-337.
47 Brada M, Rajan B, Traish D, et al. The long-term efficacy of conservative surgery and radiotherapy in the control of pituitary adenomas. Clin Endocrinol (Oxf). 1993;38:571-578.
48 Breen P, Flickinger JC, Kondziolka D, et al. Radiotherapy for nonfunctional pituitary adenoma: analysis of long-term tumor control. J Neurosurg. 1998;89:933-938.
49 Grigsby PW, Simpson JR, Emami BN, et al. Prognostic factors and results of surgery and postoperative irradiation in the management of pituitary adenomas. Int J Radiat Oncol Biol Phys. 1989;16:1411-1417.
50 McCollough WM, Marcus RBJr, Rhoton ALJr, et al. Long-term follow-up of radiotherapy for pituitary adenoma: the absence of late recurrence after greater than or equal to 4500 cGy. Int J Radiat Oncol Biol Phys. 1991;21:607-614.
51 Estrada J, Boronat M, Mielgo M, et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med. 1997;336:172-177.
52 Howlett TA, Plowman PN, Wass JA, et al. Megavoltage pituitary irradiation in the management of Cushing’s disease and Nelson’s syndrome: long-term follow-up. Clin Endocrinol (Oxf). 1989;31:309-323.
53 Minniti G, Osti M, Jaffrain-Rea ML, et al. Long-term follow-up results of postoperative radiation therapy for Cushing’s disease. J Neurooncol. 2007;84:79-84.
54 Eastman RC, Gorden P, Glatstein E, et al. Radiation therapy of acromegaly. Endocrinol Metab Clin North Am. 1992;21:693-712.
55 Biermasz NR, Dulken HV, Roelfsema F. Postoperative radiotherapy in acromegaly is effective in reducing GH concentration to safe levels. Clin Endocrinol (Oxf). 2000;53:321-327.
56 Hughes MN, Llamas KJ, Yelland ME, et al. Pituitary adenomas: long-term results for radiotherapy alone and post-operative radiotherapy. Int J Radiat Oncol Biol Phys. 1993;27:1035-1043.
57 Johnston DG, Hall K, Kendall-Taylor P, et al. The long-term effects of megavoltage radiotherapy as sole or combined therapy for large prolactinomas: studies with high definition computerized tomography. Clin Endocrinol (Oxf). 1986;24:675-685.
58 Brada M, Ford D, Ashley S, et al. Risk of second brain tumour after conservative surgery and radiotherapy for pituitary adenoma. BMJ. 1992;304:1343-1346.
59 Becker G, Kocher M, Kortmann RD, et al. Radiation therapy in the multimodal treatment approach of pituitary adenoma. Strahlenther Onkol. 2002;178:173-186.
60 Flickinger JC, Nelson PB, Martinez AJ, et al. Radiotherapy of nonfunctional adenomas of the pituitary gland. Results with long-term follow-up. Cancer. 1989;63:2409-2414.
61 Tsang RW, Brierley JD, Panzarella T, et al. Radiation therapy for pituitary adenoma: treatment outcome and prognostic factors. Int J Radiat Oncol Biol Phys. 1994;30:557-565.
62 Landolt AM, Lomax N. Gamma knife radiosurgery for prolactinomas. J Neurosurg. 2000;93(Suppl 3):14-18.
63 Pollock BE, Nippoldt TB, Stafford SL, et al. Results of stereotactic radiosurgery in patients with hormone-producing pituitary adenomas: factors associated with endocrine normalization. J Neurosurg. 2002;97:525-530.
64 Pollock BE, Jacob JT, Brown PD, et al. Radiosurgery of growth hormone-producing pituitary adenomas: factors associated with biochemical remission. J Neurosurg. 2007;106:833-838.
65 Mitsumori M, Shrieve DC, Alexander E3rd, et al. Initial clinical results of LINAC-based stereotactic radiosurgery and stereotactic radiotherapy for pituitary adenomas. Int J Radiat Oncol Biol Phys. 1998;42:573-580.
66 Kim SH, Huh R, Chang JW, et al. Gamma knife radiosurgery for functioning pituitary adenomas. Stereotact Funct Neurosurg. 1999;72(Suppl 1):101-110.
67 Petrovich Z, Yu C, Giannotta SL, et al. Gamma knife radiosurgery for pituitary adenoma: early results. Neurosurgery. 2003;53:51-59.
68 Pan L, Zhang N, Wang EM, et al. Gamma knife radiosurgery as a primary treatment for prolactinomas. J Neurosurg. 2000;93(Suppl 3):10-13.
69 Castinetti F, Nagai M, Dufour H, et al. Gamma knife radiosurgery is a successful adjunctive treatment in Cushing’s disease. Eur J Endocrinol. 2007;156:91-98.
70 Choi JY, Chang JH, Chang JW, et al. Radiological and hormonal responses of functioning pituitary adenomas after gamma knife radiosurgery. Yonsei Med J. 2003;44:602-607.
71 Devin JK, Allen GS, Cmelak AJ, et al. The efficacy of linear accelerator radiosurgery in the management of patients with Cushing’s disease. Stereotact Funct Neurosurg. 2004;82:254-262.
72 Hoybye C, Grenback E, Rahn T, et al. Adrenocorticotropic hormone-producing pituitary tumors: 12- to 22-year follow-up after treatment with stereotactic radiosurgery. Neurosurgery. 2001;49:284-291.
73 Izawa M, Hayashi M, Nakaya K, et al. Gamma knife radiosurgery for pituitary adenomas. J Neurosurg. 2000;93(Suppl 3):19-22.
74 Jagannathan J, Sheehan JP, Pouratian N, et al. Gamma knife surgery for Cushing’s disease. J Neurosurg. 2007;106:980-987.
75 Kobayashi T, Kida Y, Mori Y. Gamma knife radiosurgery in the treatment of Cushing disease: long-term results. J Neurosurg. 2002;97:422-428.
76 Mokry M, Ramschak-Schwarzer S, Simbrunner J, et al. A six year experience with the postoperative radiosurgical management of pituitary adenomas. Stereotact Funct Neurosurg. 1999;72(Suppl 1):88-100.
77 Pollock BE, Young WF Jr. Stereotactic radiosurgery for patients with ACTH-producing pituitary adenomas after prior adrenalectomy. Int J Radiat Oncol Biol Phys. 2002;54:839-841.
78 Sheehan JM, Vance ML, Sheehan JP, et al. Radiosurgery for Cushing’s disease after failed transsphenoidal surgery. J Neurosurg. 2000;93:738-742.
79 Fukuoka S, Ito T, Takanashi M, et al. Gamma knife radiosurgery for growth hormone-secreting pituitary adenomas invading the cavernous sinus. Stereotact Funct Neurosurg. 2001;76:213-217.
80 Ikeda H, Jokura H, Yoshimoto T. Transsphenoidal surgery and adjuvant gamma knife treatment for growth hormone-secreting pituitary adenoma. J Neurosurg. 2001;95:285-291.
81 Morange-Ramos I, Regis J, Dufour H, et al. Gamma-knife surgery for secreting pituitary adenomas. Acta Neurochir (Wien). 1998;140:437-443.
82 Roberts BK, Ouyang DL, Lad SP, et al. Efficacy and safety of CyberKnife radiosurgery for acromegaly. Pituitary. 2007;10:19-25.
83 Jagannathan J, Sheehan JP, Pouratian N, Laws ER, Steiner L, Vance M. Gamma knife radiosurgery for acromegaly: outcomes after failed transspenoidal surgery. Neurosurgery. 2008;62(6):1262-1270.
84 Vik-Mo EO, Oksnes M, Pedersen PH, et al. Gamma knife stereotactic radiosurgery for acromegaly. Eur J Endocrinol. 2007;157(3):255-263.