[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 27 Pituitary Tumors
Etiology and Epidemiology
Etiology
The etiology of most pituitary tumors is not known and has been a source of debate. Based on two rat pituitary models, two primary modes of pituitary oncogenesis have been suggested.1 The first model, known as the hyperplasia-adenoma sequence, suggests a hormone-dependent pathway. Another model suggests de novo occurrence without the development of hyperplasia, possibly because of a genetic alteration. In this model, dysregulation of hormone or growth factor signaling leads to hyperplasia and tumor formation; the defect has been found especially in transgenic and knockout animal models, although there is strong evidence implicating overexpression of transforming growth factor–alpha (TGF-α), fibroblast growth factor (FGF), and fibroblast growth factor receptors (FGFRs) in tumor formation.2
For some patients with pituitary adenomas, a genetic predisposition has been described with four genes known to be associated with familial pituitary tumor syndromes: MEN1, CDKN1B, PRKAR1A, and AIP.3 Of patients with MEN1, which is an autosomal dominant disease characterized by tumors of the pancreatic islet cells, parathyroid glands, and pituitary gland, 40% will develop pituitary adenomas the great majority of which are prolactinomas. Pituitary tumors associated with the MEN1 syndrome have demonstrated loss of heterozygosity of chromosome 11q13 (locale of the MEN1 gene), which has been implicated in malignant progression of pituitary adenomas.4 Mutation of this gene does not appear to increase the risk for sporadic pituitary adenomas.5 In Carney’s complex, a rare inherited condition characterized by endocrine overactivity, schwannomas, abnormal skin pigmentation, and myxomas, the implicated genetic defect is loss of function of PRKAR1A, the gene for which is located on chromosome 17q23-24.6 Additional genetic alterations are found in McCune-Albright syndrome, a genetic disorder of bones, skin pigmentation, and hormones associated with premature puberty. Activating or gain-of-function GNAS1 mutations (of the guanine nucleotide-binding protein [G protein], alpha-stimulating activity polypeptide 1 gene on chromosome 20q13.1) are seen. Patients with McCune-Albright syndrome are present in the mosaic state, resulting from a postzygotic somatic mutation appearing early in the course of development that yields a monoclonal population of mutated cells within variously affected tissues. In isolated familial somatotropinomas (i.e., the occurrence of two or more cases of acromegaly in a family without a history of MEN or Carney’s complex), germline alterations in the aryl hydrocarbon receptor interacting gene (AIP gene) have been identified.7,8
Epidemiology
Although 10% of normal adults have a pituitary abnormality detectable on MRI, pituitary tumors account for only 10% to 15% of all diagnosed primary brain tumors. The frequency of pituitary adenomas varies greatly, although recent community-based studies from Belgium and the United Kingdom suggest that the prevalence is higher than has been historically noted.9,10
Based on a meta-analysis, the overall estimated prevalence of pituitary adenomas was 16.7%, with 14.4% observed in autopsy series and 22.5% in radiographic series.11 Over time, different criteria have been used for distinguishing hyperplasia from pituitary adenomas. The broader application of immunohistochemical staining for pituitary hormones and the differences in slice thicknesses for MRI may account for the wide variation in incidence that is reported in the literature.
Biologic Characteristics and Molecular Biology
Despite the screening of common oncogenes and tumor suppressor genes for pituitary adenomas, the initial transforming event for tumorigenesis has not been identified. Based on X chromosome inactivation analysis, it is apparent that most pituitary adenomas derive from a monoclonal expansion, which suggests that general principles of tumorigenesis may apply to pituitary adenoma formation.12 The MAPK (mainly Ras/extracellular signal-regulated protein kinase [ERK]) and the PI3K/Akt signaling pathways appear to have putative roles in pituitary tumorigenesis because activity of these pathways is increased with pituitary tumors. Although its role is not fully understood, a docking protein in the endothelial growth factor receptor (EGFR) pathway, EGF pathway substrate number 8 (Eps8), is overexpressed (5.9 times higher) in pituitary tumors compared with normal anterior pituitary gland tissue.13 Because pituitary tumors are mostly benign, a protective mechanism to restrict uninhibited growth of tumor cells is probably active. The concept of oncogene-induced senescence is one possible mechanism. High pituitary p21Cip1/WAF1 levels appear to promote senescence and restrict tumor growth.14
For somatotroph (growth hormone) adenomas, an activating mutation of the alpha subunit of the guanine nucleotide stimulatory protein (Gs-alpha) gene is found in about 40% of patients, which results in activation of adenylyl cyclase.15 Overexpression of the pituitary tumor transforming gene (PTTG) is also seen in most human somatotroph adenomas.16 Mutations of fibroblast growth factor–4 (FGF-4) have led to development of lactotroph adenomas in transgenic mice.17
Anatomy, Pathways of Spread, and Pathology
Anatomy and Pathways of Spread
The pituitary gland is a midline intracranial organ that measures approximately 8 mm in the anteroposterior axis, 10 mm in the transverse axis, and 6 to 8 mm in the superior-inferior axis. It occupies the cavity in the sphenoid bone known as the sella turcica. The pituitary gland is separated from the structures above it (i.e., the optic chiasm, hypothalamus, anterior cerebral arteries, and floor of the third ventricle) by the sellar diaphragm, which is formed by a circular fold of dura. Immediately anterosuperior to the diaphragm sella lies the optic chiasm. The pituitary stalk (infundibulum) crosses the sellar diaphragm and connects the hypothalamus to the pituitary gland. Anteriorly, the tuberculum sellae lies in the floor of the sella and extends laterally to form the anterior clinoid processes. The posterior aspect of the sella forms the dorsum sellae and extends laterally to form the posterior clinoid processes. Laterally, the cavernous sinuses contain cranial nerves III, IV, V1 (ophthalmic nerve), V2 (maxillary nerve), and VI and the internal carotid artery. Given the proximity of the pituitary to these various structures, parasellar tumors can affect cranial nerves III, IV, V, and VI, and suprasellar tumor extension can lead to bitemporal hemianopsia via compression of the optic chiasm. Figure 27-1 demonstrates the complex anatomy of the parasellar region.
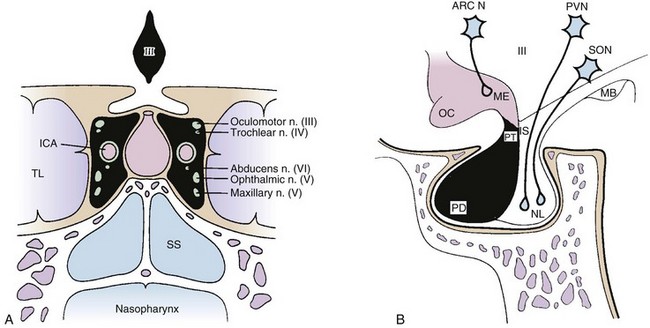
The pituitary has two distinct embryologic origins; the first gives rise to the anterior and intermediate pituitary lobes, and the second gives rise to the posterior pituitary lobe. The anterior lobe (adenohypophysis) and intermediate lobe are derived from Rathke’s pouch, which is an evagination of ectodermal tissue from the primitive oral cavity (pharyngeal pouches). The posterior lobe (neurohypophysis) and stalk are derived from the diencephalon and, therefore, have a neuroectodermal origin. The anterior pituitary lobe accounts for most of the pituitary gland and produces at least six hormones: prolactin (PRL), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), growth hormone (GH), and thyroid-stimulating hormone (TSH).18 The intermediate lobe produces melanocyte-stimulating hormone (MSH). The posterior lobe stores and releases two hormones, vasopressin (antidiuretic hormone [ADH]) and oxytocin, which are produced by the hypothalamus.
Pituitary tumors are generally classified as microadenomas or macroadenomas depending on the diameter of the tumor as described by Hardy.19 A microadenoma is a tumor less than 1 cm in diameter, whereas a macroadenoma has a diameter of 1 cm or greater. Incidental microadenomas have been observed with a frequency of 3% to 25% in large, unselected autopsy series.20 Microadenomas are more frequently seen in females, whereas macroadenomas occur with equal frequency in males and females. Macroadenomas are more common than microadenomas. For lesions less than 3 mm in diameter, the term picoadenoma has been used.21
Pathology
According to the World Health Organization (WHO) classification of pituitary tumors, these tumors are defined as neoplasms located in the sella turcica.22 This classification is based on structural similarities of the normal parenchymal cells and the immunohistochemical demonstration of hormone secretion. As a result, pituitary adenomas may express more than one or two hormones. The classification includes a new entity designating a borderline adenoma or adenoma of uncertain behavior.22 The atypical adenoma is defined as an invasive tumor with an elevated mitotic index (MIB-1-labeling index greater than 3%) and extensive nuclear immunostaining for TP53.22
Most lesions are easily identified on routine histopathologic testing, but metastatic well-differentiated neuroendocrine carcinoma can present a diagnostic challenge.23 For patients with acromegaly or Cushing’s disease, hyperplasia may be clinically indistinguishable from an adenoma. Differentiation of hyperplasia from adenoma can be made using a reticulin stain. Normal pituitary tissue is composed of small acini of pituitary cells surrounded by an intact reticulin network. In hyperplasia, the architecture of the acini is maintained, but the acini are increased in size, and the reticulin stain demonstrates an intact network. Pituitary adenomas are characterized by a complete disruption of the reticulin fiber network.
The pathologic hallmark of pituitary adenomas is the monotonous and monomorphous proliferation of neoplastic cells that replace the normal acinar pattern in the pituitary lobe. Growth hormone–producing cells are seen with greater frequency anteriorly in the lateral aspects of the pituitary. Prolactin-producing cells are distributed throughout the pituitary but have a greater density in the posterior lateral aspect of the gland. Adrenocorticotropic hormone–producing cells are present in the median wedge. Thyroid-stimulating hormone–producing cells are seen in the anterior aspect of the median wedge. Gonadotropin-producing cells are distributed throughout the anterior pituitary. Nonfunctioning adenomas typically have solid sheets, nests, and sinusoidal patterns and are interrupted by pseudopapillae and striking pseudorosettes around vascular channels. Overlap of the distribution of these functional hormonal secretory cells does exist, and they do not reside in well-demarcated zones of the pituitary. Mitoses are seen in 3.9% of noninvasive adenomas, 21.4% of invasive adenomas, and 66.7% of carcinomas.24
The usefulness of labeling-index and TP53 immunostaining is not clear, because these assessments may not correlate with tumor behavior. Invasive pituitary adenomas usually exhibit a higher Ki-67 proliferation index.25 Markers such as proliferating cell nuclear antigen (PCNA), Ki-67/MIB-1, and antiapoptotic Bcl-2 have not demonstrated consistent correlation with tumor invasiveness or recurrence.26 Galectin-3, a β-galactoside-binding protein, may play a role in pituitary tumor progression, and immunostaining for this is also available.27
The pituitary is composed of at least six distinct cell types, which are responsible for the production of at least one hormone. Advances in molecular biology have identified three major pathways (T pit; Pit-1; and SF-1, GATA-2, ER) of cytodifferentiation of adenohypophyseal cells.28,29 These pathways are determined by a complex pattern of transcription expression, which can help classify the adenomas.30 In situations where histologic and immunohistochemical profiles are atypical, electron microscopy can be helpful in achieving accurate classification.
Clinical Manifestations, Patient Evaluation, and Staging
Clinical Manifestations
Patients with pituitary tumors can have a number of different symptoms and presentations, depending on the size and mass effect of the tumor and hormonal abnormalities. Headaches can occur with a lesion of any size. When the tumor extends superiorly into the suprasellar region, visual loss can occur, including bitemporal or homonymous hemianopsia, superior or inferior field cut deficits, and central scotoma. Extension into the cavernous sinus can lead to cranial nerve dysfunction (commonly, palsies of cranial nerves III and IV). Cranial nerves V and VI may also be affected. Lateral extension into the temporal lobe may cause seizures. The clinical presentations and endocrine abnormalities can vary, depending on the age and sex of the patient. Patients with large adenomas may have compromised pituitary function, which can lead to hypogonadism, secondary hypothyroidism, and adrenal insufficiency.31 Diabetes insipidus is extremely rare and most commonly is a manifestation of an invasive and infiltrative tumor (such as a metastasis), an inflammatory condition (such as neurosarcoidosis), or a lesion that arises from or is attached to the infundibulum or hypothalamus (such as a craniopharyngioma).32 If the gland acutely infarcts or hemorrhages, pituitary apoplexy may result, which may require urgent surgical decompression.
Patient Evaluation
Laboratory Assessment
Initial tests can determine a pituitary deficiency and diagnose the secretory status of the adenoma. Screening studies should include thyroid-stimulating hormone, free thyroxine (T4), adrenocorticotropic hormone, cortisol, prolactin, somatomedin C (insulin-like growth factor–1 [IGF-1]), luteinizing hormone, follicle-stimulating hormone, alpha subunit, and, in men, testosterone. In addition to endocrine evaluation, a complete blood count, blood chemistry assessment, and urinalysis should be obtained. Because physiologic hormonal variations in the blood and urine levels can occur, the interpretation of these results should be considered based on diurnal variations, age and gender of the patient, and pregnancy and menopausal status. The conditions and timing under which the samples are obtained also influence interpretations of the results. Because the diagnosis of Cushing’s disease can be difficult, multiple tests performed over time and at different times during the day may be needed for this diagnosis to be conclusive.33
For patients with prolactinoma who also have macroadenoma, the prolactin level is usually greater than 200 ng/mL. Other common conditions that have been associated with hyperprolactinemia include end-stage renal disease, renal insufficiency, depression, primary hypothyroidism, acquired immunodeficiency syndrome, sarcoidosis, and nonalcoholic cirrhosis.34 On the other hand, pituitary stalk compression from tumor may also cause elevated prolactin levels in the range of 150 ng/mL or less. Because antidepressants, protease inhibitors, verapamil, and phenothiazines may elevate prolactin levels, carefully review the medications that the patient is taking. A pregnancy test is mandatory for women with amenorrhea or hyperprolactinemia. Elevated serum prolactin levels greater than 300 ng/mL are usually diagnostic of pituitary adenoma, and levels higher than 100 ng/mL in nonpregnant patients often are associated with pituitary adenoma.
For patients with acromegaly, most cases result from excess secretion of growth hormone by a pituitary tumor.35 The definitive test is measurement of growth hormone response to 75 or 100 g of oral glucose (oral glucose tolerance test). This glucose load should normally cause marked suppression of the growth hormone release to a level below 2 ng/mL. To ensure accuracy of the test, these measurements of serum glucose and growth hormone must be taken every 30 minutes for 2 hours. The IGF-1 value is also elevated after adjusting for sex and age variation. IGF-1 levels provide the best intermittent method for monitoring response to treatment, although dynamic testing of growth hormone kinetics with the oral glucose tolerance test (OGTT) is also frequently used.
Imaging
MRI of the brain with gadolinium is the imaging test of choice, given its superior resolution compared with CT scanning (Fig. 27-2). Thin slices (at 2- to 3-mm intervals) obtained before and after gadolinium contrast administration with images in the coronal, axial, and sagittal planes provide detailed information for the initial diagnosis and allow detection of small lesions. The posterior lobe of the pituitary has a high signal intensity on T1-weighted images (posterior pituitary bright spot) that distinguishes it from the anterior lobe, which has a signal intensity similar to that of white matter. Dynamic coronal imaging techniques after contrast administration enhance normal pituitary tissue earlier and more intensely and help delineate adenoma tissue, which tends to enhance later. For patients who have undergone previous surgery, fat suppression techniques can help differentiate surgical fat grafts from tumor tissue. For patients undergoing stereotactic radiosurgery (SRS), thin-slice (1-mm) imaging with contrast medium is obtained to define the tumor and optic apparatus. For hypersecretory adenomas, positron emission tomography (PET) imaging with co-registration may be valuable.36
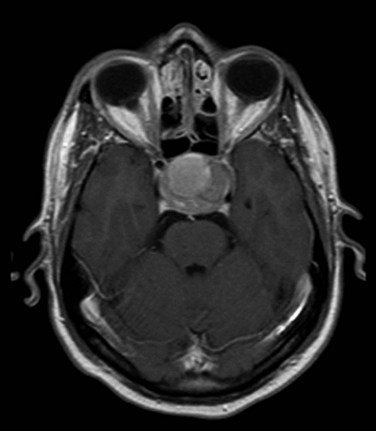
For patients with suspected Cushing’s disease, thin-slice images of 1-mm thickness have greater sensitivity; even so, the tumor may not be detected in about 50% of patients.37 Spoiled gradient recalled acquisition sequences may have superior sensitivity (80%) compared with conventional spin echo images following contrast enhancement.38 The degree of contrast enhancement does not differentiate one sellar mass from another. A clear distinction between an intrasellar mass and normal pituitary tissue is not consistent with a pituitary adenoma.
Staging
Classification of detectable or symptomatic pituitary tumors can be based on endocrine function, clinical presentation, and anatomic extent of disease. These tumors can also be broadly classified as functional or nonfunctional based on their secretory activity. As adenomas enlarge, they can extend into suprasellar, parasellar, or infrasellar structures. To aid with surgical and imaging assessment, Hardy and Verzina39 developed a classification system. Wilson’s40 modification of this classification system incorporates imaging and intraoperative findings of sellar destruction (grade) and extrasellar extension (stage). Others have defined giant adenoma as a lesion with extension beyond the sella and suprasellar space or size greater than 4 cm.41,42 No universally accepted staging system exists for pituitary tumors.
Primary Therapy
Surgery
Advances in transsphenoidal approaches with microsurgical techniques have made this procedure the initial treatment of choice for patients with nonfunctioning pituitary adenomas, acromegaly, and Cushing’s disease. Surgery provides immediate decompression for patients with progressive visual loss or pituitary hemorrhage. For patients with prolactinoma who do not tolerate or respond to medical therapy, surgery can be considered. Surgery has also been useful in reducing the tumor bulk for combined management with radiation therapy or medical therapy. For patients undergoing SRS, surgery may play an important role because a distance of 2 to 5 mm is needed between the tumor and the optic apparatus to deliver a sufficient dose to the tumor and to minimize the risk for optic neuropathy. Reoperation results in lower rates of success compared with the initial surgery.43
The transsphenoidal approach has become less invasive with development of a newer approach, endoscopic, minimally invasive pituitary surgery (MIPS). This technique can result in shorter hospitalization times.44,45 With MIPS, nasal or intraoral incisions are not needed and nasal speculums and nasal packing need not be used. There appears to be a decrease in complication rates compared with traditional sublabial transseptal approaches. MIPS is performed jointly with otorhinolaryngologists. It may also be used to complement microsurgical approaches.
Based on a questionnaire study of 958 neurosurgeons reporting on their own experience with transsphenoidal surgery, the risk for complications was inversely proportional to the surgeon’s level of experience.46 Reported complication rates among experienced surgeons include perioperative mortality (<1%), central diabetes insipidus (2%), cerebrospinal rhinorrhea (2%), and meningitis (2%). Transient central diabetes insipidus is very common and reported to occur in 22% of patients at a very active pituitary surgery center.47 These complications are more common in patients with macroadenomas. Less experienced surgeons are more likely to cause the above complications.45,48
Medical Therapy
Medical therapy can be used in several situations for patients with growth hormone–secreting tumors. Somatostatin analogs such as octreotide or lanreotide can be used to lower elevated growth hormone levels before surgery, for patients with persistent growth hormone and IGF-1 elevation after surgery, and for patients who continue to receive medical therapy until radiation therapy or radiosurgery has taken full effect. Pegvisomant, a growth hormone receptor antagonist, appears to be the most effective medical therapy for IGF-1 normalization.49 The cost of this therapy is, however, substantial (>$50,000 per year).
Medical treatment of hormonal deficiencies resulting from the pituitary tumor is best managed by an endocrinologist. Hypopituitarism can have variable clinical manifestations, and management should be individualized to minimize the impact of this condition. Depending on the deficiency, glucocorticoids, gonadal steroids, and thyroid hormones may need to be initiated. Commonly used medicines include hydrocortisone and cortisone acetate for glucocorticoid replacement and L-thyroxine for hypothyroidism. Gonadal steroids used include estrogen and progestin for women and testosterone for men. Careful optimization of medication supplementation, especially to avoid supraphysiologic doses of steroids, appears to be important in minimizing the effect of hypopituitarism.50
Radiation Therapy and Stereotactic Radiosurgery
The lack of prospective, randomized data comparing different radiation delivery methods has resulted in strong proponents for fractionated versus radiosurgical approaches.51,52,53 Most of the literature is based on single institutional results .
.
Advances in radiation therapy have led to modern radiation techniques that allow more focused delivery of radiation to the tumor and decreased dose to the normal brain. Sophisticated treatment planning systems, incorporation of CT and MRI studies, stereotactic guidance, radiation approaches such as intensity-modulated radiation therapy and image-guided radiation therapy, micro-multileaf collimators, and relocatable frames or masks have improved precision.53,54,55 Results with radiation therapy, including specifics regarding dosing and margins, will be discussed in greater detail later in the chapter.
Stereotactic radiosurgery (SRS) delivers a highly focused, single, large fraction of ionizing radiation to a small intracranial target volume (Fig. 27-3). Frame-based systems (e.g., proton beam, helium ion, Gamma Knife, and linear accelerator SRS techniques) and frameless systems (e.g., CyberKnife and linear accelerator-based units) have been used. Given the sharper dose gradient of SRS versus fractionated approaches, the potential for radiation injury to normal brain and vessels and secondary tumor formation may be less with SRS.
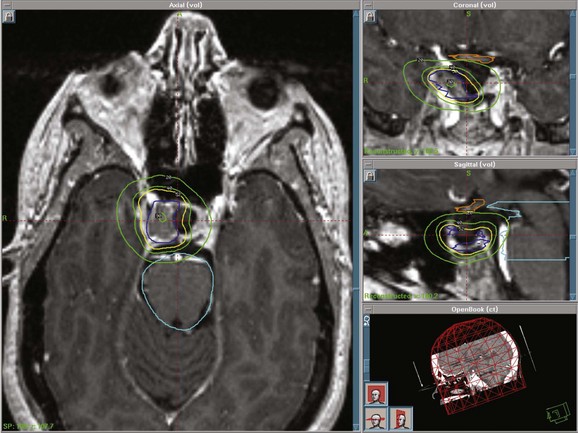
The optic apparatus (optic chiasm and nerves) is the major dose-limiting critical structure in SRS and fractionated treatment planning. A single-fraction dose to the optic chiasm should be limited to 8 to 9 Gy to minimize damage to the visual pathways. A retrospective review to evaluate the risk of clinically significant radiation optic neuropathy (RON) for patients having undergone SRS for tumors adjacent to the optic apparatus showed that 1.9% developed RON with a median maximum radiation dose to the optic nerve of 10 Gy.56 RON developed at a median of 48 months after radiosurgery. The authors suggested a clinically significant risk of RON of 1.1% for those who receive 12 Gy or less to a small segment of the optic apparatus. Accurate localization of the target volume and the optic chiasm using MRI is essential. For patients undergoing fractionated treatment, the dose to the chiasm should not exceed 54 Gy and the dose per fraction should be 1.8 Gy/day. It is important to minimize the hot spot around the optic nerves and chiasm.
Unlike the optic nerves, the cranial nerves in the cavernous sinus are more resistant to radiation effects.57 This may be a result of the increased sensitivity of sensory nerves such as the optic and acoustic nerves compared with the nonsensory nerves of the parasellar region. After repeat radiosurgery, nonsensory cranial neuropathy, however, has been reported.58
Treatment Strategies For Specific Tumors
Nonfunctioning Adenoma
Nonfunctioning adenomas are the most frequent type of macroadenoma and typically present with hypopituitarism and visual complaints such as decrease in visual acuity and visual field defects.59,60 They account for approximately 30% of all pituitary adenomas and often extend beyond the sella turcica. Other presenting symptoms may include headaches from local mass effect or obstruction of the third ventricle causing hydrocephalus, cranial nerve deficits from cavernous sinus involvement, and hypopituitarism resulting from compression of the pituitary gland. Evaluation should include visual field testing, brain MRI scanning, and an endocrine workup.
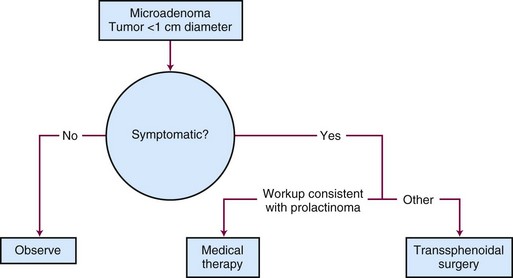
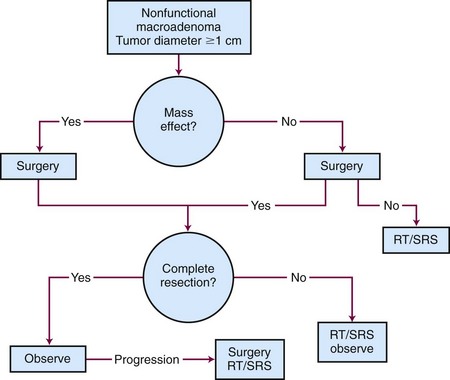
Generalized, schematic treatment algorithms for a nonfunctioning pituitary microadenoma and macroadenoma are outlined in Figures 27-4 and 27-5, respectively.
Surgery
Management of a nonfunctioning adenoma is influenced by the size of the tumor, symptoms and physical examination findings, and associated endocrine disorders. For a microadenoma or asymptomatic nonfunctioning adenoma, observation is generally recommended. For a symptomatic tumor or one with mass effect causing symptoms such as visual impairment, surgery is the best option because it provides pathologic tissue confirmation, immediate debulking of the tumor, and amelioration of the mass effect in 90% to 95% of patients. The results with surgery are quite variable, with recurrence rates ranging from 10% to 18% at 5 to 6 years and 44% at 10 years.61,62
Radiation Therapy Options
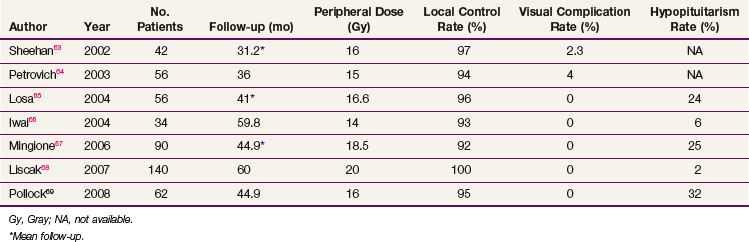
If the tumor cannot be completely removed, surgical decompression followed by radiation therapy or SRS can provide excellent results. For patients with recurrent tumors or those who are not good surgical candidates, fractionated radiation therapy or SRS can be considered, depending on the proximity of the tumor to the optic apparatus. Table 27-1 summarizes the most recent studies of SRS using doses ranging from 14 to 18.5 Gy.63–68,69 With these doses, the local control rates are excellent (92% to 100%). The tumor volume in over half of these patients decreases on follow-up MRIs. Hypopituitarism occurs in up to 32% of patients. More recent series have not reported visual complications, especially when the maximum dose to the optic apparatus is kept below 8 to 9 Gy.
Numerous retrospective studies have demonstrated excellent local control rates with conventional fractionated radiation therapy (45 Gy in 25 fractions). The largest series from Brada and colleagues70 of 252 patients reported local control rates of 97% and 92% at 10 and 20 years, respectively. Another large study from Sasaki and associates71 demonstrated a 98% local control rate at 10 years. No trials have compared fractionated radiation therapy with SRS.
Prolactinoma
Prolactinomas are the most common functioning pituitary tumors, with the vast majority occurring as microadenomas in women. Symptoms may include amenorrhea, galactorrhea, and infertility in women and decrease in libido, infertility, and visual disturbances in men. Diagnosis is typically made from sustained elevation of serum prolactin levels above the normal range. MRI of the brain can help confirm the diagnosis, although a negative MRI can be seen in women with microadenomas.72
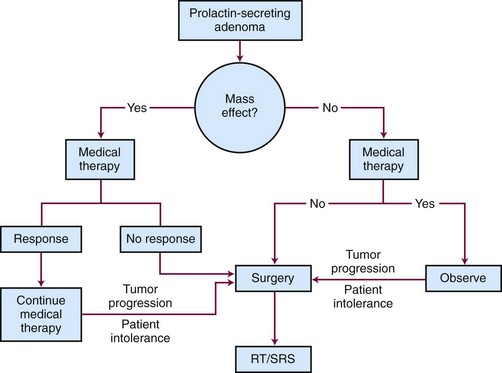
Goals of treatment include restoration of sexual and reproductive function, normalization of prolactin levels, control of galactorrhea, and improvement of neurologic symptoms. It is important that other causes of increased prolactin levels such as effects of medicines, effects of pregnancy, and renal insufficiency are considered in the differential diagnosis. Treatment options include observation, medical therapy, surgery, and irradiation.34 A generalized schematic treatment algorithm for a prolactinoma is outlined in Figure 27-6.
Surgery
Surgery is standard second-line therapy and is reserved for patients who are noncompliant with medical therapy, cannot tolerate medical therapy, have rapid progressive vision loss, or are refractory to medical therapy. Based on a review of 50 surgical series of over 4000 patients, the curative surgical resection rates for microprolactinomas and macroprolactinomas was 74.7% and 33.9%, respectively, using a normalization of prolactin levels before 12-week follow-up as the definition of remission.73 Biochemical recurrence, however, occurs in 20% of patients within the first year. The best predictor for persistent cure is a prolactin level less than or equal to 5 ng/mL on postoperative day 1. The long-term surgical cure rates for microprolactinomas and macroprolactinomas are 50% to 60% and 25%, respectively, where a cure is considered normal prolactin levels. Surgery is also the treatment of choice for women with a large adenoma who want to become pregnant.
Medical Therapy
The initial treatment of choice for patients with prolactinoma is medical therapy with a dopamine agonist such as bromocriptine or cabergoline because these drugs decrease secretion and tumor size in most patients. Medical therapy can rapidly normalize prolactin levels and reduce tumor size in 80% to 90% of patients.73 Bromocriptine, which is taken two or three times per day with meals, is associated with a greater number of side effects (e.g., nausea, vomiting, fatigue, and orthostatic hypotension) compared with cabergoline, which is taken once or twice a week. In a double-blinded study of 459 female patients comparing bromocriptine with cabergoline over 8 weeks, normal prolactin levels were achieved in 59% and 83% of patients, respectively.74 Cabergoline was better tolerated, with 3% discontinuing its use, versus 12% who stopped taking bromocriptine. Because cabergoline has been associated with an increased risk for cardiac valve thickening in patients with Parkinson’s disease, who take much higher doses compared with prolactinoma patients, this potential risk needs to be reviewed and carefully monitored.75 For some patients, medical therapy may be gradually withdrawn every 2 to 3 years to assess remission. This strategy is most effective for patients with low prolactin levels and small tumors. Approximately 8% of patients with prolactinoma are intolerant to or unresponsive to medical treatment.
Radiation Therapy Options
The results with conventional radiotherapy from the late 1970s to the early 1990s have been modest. One of the earliest studies from Sheline and colleagues76 of 28 patients demonstrated a prolactin normalization rate of 29%. Littley and associates77 found a decrease in serum prolactin levels in all 58 patients treated with EBRT and a 50% probability of prolactin level reduction to 500 mU/L in 10 years. Patients more likely to achieve normalization of serum prolactin levels after EBRT had smaller tumors, pretreatment prolactin levels higher than 6000 mU/L, and tumors with positive immunostaining for prolactin. Tsagarakis78 reported normal serum prolactin levels at a mean of 8.5 years after EBRT in 18 of 36 patients. Pituitary irradiation can lead to hypothalamic dysfunction and impairment of dopamine secretion, which causes hyperprolactinemia. This may explain the mildly elevated prolactin levels (usually, <50 mU/L) in some patients after radiotherapy of pituitary adenomas.
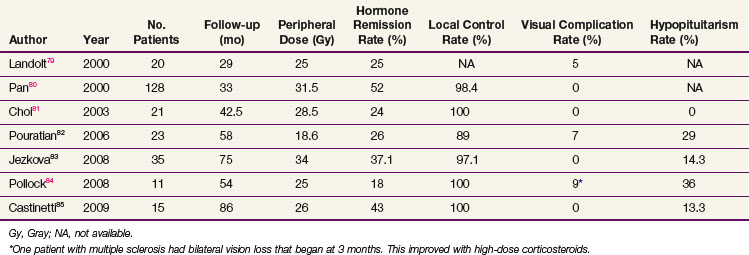
SRS has also been used after failure of medical therapy and/or surgical treatment. Table 27-2 summarizes recent publications on the results of SRS for prolactinomas with normalization rates for prolactin ranging from 18% to 52%.* The best result with SRS was from Pan, who reported a 52% endocrine cure rate in 128 patients in whom SRS was used as first-line treatment.80 The median prescription dose was 31.2 Gy, which is higher than in most other reported series.
Because it may take some time for normalization of prolactin levels to occur, the use of dopamine agonists may be required. Landolt79 reported worse outcomes for patients receiving dopamine agonists at the time of SRS. Pouratian82 also reported worse outcomes in patients receiving antisecretory medication at the time of SRS. Antisecretory medications may alter the cell cycle, which makes the tumor cells less radiosensitive. As a result, a 2-month break between medical therapy and SRS has been suggested.79
Acromegaly
Acromegaly is a rare condition of growth hormone hypersecretion that leads to metabolic and functional disturbances of multiple organs (i.e., cardiovascular, musculoskeletal, and respiratory dysfunction) and possibly an increased risk for colorectal cancer.35 In addition, characteristic bony enlargement occurs in the frontal bones, hands, feet, spine, nose, and mandible. As a result, early diagnosis and treatment are essential to minimize the impact on life expectancy and morbidity. Unfortunately, the gradual onset of symptoms and changes as a result of excessive growth hormone secretion causes delayed diagnosis.
Although surgery is the initial treatment of choice, a multidisciplinary approach is often needed given the complexity of this disease.86 The primary goals of therapy are to normalize IGF-1 levels to within the reference range for the patient’s age and gender, to lower the serum growth hormone levels to less than 1 ng/mL in response to an oral glucose challenge, to preserve pituitary function, and to avoid tumor growth. Normalization of IGF-1 levels is a better criterion for cure than growth hormone levels because some acromegalic patients can have normal growth hormone levels.87,88 Untreated or incompletely treated acromegaly is associated with significant morbidity and excess mortality, which may be as high as three times that of the general population.50,86 The treatment options for acromegaly include surgery, irradiation, or medical therapy or a combination of treatment approaches. The treatment algorithm for acromegaly is outlined in Figure 27-7.
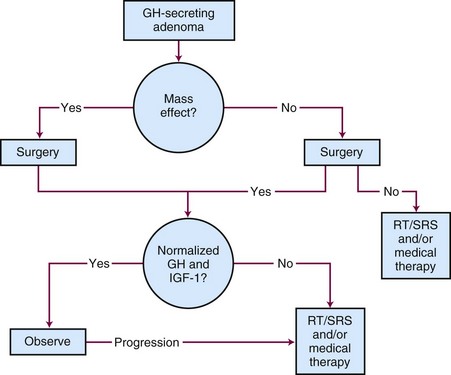
Surgery
The initial treatment of choice for acromegaly is surgery.86,89 Goals of surgery include alleviation of mass effect from suprasellar extension and complete removal of the growth hormone-secreting adenoma. Because the criteria used for biochemical cure have changed with time and the duration of follow-up varies, surgical series are not easily comparable. When surgery is performed by experienced pituitary neurosurgeons, the growth hormone levels fall to normal in 80% to 90% of patients with microadenomas.90,91 The success rate is lower in patients with higher preoperative growth hormone concentrations and/or macroadenomas (<50%).92 Recurrence rates after surgery are estimated to be approximately 20%.93,94 Using modern criteria for cure, the surgical remission rate for normal serum IGF-1 and glucose-suppressed growth hormone concentrations of less than 1 ng/mL were 70% and 61%, respectively.95 The largest series evaluated 506 patients who underwent transsphenoidal surgery. Overall remission was 57.3% as defined by normalization of IGF-1 levels.96 The results for microadenomas and macroadenomas were 75% and 50%, respectively.
Medical Therapy
Three different types of medicines (somatostatin analogs, dopamine agonists, and a growth hormone receptor antagonist) are used to treat patients with acromegaly who have failed surgery, are not surgical candidates, or are refractory to radiation options. Given the latency for normalization of IGF-1 and growth hormone levels after irradiation, medical therapy is also used as an adjunct after radiation treatments. The most commonly used medicines are the somatostatin analogs (octreotide and lanreotide) rather than somatostatin, given the much higher potency and half-life of the analogs. These analogs bind to somatostatin receptors subtypes 2 and 5, reduce IGF-1 levels in 38% to 66% of patients, and lead to tumor shrinkage in 30% to 45% of patients.97–99 Response is best predicted by determining whether the subtype of somatotroph adenoma is sparsely or densely granulated.100 The most common side effects of somatostatin analogs include nausea, abdominal cramping, diarrhea, and gallstones. A disadvantage of these agents is the need for lifelong dependence on them. Dopamine agonists such as cabergoline are generally not effective and are not used frequently unless a patient is unable to tolerate somatostatin analogs.
Pegvisomant, a growth hormone receptor antagonist that is administered daily via the subcutaneous route, has been tested in a placebo-controlled randomized trial and shown to normalize IGF-1 levels in 90% to 97% of patients.101,102 Side effects include flu-like symptoms, nausea, diarrhea, and abnormal liver function tests. Because pegvisomant does not inhibit growth hormone secretion or tumor growth, it is not used as first-line therapy. Pegvisomant requires lifelong treatment, may cause elevated aminotransaminases in 20% of patients, and may carry a small risk for increase in tumor size. In addition, pegvisomant is extremely expensive compared with other medical therapies.
Radiation Therapy Options
Irradiation can be used in the definitive or adjuvant setting for patients with acromegaly. Given the relatively long lag time for normalization of the IGF-1 and growth hormone levels, it is important that these patients be followed closely after treatment, with blood work every 3 to 4 months and yearly MRIs. The published reports on radiation therapy and SRS have typically used varying definitions of growth hormone levels. Earlier publications, which defined remission as growth hormone levels below 5 to 10 ng/mL, reported remission rates of 80% to 100%.103–105 Using modern definitions for normalization of both growth hormone levels (<2 ng/mL) and IGF-1 levels, the results for radiation therapy and SRS are less impressive.
Table 27-3 summarizes the recent radiation therapy results.106,107,108,109 The rate of hormone remission and hypopituitarism at 10 years is approximately 50% to 60% and 60% to 80%, respectively. The largest study, a retrospective review of 884 patients with acromegaly undergoing radiotherapy, demonstrated that 63% of patients had normalization of IGF-1 levels by 10 years after radiation therapy.108 In this series, new hormone deficiencies that occurred 10 years after irradiation were 18% for luteinizing hormone and follicle-stimulating hormone, 15% for adrenocorticotropic hormone, and 27% for thyroid-stimulating hormone. Variability in the success rates in normalization of IGF-1 levels for patients undergoing radiation therapy may be a result of differences in pretreatment serum growth hormone concentrations. Within the first 2 years, the mean growth hormone values drop to 50% to 70% of their maximal values, followed by a slow decrease over years.110 Higher doses of EBRT may be considered when treating patients with acromegaly (50.4 to 54 Gy vs. 45 Gy for nonfunctional tumors).
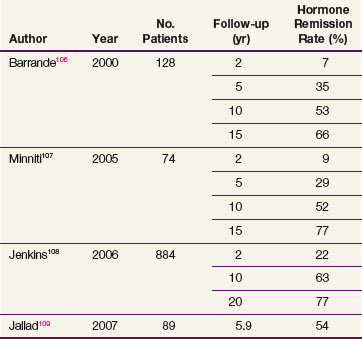
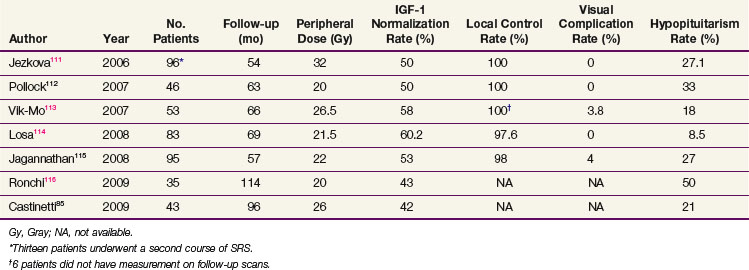
Table 27-4 summarizes the recent radiosurgery results that have used IGF-1 level normalization as the definition for cure.* Using peripheral doses of 20 to 32 Gy, IGF-1 level normalization rates range from 42% to 60%. The local control rates are excellent, suggesting that radiosurgery could possibly also have a definitive role in this condition, a hypothesis that will be tested in a developing Radiation Therapy Oncology Group (RTOG) trial. Mean time to remission ranges from 24 months to 36 months for patients undergoing SRS. Although the time to remission appears to be faster with SRS compared with radiation therapy, the ideal SRS candidate should have a well-defined target volume that is far enough away from the optic chiasm, and this factor limits the use of SRS.117
Medication may be used as an adjuvant treatment after radiation therapy to help lower the growth hormone and IGF-1 levels. Given the concern for the potential suppressive effects of somatostatin analogs, which may compromise the effectiveness of radiation therapy, some centers do not recommend using these medications for, typically, 6 to 8 weeks before and after radiation treatment at some centers.112,118
Cushing’s Disease
Cushing’s disease results from an adrenocorticotropic hormone-secreting adenoma of the anterior pituitary, which is responsible for 70% to 80% of adrenocorticotropic hormone-dependent Cushing’s syndrome cases in adults. Given the many nonspecific symptoms and signs, the diagnosis is often delayed. This syndrome, which is associated with increased morbidity and age-corrected mortality, occurs more commonly in women and is a heterogeneous disorder. Clinical findings include truncal obesity, hirsutism, acne, easy bruisability, muscle weakness, and moon facies. These patients can have menstrual irregularities, gonadal dysfunction, hypertension, diabetes mellitus, and osteoporosis. Tests used to diagnose Cushing’s disease include a 24-hour urine collection to measure urinary free cortisol levels, midnight testing of adrenocorticotropic hormone levels, and the dexamethasone suppression test.33 Untreated or incompletely treated hypercortisolism is associated with significant morbidity and excess mortality, which may be as high as two to three times the rates of the general population.50
The treatment options for Cushing’s disease include surgery, irradiation, or medical therapy or a combination of treatment approaches.119 Most centers define remission as urinary free cortisol levels in the normal range with resolution of clinical findings or a series of normal serum cortisol levels obtained throughout the day.120 A generalized, schematic treatment algorithm for Cushing’s disease is outlined in Figure 27-8.
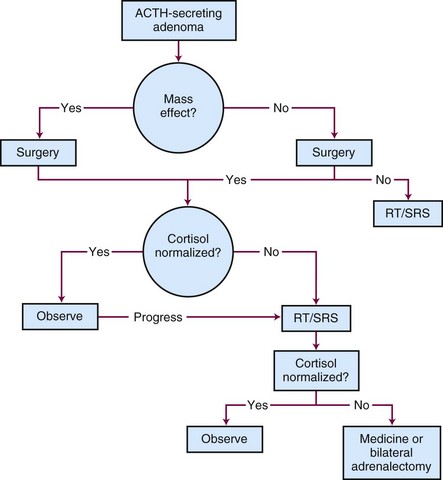
Surgery
Surgical removal of the pituitary adenoma is the best initial treatment for Cushing’s disease because it can rapidly correct the hypersecretion of adrenocorticotropic hormone. Because most patients have microadenomas, 25% to 50% of patients with Cushing’s disease will have no discernible lesion on MRI of the brain; no tumor may be identified in up to 50% of these cases at the time of surgical exploration.121,122 For patients with no discernible lesion who undergo surgery, inferior petrosal sinus sampling may help improve the outcome. Ideally, an experienced neurosurgeon should remove the tumor in its entirety, leaving the normal pituitary intact. Although an abnormality may not always be seen on MRI of the brain, surgery leads to hormonal control in 80% to 90% of patients with microadenomas and in up to 50% with macroadenomas.122 If a microadenoma cannot be identified in an adult patient in whom fertility is not an issue, 80% to 90% of the pituitary should be removed, with a small amount left attached to the stalk.123
For most patients, the surgical approach is transsphenoidal. A series from Cavagnini of 300 patients with a follow-up time of 10 years demonstrated a remission rate of 70% and a recurrence rate of 15%.124 In another study, low adrenocorticotropic hormone levels (<10 ng/mL) during the early postoperative period correlated with remission in 97.9% of cases, whereas an adrenocorticotropic hormone level of more than 30 ng/mL correlated with a remission rate of only 18%.125 A more recent series from Patil and colleagues126 noted a 25% recurrence rate at 5 years for patients who achieved initial remission after transsphenoidal surgery. Given the varied results, experience with transsphenoidal surgery for Cushing’s disease appears to be an important factor in terms of remission and recurrence.
Medical Therapy
Medical management is reserved for patients who do not respond to surgery or irradiation options; lifelong therapy is needed. Medicines used to inhibit steroidogenesis include ketoconazole, aminoglutethimide, metyrapone, mitotane, and etomidate.127 Ketoconazole is the best tolerated drug and is effective as monotherapy in up to 70% of patients. Because this drug can be hepatotoxic, it is important to monitor liver function tests. Metyrapone, an 11 alpha-hydroxylase inhibitor, is often used as an adjuvant to ketoconazole or radiation therapy. Etomidate can reduce cortisol levels within hours and is used for acute control of elevated cortisol levels. Mitotane causes adrenal suppression of cortisol secretion and is cytotoxic to adrenocortical cells. It is typically used for patients who have failed multiple medical therapies. Until the effects of irradiation have normalized the adrenocorticotropic hormone levels, hypercortisolism can be decreased by use of the above medications.
Radiation Therapy Options
Irradiation is typically used as an adjuvant for patients who are not cured after surgery. It can also be used definitively for patients unable to undergo surgery. For children, some consider this as the initial therapy because the cure rates are similar to those of surgery.128,129 Both radiation therapy and SRS have been used to treat patients with Cushing’s disease. For patients who are treated with EBRT, doses of 50 to 54 Gy may be considered. One study from Estrada used postoperative irradiation (mean dose, 50 Gy) and reported an 83% remission rate at 5 years.130 Recently, Minniti and colleagues131 reported on 40 patients with Cushing’s disease who received 45 Gy of EBRT. The normalization rate of cortisol levels was 78% at 5 years and 84% at 10 years. Other EBRT series have yielded remission rates ranging from 46% to 56%.132,133
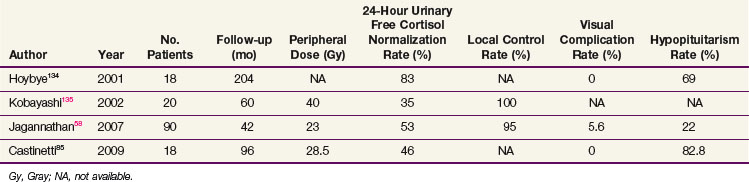
Most of the SRS series are small and report endocrine cure rates that are similar to the EBRT results. Recent series have reported remission rates of 35% to 83%,58,85,134,135 which are listed in Table 27-5. As with other secretory tumors, the peripheral dose used is higher (>20 Gy). The largest series from Jagannathan58 reported on 90 patients with a median follow-up of 45 months. In this series, a 54% remission rate was achieved. The timing of hormone normalization can be quite variable (2 months to 8 years), although most patients will achieve remission within the first 2 years. Patients with persistent disease should be considered for adrenalectomy or repeat SRS, which has been associated with higher rates of cranial nerve damage.58 No trials have compared SRS with fractionated treatments.
Nelson’s Syndrome
Some Cushing’s disease patients do not achieve remission after surgery or irradiation and may require bilateral adrenalectomy as salvage treatment. As a result, some patients may develop Nelson’s syndrome, which is characterized by hyperpigmentation, rapid growth of the adenoma, and invasion of the tumor into the parasellar regions.136 The few reports of SRS for Nelson’s syndrome have resulted in endocrine remission rates of 36% or less.137,138
Thyroid-Stimulating Hormone–Secreting Adenomas
Surgical removal of the rarely seen thyroid-stimulating hormone-secreting adenoma (0.5% of all pituitary tumors) is the best treatment option after hyperthyroidism has been controlled with medications. Because these tumors are usually large at diagnosis and can invade the sella or extend beyond it, complete removal can be difficult; for this reason, remission rates may be only 50% after surgery.139 When the tumor cannot be completely removed or the patient is not a surgical candidate, irradiation options can be considered. Somatostatin analogs or dopamine agonists have been used to decrease thyroid-stimulating hormone hypersecretion. The use of octreotide results in normalization of thyroid-stimulating hormone levels in 79% of patients.140
Pituitary Carcinomas
Pituitary carcinomas are rare (0.2% of all pituitary tumors). They have clinical features of pituitary adenomas, with most secreting prolactin or adrenocorticotropic hormone. Because the malignant nature of these tumors cannot be clearly determined by pathologic testing, the diagnosis is a clinical one based on findings of subarachnoid spread, brain involvement, or systemic metastases from a pituitary tumor.141 The overall prognosis for these tumors is poor despite aggressive treatments that have included radiation therapy. Rare examples of persistent response to chemotherapy (temozolomide) have been reported.142
Locally Advanced Disease and Palliation
Repeated Course of Irradiation
If tumors recur after fractionated radiation therapy, a repeat course of fractionated treatments may be considered after careful consideration of alternative treatment options, the interval that has elapsed since the first course of radiation therapy, and the details of the prior radiation treatment (e.g., technique used, dose given, and fractionation schedule used). Schoenthaler reported on 15 patients who were reirradiated with a median dose of 42 Gy in 1.8- to 2-Gy fractions.143 Local control was achieved in 12 patients. All patients developed hypopituitarism from the initial or repeat course of radiation therapy. Two patients later developed pituitary carcinoma and temporal lobe radionecrosis. Flickinger and colleagues144 reported on 10 patients with suprasellar or pituitary tumors who were reirradiated with doses ranging from 35 Gy to 49.6 Gy in 1.8- to 2-Gy fractions. Six patients had pituitary tumors. One patient developed optic neuropathy a little more than 1 year after completing the therapy. The study authors suggest that a 40% estimation of the original radiation dose effect is a reasonable guideline to account for prior radiation therapy.
Palliation of Metastases
Metastases to the pituitary occur infrequently and most commonly arise from breast and lung cancers.145 The reported rates of breast cancer metastasizing to the pituitary range from 6% to 8%. The most commonly reported symptoms include diabetes insipidus, anterior pituitary dysfunction, visual field defects, and headaches. Most metastases occur in the neurohypophyseal region, although breast cancer metastases have a predilection for the adenohypophyseal region. Because these are invasive tumors, surgery is difficult. SRS (median dose, 13 Gy) has been used for select patients with a progression-free survival rate of 66.7%, a 12-month survival rate of 18%, and a median survival time of 5.2 months.146
Irradiation Techniques and Tolerance
Techniques of Irradiation
Before the advent of sophisticated radiation delivery techniques, imaging techniques, and treatment planning efforts, the field arrangement for pituitary tumors included a two-field technique of opposed lateral views, a three-field technique of two opposed lateral views, and an anterior vertex field. Bilateral 110-degree coronal arcs with a 30-degree wedge and 330-degree rotational arcs were also used. Comparison of these various techniques demonstrated that the three-field technique delivered less dose to the temporal lobes than the two-field technique, the rotational techniques were superior to the stationary field techniques in terms of sparing the temporal lobe, and the four-field noncoplanar arc technique delivered less dose to the frontal lobe.147 When comparing the three-field coplanar technique to the four-field noncoplanar arrangement, the mean volume of normal brain receiving dose was higher with the coplanar techniques.148
Modern techniques of fractionated radiation therapy and SRS have capitalized on improvements in immobilization, imaging, planning, and treatment delivery. Using a fixed head frame, SRS is delivered in one fraction using a linear accelerator, cobalt unit (Gamma Knife), or protons. The vast majority of published results with SRS are with the Gamma Knife. Very few publications exist on the use of linear accelerator-based SRS, proton beam therapy, or CyberKnife therapy.* The use of SRS is limited to smaller tumors (<4 cm in diameter) that are 2 to 5 mm away from the optic apparatus.
Given the success of SRS in treating pituitary tumors, fractionated irradiation with highly conformal fields is being used more frequently for patients who are not ideal candidates for SRS or in centers that do not have access to SRS. The use of relocatable frames and precision thermoplastic masks and the verification of proper positioning prior to treatment have replaced conventional fractionated approaches at most centers. This precision allows for sparing of more normal brain tissue. As a result, the treatment margin around the tumor can be decreased to 3 to 5 mm. Minniti and colleagues53 recently reviewed the results of eight studies with 490 patients with functioning or nonfunctioning pituitary adenomas treated with stereotactic conformal radiotherapy. With a median follow-up of 39 months, tumor control was 98%. The preliminary results suggest that conformal radiation techniques are equivalent to those of conventional radiation therapy. The potential difference in biologic effectiveness of SRS versus fractionated courses has not been tested in a prospective manner.
Treatment Morbidity
Neurologic intolerance may be seen with either fractionated EBRT or SRS. For patients undergoing conventional EBRT with older irradiation techniques, the incidence of optic neuropathy is 1% to 3% and the risk of radiation necrosis is 0% to 2%.155,156 Using modern radiation approaches and MRI-based planning, the risk for radiation necrosis and optic neuropathy should be 1% or less. A review of 34 studies of SRS for pituitary adenomas reported a 1% risk of decrease in visual acuity and a 1.3% risk for other cranial nerve neuropathies (trigeminal, oculomotor, trochlear, and/or abducens nerve) after SRS.52
Vascular intolerance can also be found in EBRT and SRS series. An increased incidence for cerebrovascular accidents (1.7 to 2.8 times increase) has been reported for patients undergoing radiation therapy, although the relative contribution from irradiation remains to be determined.157,158 The risk of injury to the cavernous portion of the carotid artery is rare with a few case reports.159
The effect of irradiation on neurocognitive status is not clear given the effects of other therapies, such as medications and surgery, and of the tumor.160 It appears that patients who underwent surgery prior to radiation therapy may be at a higher risk for cognitive deficits.161,162
A cumulative incidence of the development of meningiomas and gliomas after the treatment of pituitary adenoma at 20 years is 2%.163 Loeffler and colleagues164 reported on two patients with pituitary adenomas who developed new tumors following SRS. He concluded that the risk for new tumors after SRS appears to be significantly less than that after fractionated EBRT. Another study of 5000 Gamma Knife radiosurgery patients followed for more than 10 years reported no increased risk of malignant disease.165
Treatment Algorithms, Challenges, Controversies, Future Possibilities, and Clinical Trials
The optimal management of pituitary neoplasms requires a multidisciplinary and individualized approach because these tumors are diverse, are associated with morbidity and mortality, and present a challenge regarding the best treatment approach. Because the treatment management strategy varies depending on a number of factors, including the type of tumor (secreting tumor vs. nonsecreting tumor), the imaging characteristics, and the clinical presentation and symptoms, a multidisciplinary approach ensures that the most effective therapy is being considered for each patient (see treatment algorithm by five specific tumor types within prior section). Because surgical treatment, medical therapy, and irradiation are all effective options, choosing the optimal treatment for an individual patient may be controversial and challenging. The patient’s age, medical condition, compliance with medicine, treatment tolerance, and preference should also influence the decision, as should an assessment of the potential long-term effects of hypopituitarism on excess morbidity and mortality.50 Close collaboration among the various disciplines is necessary for comprehensive long-term care. Clinical findings, endocrine evaluation, and MRI findings can establish the diagnosis of pituitary tumors in most patients and help define the initial treatment decisions.

3 Elston MS, McDonald KL, Clifton-Bligh RJ, Robinson BG. Familial pituitary tumor syndromes. Nat Rev Endocrinol. 2009;5:453-461.
11 Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas. A systematic review. Cancer. 2004;101:613-619.
22 Lloyd RJ, Kovacs K, Young WFJr, et al. Tumours of the pituitary gland. In: DeLellis RA, Lloyd RV, Heitz PU, Eng C, editors. Pathology and genetics of tumours of endocrine organs. Lyon, France: International Agency for Research and Cancer (IARC); 2004:9-48.
33 Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome. An endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:1526-1540.
34 Klibanski A. Clinical practice. Prolactinomas. N Engl J Med. 2010;362:1219-1226.
35 Melmed S. Medical progress. Acromegaly. N Engl J Med. 2006;355:2558-2573.
46 Ciric I, Ragin A, Baumgartner C, Pierce D. Complications of transsphenoidal surgery. Results of the national survey, review of the literature, and personal experience. Neurosurgery. 1997;40:225-237.
48 Barker FG2nd, Klibanski A, Swearingen B. Transsphenoidal surgery for pituitary tumors in the United States, 1996-2000. Mortality, morbidity, and the effects of hospital and surgeon volume. J Clin Endocrinol Metab. 2003;88:4709-4719.
50 Sherlock M, Ayuk J, Tomlinson JW, et al. Mortality in patients with pituitary disease. Endocr Rev. 2010;31:301-342.
52 Laws ER, Sheehan JP, Sheehan JM, et al. Stereotactic radiosurgery for pituitary adenomas. A review of the literature. J Neurooncol. 2004;69:257-272.
53 Minniti G, Gilbert DC, Brada M. Modern techniques for pituitary radiotherapy. Rev Endocr Metab Disord. 2009;10:135-144.
56 Stafford SL, Pollock BE, Leavitt JA, et al. A study on the radiation tolerance of the optic nerves and chiasm after stereotactic radiosurgery. Int J Radiat Oncol Biol Phys. 2003;55:1177-1181.
62 Dekkers OM, Pereira AM, Roelfsema F, et al. Observation alone after transsphenoidal surgery for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab. 2006;91:1796-1801.
69 Pollock BE, Cochran J, Natt N, et al. Gamma knife radiosurgery for patients with nonfunctioning pituitary adenomas. Results from a 15-year experience. Int J Radiat Oncol Biol Phys. 2008;70:1325-1329.
70 Brada M, Rajan B, Traish D, et al. The long-term efficacy of conservative surgery and radiotherapy in the control of pituitary adenomas. Clin Endocrinol (Oxf). 1993;38:571-578.
71 Sasaki R, Murakami M, Okamoto Y, et al. The efficacy of conventional radiation therapy in the management of pituitary adenomas. Int J Radiat Oncol Biol Phys. 2000;47:1337-1345.
73 Gillam MP, Molitch ME, Lombardi G, et al. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27:485-534.
74 Webster J, Piscitelli G, Polli A, et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group. N Engl J Med. 1994;331:904-909.
82 Pouratian N, Sheehan J, Jagannathan J, et al. Gamma knife radiosurgery for medically and surgically refractory prolactinomas. Neurosurgery. 2006;59:255-266.
83 Ježková J, Hána V, Kršek M, et al. Use of the Leksell gamma knife in the treatment of prolactinoma patients. Clin Endocrinol (Oxf). 2009;70:732-741.
85 Castinetti F, Nagai M, Morange I, et al. Long-term results of stereotactic radiosurgery in secretory pituitary adenomas. J Clin Endocrinol Metab. 2009;94:3400-3407.
86 Melmed S, Colao A, Barkan A, et al. Guidelines for acromegaly management. An update. J Clin Endocrinol Metab. 2009;94:1509-1517.
88 Giustina A, Chanson P, Bronstein MD, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95:3141-3148.
89 Melmed S, Casanueva F, Cavagnini F, et al. Consensus statement. Medical management of acromegaly. Eur J Endocrinol. 2005;153:737-740.
91 Erturk E, Tuncel E, Kiyici S, et al. Outcome of surgery for acromegaly performed by different surgeons. Importance of surgical experience. Pituitary. 2005;8:93-97.
101 Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000;342:1171-1177.
107 Minniti G, Jaffrain-Rea ML, Osti M, et al. The long-term efficacy of conventional radiotherapy in patients with GH-secreting pituitary adenomas. Clin Endocrinol (Oxf). 2005;62:210-216.
108 Jenkins PJ, Bates P, Carson MN, et al. Conventional pituitary irradiation is effective in lowering serum growth hormone and insulin-like growth factor-I in patients with acromegaly. J Clin Endocrinol Metab. 2006;91:1239-1245.
112 Pollock BE, Jacob JT, Brown PD, et al. Radiosurgery of growth hormone-producing pituitary adenomas. Factors associated with biochemical remission. J Neurosurg. 2007;106:833-838.
115 Jagannathan J, Pouratian N, Laws ERJr, et al. Gamma knife radiosurgery for acromegaly: outcomes after failed transsphenoidal surgery. Neurosurgery. 2008;62:1262-1270.
117 Castinetti F, Morange I, Dufour H, et al. Radiotherapy and radiosurgery in acromegaly. Pituitary. 2009;12:3-10.
118 Landolt AM, Haller D, Lomax N, et al. Octreotide may act as a radioprotective agent in acromegaly. J Clin Endocrinol Metabol. 2000;85:1287-1289.
119 Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome. A consensus statement. J Clin Endocrinol Metab. 2008;93:2454-2462.
123 Pouratian N, Prevedello DM, Jagannathan J, et al. Outcomes and management of patients with Cushing’s disease without pathological confirmation of tumor resection after transsphenoidal surgery. J Clin Endocrinol Metab. 2007;92:3383-3388.
126 Patil CG, Prevedello DM, Lad SP, et al. Late recurrences of Cushing’s disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab. 2008;93:358-362.
127 Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs. 2009;14:661-671.
131 Minniti G, Osti M, Jaffrain-Rea ML, et al. Long-term follow-up results of postoperative radiation therapy for Cushing’s disease. J Neurooncol. 2007;84:79-84.
134 Höybye C, Grenbäck E, Rähn T, et al. Adrenocorticotropic hormone producing pituitary tumors: 12- to 22-year follow-up after treatment with stereotactic radiosurgery. Neurosurgery. 2001;49:284-292.
137 Mauermann WJ, Sheehan JP, Chernavvsky DR, et al. Gamma Knife surgery for adrenocorticotropic hormone-producing pituitary adenomas after bilateral adrenalectomy. J Neurosurg. 2007;106:988-993.
141 Pernicone PJ, Scheithauer BW, Sebo TJ, et al. Pituitary carcinoma. A clinicopathologic study of 15 cases. Cancer. 1997;79:804-812.
143 Schoenthaler R, Albright NA, Wara WM, et al. Re-irradiation of pituitary adenoma. Int J Radiat Oncol Biol Phys. 1992;24:307-314.
147 Sohn JW, Dalzell JG, Suh JH, et al. Dose-volume histogram analysis of techniques for irradiating pituitary adenomas. Int J Radiat Oncol Biol Phys. 1995;32:831-837.
148 Perks JR, Jalali R, Cosgrove VP, et al. Optimization of stereotactically guided conformal treatment planning of sellar and parasellar tumors, based on normal brain dose volume histograms. Int J Radiat Oncol Biol Phys. 1999;45:507-513.
149 Voges J, Kocher M, Runge M, et al. Linear accelerator radiosurgery for pituitary macroadenomas. A 7-year follow-up study. Cancer. 2006;107:1355-1364.
151 Petit JH, Biller BM, Coen JJ, et al. Proton stereotactic radiosurgery in management of persistent acromegaly. Endocr Pract. 2007;13:726-734.
154 Tuniz F, Soltys SG, Choi CY, et al. Multisession cyberknife stereotactic radiosurgery of large, benign cranial base tumors: preliminary study. Neurosurgery. 2009;65:898-907.
157 Erfurth EM, Bülow B, Svahn-Tapper G, et al. Risk factors for cerebrovascular deaths in patients operated and irradiated for pituitary tumors. J Clin Endocrinol Metab. 2002;87:4892-4899.
158 Brada M, Burchell L, Ashley S, et al. The incidence of cerebrovascular accidents in patients with pituitary adenomas. Int J Radiat Oncol Biol Phys. 1999;45:693-698.
163 Brada M, Ford D, Ashley S, et al. Risk of second brain tumor after conservative surgery and radiotherapy for pituitary adenoma. Br Med J. 1992;304:1343-1346.
164 Loeffler JS, Niemierko A, Chapman PH. Second tumors after radiosurgery. Tip of the iceberg or a bump in the road? Neurosurgery. 2003;52:1436-1440.
165 Rowe J, Grainger A, Walton L, et al. Risk for malignancy after gamma knife stereotactic radiosurgery. Neurosurgery. 2007;60:60-66.
1 Tada M, Kobayashi H, Moriuchi T. Molecular basis of pituitary oncogenesis. Neuro-Oncology. 1999;45:83-96.
2 Asa SL, Ezzat S. The pathogenesis of pituitary tumors. Annu Rev Pathol. 2009;4:97-126.
3 Elston MS, McDonald KL, Clifton-Bligh RJ, Robinson BG. Familial pituitary tumor syndromes. Nat Rev Endocrinol. 2009;5:453-461.
4 Bates AS, Farrell WE, Bicknell EJ, et al. Allelic deletion in pituitary adenomas reflects aggressive biologic activity and has potential value as a prognostic marker. J Clin Endocrinol Metab. 1997;82:818-824.
5 Crabtree JS, Scacheri PC, Ward JM, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98:1118-1123.
6 Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89-92.
7 Georgitsi M, Raitila A, Karhu A, et al. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proc Natl Acad Sci U S A. 2007;104:4101-4105.
8 Vierimaa O, Georgitsi M, Lehtonen R, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312:1228-1230.
9 Daly AF, Rixhon M, Adams C, et al. High prevalence of pituitary adenomas. A cross- sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006;91:4769-4775.
10 Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas. A community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72:377-382.
11 Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas. A systematic review. Cancer. 2004;101:613-619.
12 Spada A, Lania A, Mantovani G. Hormonal signaling and pituitary adenomas. Neuroendocrinology. 2007;85:101-109.
13 Xu M, Shorts-Cary L, Knox AJ, et al. Epidermal growth factor receptor pathway substrate 8 (eps8) is overexpressed in human pituitary tumors. Role in proliferation and survival. Endocrinology. 2009;150:2064-2071.
14 Chesnokova V, Zonis S, Kovacs K, et al. p21(Cip1) restrains pituitary tumor growth. Proc Natl Acad Sci U S A. 2008;105:17498-17503.
15 Hayward BE, Barlier A, Korbonits M, et al. Imprinting of the G(s) α gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. 2001;107:R31-R36.
16 Zhang X, Horwitz GA, Heaney AP, et al. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab. 1999;84:761-767.
17 Ezzat S, Zheng L, Zhu XF, et al. Targeted expression of a human pituitary tumor-derived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J Clin Invest. 2002;109:69-78.
18 Asa SL, Kovacs K, Shlomad S. The hypothalamic-pituitary axis. In: Melmed S, editor. The pituitary. Cambridge: Blackwell Scientific; 1995:p 3.
19 Hardy J. Transsphenoidal surgery of hypersecreting pituitary tumors. In: Kohler PO, Ross FT, editors. Diagnosis and treatment of pituitary tumors. New York: Elsevier; 1973:179.
20 Teramoto A, Hirakawa K, Sanno N, et al. Incidental pituitary lesions in 1000 unselected autopsy specimens. Radiology. 1994;193:161-164.
21 Bonneville JF, Bonneville F, Cattin F. Magnetic resonance imaging of pituitary adenomas. Eur Radiol. 2005;15:543-548.
22 Lloyd RJ, Kovacs K, Young WFJr, et al. Tumours of the pituitary gland. In: DeLellis RA, Lloyd RV, Heitz PU, Eng C, editors. Pathology and genetics of tumours of endocrine organs. Lyon, France: International Agency for Research and Cancer (IARC); 2004:9-48.
23 Branch CLJr, Laws ER Jr. Metastatic tumors of the sella turcica masquerading as primary pituitary tumors. J Clin Endocrinol Metab. 1987;65:469-474.
24 Pernicone PJ, Scheithauer BW. Invasive pituitary adenoma and pituitary carcinoma. In: Thapar K, Kovacs K, Scheithauer BW, Lloyd RV, editors. Diagnosis and management of pituitary tumors. totowa. New Jersey: Humana Press; 2001:369-386.
25 Saeger W, Lüdecke B, Lüdecke DK. Comparison of proliferation markers and clinical tumor growth in inactive adenomas. Endocr Pathol. 2004;15:264-265.
26 Amar AP, Hinton DR, Krieger MD, Weiss MH. Invasive pituitary adenomas. Significance of proliferation parameters. Pituitary. 1999;2:117-122.
27 Riss D, Jin L, Qian X, et al. Differential expression of galectin-3 in pituitary tumors. Cancer Res. 2003;63:2251-2255.
28 Asa SL, Ezzat S. The cytogenesis and pathogenesis of pituitary adenomas. Endocr Rev. 1998;19:798-827.
29 Asa SL, Ezzat S. Molecular basis of pituitary development and cytogenesis. Front Horm Res. 2004;32:1-19.
30 Zhu X, Gleiberman AS, Rosenfeld MG. Molecular physiology of pituitary development. Signaling and transcriptional networks. Physiol Rev. 2007;87:933-963.
31 Loh JA, Verbalis JG. Disorders of water and salt metabolism associated with pituitary disease. Endocrinol Metab Clin North Am. 2008;37:213-234.
32 Gopan T, Toms SA, Prayson RA, et al. Symptomatic pituitary metastases from renal cell carcinoma. Pituitary. 2007;10:251-259.
33 Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome. An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526-1540.
34 Klibanski A. Clinical practice. Prolactinomas. N Engl J Med. 2010;362:1219-1226.
35 Melmed S. Medical progress. Acromegaly. N Engl J Med. 2006;355:2558-2573.
36 Levivier M, Massager N, Wikler D, et al. Integration of functional imaging in radiosurgery. The example of PET scan. Prog Neurol Surg. 2007;20:68-81.
37 Bakiri F, Tatai S, Aouali R, et al. Treatment of Cushing’s disease by transsphenoidal, pituitary microsurgery. Prognosis factors and long-term follow-up. J Endocrinol Invest. 1996;19:572-580.
38 Esposito F, Dusick JR, Cohan P, et al. Clinical review. Early morning cortisol levels as a predictor of remission after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab. 2006;91:7-13.
39 Hardy J, Verzina JL. Transsphenoidal neurosurgery of intracranial neoplasm. In: Thompson RA, Green JR, editors. Advances in neurology. New York: Raven Press; 1976:261. (updated at the International Pituitary Conference, Zurich, 1987)
40 Wilson CB. Neurosurgical management of large and invasive pituitary tumors. In: Tinadall GT, Collins WF, editors. Clinical management of pituitary disorders. New York: Raven Press; 1979:335.
41 Laws ER, Jane JAJr. Neurosurgical approach to treating pituitary adenomas. Growth Horm IGF Res. 2005;15(Suppl A):S36-S41.
42 Socin HV, Chanson P, Delemer B, et al. The changing spectrum of TSH-secreting pituitary adenomas. Diagnosis and management in 43 patients. Eur J Endocrinol. 2003;148:433-442.
43 Benveniste RJ, King WA, Walsh J, et al. Repeated transsphenoidal surgery to treat recurrent or residual pituitary adenoma. J Neurosurg. 2005;102:1004-1012.
44 White DR, Sonnenburg RE, Ewend MG, et al. Safety of minimally invasive pituitary surgery (MIPS) compared with a traditional approach. Laryngoscope. 2004;114:1945-1948.
45 Atkinson AB, Kennedy A, Wiggman MI, et al. Long-term remission rates after pituitary surgery for Cushing’s disease: the need for long-term surveillance. Clin Endocrinol (Oxf). 2005;63:549-559.
46 Ciric I, Ragin A, Baumgartner C, Pierce D. Complications of transsphenoidal surgery. Results of the national survey, review of the literature, and personal experience. Neurosurgery. 1997;40:225-237.
47 Nemergut EC, Zuo Z, Jane JAJr, et al. Predictors of diabetes insipidus after transsphenoidal surgery. A review of 881 patients. J Neurosurg. 2005;103:448-454.
48 Barker FG2nd, Klibanski A, Swearingen B. Transsphenoidal surgery for pituitary tumors in the United States, 1996-2000. Mortality, morbidity, and the effects of hospital and surgeon volume. J Clin Endocrinol Metab. 2003;88:4709-4719.
49 Stewart PM. Pegvisomant. An advance in clinical efficacy in acromegaly. Eur J Endocrinol. 2003;148(Suppl 2):S27-S32.
50 Sherlock M, Ayuk J, Tomlinson JW, et al. Mortality in patients with pituitary disease. Endocr Rev. 2010;31:301-342.
51 Jalali R, Brada M. Radiosurgery for pituitary adenoma. Crit Rev Neurosurg. 1999;9:167-173.
52 Laws ER, Sheehan JP, Sheehan JM, et al. Stereotactic radiosurgery for pituitary adenomas. A review of the literature. J Neurooncol. 2004;69:257-272.
53 Minniti G, Gilbert DC, Brada M. Modern techniques for pituitary radiotherapy. Rev Endocr Metab Disord. 2009;10:135-144.
54 Mackley HB, Reddy CA, Lee SY, et al. Intensity-modulated radiotherapy for pituitary adenomas. The preliminary report of the Cleveland Clinic experience. Int J Radiat Oncol Biol Phys. 2007;67:232-239.
55 Kumar S, Burke K, Nalder C, et al. Treatment accuracy of fractionated stereotactic radiotherapy. Radiother Oncol. 2005;74:53-59.
56 Stafford SL, Pollock BE, Leavitt JA, et al. A study on the radiation tolerance of the optic nerves and chiasm after stereotactic radiosurgery. Int J Radiat Oncol Biol Phys. 2003;55:1177-1181.
57 Tishler RB, Loeffler JS, Lunsford LD, et al. Tolerance of cranial nerves in the cavernous sinus to radiosurgery. Int J Radiat Oncol Biol Phys. 1993;27:215-221.
58 Jagannathan J, Sheehan JP, Pouratian N, et al. Gamma Knife surgery for Cushing’s disease. Neurosurgery. 2007;106:980-987.
59 Snyder PJ. Clinically nonfunctioning pituitary adenomas. Endocrinol Metab Clin North Am. 1993;22:163-175.
60 Wang H, Sun W, Fu Z, et al. The pattern of visual impairment in patients with pituitary adenoma. J Int Med Res. 2008;36:1064-1069.
61 Turner HE, Stratton IM, Byrne JV, et al. Audit of selected patients with nonfunctioning pituitary adenomas treated without irradiation. A follow-up study. Clin Endocrinol (Oxf). 1999;51:281-284.
62 Dekkers OM, Pereira AM, Roelfsema F, et al. Observation alone after transsphenoidal surgery for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab. 2006;91:1796-1801.
63 Sheehan JP, Kondziolka D, Flickinger J, Lunsford LD. Radiosurgery for residual or recurrent nonfunctioning pituitary adenoma. J Neurosurg. 2002;97(5 Suppl):408-414.
64 Petrovich Z, Yu C, Giannotta SL, Apuzzo ML. Gamma knife radiosurgery for pituitary adenoma. Early results. Neurosurgery. 2003;53:51-59.
65 Losa M, Valle M, Mortini P, et al. Gamma knife surgery for treatment of residual nonfunctioning pituitary adenomas after surgical debulking. J Neurosurg. 2004;100(3):438-444.
66 Iwai Y, Yamanaka K, Yoshioka K. Radiosurgery for nonfunctioning pituitary adenomas. Neurosurgery. 2005;56(4):699-705.
67 Mingione V, Yen CP, Vance ML, et al. Gamma surgery in the treatment of nonsecretory pituitary macroadenoma. J Neurosurg. 2006;104(6):876-883.
68 Liscak R, Vladyka V, Marek J, et al. Gamma knife radiosurgery for endocrine-inactive pituitary adenomas. Acta Neurochir (Wien). 2007;149(10):999-1006.
69 Pollock BE, Cochran J, Natt N, et al. Gamma knife radiosurgery for patients with nonfunctioning pituitary adenomas. Results from a 15-year experience. Int J Radiat Oncol Biol Phys. 2008;70:1325-1329.
70 Brada M, Rajan B, Traish D, et al. The long-term efficacy of conservative surgery and radiotherapy in the control of pituitary adenomas. Clin Endocrinol (Oxf). 1993;38:571-578.
71 Sasaki R, Murakami M, Okamoto Y, et al. The efficacy of conventional radiation therapy in the management of pituitary adenomas. Int J Radiat Oncol Biol Phys. 2000;47:1337-1345.
72 Naidich MJ, Russell EJ. Current approaches to imaging of the sellar region and pituitary. Endocrinol Metab Clin North Am. 1999;28:45-79.
73 Gillam MP, Molitch ME, Lombardi G, et al. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27:485-534.
74 Webster J, Piscitelli G, Polli A, et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group. N Engl J Med. 1994;331:904-909.
75 Valassi E, Klibanski A, Biller BM. Potential cardiac valve effects of dopamine agonists in hyperprolactinemia. J Clin Endocrinol Metab. 2010;95:1025-1033.
76 Sheline G, Grossman A, Jones AE, et al. Radiation Therapy for Prolactinomas. New York: Raven Press; 1984.
77 Littley MD, Shalet SM, Reid H, et al. The effect of external pituitary irradiation on elevated serum prolactin levels in patients with pituitary macroadenomas. Q J Med. 1991;81:985-998.
78 Tsagarakis S, Grossman A, Plowman PN, et al. Megavoltage pituitary irradiation in the management of prolactinomas. Long-term follow-up. Clin Endocrinol. 1991;34:399-406.
79 Landolt AM, Lomax N. Gamma Knife radiosurgery for prolactinomas. J Neurosurg. 2000;93(Suppl 3):14-18.
80 Pan L, Zhang N, Wang EM, et al. Gamma knife radiosurgery as a primary treatment for prolactinomas. J Neurosurg. 2000;93(Suppl 3):10-13.
81 Choi JY, Chang JH, Chang JW, et al. Radiological and hormonal responses of functioning pituitary adenomas after gamma knife radiosurgery. Yonsei Med J. 2003;44:602-607.
82 Pouratian N, Sheehan J, Jagannathan J, et al. Gamma knife radiosurgery for medically and surgically refractory prolactinomas. Neurosurgery. 2006;59:255-266.
83 Ježková J, Hána V, Kršek M, et al. Use of the Leksell gamma knife in the treatment of prolactinoma patients. Clin Endocrinol (Oxf). 2009;70:732-741.
84 Pollock BE, Brown PD, Nippoldt TB, Young WF Jr. Pituitary tumor type affects the chance of biochemical remission after radiosurgery of hormone-secreting pituitary adenomas. Neurosurgery. 2008;62:1271-1278.
85 Castinetti F, Nagai M, Morange I, et al. Long-term results of stereotactic radiosurgery in secretory pituitary adenomas. J Clin Endocrinol Metab. 2009;94:3400-3407.
86 Melmed S, Colao A, Barkan A, et al. Guidelines for acromegaly management. An update. J Clin Endocrinol Metab. 2009;94:1509-1517.
87 Dimaraki EV, Jaffe CA, DeMott-Friberg R, et al. Acromegaly with apparently normal GH secretion. Implications for diagnosis and follow-up. J Clin Endocrinol Metab. 2002;87:3537-3542.
88 Giustina A, Chanson P, Bronstein MD, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95:3141-3148.
89 Melmed S, Casanueva F, Cavagnini F, et al. Consensus statement. Medical management of acromegaly. Eur J Endocrinol. 2005;153:737-740.
90 Swearingen B, Barker FG2nd, Katznelson L, et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998;83:3419-3426.
91 Erturk E, Tuncel E, Kiyici S, et al. Outcome of surgery for acromegaly performed by different surgeons. Importance of surgical experience. Pituitary. 2005;8:93-97.
92 Freda PU, Wardlaw SL, Post KD. Long-term endocrinological follow-up evaluation in 115 patients who underwent transsphenoidal surgery for acromegaly. J Neurosurg. 1998;89:353-358.
93 Biermasz NR, van Dulen H, Roelfsema F. Ten-year follow-up results of transsphenoidal microsurgery in acromegaly. J Clin Endocrinol Metab. 2000;85:4596-4602.
94 Krieger MD, Couldwell WT, Weiss MH. Assessment of long-term remission of acromegaly after surgery. J Neurosurg. 2003;98:719-724.
95 Kreutzer J, Vance ML, Lopes MBS, Laws ER Jr. Surgical management of GH-secreting pituitary adenomas. An outcome study using modern remission criteria. J Clin Endocrinol Metab. 2001;86:4072-4077.
96 Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical cure. Eur J Endocrinol. 2005;152:379-387.
97 Bevan JS, Atkin SL, Atkinson AB, et al. Primary medical therapy for acromegaly. An open, prospective, multicenter study of the effects of subcutaneous and intramuscular slow-release octreotide on growth hormone, insulin-like growth factor-I, and tumor size. J Clin Endocrinol Metab. 2002;87:4554-4563.
98 Freda PU. Somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2002;87:3013-3018.
99 Melmed S, Sternberg R, Cook D, et al. A critical analysis of pituitary tumor shrinkage during primary medical therapy in acromegaly. J Clin Endocrinol Metab. 2005;90:4405-4410.
100 Bhayana S, Booth GL, Asa SL, et al. The implication of somatotroph adenoma phenotype to somatostatin analog responsiveness in acromegaly. J Clin Endocrinol Metab. 2005;90:6290-6295.
101 Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000;342:1171-1177.
102 van der Lely AJ, Hutson RK, Trainer PJ, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet. 2001;358:1754-1759.
103 Eastman RC, Gorden P, Glatstein E, Roth J. Radiation therapy of acromegaly. Endocrinol Metab Clin North Am. 1992;21:693-712.
104 Bates AS, Van’t Hoff W, Jones JM, Clayton RN. An audit of outcomes of treatment in acromegaly. Q J Med. 1993;86:293-299.
105 Zhang N, Pan L, Wang EM, et al. Radiosurgery for growth hormone-producing pituitary adenomas. J Neurosurg. 2000;93(Suppl 3):6-9.
106 Barrande G, Pittino-Lungo M, Coste J, et al. Hormonal and metabolic effects of radiotherapy in acromegaly. Long-term results in 128 patients followed in a single center. J Clin Endocrinol Metab. 2000;85:3779-3785.
107 Minniti G, Jaffrain-Rea ML, Osti M, et al. The long-term efficacy of conventional radiotherapy in patients with GH-secreting pituitary adenomas. Clin Endocrinol (Oxf). 2005;62:210-216.
108 Jenkins PJ, Bates P, Carson MN, et al. Conventional pituitary irradiation is effective in lowering serum growth hormone and insulin-like growth factor-I in patients with acromegaly. J Clin Endocrinol Metab. 2006;91:1239-1245.
109 Jallad RS, Musolino NR, Salgado LR, et al. Treatment of acromegaly. Is there still a place for radiotherapy? Pituitary. 2007;10:53-59.
110 Jaffe CA. Reevaluation of conventional pituitary irradiation in the therapy of acromegaly. Pituitary. 1999;2:55-62.
111 Jezkova J, Marek J, Hana V, et al. Gamma knife radiosurgery for acromegaly. Long-term experience. Clin Endocrinol (Oxf). 2006;64:588-595.
112 Pollock BE, Jacob JT, Brown PD, et al. Radiosurgery of growth hormone-producing pituitary adenomas. Factors associated with biochemical remission. J Neurosurg. 2007;106:833-838.
113 Vik-Mo EO, Oksnes M, Pedersen PH, et al. Gamma knife stereotactic radiosurgery for acromegaly. Eur J Endocrinol. 2007;157:255-263.
114 Losa M, Gioia L, Picozzi P, et al. The role of stereotactic radiotherapy in patients with growth hormone secreting pituitary adenoma. J Clin Endocrinol Metab. 2008;93:2546-2552.
115 Jagannathan J, Pouratian N, Laws ERJr, et al. Gamma knife radiosurgery for acromegaly: outcomes after failed transsphenoidal surgery. Neurosurgery. 2008;62:1262-1270.
116 Ronchi CL, Attanasio R, Verrua E, et al. Efficacy and tolerability of gamma knife radiosurgery in acromegaly. A 10-year follow-up study. Clin Endocrinol (Oxf). 2009;71:846-852.
117 Castinetti F, Morange I, Dufour H, et al. Radiotherapy and radiosurgery in acromegaly. Pituitary. 2009;12:3-10.
118 Landolt AM, Haller D, Lomax N, et al. Octreotide may act as a radioprotective agent in acromegaly. J Clin Endocrinol Metabol. 2000;85:1287-1289.
119 Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome. A consensus statement. J Clin Endocrinol Metab. 2008;93:2454-2462.
120 Nieman LK. Medical therapy of Cushing’s disease. Pituitary. 2002;5:77-82.
121 Weiss MH, Couldwell WT. Gamma knife radiosurgery for Cushing’s disease. J Neurosurg. 2007;106:976-977.
122 Bochicchio D, Losa M, Buchfelder M. Factors influencing the immediate and late outcome of Cushing’s disease treated by transsphenoidal surgery. A retrospective study by the European Cushing’s Disease Survey Group. J Clin Endocrinol Metab. 1995;80:3114-3120.
123 Pouratian N, Prevedello DM, Jagannathan J, et al. Outcomes and management of patients with Cushing’s disease without pathological confirmation of tumor resection after transsphenoidal surgery. J Clin Endocrinol Metab. 2007;92:3383-3388.
124 Cavagnini F, Pecori Giraldi F. Epidemiology and follow-up of Cushing’s disease. Ann Endocrinol (Paris). 2001;62:168-172.
125 Flitsch J, Knappe UJ, Ludecke DK. The use of postoperative ACTH levels as a marker for successful transsphenoidal microsurgery in Cushing’s disease. Zentralbl Neurochir. 2003;64:6-11.
126 Patil CG, Prevedello DM, Lad SP, et al. Late recurrences of Cushing’s disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab. 2008;93:358-362.
127 Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs. 2009;14:661-671.
128 Devoe DJ, Miller WL, Conte FA, et al. Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab. 1997;82:3196-3202.
129 Jennings AS, Liddle GW, Orth DN. Results of treating childhood Cushing’s disease with pituitary irradiation. N Engl J Med. 1977;297:957-962.
130 Estrada J, Boronat M, Mielgo M, et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med. 1997;336:215-217.
131 Minniti G, Osti M, Jaffrain-Rea ML, et al. Long-term follow-up results of postoperative radiation therapy for Cushing’s disease. J Neurooncol. 2007;84:79-84.
132 Littley MD, Shalet SM, Beardwell CG, et al. Long-term follow-up of low-dose external pituitary irradiation for Cushing’s disease. Clin Endocrinol (Oxf). 1990;33:445-455.
133 Tsang RW, Brierley JD, Panzarella T, et al. Radiation therapy for pituitary adenoma. Treatment outcome and prognostic factors. Int J Radiat Oncol Biol Phys. 1994;30:557-565.
134 Höybye C, Grenbäck E, Rähn T, et al. Adrenocorticotropic hormone producing pituitary tumors: 12- to 22-year follow-up after treatment with stereotactic radiosurgery. Neurosurgery. 2001;49:284-292.
135 Kobayashi T, Kida Y, Mori Y. Gamma knife radiosurgery in the treatment of Cushing disease. Long-term results. J Neurosurg. 2002;97(Suppl. 5):422-428.
136 Nelson DH, Meakin JW, Thorn GW. ACTH-producing pituitary tumors following adrenalectomy for Cushing’s syndrome. Ann Intern Med. 1960;52:560-569.
137 Mauermann WJ, Sheehan JP, Chernavvsky DR, et al. Gamma Knife surgery for adrenocorticotropic hormone-producing pituitary adenomas after bilateral adrenalectomy. J Neurosurg. 2007;106:988-993.
138 Pollock BE, Young WFJr. Stereotactic radiosurgery for patients with ACTH-producing pituitary adenomas after prior adrenalectomy. Int J Radiat Oncol Biol Phys. 2002;54:839-841.
139 Clarke MJ, Erickson D, Castro R, et al. Thyroid-stimulating hormone pituitary adenomas. J Neurosurg. 2008;109:17-22.
140 Beck-Peccoz P, Brucker-Davis F, Persani L, et al. Thyrotropin-secreting pituitary tumors. Endocr Rev. 1996;17:610-638.
141 Pernicone PJ, Scheithauer BW, Sebo TJ, et al. Pituitary carcinoma. A clinicopathologic study of 15 cases. Cancer. 1997;79:804-812.
142 Fadul CE, Kominsky AL, Meyer LP, et al. Long-term response of pituitary carcinoma to temozolomide. Report of two cases. J Neurosurg. 2006;105:621-626.
143 Schoenthaler R, Albright NA, Wara WM, et al. Re-irradiation of pituitary adenoma. Int J Radiat Oncol Biol Phys. 1992;24:307-314.
144 Flickinger JC, Deutsch M, Lunsford LD. Repeat megavoltage irradiation of pituitary and suprasellar tumors. Int J Radiat Oncol Biol Phys. 1989;17:171-175.
145 Fassett DR, Couldwell WT. Metastases to the pituitary gland. Neurosurg Focus. 2004;16:E8.
146 Kano H, Niranjan A, Kondziolka D, et al. Stereotactic radiosurgery for pituitary metastases. Surg Neurol. 2009;72:248-255.
147 Sohn JW, Dalzell JG, Suh JH, et al. Dose-volume histogram analysis of techniques for irradiating pituitary adenomas. Int J Radiat Oncol Biol Phys. 1995;32:831-837.
148 Perks JR, Jalali R, Cosgrove VP, et al. Optimization of stereotactically guided conformal treatment planning of sellar and parasellar tumors, based on normal brain dose volume histograms. Int J Radiat Oncol Biol Phys. 1999;45:507-513.
149 Voges J, Kocher M, Runge M, et al. Linear accelerator radiosurgery for pituitary macroadenomas. A 7-year follow-up study. Cancer. 2006;107:1355-1364.
150 Chen CC, Chapman P, Petit J, Loeffler J. Proton radiosurgery in neurosurgery. Neurosurg Focus. 2007;23:E5.
151 Petit JH, Biller BM, Coen JJ, et al. Proton stereotactic radiosurgery in management of persistent acromegaly. Endocr Pract. 2007;13:726-734.
152 Aghi MK, Petit J, Chapman P, et al. Management of recurrent and refractory Cushing’s disease with reoperation and/or proton beam radiosurgery. Clin Neurosurg. 2008;55:141-144.
153 Adler JR, Gibbs IC, Puataweepong P, Chang SD. Visual field preservation after multisession Cyberknife radiosurgery for perioptic lesions. Neurosurgery. 2006;59:244-254.
154 Tuniz F, Soltys SG, Choi CY, et al. Multisession cyberknife stereotactic radiosurgery of large, benign cranial base tumors: preliminary study. Neurosurgery. 2009;65:898-907.
155 Tsang RW, Brierley JD, Panzarella T, et al. Radiation therapy for pituitary adenoma. Treatment outcome and prognostic factors. Int J Radiat Oncol Biol Phys. 1994;30:557-565.
156 Becker G, Kocher M, Kortmann RD, et al. Radiation therapy in the multimodal treatment approach of pituitary adenomas. Strahlenther Onkol. 2002;178:173-186.
157 Erfurth EM, Bülow B, Svahn-Tapper G, et al. Risk factors for cerebrovascular deaths in patients operated and irradiated for pituitary tumors. J Clin Endocrinol Metab. 2002;87:4892-4899.
158 Brada M, Burchell L, Ashley S, et al. The incidence of cerebrovascular accidents in patients with pituitary adenomas. Int J Radiat Oncol Biol Phys. 1999;45:693-698.
159 Lim YJ, Leem W, Park JT, et al. Cerebral infraction with ICA occlusion after Gamma Knife radiosurgery for pituitary adenoma. A case report. Stereotact Funct Neurosurg. 1999;72(Suppl 1):132-139.
160 Tooze A, Gittoes NJ, Jones CA, et al. Neurocognitive consequences of surgery and radiotherapy for tumours of the pituitary. Clin Endocrinol (Oxf). 2009;70:503-511.
161 McCord MW, Buatti JM, Fennell EM, et al. Radiotherapy for pituitary adenomas. Long-term outcome and sequelae. Int J Radiat Oncol Biol Phys. 1997;39:437-444.
162 Noad R, Narayanan KR, Howlett T, et al. Evaluation of the effect of radiotherapy for pituitary tumours on cognitive function and quality of life. Clin Oncol (R Coll Radiol). 2004;16:233-237.
163 Brada M, Ford D, Ashley S, et al. Risk of second brain tumor after conservative surgery and radiotherapy for pituitary adenoma. Br Med J. 1992;304:1343-1346.
164 Loeffler JS, Niemierko A, Chapman PH. Second tumors after radiosurgery. Tip of the iceberg or a bump in the road? Neurosurgery. 2003;52:1436-1440.
165 Rowe J, Grainger A, Walton L, et al. Risk for malignancy after gamma knife stereotactic radiosurgery. Neurosurgery. 2007;60:60-66.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 27 Pituitary Tumors
Etiology and Epidemiology
Etiology
The etiology of most pituitary tumors is not known and has been a source of debate. Based on two rat pituitary models, two primary modes of pituitary oncogenesis have been suggested.1 The first model, known as the hyperplasia-adenoma sequence, suggests a hormone-dependent pathway. Another model suggests de novo occurrence without the development of hyperplasia, possibly because of a genetic alteration. In this model, dysregulation of hormone or growth factor signaling leads to hyperplasia and tumor formation; the defect has been found especially in transgenic and knockout animal models, although there is strong evidence implicating overexpression of transforming growth factor–alpha (TGF-α), fibroblast growth factor (FGF), and fibroblast growth factor receptors (FGFRs) in tumor formation.2
For some patients with pituitary adenomas, a genetic predisposition has been described with four genes known to be associated with familial pituitary tumor syndromes: MEN1, CDKN1B, PRKAR1A, and AIP.3 Of patients with MEN1, which is an autosomal dominant disease characterized by tumors of the pancreatic islet cells, parathyroid glands, and pituitary gland, 40% will develop pituitary adenomas the great majority of which are prolactinomas. Pituitary tumors associated with the MEN1 syndrome have demonstrated loss of heterozygosity of chromosome 11q13 (locale of the MEN1 gene), which has been implicated in malignant progression of pituitary adenomas.4 Mutation of this gene does not appear to increase the risk for sporadic pituitary adenomas.5 In Carney’s complex, a rare inherited condition characterized by endocrine overactivity, schwannomas, abnormal skin pigmentation, and myxomas, the implicated genetic defect is loss of function of PRKAR1A, the gene for which is located on chromosome 17q23-24.6 Additional genetic alterations are found in McCune-Albright syndrome, a genetic disorder of bones, skin pigmentation, and hormones associated with premature puberty. Activating or gain-of-function GNAS1 mutations (of the guanine nucleotide-binding protein [G protein], alpha-stimulating activity polypeptide 1 gene on chromosome 20q13.1) are seen. Patients with McCune-Albright syndrome are present in the mosaic state, resulting from a postzygotic somatic mutation appearing early in the course of development that yields a monoclonal population of mutated cells within variously affected tissues. In isolated familial somatotropinomas (i.e., the occurrence of two or more cases of acromegaly in a family without a history of MEN or Carney’s complex), germline alterations in the aryl hydrocarbon receptor interacting gene (AIP gene) have been identified.7,8
Epidemiology
Although 10% of normal adults have a pituitary abnormality detectable on MRI, pituitary tumors account for only 10% to 15% of all diagnosed primary brain tumors. The frequency of pituitary adenomas varies greatly, although recent community-based studies from Belgium and the United Kingdom suggest that the prevalence is higher than has been historically noted.9,10
Based on a meta-analysis, the overall estimated prevalence of pituitary adenomas was 16.7%, with 14.4% observed in autopsy series and 22.5% in radiographic series.11 Over time, different criteria have been used for distinguishing hyperplasia from pituitary adenomas. The broader application of immunohistochemical staining for pituitary hormones and the differences in slice thicknesses for MRI may account for the wide variation in incidence that is reported in the literature.
Biologic Characteristics and Molecular Biology
Despite the screening of common oncogenes and tumor suppressor genes for pituitary adenomas, the initial transforming event for tumorigenesis has not been identified. Based on X chromosome inactivation analysis, it is apparent that most pituitary adenomas derive from a monoclonal expansion, which suggests that general principles of tumorigenesis may apply to pituitary adenoma formation.12 The MAPK (mainly Ras/extracellular signal-regulated protein kinase [ERK]) and the PI3K/Akt signaling pathways appear to have putative roles in pituitary tumorigenesis because activity of these pathways is increased with pituitary tumors. Although its role is not fully understood, a docking protein in the endothelial growth factor receptor (EGFR) pathway, EGF pathway substrate number 8 (Eps8), is overexpressed (5.9 times higher) in pituitary tumors compared with normal anterior pituitary gland tissue.13 Because pituitary tumors are mostly benign, a protective mechanism to restrict uninhibited growth of tumor cells is probably active. The concept of oncogene-induced senescence is one possible mechanism. High pituitary p21Cip1/WAF1 levels appear to promote senescence and restrict tumor growth.14
For somatotroph (growth hormone) adenomas, an activating mutation of the alpha subunit of the guanine nucleotide stimulatory protein (Gs-alpha) gene is found in about 40% of patients, which results in activation of adenylyl cyclase.15 Overexpression of the pituitary tumor transforming gene (PTTG) is also seen in most human somatotroph adenomas.16 Mutations of fibroblast growth factor–4 (FGF-4) have led to development of lactotroph adenomas in transgenic mice.17
Anatomy, Pathways of Spread, and Pathology
Anatomy and Pathways of Spread
The pituitary gland is a midline intracranial organ that measures approximately 8 mm in the anteroposterior axis, 10 mm in the transverse axis, and 6 to 8 mm in the superior-inferior axis. It occupies the cavity in the sphenoid bone known as the sella turcica. The pituitary gland is separated from the structures above it (i.e., the optic chiasm, hypothalamus, anterior cerebral arteries, and floor of the third ventricle) by the sellar diaphragm, which is formed by a circular fold of dura. Immediately anterosuperior to the diaphragm sella lies the optic chiasm. The pituitary stalk (infundibulum) crosses the sellar diaphragm and connects the hypothalamus to the pituitary gland. Anteriorly, the tuberculum sellae lies in the floor of the sella and extends laterally to form the anterior clinoid processes. The posterior aspect of the sella forms the dorsum sellae and extends laterally to form the posterior clinoid processes. Laterally, the cavernous sinuses contain cranial nerves III, IV, V1 (ophthalmic nerve), V2 (maxillary nerve), and VI and the internal carotid artery. Given the proximity of the pituitary to these various structures, parasellar tumors can affect cranial nerves III, IV, V, and VI, and suprasellar tumor extension can lead to bitemporal hemianopsia via compression of the optic chiasm. Figure 27-1 demonstrates the complex anatomy of the parasellar region.
The pituitary has two distinct embryologic origins; the first gives rise to the anterior and intermediate pituitary lobes, and the second gives rise to the posterior pituitary lobe. The anterior lobe (adenohypophysis) and intermediate lobe are derived from Rathke’s pouch, which is an evagination of ectodermal tissue from the primitive oral cavity (pharyngeal pouches). The posterior lobe (neurohypophysis) and stalk are derived from the diencephalon and, therefore, have a neuroectodermal origin. The anterior pituitary lobe accounts for most of the pituitary gland and produces at least six hormones: prolactin (PRL), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), growth hormone (GH), and thyroid-stimulating hormone (TSH).18 The intermediate lobe produces melanocyte-stimulating hormone (MSH). The posterior lobe stores and releases two hormones, vasopressin (antidiuretic hormone [ADH]) and oxytocin, which are produced by the hypothalamus.
Pituitary tumors are generally classified as microadenomas or macroadenomas depending on the diameter of the tumor as described by Hardy.19 A microadenoma is a tumor less than 1 cm in diameter, whereas a macroadenoma has a diameter of 1 cm or greater. Incidental microadenomas have been observed with a frequency of 3% to 25% in large, unselected autopsy series.20 Microadenomas are more frequently seen in females, whereas macroadenomas occur with equal frequency in males and females. Macroadenomas are more common than microadenomas. For lesions less than 3 mm in diameter, the term picoadenoma has been used.21
Pathology
According to the World Health Organization (WHO) classification of pituitary tumors, these tumors are defined as neoplasms located in the sella turcica.22 This classification is based on structural similarities of the normal parenchymal cells and the immunohistochemical demonstration of hormone secretion. As a result, pituitary adenomas may express more than one or two hormones. The classification includes a new entity designating a borderline adenoma or adenoma of uncertain behavior.22 The atypical adenoma is defined as an invasive tumor with an elevated mitotic index (MIB-1-labeling index greater than 3%) and extensive nuclear immunostaining for TP53.22
Most lesions are easily identified on routine histopathologic testing, but metastatic well-differentiated neuroendocrine carcinoma can present a diagnostic challenge.23 For patients with acromegaly or Cushing’s disease, hyperplasia may be clinically indistinguishable from an adenoma. Differentiation of hyperplasia from adenoma can be made using a reticulin stain. Normal pituitary tissue is composed of small acini of pituitary cells surrounded by an intact reticulin network. In hyperplasia, the architecture of the acini is maintained, but the acini are increased in size, and the reticulin stain demonstrates an intact network. Pituitary adenomas are characterized by a complete disruption of the reticulin fiber network.
The pathologic hallmark of pituitary adenomas is the monotonous and monomorphous proliferation of neoplastic cells that replace the normal acinar pattern in the pituitary lobe. Growth hormone–producing cells are seen with greater frequency anteriorly in the lateral aspects of the pituitary. Prolactin-producing cells are distributed throughout the pituitary but have a greater density in the posterior lateral aspect of the gland. Adrenocorticotropic hormone–producing cells are present in the median wedge. Thyroid-stimulating hormone–producing cells are seen in the anterior aspect of the median wedge. Gonadotropin-producing cells are distributed throughout the anterior pituitary. Nonfunctioning adenomas typically have solid sheets, nests, and sinusoidal patterns and are interrupted by pseudopapillae and striking pseudorosettes around vascular channels. Overlap of the distribution of these functional hormonal secretory cells does exist, and they do not reside in well-demarcated zones of the pituitary. Mitoses are seen in 3.9% of noninvasive adenomas, 21.4% of invasive adenomas, and 66.7% of carcinomas.24
The usefulness of labeling-index and TP53 immunostaining is not clear, because these assessments may not correlate with tumor behavior. Invasive pituitary adenomas usually exhibit a higher Ki-67 proliferation index.25 Markers such as proliferating cell nuclear antigen (PCNA), Ki-67/MIB-1, and antiapoptotic Bcl-2 have not demonstrated consistent correlation with tumor invasiveness or recurrence.26 Galectin-3, a β-galactoside-binding protein, may play a role in pituitary tumor progression, and immunostaining for this is also available.27
The pituitary is composed of at least six distinct cell types, which are responsible for the production of at least one hormone. Advances in molecular biology have identified three major pathways (T pit; Pit-1; and SF-1, GATA-2, ER) of cytodifferentiation of adenohypophyseal cells.28,29 These pathways are determined by a complex pattern of transcription expression, which can help classify the adenomas.30 In situations where histologic and immunohistochemical profiles are atypical, electron microscopy can be helpful in achieving accurate classification.
Clinical Manifestations, Patient Evaluation, and Staging
Clinical Manifestations
Patients with pituitary tumors can have a number of different symptoms and presentations, depending on the size and mass effect of the tumor and hormonal abnormalities. Headaches can occur with a lesion of any size. When the tumor extends superiorly into the suprasellar region, visual loss can occur, including bitemporal or homonymous hemianopsia, superior or inferior field cut deficits, and central scotoma. Extension into the cavernous sinus can lead to cranial nerve dysfunction (commonly, palsies of cranial nerves III and IV). Cranial nerves V and VI may also be affected. Lateral extension into the temporal lobe may cause seizures. The clinical presentations and endocrine abnormalities can vary, depending on the age and sex of the patient. Patients with large adenomas may have compromised pituitary function, which can lead to hypogonadism, secondary hypothyroidism, and adrenal insufficiency.31 Diabetes insipidus is extremely rare and most commonly is a manifestation of an invasive and infiltrative tumor (such as a metastasis), an inflammatory condition (such as neurosarcoidosis), or a lesion that arises from or is attached to the infundibulum or hypothalamus (such as a craniopharyngioma).32 If the gland acutely infarcts or hemorrhages, pituitary apoplexy may result, which may require urgent surgical decompression.
Patient Evaluation
Laboratory Assessment
Initial tests can determine a pituitary deficiency and diagnose the secretory status of the adenoma. Screening studies should include thyroid-stimulating hormone, free thyroxine (T4), adrenocorticotropic hormone, cortisol, prolactin, somatomedin C (insulin-like growth factor–1 [IGF-1]), luteinizing hormone, follicle-stimulating hormone, alpha subunit, and, in men, testosterone. In addition to endocrine evaluation, a complete blood count, blood chemistry assessment, and urinalysis should be obtained. Because physiologic hormonal variations in the blood and urine levels can occur, the interpretation of these results should be considered based on diurnal variations, age and gender of the patient, and pregnancy and menopausal status. The conditions and timing under which the samples are obtained also influence interpretations of the results. Because the diagnosis of Cushing’s disease can be difficult, multiple tests performed over time and at different times during the day may be needed for this diagnosis to be conclusive.33
For patients with prolactinoma who also have macroadenoma, the prolactin level is usually greater than 200 ng/mL. Other common conditions that have been associated with hyperprolactinemia include end-stage renal disease, renal insufficiency, depression, primary hypothyroidism, acquired immunodeficiency syndrome, sarcoidosis, and nonalcoholic cirrhosis.34 On the other hand, pituitary stalk compression from tumor may also cause elevated prolactin levels in the range of 150 ng/mL or less. Because antidepressants, protease inhibitors, verapamil, and phenothiazines may elevate prolactin levels, carefully review the medications that the patient is taking. A pregnancy test is mandatory for women with amenorrhea or hyperprolactinemia. Elevated serum prolactin levels greater than 300 ng/mL are usually diagnostic of pituitary adenoma, and levels higher than 100 ng/mL in nonpregnant patients often are associated with pituitary adenoma.
For patients with acromegaly, most cases result from excess secretion of growth hormone by a pituitary tumor.35 The definitive test is measurement of growth hormone response to 75 or 100 g of oral glucose (oral glucose tolerance test). This glucose load should normally cause marked suppression of the growth hormone release to a level below 2 ng/mL. To ensure accuracy of the test, these measurements of serum glucose and growth hormone must be taken every 30 minutes for 2 hours. The IGF-1 value is also elevated after adjusting for sex and age variation. IGF-1 levels provide the best intermittent method for monitoring response to treatment, although dynamic testing of growth hormone kinetics with the oral glucose tolerance test (OGTT) is also frequently used.
Imaging
MRI of the brain with gadolinium is the imaging test of choice, given its superior resolution compared with CT scanning (Fig. 27-2). Thin slices (at 2- to 3-mm intervals) obtained before and after gadolinium contrast administration with images in the coronal, axial, and sagittal planes provide detailed information for the initial diagnosis and allow detection of small lesions. The posterior lobe of the pituitary has a high signal intensity on T1-weighted images (posterior pituitary bright spot) that distinguishes it from the anterior lobe, which has a signal intensity similar to that of white matter. Dynamic coronal imaging techniques after contrast administration enhance normal pituitary tissue earlier and more intensely and help delineate adenoma tissue, which tends to enhance later. For patients who have undergone previous surgery, fat suppression techniques can help differentiate surgical fat grafts from tumor tissue. For patients undergoing stereotactic radiosurgery (SRS), thin-slice (1-mm) imaging with contrast medium is obtained to define the tumor and optic apparatus. For hypersecretory adenomas, positron emission tomography (PET) imaging with co-registration may be valuable.36
For patients with suspected Cushing’s disease, thin-slice images of 1-mm thickness have greater sensitivity; even so, the tumor may not be detected in about 50% of patients.37 Spoiled gradient recalled acquisition sequences may have superior sensitivity (80%) compared with conventional spin echo images following contrast enhancement.38 The degree of contrast enhancement does not differentiate one sellar mass from another. A clear distinction between an intrasellar mass and normal pituitary tissue is not consistent with a pituitary adenoma.
Staging
Classification of detectable or symptomatic pituitary tumors can be based on endocrine function, clinical presentation, and anatomic extent of disease. These tumors can also be broadly classified as functional or nonfunctional based on their secretory activity. As adenomas enlarge, they can extend into suprasellar, parasellar, or infrasellar structures. To aid with surgical and imaging assessment, Hardy and Verzina39 developed a classification system. Wilson’s40 modification of this classification system incorporates imaging and intraoperative findings of sellar destruction (grade) and extrasellar extension (stage). Others have defined giant adenoma as a lesion with extension beyond the sella and suprasellar space or size greater than 4 cm.41,42 No universally accepted staging system exists for pituitary tumors.
Primary Therapy
Surgery
Advances in transsphenoidal approaches with microsurgical techniques have made this procedure the initial treatment of choice for patients with nonfunctioning pituitary adenomas, acromegaly, and Cushing’s disease. Surgery provides immediate decompression for patients with progressive visual loss or pituitary hemorrhage. For patients with prolactinoma who do not tolerate or respond to medical therapy, surgery can be considered. Surgery has also been useful in reducing the tumor bulk for combined management with radiation therapy or medical therapy. For patients undergoing SRS, surgery may play an important role because a distance of 2 to 5 mm is needed between the tumor and the optic apparatus to deliver a sufficient dose to the tumor and to minimize the risk for optic neuropathy. Reoperation results in lower rates of success compared with the initial surgery.43
The transsphenoidal approach has become less invasive with development of a newer approach, endoscopic, minimally invasive pituitary surgery (MIPS). This technique can result in shorter hospitalization times.44,45 With MIPS, nasal or intraoral incisions are not needed and nasal speculums and nasal packing need not be used. There appears to be a decrease in complication rates compared with traditional sublabial transseptal approaches. MIPS is performed jointly with otorhinolaryngologists. It may also be used to complement microsurgical approaches.
Based on a questionnaire study of 958 neurosurgeons reporting on their own experience with transsphenoidal surgery, the risk for complications was inversely proportional to the surgeon’s level of experience.46 Reported complication rates among experienced surgeons include perioperative mortality (<1%), central diabetes insipidus (2%), cerebrospinal rhinorrhea (2%), and meningitis (2%). Transient central diabetes insipidus is very common and reported to occur in 22% of patients at a very active pituitary surgery center.47 These complications are more common in patients with macroadenomas. Less experienced surgeons are more likely to cause the above complications.45,48
Medical Therapy
Medical therapy can be used in several situations for patients with growth hormone–secreting tumors. Somatostatin analogs such as octreotide or lanreotide can be used to lower elevated growth hormone levels before surgery, for patients with persistent growth hormone and IGF-1 elevation after surgery, and for patients who continue to receive medical therapy until radiation therapy or radiosurgery has taken full effect. Pegvisomant, a growth hormone receptor antagonist, appears to be the most effective medical therapy for IGF-1 normalization.49 The cost of this therapy is, however, substantial (>$50,000 per year).
Medical treatment of hormonal deficiencies resulting from the pituitary tumor is best managed by an endocrinologist. Hypopituitarism can have variable clinical manifestations, and management should be individualized to minimize the impact of this condition. Depending on the deficiency, glucocorticoids, gonadal steroids, and thyroid hormones may need to be initiated. Commonly used medicines include hydrocortisone and cortisone acetate for glucocorticoid replacement and L-thyroxine for hypothyroidism. Gonadal steroids used include estrogen and progestin for women and testosterone for men. Careful optimization of medication supplementation, especially to avoid supraphysiologic doses of steroids, appears to be important in minimizing the effect of hypopituitarism.50
Radiation Therapy and Stereotactic Radiosurgery
The lack of prospective, randomized data comparing different radiation delivery methods has resulted in strong proponents for fractionated versus radiosurgical approaches.51,52,53 Most of the literature is based on single institutional results.
Advances in radiation therapy have led to modern radiation techniques that allow more focused delivery of radiation to the tumor and decreased dose to the normal brain. Sophisticated treatment planning systems, incorporation of CT and MRI studies, stereotactic guidance, radiation approaches such as intensity-modulated radiation therapy and image-guided radiation therapy, micro-multileaf collimators, and relocatable frames or masks have improved precision.53,54,55 Results with radiation therapy, including specifics regarding dosing and margins, will be discussed in greater detail later in the chapter.
Stereotactic radiosurgery (SRS) delivers a highly focused, single, large fraction of ionizing radiation to a small intracranial target volume (Fig. 27-3). Frame-based systems (e.g., proton beam, helium ion, Gamma Knife, and linear accelerator SRS techniques) and frameless systems (e.g., CyberKnife and linear accelerator-based units) have been used. Given the sharper dose gradient of SRS versus fractionated approaches, the potential for radiation injury to normal brain and vessels and secondary tumor formation may be less with SRS.
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
