[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 18
Pituitary insufficiency
1. What is pituitary insufficiency?
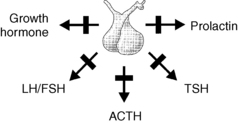
Pituitary insufficiency is a syndrome characterized by one or more anterior pituitary hormone deficiencies as a result of aplasia or hypoplasia, destruction, infiltration, compression, or displacement of the hypothalamus and/or pituitary gland (Fig. 18-1). Pituitary insufficiency can be congenital or acquired; familial or sporadic; partial or complete; and transient (reversible) or permanent. Posterior pituitary failure, characterized by decreased concentrations of antidiuretic hormone, is referred as central diabetes insipidus.
2. Is diabetes insipidus a manifestation of pituitary insufficiency?
Most patients with anterior pituitary insufficiency do not have concomitant posterior pituitary failure. In those in whom central diabetes insipidus is also present, hypophysitis, metastatic cancer, or sarcoidosis should be suspected.
3. How common is hypopituitarism in the general population?
According to a population study conducted in northwestern Spain, the annual incidence of hypopituitarism is 42 cases per million, and the prevalence ranges from 290 to 455 cases per million.
4. What causes pituitary insufficiency?
Almost any disease that disturbs the normal interaction between the hypothalamus and the pituitary gland can cause hypopituitarism. The most common etiology of pituitary insufficiency is pituitary damage associated with a pituitary adenoma and/or the effect of its treatment (surgery and/or radiation therapy). Among patients with pituitary macroadenomas, about one third have one or more pituitary hormone deficiencies. Other frequent causes are shown in Box 18-1.
5. How does a patient with pituitary insufficiency present?
The clinical manifestations of hypopituitarism depend on the extent and severity of the specific pituitary hormone deficiency. If the onset is acute, the patient may be critically ill and present with hypotension and shock, obtundation, and even coma. However, if the onset is chronic and the pituitary deficiency is mild, the patient may complain only of fatigue and malaise.





6. Are there any signs on physical examination that may suggest pituitary insufficiency?
Aside from delayed relaxation of tendon reflexes in hypothyroid patients, there are no specific or pathognomonic findings on physical exam. Physical findings for specific pituitary hormone deficiencies are as follows:





7. How is hypopituitarism diagnosed?
In the setting of typical manifestations of hypopituitarism, measurement of basal serum hormone levels may be all that is needed. On the other hand, dynamic endocrine testing is usually indicated in patients with more subtle manifestations.
 ACTH deficiency: Nonspecific laboratory findings include hyponatremia, normochromic, normocytic anemia, and eosinophilia. A morning serum cortisol level lower than 3 μg/dL and/or a peak serum cortisol level lower than 20 μg/dL 30 or 60 minutes after the administration of 250 μg of ACTH and an inappropriately low or normal plasma ACTH level are consistent with central adrenal insufficiency.
ACTH deficiency: Nonspecific laboratory findings include hyponatremia, normochromic, normocytic anemia, and eosinophilia. A morning serum cortisol level lower than 3 μg/dL and/or a peak serum cortisol level lower than 20 μg/dL 30 or 60 minutes after the administration of 250 μg of ACTH and an inappropriately low or normal plasma ACTH level are consistent with central adrenal insufficiency.



 PRL deficiency: Low or undetectable serum PRL concentration.
PRL deficiency: Low or undetectable serum PRL concentration.
8. When should pituitary magnetic resonance imaging (MRI) be ordered in someone with hypopituitarism?
Any patient with two or more pituitary hormone deficiencies, unexplained or significant hyperprolactinemia, or symptoms of tumor mass effect should undergo dedicated pituitary MRI with and without contrast agent. Patients with isolated functional hypopituitarism do not usually need imaging studies. Men with isolated central hypogonadism with a serum testosterone level lower than 100 ng/mL should undergo imaging.
9. How common is pituitary insufficiency after traumatic brain injury?
The annual incidence of traumatic brain injury in industrialized countries is 180 to 250 cases per 100,000 people. Longitudinal studies that have included patients with different degrees of severity of traumatic brain injury have shown that pituitary insufficiency is common after head trauma. Although some of these deficiencies resolve within weeks or months after the cranial trauma, some new deficiencies may be detected during follow-up. Approximately 25% to 50% of patients have at least one anterior pituitary hormone deficiency a year after suffering traumatic brain injury. GH and gonadotropin deficiencies are the two most common disturbances. The causative mechanism seems to be hemorrhagic infarction of the pituitary and/or hypothalamus as a result of direct damage to these structures, increased intracranial pressure, hypoxia, or bleeding.
10. How soon after radiation therapy should pituitary insufficiency be expected?
Any radiation fields that impact the hypothalamic-pituitary area can cause neuroendocrine dysfunction. Irradiation for sellar and parasellar tumors, primary brain tumors, nasopharyngeal carcinoma, acute lymphoid leukemia, and tumors of the skull base have been shown to compromise pituitary function. Depending on the radiation dose and the presence of preexisting pituitary disease, it may take from several months to years for pituitary insufficiency to develop. The 5-year cumulative incidence of GH deficiency, gonadotropin deficiency, ACTH deficiency, and TSH deficiency in patients with pituitary tumors (radiation dose: 30-50 Gray [Gy]) and patients with nasopharyngeal carcinoma (radiation dose: > 60 Gy) were 100%, 57%, 61%, and 27.5%; and 63.5%, 31%, 27%, and 15%, respectively. Regular testing is mandatory to ensure timely diagnosis and early treatment.
11. What is Sheehan’s syndrome?
Sheehan’s syndrome is a form of panhypopituitarism that occurs after delivery as a consequence of infarction in the adenohypophysis due to massive uterine hemorrhage and hypovolemia. Typically women have an inability to lactate for the newborn, persistent amenorrhea, and symptoms of hypocortisolemia and hypothyroidism. Pituitary imaging studies show an atrophic pituitary gland and sometimes an empty sella. Pathologic studies have shown replacement of organizing necrotic areas by a fibrous scar. The pituitary gland cannot regenerate; new cells do not form to replace the necrotized cells. With modernization of medicine and improved obstetric care, the incidence of Sheehan’s syndrome has plummeted in industrialized countries.
12. What is pituitary apoplexy?
Pituitary apoplexy is the abrupt destruction of most of the anterior pituitary cells as a result of an acute hemorrhage and/or infarction within an unrecognized pituitary adenoma. Precipitating factors for this complication include anticoagulation therapy, bleeding disorders, head trauma, diabetes mellitus, and radiation therapy. Patients usually present with severe headaches, obtundation, ophthalmoplegia, visual loss, hypotension, and shock. Biochemical panhypopituitarism and acute hemorrhage within a pituitary adenoma on pituitary MRI confirm the diagnosis. Rapid glucocorticoid and thyroid replacement therapies and surgical decompression, if needed, are life-saving. Of note, small hemorrhages within known pituitary adenomas may be incidentally detected during monitoring pituitary MRI.
Hypophysitis is chronic inflammation of the pituitary gland that has been classified according to the anatomic location of the pituitary involvement, cause, and histopathologic appearance. On the basis of clinical, radiologic, and pathologic findings, it is divided into adenohypophysitis, infundibuloneurohypophysitis, and panhypophysitis. Depending on the etiology, it can be classified as primary or secondary, the latter having a clear cause. According to pathology, it can be lymphocytic, granulomatous, or other less common forms. Lymphocytic hypophysitis is the most common and is characterized by a marked infiltration of lymphocytes that populate the pituitary gland both diffusely and occasionally in focal clusters. Lymphocytes are accompanied by scattered plasma cells, eosinophils, and fibroblasts, and in later disease stages by fibrosis. Lymphocytic hypophysitis is three times more common in women and uniquely manifests in association with pregnancy and the postpartum period in about 40% of affected women.
14. What is empty sella syndrome?
An empty sella turcica occurs as a result of intrasellar herniation of the suprasellar subarachnoid space with compression of the pituitary gland, producing in many cases a remodeling of the sella resulting from a combination of an incomplete diaphragma sella and increased cerebrospinal fluid (CSF) pressure. It is classified as primary or secondary. Primary empty sella syndrome is more common in obese multiparous women, who may complain of headaches and have hypertension; pituitary function is usually normal. Secondary empty sella syndrome is due to pituitary diseases, surgery, or irradiation. The predominant clinical finding in patients with this disorder is a visual abnormality, occurring from arachnoidal adhesions and traction on the optic apparatus. Some patients can also have mild hyperprolactinemia due to stretching of the pituitary stalk.
15. Describe functional causes of pituitary insufficiency.
It is important to exclude functional hypopituitarism in the patient presenting with an isolated pituitary deficiency. Patients who are receiving high doses of oral glucocorticoids for more than 6 weeks, are having frequent articular or epidural glucocorticoid injections, or are using high doses of potent inhaled glucocorticoids may demonstrate transient adrenal atrophy. As expected, these patients present with cushingoid features. Body builders using anabolic steroids can present with central hypogonadism. Hypothalamic amenorrhea can be caused by anorexia nervosa and strenuous exercise. Decreased testosterone or estrogen concentrations can normalize after correction of hyperprolactinemia with dopamine agonists. Critical illness and high-dose narcotics may suppress both the hypothalamic-pituitary-adrenal and hypothalamic-pituitary-gonadal axes.
16. Describe the treatment for pituitary insufficiency.
 ACTH deficiency (central adrenal insufficiency): Hydrocortisone 15-25 mg/day, in two or three divided doses, or prednisone 5 mg per day, are commonly used to replace cortisol. Because ACTH is not the main determinant of aldosterone secretion, patients with central adrenal insufficiency do not need fludrocortisone. Some women with decreased libido and muscle weakness may benefit from taking 25 to 50 mg/day of dehydroepiandrosterone (DHEA). Patients should be educated about doubling the dose of hydrocortisone or prednisone in case of intercurrent or febrile illnesses.
ACTH deficiency (central adrenal insufficiency): Hydrocortisone 15-25 mg/day, in two or three divided doses, or prednisone 5 mg per day, are commonly used to replace cortisol. Because ACTH is not the main determinant of aldosterone secretion, patients with central adrenal insufficiency do not need fludrocortisone. Some women with decreased libido and muscle weakness may benefit from taking 25 to 50 mg/day of dehydroepiandrosterone (DHEA). Patients should be educated about doubling the dose of hydrocortisone or prednisone in case of intercurrent or febrile illnesses.



17. Can treatment of one pituitary deficiency unmask others?
The effect of GH on interconversion of T4 to triiodothyronine (T3) results in masking of TSH deficiency in the GH-deficient state with subsequent lowering of T4 during GH replacement therapy.
Increased activity of 11β-hydroxysteroid dehydrogenase type 1 in GH deficiency results in an alteration of the set point of cortisol-to-cortisone interconversion in individual tissues in favor of cortisol. Thus, GH replacement, by increasing the conversion from cortisol to cortisone, may expose occult central adrenal insufficiency in patients with borderline ACTH reserve. Gonadal steroids influence GH-mediated hepatic IGF-1 generation. Oral estrogens decrease it, and DHEA sulfate increases it; therefore, women taking the former need higher doses of GH to achieve a normal IGF-1 level, whereas those taking DHEA need lower doses. Commencement of glucocorticoid replacement therapy can unmask central diabetes insipidus. Finally, because thyroid hormone increases the metabolic clearance rate of cortisol, initiating thyroid hormone replacement in a patient with coexistent but unrecognized adrenal insufficiency can precipitate an adrenal crisis.
18. Is life expectancy altered by hypopituitarism?
Rates of all-cause mortality and vascular death are higher in patients with hypopituitarism than in age- and sex-matched controls. A metaanalysis concluded that the standardized mortality ratio associated with hypopituitarism in men is 2.06 [95% confidence interval (CI), 1.94-2.20] and in women 2.80 (95% CI, 2.59-3.02). Pituitary irradiation seems to impose an increased risk of death from cerebrovascular disease.
 KEY POINTS 1
KEY POINTS 1
1. The best test to confirm adequacy of thyroid replacement therapy in patients with central hypothyroidism is a free T4 concentration in the mid-normal range.
2. A dedicated pituitary MRI (more slices through the sellar and parasellar regions), with and without contrast agent, rather than a brain MRI, should be ordered if indicated, in patients with hypopituitarism.
3. Traumatic brain injury and subarachnoid hemorrhage are increasingly recognized as causes of hypopituitarism.
 WEBSITES
WEBSITESAimaretti, G, Ambrosio, MR, Di Somma, C, et al, Residual pituitary function after brain injury-induced hypopituitarism. a prospective 12-month study. J Clin Endocrinol Metab 2005;90:6085–6092.
Appelman-Dijkstra, NM, Kokshoorn, NE, Dekkers, OM, et al, Pituitary dysfunction in adult patients after cranial radiotherapy. systematic review and meta-analysis. J Clin Endocrinol Metab 2011;96:2330–2340.
Darzy, KH, Shalet, SM. Hypopituitarism following radiotherapy. Pituitary. 2009;12:40–50.
Kovacs, K. Sheehan syndrome. Lancet. 2003;361:520–522.
Nawar, RN, Abdel-Mannan, D, Selman, WR, et al, Pituitary tumor apoplexy. a review. J Intensive Care Med 2008;23:75–90.
Nielsen, EH, Lindholm, J, Laurberg, P, Excess mortality in women with pituitary disease. a meta-analysis. Clin Endocrinol (Oxf) 2007;67:693–697.
Orrego, JJ, Barkan, AL. Testing of hypothalamic-pituitary axis. In: Shepard MC, Stewart PM, eds. Pituitary disease. 1st ed. Netherlands: Kluwer Academic Publishers; 2002:247–267.
Regal, M, Paramo, C, Sierra, SM, et al. Prevalence and incidence of hypopituitarism in an adult Caucasian population in northwestern Spain. Clin Endocrinol (Oxf). 2001;55:735–740.
Schneider, HJ, Aimaretti, G, Kreitschmann-Andermahr, I, et al. Hypopituitarism. Lancet. 2007;369:1461–1470.
Sherlock, M, Ayuk, J, Tomlinson, JW, et al. Mortality in patients with pituitary disease. Endocrine Reviews. 2010;31:301–342.
Toogood, AA, Stewart, PM, Hypopituitarism. clinical features, diagnosis, and management. Endocrinol Metab Clin N Am 2008;37:235–261.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 18
Pituitary insufficiency
1. What is pituitary insufficiency?
Pituitary insufficiency is a syndrome characterized by one or more anterior pituitary hormone deficiencies as a result of aplasia or hypoplasia, destruction, infiltration, compression, or displacement of the hypothalamus and/or pituitary gland (Fig. 18-1). Pituitary insufficiency can be congenital or acquired; familial or sporadic; partial or complete; and transient (reversible) or permanent. Posterior pituitary failure, characterized by decreased concentrations of antidiuretic hormone, is referred as central diabetes insipidus.
2. Is diabetes insipidus a manifestation of pituitary insufficiency?
Most patients with anterior pituitary insufficiency do not have concomitant posterior pituitary failure. In those in whom central diabetes insipidus is also present, hypophysitis, metastatic cancer, or sarcoidosis should be suspected.
3. How common is hypopituitarism in the general population?
According to a population study conducted in northwestern Spain, the annual incidence of hypopituitarism is 42 cases per million, and the prevalence ranges from 290 to 455 cases per million.
4. What causes pituitary insufficiency?
Almost any disease that disturbs the normal interaction between the hypothalamus and the pituitary gland can cause hypopituitarism. The most common etiology of pituitary insufficiency is pituitary damage associated with a pituitary adenoma and/or the effect of its treatment (surgery and/or radiation therapy). Among patients with pituitary macroadenomas, about one third have one or more pituitary hormone deficiencies. Other frequent causes are shown in Box 18-1.
5. How does a patient with pituitary insufficiency present?
The clinical manifestations of hypopituitarism depend on the extent and severity of the specific pituitary hormone deficiency. If the onset is acute, the patient may be critically ill and present with hypotension and shock, obtundation, and even coma. However, if the onset is chronic and the pituitary deficiency is mild, the patient may complain only of fatigue and malaise.
6. Are there any signs on physical examination that may suggest pituitary insufficiency?
Aside from delayed relaxation of tendon reflexes in hypothyroid patients, there are no specific or pathognomonic findings on physical exam. Physical findings for specific pituitary hormone deficiencies are as follows:
7. How is hypopituitarism diagnosed?
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
