[level-membership-for-neonatal-and-perinatal-medicine-category]
Pituitary, adrenal, and thyroid function
Pituitary, adrenal, and thyroid function and the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-thyroid (HPT) axes are critical for normal function and adaptation during pregnancy, growth, and development of the fetus, and adaptation of the newborn to the extrauterine environment. HPA axis function in the mother and fetus are closely interrelated with placental function. The hormones of the HPA and HPT axes (see Figure 2-7) are necessary for many body functions, for development of the central nervous system (CNS) and other growth processes, and for reproductive function. Disorders of these systems are associated with infertility, alterations in normal changes at puberty, and complications of pregnancy. Concentrations of HPA and HPT axis hormones are altered in pregnant women and neonates. In the neonate marked changes in adrenal and thyroid function occur with birth. This chapter examines changes in the glands and hormones of the HPA and HPT axes during pregnancy; development of neuroendocrine function in the fetus and neonate; and implications for the mother, fetus, and neonate.
Maternal physiologic adaptations
Antepartum period
Hypothalamic-pituitary-adrenal axis
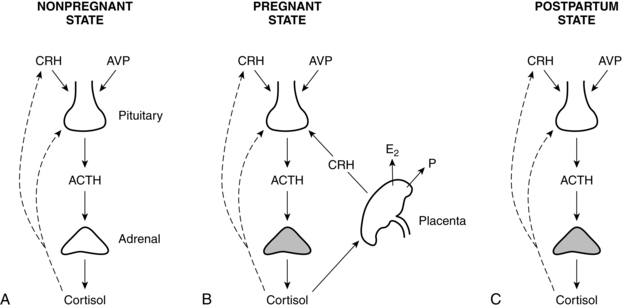
Marked changes occur in the hypothalamic-pituitary-adrenal (HPA) axis during pregnancy, resulting in a state of increased HPA function.77,113 (See Figure 2-7 for an illustration of the HPA axis.) These changes are mediated primarily by placental hormones, including placental ACTH, GH, and CRH. Maternal hypothalamic-pituitary function is discussed further in Chapter 2 in conjunction with the hypothalamic-pituitary-ovarian axis. Changes in the hypothalamus and pituitary during pregnancy are summarized in Box 19-1 on page 628.
Anterior pituitary function.
The anterior pituitary is composed of six cell types, each of which produces different hormones. These types of cells and their major hormones include lactotroph (prolactin), corticotroph (pro-opiomelanocortin [POMC] and its derivatives including ACTH, β-endorphin, and β-lipotropin), somatotroph (GH), gonadotroph (follicle-stimulating hormone [FSH] and luteinizing hormone [LH]), and cells producing thyroid-stimulating hormone (TSH) (thyrotropin). The anterior pituitary gland increases in size and weight (from an average of 660 mg in the nonpregnant woman to 760 mg or greater during pregnancy) due to an estrogen-induced increase in the lactotroph cells.32,35,86,93,97,151 The anterior pituitary also develops a more convex, dome-shaped surface, which may cause it to bulge upward in some women, compressing the optic chiasma.32 For these women, this can result in a transient hemianopia. Changes in pituitary function during pregnancy are summarized in Box 19-1 on page 628.
In the nonpregnant woman, the prolactin-producing lactotroph cells make up approximately 20% of the anterior pituitary; this increases to around 60% during pregnancy.151 Prolactin isoforms increase during pregnancy, with the nonglycosylated forms exceeding the N-linked glycosylated form that is most common in nonpregnant women.32,151 The nonglycosylated form may be more bioactive and function to prepare the breast for lactation (see Chapter 5). Prolactin increases 10-fold during pregnancy to peak at delivery at 140 ng/mL (6068 pmol/L).32,35,74,97 Most of the increase in maternal prolactin is from the maternal anterior pituitary, although prolactin is also produced by the maternal decidua. Decidual prolactin is found primarily in amniotic fluid; little enters the maternal circulation. Prolactin levels in the amniotic fluid peak in the second trimester at 6000 ng/mL (260,886 pmol/L).32,35,151
Corticotroph cells do not change in size during pregnancy.151 However, ACTH secretion and plasma ACTH levels increase progressively, peaking during the intrapartum period (Figure 19-1), from approximately 10 pg/mL (2.2 pmol/L) in nonpregnant women to 50 pg/mL (11 pmol/L) at term.32,35,97,151 ACTH secretion is stimulated by CRH and, in turn, stimulates release of cortisol by the adrenal gland. Changes in ACTH parallel the increase in free and total cortisol (see Figure 19-1).35,86 During pregnancy, the adrenal gland is more responsive to ACTH with a blunted HPA axis response to exogenous glucocorticoids.77 There is a two- to fourfold increase in ACTH from the first to third trimesters, in spite of the increased bound and free plasma cortisol.97 The increase in ACTH in the face of increased cortisol suggests a change in the set point for cortisol release that alters the ACTH-cortisol feedback loop.44,97 ACTH maintains its diurnal variation during pregnancy, although this cycling may be blunted.35,43,77,86,97 Placental ACTH increases in the second and third trimesters and it is unclear how much of increased maternal serum ACTH comes from the maternal anterior pituitary versus the placenta.93 The effects of ACTH on the adrenal gland and changes in cortisol during pregnancy are described further under Adrenal Function.
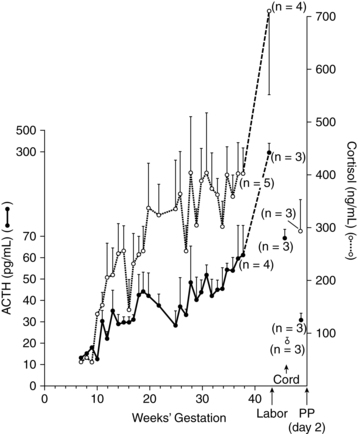
The increased ACTH secretion during pregnancy is thought to be due primarily to increased placental (and some decidual and fetal membrane) CRH, rather than maternal hypothalamic CRH (Figure 19-2, B).32,35,77,86,151,159 Other factors that may contribute to this increase are the decreased pituitary gland sensitivity to cortisol feedback, enhanced pituitary responsiveness to corticotropin-releasing factors such as vasopressin, and CRH.77,159 Maternal serum CRH increases markedly beginning by 8 to 10 weeks and rises from prepregnant values of 10 to 100 pg/mL (47.6 to 476 pmol/L) to 300 to 1000 pg/mL (1429 to 4762 pmol/L) by the third trimester.86,151,159 CRH-binding protein (CRH-BP) decreases the bioactivity of CRH throughout most of gestation. CRH-BP levels are similar to nonpregnant levels until the third trimester and then fall by two thirds during the last 6 weeks in preparation for birth. CRH has multiple roles in establishing and maintaining pregnancy and is produced by placental and uterine tissues in addition to the hypothalamus.159 CRH plays an important role in the onset of parturition; increased CRH prior to term plays a role in preterm labor onset (see Chapter 4).159
Somatotroph and gonadotroph cells of the anterior pituitary decrease during pregnancy.6 Hypothalamic gonadotropin-releasing hormone (GnRH) is suppressed in pregnancy by the elevated CRH, β-endorphins, and cortisol, with a blunted response of the pituitary to GnRH and low LH and FSH levels by 6 to 7 weeks.159 By midpregnancy, LH and FSH levels are undetectable.32,35,97,151 Gonadotropin function is described in Chapter 2. β-endorphins during pregnancy and the intrapartum period are described in Chapter 15.
Pituitary GH (GH-N) decreases after the first trimester as placental GH (GH-V) increases.35,93,97 GH-V is a GH variant that stimulates bone growth and regulates maternal insulin-like growth factor-I (IGF-I), which in turn alters maternal metabolism, stimulating gluconeogenesis and lipolysis.93 GH-V is secreted continuously, as opposed to the pulsatile secretion characteristic of GH-N.5 GH-N is the main maternal GH until 15 to 20 weeks; then it decreases and becomes undetectable by term. GH-V increases progressively from 15 to 20 weeks until term.97 GH-V stimulates IGF-I, with negative feedback suppression of maternal GH-N.5,32
Posterior pituitary function.
The major posterior pituitary hormones are arginine vasopressin (AVP) and oxytocin. Posterior pituitary function changes are associated with osmoregulatory changes and parturition. AVP levels are within normal ranges during pregnancy; however, the threshold at which AVP is secreted is reset so that AVP is secreted at a lower plasma osmolality during pregnancy (see Chapter 11) with a decline in plasma osmolality by 5 to 10 mOsm/kg below the nonpregnant mean of 285 mOsm/kg.97,98 AVP also modulates ACTH release.43 Oxytocin levels progressively increase during pregnancy, with further increase at term (see Chapter 4) and with lactation (see Chapter 5).35,97
Adrenal function.
Pregnancy is characterized by a transient hypercortisolism that begins by at least 12 weeks (see Figure 19-1).59,86 Both total serum cortisol (the primary glucocorticoid) and free cortisol increase, with total cortisol peaking at levels three- to eightfold times higher by term.97,127 Salivary cortisol increases twofold by 25 weeks and then plateaus to term.21 The pregnant woman does not demonstrate signs of hypercortisolism because the free cortisol fraction is still within normal range.21 Urinary cortisol increases at least 180% by term.97 The diurnal secretion of cortisol—with higher levels in the morning versus evening—is blunted but maintained.77,86,97 Physiologic responses to stress are maintained during pregnancy, but appear to be blunted.21,159
The increase in plasma cortisol parallels the increase in ACTH (see Figure 19-1). The increase in total cortisol is primarily due to an estrogen-stimulated increase in cortisol-binding globulin (CBG) that increases to two- to threefold during pregnancy.97 Increased CBG reduces liver catabolism and clearance of cortisol, yielding a twofold increase in cortisol half-life.86 The increase in free cortisol is also due in part to displacement of cortisol from CBG by progesterone.35
The adrenal gland becomes hypertrophic as the zona fasciculata (site of glucocorticoid production) widens, with no changes in the size of the zona glomerulosa or reticularis (see Box 19-2 on page 630).35 Levels of aldosterone (the primary mineralocorticoid) also are markedly increased in pregnancy (see Chapter 11). Synthesis of androgens and dehydroepiandrosterone sulfate (DHEA-S) by the zona reticularis also increases, but less so than the glucocorticoids and mineralocorticoids. An increase in sex hormone binding globulin during pregnancy increases total and protein bound testosterone levels. Levels of free testosterone are low to normal to 28 weeks and then increase along with levels of androstenedione.97 Maternal serum DHEA-S levels remain low due to placental uptake and metabolism.44
Hypothalamic-pituitary-thyroid axis
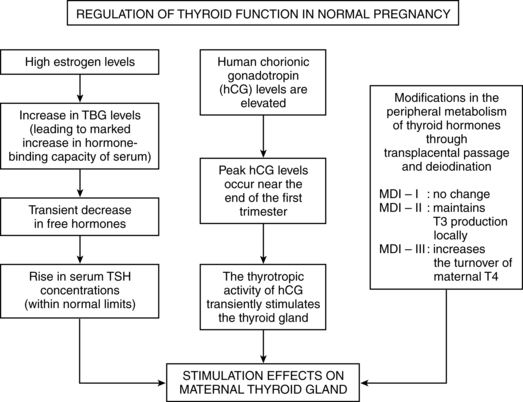
Marked changes are also seen in the HPT axis during pregnancy. These changes occur primarily in the first half of gestation so that the woman achieves a new steady state in HPT function by midgestation that is maintained until delivery.62,71 The net result of changes in thyroid function during pregnancy is to increase the availability of thyroid hormones by 40% to 100%.142 Morreale de Escobar et al. summarized the major changes in thyroid function during pregnancy: (1) Increased estrogen leads to a two- to threefold increase in thyroid-binding globulin production by the liver. This decreases levels of free thyroid hormone and stimulates the hypothalamic-pituitary-thyroid (HPT) axis; (2) increased human chorionic gonadotropin (hCG), which has a structure similar to thyroid-stimulating hormone (TSH), stimulates increased T3 and T4. This leads to negative feedback to the pituitary gland and a decrease in TSH (especially during weeks 8 to 14 when hCG is peaking); (3) peripheral metabolism of thyroid hormones increases in the second and third trimesters due to increased production of type II and III monodeiodinases (see Box 19-3 below) by the placenta.95
Adaptations during pregnancy related to thyroid physiology mimic hyperthyroidism. The pregnant woman can be described as being in a state of euthyroid hyperthyroxinemia, however, since thyroid function per se does not change during pregnancy.95 Thyroid hormone changes are important in supporting the altered carbohydrate, protein, and lipid metabolism of pregnancy and changes in basal metabolic rate (see Chapter 16).79 The factors primarily responsible for changes in HPT axis function during pregnancy are the elevated hCG and thyroid-binding globulin (TBG) levels and the increased urinary iodide excretion that lowers maternal plasma iodine.40,43 Box 19-3 on page 630, Box 19-4 below and Figure 19-3 review thyroid hormone production and regulation. Figure 19-4 illustrates regulation of maternal thyroid hormone function during pregnancy.
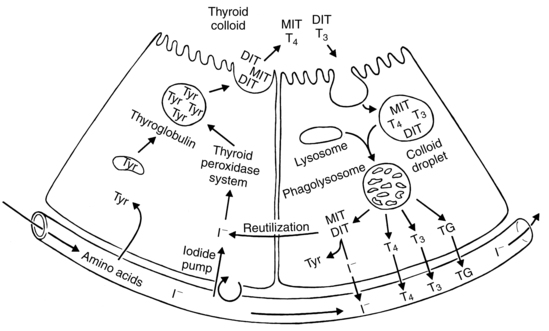
Thyroid hormones are transported in the blood bound to binding proteins, such as TBG, albumin, and transthyretin (TTR; formerly called thyroxine-binding prealbumin). In the nonpregnant individual, about two thirds of the T4 is bound to TBG, increasing to 75% or greater during pregnancy.34,43 Under the influence of estrogen, hepatic synthesis and sialylation of TBG increase twofold to threefold beginning within a few weeks after fertilization and plateau from midgestation to delivery.34,71,99,100,144 Increased sialylation increases the half-life of TBG. The ability of TBG to bind thyroxine doubles during this period; TTR, also influenced by estrogen, decreases.43 These changes increase serum TBG levels, decrease the percent of T4 bound to TTR and increase total T4 and T3.100 The increased TBG is accompanied by 10% to 15% decrease in free T4 and free T3 if iodine is sufficient; if iodine is inadequate, T4 levels increase.99
A transient increase in free T4 is reported in the first trimester, related to the increase in hCG, with a decrease in the second and third trimesters.71,72 Variations in findings are most often due to differences in the iodine status of the populations studied and measurement techniques.73 Free T3 changes parallel those of free T4.72 Concentrations of free T3 and free T4, although low, remain within normal physiologic limits.72,99 The basis for these changes is thought to be primarily related to the interaction of estrogen, TSH, and thyroid-binding proteins.72
In contrast to changes in free T3 and free T4, total T3 and T4 increase, peaking at 10 to 15 weeks’ gestation and then plateauing at levels 40% to 100% higher than nonpregnant values.142,144 This increase is primarily due to increases in TBG and hCG, and possibly to an increase in the production of MDI-III (see Box 19-3 on page 630) by the placenta.71 This enzyme converts T4 to reverse T3 (rT3), an inactive compound, and T3 to T2, another inactive compound.98 Levels of T3 are elevated because of the increased availability of T4 for deiodination to T3 in peripheral tissue rather than due to increased T3 production per se.
The increased T3 and T4 are also related to increased TSH bioactivity stimulated by hCG, which peaks at about the same time.58 hCG has a mild TSH-like activity that increases secretion of T4.34,100 hCG is a partial inhibitor of the pituitary gland and has β subunits similar to TSH (as well as luteinizing and follicle-stimulating hormone). Thus hCG has thyrotropic activity and can activate TSH receptors.34,100 Serum hCG is positively correlated with free T4 and inversely correlated with TSH levels in early pregnancy.40,73 Elevated T3 and T4 suppress endogenous TSH secretion by the anterior pituitary.98 TSH decreases transiently from 8 to 14 weeks’ gestation at the time of the hCG peak, progressively returning to prepregnancy levels by term.34,100,151 Occasionally some otherwise healthy pregnant women will experience a transient hyperthyroxinemia (generally without clinical signs) associated with higher than usual hCG levels in the first trimester and a greater fall in TSH.43,72,99 The increase in total T3 and T4 is less than the increase in TBG, resulting in a decreased T4/TBG ratio, leading to a state of relative hypothyroxinemia during pregnancy.
Pregnancy is characterized by significant changes in iodide metabolism with increased renal iodide clearance and increased iodine needed to make thyroid hormones.38,71 The increased total T4 and T3 stimulate increased serum protein-bound iodine (PBI).38 Because circulating levels of free thyroid hormone are not significantly altered, increased PBI does not reflect maternal hyperthyroidism. Thyroid iodine uptake increases because of a decrease in the total body iodine pool. This pool is altered because of increased renal iodide loss secondary to the increased renal blood flow and glomerular filtration rate (see Chapter 11) and placental transfer of iodine to the fetus.43,71,99,144
The thyroid compensates for the increased loss of iodine by hyperplasia and increased plasma iodine clearance, reducing plasma iodine levels.88,99 Iodine is stored in the colloid of the thyroid gland follicles (see Figure 19-3). With TSH stimulation, thyroglobulin is catabolized to form T4 and T3.99 Serum thyroglobulin increases in the first trimester but is most marked in later pregnancy. This increase is associated with an increase in thyroid volume, especially in areas of low iodine intake.43,72,99 The degree of hyperplasia is related to the degree of imbalance between iodine needs versus iodine intake and stores (see Iodine Needs during Pregnancy).72 Mild thyroid hyperplasia (10% to 15% increase), due primarily to increased vascularity, is seen in areas such as North America, where diets are generally thought to be iodine sufficient.38,99,100 Therefore moderate to marked thyroid enlargement in these women cannot be considered normal and requires further evaluation. Women who live in iodine-poor areas have an increase in thyroid volume of 15% to 30% during pregnancy.36,99 Goiter is generally not a risk if iodine levels are greater than 0.08 mcg/dL (0.006 μmol/L); in North America, levels average 0.3 mcg/dL (0.023 μmol/L). Goiter is a significant risk in areas with low iodine intake.
Intrapartum period
The HPA and HPT axes undergo further alterations during the intrapartum period. CRH appears to be a trigger in the initiation of labor, and activation of the HPA axis may serve as a “biologic clock” timing the length of gestation.99 Maternal plasma CRH, ACTH, β-endorphin, and cortisol levels increase up to sevenfold with labor onset and during labor (see Chapter 4).20,35,77,86,93,172 ACTH reaches its highest level, increasing from 50 pg/mL (11 pmol/L) at term to 300 pg/mL (66 pmol/L) in labor (see Figure 19-1).32,77,151 Further increases in ACTH and cortisol are seen in women with poor progress in labor.31 Low β-endorphin levels at term have been associated with an increased need for pain medication during labor, although a causal relationship is unclear.20
Levels of total and free T3 increase during labor. This change probably reflects the energy demands of labor on the maternal system. T3 and T4 have similar functions, but T3 is three to five times more active. T3 and T4 increase intracellular enzymes (increased cellular metabolism), the number and activity of mitochondria (to provide energy for cellular enzyme systems), and Na-K adenosine triphosphatase (ATPase) (because of the increased energy use for myometrial contractions).47
Postpartum period
The alterations in the HPA (see Figure 19-2, C) and HPT axes during pregnancy are reversed during the postpartum period. CRH levels drop rapidly with removal of the placenta and placental CRH.151 Maternal ACTH and cortisol levels decrease rapidly in the immediate postpartum period and reach nonpregnant values by 1 to 4 days postpartum.35,77,86 The HPA axis is depressed with a reduction in hypothalamic CRH for 3 to 6 weeks, returning to normal levels by 12 weeks.86 This depression of the HPA axis may play a role in postpartum mood disorders or in exacerbation of autoimmune disorders in the postpartum period.1,62 Although ACTH secretion may be suppressed, total serum cortisol is within normal limits, probably secondary to a mild hypertrophy of the adrenal cortex.86 Hyperplasia of the lactotrophs of the anterior pituitary gland peaks in the first 3 days postpartum. This tissue decreases in size by 1 month in nonlactating and more slowly in lactating women, but never returns to nulliparous size.32,93,97 Prolactin falls at delivery and returns to nonpregnant values by 3 months; in breastfeeding women, prolactin increases after delivery.32,35,151 β-endorphins decrease by 24 hours after birth and are higher in colostrum than in maternal plasma.173 Serum growth hormone levels may remain elevated for several months.32 Postpartum changes in prolactin, FSH, and LH are described in Chapter 5.
After delivery with removal of the placenta and reduction in estrogen, hepatic synthesis of TBG decreases, as does the renal excretion of iodine. As a result, the metabolic alterations in thyroid processes gradually reverse over 4 to 6 weeks, although they may persist for up to 6 to 12 weeks.43,144 Thyroid-releasing hormone (TRH) is a (minor) stimulus of prolactin release and has been used to induce relactation.70 Free T4 may be low and TSH elevated in the first 3 to 4 days, which may confound assessment of thyroid function.65 Transient disorders in thyroid function are seen in some postpartum women (see Postpartum Thyroid Disorders).
Thyroid hormones are secreted in breast milk. Levels are low initially and then rise. Breast milk T4 and T3 have been reported to delay the development of hypothyroidism in some infants with this disorder.131
Clinical implications for the pregnant woman and her fetus
Changes in the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-thyroid (HPT) axes are critical for maintenance of pregnancy. In addition, the maternal and fetal HPA axes and the interrelationship between maternal and fetal-placental function are essential for initiation of labor (see Chapter 4). Alterations in the HPA axis with infection or stress may lead to preterm labor. These risks are discussed further in Chapter 4. Thyroid disorders are more common in women and are not uncommon in pregnant women, being the second most common endocrine disorder (after diabetes mellitus) seen during pregnancy. Disorders of the adrenal and pituitary gland, such as prolactinomas, which may increase in size during pregnancy due the stimulating effects of elevated prolactin levels, and Cushing disease, are uncommon.78,93,97,98 Cushing disease tends to be exacerbated during pregnancy with remission postpartum and is more often due to adrenal lesions than excess adrenocorticotropin (ACTH).78 Diagnosis of thyroid dysfunction during pregnancy may be more difficult in that symptoms of thyroid disorders often mimic some of the usual physiologic changes of pregnancy, and radioactive iodine tests cannot be used because of fetal risks. Implications of alterations in thyroid function and changes in laboratory tests during pregnancy are discussed in this section, along with disorders of thyroid function unique to the postpartum period.
Iodine needs during pregnancy
Adequate iodine stores and nutrition are essential for normal function of the maternal and fetal thyroid.23,36,39 Iodine needs increase during pregnancy due to increased renal loss and increased placental—and thus fetal—uptake.30,36,131 Dietary iodine is converted to iodide; 20% of this is absorbed by the thyroid gland, the rest is cleared by the kidney. During pregnancy renal iodide clearance doubles due to the increased renal blood flow and glomerular filtration rate (see Chapter 11).99 The World Health Organization recommends an iodine intake of 250 μg/day during pregnancy and lactation; others recommend 200 or up to 300 μg/day.30,76,88,131,174,175 Iodine needs can be met by iodine in prenatal vitamins and use of iodinated salt.43 Mean urinary iodine excretion, used to assess iodine deficiency, is insufficient if less than 150 μg/L during pregnancy or less than 100 μg/L during lactation.30
Iodine deficiency is lowest in North America, higher in Europe, with Southeast Asia accounting for nearly one fourth of the worldwide population with insufficient iodine uptake.88 Iodine stores are low in developing countries and have been falling in many developed countries, for example, 50% to 65% of women in western and central Europe were found to be iodine deficient (versus 11% of women in the United States).36,175 Recent reports suggest a decline in iodine sufficiency in childbearing women in the United States.15 Women who enter pregnancy in an iodine-deficient state fall even further behind so that iodine deficiency in the first trimester tends to become more severe in later pregnancy.71,73 Iodine deficiency is the most frequent cause worldwide of preventable mental retardation and often the damage is done by the time of birth.88,175 Iodine deficiency is also associated with an increased risk of spontaneous abortion and stillbirths.88,175 Even in iodine-sufficient areas, there has been increasing concern that iodine deficiency may increase the risk of later neurodevelopmental impairment of offspring in women with marked subclinical thyroid dysfunction.72 Various professional groups have differing recommendations regarding whether all pregnant women should be routinely screened to identify subclinical hypothyroidism.1,2,9,14,30,62,167
Thyroid function tests during pregnancy
Changes in TBG and thyroid hormones during pregnancy alter parameters for many tests used to assess thyroid status. These alterations, which vary with trimester, must be considered when evaluating thyroid function in pregnant and postpartum women. Free T3 and T4 assays, rather than total values, are generally preferred because of the increased thyroxine-binding globulin (TBG).30 Glinoer and Spencer indicate that serum TSH assay “is the most sensitive index to reliably detect thyroid function abnormalities” during pregnancy.40 Reference ranges for thyroid function tests during pregnancy have been described in several studies, with variations by trimester of testing, number of fetuses, and testing method.30 Table 19-1 summarizes the changes in thyroid function during pregnancy that lead to alterations in these tests. As noted in the previous section, recommendations for routine thyroid screening during pregnancy vary among different professional groups worldwide.1,2,9,14,30,62,167
Table 19-1
Thyroid Function Tests during Pregnancy
| PHYSIOLOGIC CHANGES | RESULTING CHANGE IN THYROID ACTIVITY |
| ↑ Serum estrogens | ↑ Serum TBG |
| ↑ Serum TBG |

Modified from Brent, G.A. (1997). Maternal thyroid function: Interpretation of thyroid function test in pregnancy. Clin Obstet Gynecol, 40, 3, by Fantz, C.R., et al. (1999). Thyroid function during pregnancy. Clin Chem, 45, 2250.
Antithyroid peroxidase enzymes have been reported in 10% to 14% of women at 14 weeks’ gestation and may increase the risk of gestational thyroid dysfunction and postpartum thyroiditis.72 Most women are euthyroid but some have increased thyroid-stimulating hormone (TSH) or decreased free T4 or both.72
Thyroid function and nausea and vomiting in pregnancy
Nausea and vomiting in pregnancy (NVP) has been linked to alterations in T4, TSH, and human chorionic gonadotropin (hCG) (which has TSH-like activity). hCG stimulates receptors for both TSH (increases T4) and hCG (increases estriol and possibly NVP). NVP severity has been correlated with increased free T4 and decreased TSH, with values returning to normal pregnant ranges as the nausea and vomiting resolve.71 These findings may lead to or be a consequence of emesis during early pregnancy. NVP is discussed further in Chapter 12.
Hyperemesis gravidarum in women without any history or evidence of thyroid dysfunction has also been associated with increased free T4 and decreased TSH. These findings, seen in about 60% of women with hyperemesis, are similar to those of the euthyroid sick syndrome seen with severe illness.73 T4 levels in women with hyperemesis return to usual values in 1 to 4 weeks after resolution with or without treatment with antithyroid drugs.1,43,72,98
The pregnant woman with hyperthyroidism
A transient hyperthyroidism is seen in 10% to 20% of otherwise healthy pregnant women during the first trimester; this may be associated with increased hCG levels, multiple gestation, or NVP.34,36 This transient form of subclinical hyperthyroidism is characterized by normal free T4 and decreased TSH.
Another form of transient clinical hyperthyroidism, characterized by increased free T4 and decreased TSH, is seen less frequently and may be associated with multiple gestation, hyperemesis gravidarum, and gestational trophoblast disorders with markedly increased hCG production.34,62,72 Trophoblast disorders such as a molar pregnancy occasionally cause biochemical and, in some women (5% to 64%), clinical findings of hyperthyroidism because of the high levels of hCG secreted by the trophoblastic mass.100
Chronic hyperthyroidism occurs in 0.05% to 0.2% of pregnant women.99 The diagnosis of hyperthyroidism during pregnancy may be difficult in that many of the signs and symptoms associated with this disorder are often seen normally during pregnancy. Findings common to hyperthyroidism and certain stages of pregnancy include fatigue, heat intolerance, warm skin, emotional lability, insomnia, increased appetite, sweating, breathlessness, ankle edema, palpitations, and increased pulse pressure.30,34,43,72,99,100 Failure to gain weight with a good appetite and persistent tachycardia (greater than 100 beats/min) are most suggestive of hyperthyroidism in pregnancy.43 Increased free T3 and total T4 (greater than 15 mcg/dL [193 nmol/L]) and increased or high-normal RT3U are seen with hyperthyroidism. Alterations in thyroid function tests during pregnancy (see Table 19-1) must be considered when interpreting test results. For example, because RT3U is decreased in pregnancy, values in the nonpregnant range suggest hyperthyroidism. TSH levels less than 0.05 μU/L (mIU/mL) and increased free T4 levels greater than 11.6 mcg/dL (149 pmol/L) are diagnostic.99
Hyperthyroidism in pregnant women is almost always (85% to 95%) due to Graves’ disease.34,72,73,81 Graves’ disease is an autoimmune disorder in which thyroid-stimulating immunoglobulins (TSIs), such as TSH receptor antibody (TRAb), attach to and activate TSH receptors on the thyroid follicular cells. This leads to increased production of thyroid hormones and the clinical finding of hyperthyroidism.
Women with mild hyperthyroidism generally do well during pregnancy because increased serum TBG offsets the increased secretion of thyroid hormones. In women with Graves’ disease, their disorder may be aggravated in the first trimester (due to the increased hCG) and then improve during the third trimester, with remission and occasionally complete resolution.30,165 This improvement is related to suppression of the maternal immune system responses (see Chapter 13) by fetal cytokines with lower TSI levels and decreased thyroid hormone production. Relapse or exacerbation generally occurs postpartum, usually within several weeks of delivery, as immune system alterations and production of TBG return to prepregnancy levels.100,165
Pregnant women with hyperthyroidism usually require a caloric intake that is higher than that generally recommended during pregnancy to compensate for their increased metabolic rate. These women are also at risk for fluid loss and dehydration as a result of the diarrhea and tachycardia that often accompany hyperthyroidism.99 Women with hyperthyroidism, particularly if this disorder is poorly controlled, are at increased risk for placental abruption, miscarriage, preterm labor, fetal growth restriction, congestive heart failure, and thyroid storm.30,34,36,69,72,119 Their infants can develop transient hyperthyroidism from transplacental passage of TSIs or hypothyroidism secondary to the effects of maternal antithyroid drugs.36,119 Hyperthyroidism also alters sex steroid metabolism, sperm motility, and fertility.61,131
Transplacental passage of TSIs, especially TRAbs, leads to a transient neonatal hyperthyroidism in about 1% to 5% of infants of mothers with Graves’ disease (see Chapter 13).1,30,33,116 The risk is present even if the woman is euthyroid in pregnancy or has had thyroid ablation via surgery or radiation before pregnancy because TSIs are still present.82,100 The fetal risk increases after 20 weeks when fetal TSH receptors become responsive to TSH.81 TSH-binding inhibitory immunoglobulins may also cross the placenta, causing a transient neonatal hypothyroidism.131
Hyperthyroidism is treated with antithyroid drugs (thioamides), surgical removal, or thyroid ablation with 131I. The most common thioamides are propylthiouracil (PTU) and methimazole (Tapazole or carbimazole, which is metabolized to methimazole). Thioamides cross the placenta and can block synthesis of thyroid hormones by the fetus.119 The lower hormone levels stimulate increased TSH production, which can lead to goiter and tracheal obstruction. Infants of mothers treated with thioamides may have decreased T4 and increased TSH levels after birth. These values are generally within normal neonatal limits by 4 to 5 days of age. PTU is recommended as the first-line drug during pregnancy.1,34,36,69 Although methimazole (MMI) has the advantage of less frequent dosing and fewer tablets per dose, MMI is associated with a risk of fetal scalp defects (cutis aplasia) and possibly esophageal and choanal atresia.30,36,73,100,152 Pharmacologic effects must be monitored carefully, particularly during the third trimester in women with Graves’ disease, when remission can lead to decreased thyroid hormone production and a transient decrease in required drug dosage by 32 to 36 weeks.99 In some women, the drug can be transiently discontinued in late pregnancy.165
Pregnant women may also develop a transient subacute thyroiditis that often occurs in association with viral infections. The inflammation and destruction of thyroid tissue lead to release of stored thyroid hormone into serum and a transient hyperthyroxinemia. As the disorder resolves, the woman may develop hypothyroidism because the released thyroid hormones are used up before the thyroid gland can produce an adequate new supply. If treatment is initiated, β-blockers such as propranolol are required rather than antithyroid drugs that block thyroid hormone production, because with subacute thyroiditis the thyroid is not making hormones.43,72 Propranolol is generally not indicated for long-term treatment of pregnant women with hyperthyroidism. This drug crosses the placenta and has been associated with fetal growth restriction and impaired responses to anoxia and neonatal hypoglycemia and bradycardia.
The pregnant woman with hypothyroidism
Hypothyroidism in iodine-sufficient pregnant women is usually secondary to autoimmune disorders (after surgical removal or ablation of the thyroid with radioactive 131I for Graves’ disease and idiopathic myxedema) or Hashimoto thyroiditis.30,62 Worldwide, iodine deficiency is the most common cause of hypothyroidism and can lead to cretinism (mental retardation, deafness, other neurologic symptoms in the infant).36,48,62,100 Iodine deficiency leads to hypothyroxinemia, thyroid stimulation from thyroid-releasing hormone (TRH) and TSH feedback loops, and development of goiter.71 Women with untreated hypothyroidism have a high incidence of infertility and spontaneous abortion.30,34,43,73,124
The diagnosis of hypothyroidism during pregnancy may be missed because some of the signs and symptoms associated with this disorder—fatigue, weight gain, muscle cramps, constipation, and amenorrhea—are also seen normally during pregnancy.30,43,99 The increase in TBG in pregnancy may mask the decrease in thyroid hormones; however, levels are usually still low for pregnant norms. In addition, free T4 is low and TSH is elevated.72
Pregnancy in women with primary hypothyroidism or autoimmune thyroiditis may be complicated by an increased risk of fetal loss or prolonged pregnancy, possibly due to compromised placental blood flow or the inability of the thyroid gland to meet the metabolic demands of pregnancy. These women are also at greater risk for preterm delivery, preeclampsia, and cesarean section.30,34,36,62,92,107,148 Weight gain patterns must be carefully monitored in the woman with hypothyroidism. These women may also have difficulty with fatigue and constipation during pregnancy.
Thyroid hormone replacement doses usually need to be increased as a result of the increased TBG and T4 demand.3,49,72,100,145,158 For example, levothyroxine may need to be increased 25% to 40% to maintain normal serum TSH levels.3,34,73 Increased requirements may occur as early as the fifth week of gestation and are influenced by maternal estrogen levels, pregestation TSH levels, maternal volume of distribution, parity, and etiology of the hypothyroidism.3,4,34,80 Postpartum levothyroxine levels usually decrease to prepregnancy levels, but some women continue to have a greater requirement.30,34
Although overt hypothyroidism is associated with neurodevelopmental problems in offspring, the role of subclinical hypothyroidism is less clear.1,48,88,101,170 Subclinical hypothyroidism (elevated TSH but normal free T4) occurs in 2% to 5% of pregnancies and increases the risk of prematurity, abruption, and impaired neurologic outcome in infants.1,30,48,72,88 The risk of later neurologic problems in offspring is thought to be greatest with untreated or subclinical hypothyroidism in the first 10 to 12 weeks of gestation because the fetus is completely dependent on maternal thyroid hormone for brain development during this time.115 However, the role of subclinical hypothyroidism in infants’ later development is still an area of debate, due to inadequate data on the causal relationship between subclinical hypothyroidism and lower IQ levels.30,48,114 Routine screening is recommended for all pregnant women or for all who are high risk for this disorder by some, but not all professional organizations1,2,9,14,62,167
Postpartum thyroid disorders
The postpartum period is associated with transient thyroid disorders. Physiologic alterations and experiences of pregnancy can mask clinical findings of hypothyroidism or hyperthyroidism. As a result, these disorders may first become apparent in the postpartum period. Although less common than these transient disorders, postpartum women are also at increased risk of developing Graves’ disease, especially women older than 35 years of age.10
Postpartum thyroid disorder (PPTD) is a transient disorder seen in 6% to 9% of postpartum women.62 The incidence of PPTD is 33% to 50% in women with thyroid peroxidase antibodies in early pregnancy (versus 0.5% in those without these antibodies).62,72 PPTD is also more prevalent in women with type 1 diabetes.62 Classic PPTD accounts for about 28% of these cases and generally appears in the first 6 months (usually by 6 to 8 weeks after delivery). PPTD (Figure 19-5) is generally characterized by weeks or months (average, 2 to 4 months, but can be up to 6 months) of mild hyperthyroidism, followed by weeks or months (up to a year) of hypothyroidism, and finally a return to normal thyroid function in most women—usually by 12 months postpartum.1,43,62,72,100 Others present with only hypothyroidism or hyperthyroidism.
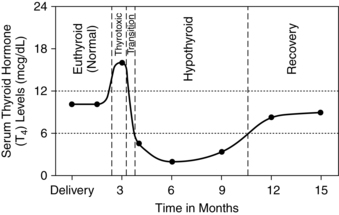
The exact cause of PPTD is unclear but it appears to be an autoimmune disorder with increased susceptibility in the postpartum woman as her immune system rapidly changes from Th2 to Th1 T-lymphocyte dominance (see Chapter 13).1,72 The initial hyperthyroid phase is characterized by thyroid cell destruction with excessive release of thyroid hormone. PPTD may reflect postpartum exacerbation of a subclinical autoimmune disorder that, with release of pregnancy-induced immunosuppression, leads to rebound of immune components with excessive production of thyroid autoantibodies.43,62 The subsequent hypothyroid phase is due to decreased thyroid hormones from excessive loss of thyroid cells during the first phase. As the thyroid cells regrow, normal function returns.
Up to 30% to 50% of women with PPTD remain hypothyroid or develop permanent hypothyroidism within 5 to 15 years.1,62 Biochemical abnormalities include elevated free T4, suppression of TSH, presence of microsomal antibodies, and a low 131I uptake.72 PPTD may be misdiagnosed as postpartum depression and often recurs with subsequent deliveries.30,72 Transient hypothyroidism is also seen occasionally during the postpartum period. Findings include fatigue, weight gain, low free T4 and elevated TSH levels, and elevated antimicrosomal and antithyroglobulin antibody titers.
Breastfeeding in women with thyroid disorders
Breastfeeding is not contraindicated in women with hypothyroidism, because thyroid hormones cross in only small amounts and the infant should receive a dose no higher than that from a euthyroid woman.58,70 Breastfeeding in women with hyperthyroidism may be a concern because of passage of antithyroid medications in breast milk.90 However, PTU is excreted in breast milk in relatively small amounts (0.025% to 0.077% of the maternal dose).70 Most sources suggest that breastfeeding is not routinely contraindicated in women on PTU who are carefully monitored, but that each woman needs to weigh the risks and benefits and the infant must be carefully monitored. Propranolol and thiouracil cross in significant amounts and are generally contraindicated in breastfeeding women.70
The mammary glands actively take up, concentrate, and secrete iodine in breast milk. Thus breastfeeding is interrupted if the woman requires thyroid uptake studies or scans involving use of 123I or 131I. If radioactive iodine is needed, 123I is preferred over 131 I, since breastfeeding generally needs to be stopped for only about 48 hours with 123 I versus for a prolonged period of time with 131I.43
Use of radioiodine and iodides
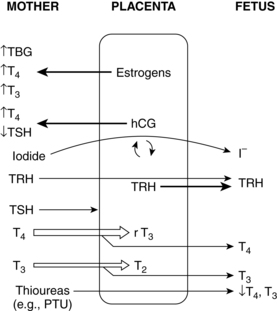
Iodine is actively transported across the placenta and taken up by the fetal thyroid (Figure 19-6). Avidity of the fetal thyroid for iodine is 20 to 50 times greater than that of the mother.43 Administration of any form of iodine to pregnant women results in significantly higher concentrations per weight in fetal tissues and can lead to development of a goiter, especially after 12 weeks’ gestation, when the fetal thyroid begins to concentrate iodine.53 Fetal risks of radioactive substances include thyroid damage, microcephaly, growth restriction, mental retardation, later malignancy, and death. As a result, radioiodine is contraindicated during pregnancy. Radioiodine also alters spermatogenesis, so a waiting period is recommended in males between therapy and fertilization.
All women should have a pregnancy test before radioactive iodine studies. If these studies are done with 123I in a woman who is later found to be pregnant, however, the risks are minimal since the dose of 123I used is generally low. The use of 131I for definitive diagnosis and therapy of Graves’ disease is associated with increased pregnancy loss and potential damage to the fetal thyroid; thus this agent is avoided during pregnancy.34
The use of nonradioactive iodides has also been reported to increase the risk of fetal goiter, risk of tracheal obstruction, and hypothyroidism. Iodides are found in Betadine-containing vaginal suppositories and douches, saturated solution of potassium iodide (SSKI), iodinated medications for asthmatics, and some contrast materials.43,131 The incidence of drug-induced fetal goiter is 1 in 10,000 and is seen primarily with daily iodide doses over 12 mcg.
Maternal-fetal endocrine relationships
Maternal, fetal, and placental HPA functions are closely interrelated. The placenta produces significant amounts of some hypothalamic and pituitary hormones such as corticotropin-releasing hormone (CRH) and growth hormone (GH). The fetus and placenta, via CRH production, influence the timing of parturition (see Chapter 4). Large quantities of steroids are produced by interaction of the mother, placenta, and fetus. Some of these substances are dependent upon the viability and well-being of the fetus and placenta, whereas others, such as progesterone, do not require the fetus to be viable for production to continue. Steroid-producing cells appear early in placental development. They can be seen first in the trophoblast tissue of the placenta; however, the syncytiotrophoblast lacks several of the key enzymes necessary for steroid metabolism. Therefore the placenta is an incomplete steroidogenic tissue and must receive precursors from exogenous sources (i.e., the fetus or maternal system) (see Chapter 3).
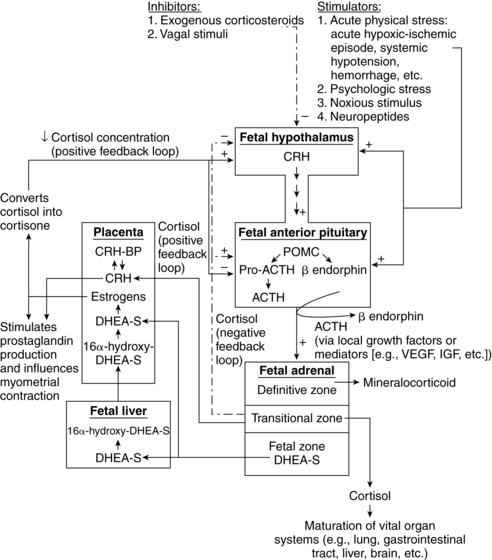
During early gestation, maternal cortisol crossing the placenta provides negative feedback to the fetal HPA axis. This suppresses fetal cortisol production. Later in pregnancy, as placental metabolic activity matures, less maternal cortisol reaches the fetus as much of this cortisol is converted to inactive cortisone in the placenta. In late pregnancy, there is less negative feedback to the fetal HPA axis and increased fetal cortisol production. Placental CRH, which increases during gestation, has a stimulatory effect on the fetal HPA axis and is stimulated by cortisol.162
The fetal HPT axis develops relatively independently of maternal influences.28,45 Maternal TRH does cross the placenta but does not seem to have a major influence on fetal pituitary or thyroid function, probably because maternal TRH levels are low and much of the maternal TRH is degraded in the placenta.29,100 TSH does not cross the placenta.131 Administration of thyroid hormone to the mother does not significantly increase fetal hormone levels.28
Maternal T4 crosses the placenta to the fetus and is critical for normal development of the central nervous system and other organ systems, especially in the first trimester before the fetal thyroid is functional when the mother is the only source of T4 (see Role of Maternal Thyroid Hormones).28,29,43,71,95,99,144 Transfer continues throughout pregnancy but is decreased in later pregnancy due to increased placental deiodination of T4 (to inactive rT3) and T3 (to T2) by MDI-III.29,131 Inadequate maternal iodine may decrease transfer of maternal T4 to the fetus.76,84,88,175 Increased T4 transfer has been reported in late gestation with fetuses with severe congenital hypothyroidism.28 Athyroid fetuses have T4 levels that are 25% to 50% of normal.131 The fetus is also dependent on the mother for iodides—either via direct transfer or breakdown of maternal T4 by the placenta (see Figure 19-6).28 The placenta is permeable to commonly used antithyroid drugs (PTU and methimazole), β-blockers (propranolol), and iodine.131 Placental permeability to TSIs can lead to development of transient neonatal hyperthyroidism in infants of women with Graves’ disease (see The Pregnant Woman with Hyperthyroidism and Chapter 13).
Maternal stress responses and fetal endocrine programming
Even though both ACTH and cortisol are elevated during pregnancy, physiologic responses (e.g., blood pressure, heart rate, and cortisol reactivity) to stress, although maintained, appear to be blunted in the pregnant woman.13,21,25 However, data are limited and wide individual differences have been reported. This blunting may be due to glucocorticoid negative feedback, possibly nitric oxide, urocortins, β-endorphins, and other endogenous opioids.13
Fetal programming refers to the effects of the fetal environment on susceptibility to later disorders. For example, the nutritional/hormonal status during fetal/early postbirth life can alter organ development including the hypothalamus and other endocrine structures. Increases in cortisol with stress in the mother can lead to increases in fetal cortisol. This is important because before birth, fetal systems such as the fetal HPA axis are programmed for postnatal function, with adverse consequences for later function if programming is altered.13,159 Normally most of the cortisol reaching the placenta is broken down into inert metabolites by placental enzymes, such as 11-β-hydroxysteroid dehydrogenase, in the syncytiotrophoblast. These enzymes convert active glucocorticoids, such as cortisol and corticosterone, to inactive metabolites.77 As a result, fetal circulating cortisol levels are maintained lower than maternal levels.
Wide individual differences have been reported in both maternal stress reactivity and in the activity and sensitivity of these placental enzymes.21,25 There is increasing evidence in both human and animal studies of the effects of prenatal stress on offspring, including prematurity, low birth weight, delivery complications, and impaired prenatal and postnatal growth and development.18,21,41,109,159,161,166 For example, the risk of early pregnancy loss with maternal stress may be mediated by alterations in glucocorticoids, progesterone/prolactin, and the immune system, leading to alterations in the early pregnancy cytokine environmental balance (see Chapter 13) needed for implantation and early pregnancy maintenance.13 Early stress may also induce epigenetic changes in gene expression that increase the risk of later metabolic, vascular, and neurodevelopmental disorders in offspring.18,89,159 Prolonged exposure to maternal stress mediators may permanently reset the HPA axis and increase the risk of adult-onset disorders such as type 2 diabetes, hypertension, obesity, coronary artery disease, end-stage renal disease, and depression.13,150,159,162
The specific mechanisms by which maternal stress affects the fetus are unclear, but probably involve changes in the fetal HPA axis from maternal increases in sympathetic nervous system and HPA activity and perhaps changes in the limbic system with stress.24 Glucocorticoids are thought to have a major role in fetal tissue programming because excess levels can inhibit growth; alter expression of receptors, enzymes, ion channels, and transporters; and alter growth factors, structural proteins, binding proteins, and signaling pathways.113 Possible mechanisms include (1) stress-induced increase in maternal cortisol results in increased fetal cortisol and changes in the feedback regulation of the fetal HPA axis; (2) hormones from the maternal HPA axis stimulate the placenta to increase CRH, which alters brain glucocorticoid receptor development and function in the fetus; and (3) increased cortisol and catecholamines seen with stress may alter uteroplacental blood flow, leading to poorer fetal growth, prematurity, and other complications.21,56,113,159,166 Figure 19-7 illustrates a proposed model of factors contributing to indirect and direct effects of stress on the fetus. Fetal programming is discussed further in Chapters 12 and 16.
Summary
Changes in the hypothalamic-pituitary-adrenal (HPA) axis during pregnancy enhance maternal adaptations and availability of nutrients to the fetus. Maternal HPA function is interrelated with placental function, because the placenta produces hypothalamic and pituitary hormones such as corticotropin-releasing hormone (CRH) and growth hormone (GH). Placental CRH plays a major role in initiation of parturition (see Chapter 4). Changes in HPA and hypothalamic-pituitary-thyroid (HPT) function can alter reproductive processes. The pregnant woman is in a state of euthyroid hyperthyroxinemia and transient hypercortisolism, although control set points are reset so the woman responds to stress in a manner that is similar to that of nonpregnant individuals. As a result, values for some endocrine function tests are altered. These changes must be considered when evaluating neuroendocrine function during pregnancy. Thyroid disorders are not uncommon in pregnant women. Knowledge of the effects of these disorders and their treatment is important to optimize fetal and neonatal outcome. Clinical recommendations related to thyroid function during pregnancy are summarized in Table 19-2.
Table 19-2
Recognize the usual changes in pituitary and adrenal function during pregnancy (pp. 627-629 and Box 19-1).
Recognize the usual changes in thyroid function during pregnancy (pp. 629-633).
Assess and monitor maternal nutrition in terms of iodine intake (p. 634).
Monitor fetal growth in women with normal and abnormal thyroid function (pp. 635-636, 638).
Know the usual parameters for thyroid function tests during pregnancy (pp. 634-635 and Table 19-1).
Recognize the signs and symptoms of thyroid dysfunction and similarities with certain pregnancy-related findings (pp. 635-636).
Counsel women with hyperthyroidism and hypothyroidism regarding the effects of their disorder on pregnancy and the fetus and of pregnancy on the thyroid disorder (pp. 635-636).
Monitor medication levels and requirements in the pregnant woman with hyperthyroidism (pp. 635-636).
Know the fetal and neonatal risks of maternal iodide and radioactive iodine administration (pp. 637-638).
Avoid the use of iodides and radioactive iodine in pregnant women (pp. 637-638).
Know the fetal and neonatal risks associated with the use of thyroid and antithyroid agents (pp. 635-638).
Recognize and monitor for transient postpartum thyroid disorders (pp. 636-637 and Figure 19-5).
Counsel women with hypothyroidism and hyperthyroidism regarding breastfeeding considerations (p. 637).
Development of hypothalamic, pituitary, adrenal, and thyroid function in the fetus
Maturation of thyroid and adrenal function is interrelated with that of the hypothalamic and pituitary glands and the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-thyroid (HPT) axes. This process can be divided into three overlapping phases: embryogenesis (phase I), hypothalamic maturation (phase II), and maturation of thyroid and adrenal system function (phase III).29 During the first phase (10 to 12 weeks), the adrenal and thyroid glands develop morphologically. Hypothalamic function matures during phase II (from 4 to 5 until 35 to 40 weeks). Thyroid-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH), and somatostatin are detected in the hypothalamus by 10 to 12 weeks (by radioimmunoassay) and in fetal blood in the third trimester.42 Phase III lasts from midgestation until term or 1 month after birth if transitional changes are considered. This stage involves increasing maturation and integration of endocrine system function.
Hypothalamus and pituitary gland
Anatomic development
The hypothalamus develops from 6 to 12 weeks from the ventral portion of the diencephalon. Hypothalamic nuclei and supraoptic track fibers develop by 12 to 14 weeks, with maturation of the hypothalamic neurons by 30 to 35 weeks.28 The anterior pituitary arises from the anterior wall of the Rathke pouch, an upward offshoot of the primitive oral cavity.45 The posterior pituitary and stalk develop from the infundibulum, a thickening on the floor of the diencephalon.135 The anterior pituitary can be seen by 4 weeks and is independent of the oral cavity by 12 weeks. During the fifth week, the primitive anterior pituitary becomes connected with the infundibulum.
Cellular differentiation within the anterior pituitary gland begins at 7 to 8 weeks, under the influence of Pit-1 (a transcription regulator) that activates expression of genes encoding growth hormone (GH) and prolactin and differentiation of the different anterior pituitary cell types.45 Thyrotropic cells appear at 12 to 13 weeks. An increase in TSH-secreting cell volume occurs by 23 weeks. Corticotroph cells can be identified by 6 weeks and express adrenocorticotropic hormone (ACTH) by 7 weeks; somatotroph can be seen by 8 weeks. TSH and gonadotropin β subunits are seen by 12 weeks. Lactotroph cells do not produce prolactin until after 24 weeks.32
Hypothalamic and pituitary function
The HPA axis is essential for regulating intrauterine homeostasis and maturation of the lungs, liver, and central nervous system; in conjunction with the placenta, this axis essential in the timing of parturition (see Chapter 4).103 The hypothalamus and pituitary gland also play major roles in regulating the principal fetal steroid-secreting glands (adrenal glands and gonads) via a negative feedback mechanism. Figure 19-7 summarizes the HPA axis in the fetus and its interaction with placental function.
The fetal hypothalamus is visible by 7 weeks’ gestation.123 The hypothalamus-pituitary axis is generally complete by 12 to 18 weeks’ gestation.123 The HPA axis is established by 20 weeks and matures throughout the remainder of gestation; integrated HPA function matures postnatally.29 The hypothalamic-pituitary portal system is functional by 10 to 12 weeks and most of the hormones of the HPA axis are present by 10 to 16 weeks’ gestation (Table 19-3).29,90,98 For example, ACTH is seen in pituitary cells by 5 to 8 weeks and is measurable in fetal plasma by 16 weeks.123 Plasma concentrations of these hormones rise for the first half of gestation. At this time the negative feedback loop comes into operation and the production and release of pituitary hormones become more finely regulated. Corticotropin-releasing hormone (CRH) from the fetus and placenta stimulates ACTH, which controls growth, differentiation, and function of the adrenal cortex in conjunction with growth factors (GFs) and other mediators.103
Table 19-3
Ontogeny of Human Fetal Hypothalamic and Pituitary Hormones
| HORMONE | AGE DETECTED (WEEKS) |
| HYPOTHALAMIC | |
| Gonadotropin-releasing hormone | 14 |
| Thyrotropin-releasing hormone | 10 |
| Somatostatin | 14 |
| Dopamine | 11 |
| Growth hormone–releasing hormone | 18 |
| Corticotropin-releasing hormone | 16 |
| PITUITARY | |
| Prolactin | 16.5 |
| Growth hormone | 10.5 |
| Adrenocorticotropin | 7 |
| Thyroid-stimulating hormone | 13 |
| Luteinizing hormone | 10.5 |
| Follicle-stimulating hormone | 10.5 |
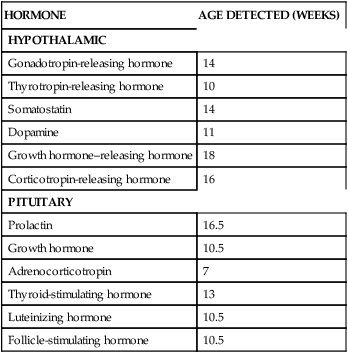
From Mesiano, S., & Jaffe, R.B. (2004). The endocrinology of human pregnancy and fetal-placental neuroendocrine development. In J.F. Strauss & R.L. Barbieri (Eds.), Yen and Jaffe’s Reproductive endocrinology: Physiology, pathophysiology, and clinical management (5th ed.). Philadelphia: Saunders.
GnRH appears by 18 weeks and increases to 30 weeks.90 Luteinizing hormone (LH) and follicle-stimulating hormone (FSH) appear in the fetal pituitary gland during the tenth week and reach their peaks at about 20 to 22 weeks’ gestation. The concentration of LH is higher than that of FSH, and females have higher levels than males.90 FSH promotes development of follicles in females; it stimulates growth of seminiferous tubules and initiates spermatogenesis in males. LH increases the synthesis and secretion of testosterone in the male embryo and stimulates steroid synthesis in ovarian cells.
Fetal growth hormone (GH) levels are higher than after birth due to immature inhibitory control and lack of GH receptors on fetal tissues.42 Increases in cortisol before delivery are thought to induce GH receptors and changes in insulin-like growth factor-I (IGF-I).
Adrenal glands
Anatomic development
The adrenal cortex arises in the fourth to fifth week from the intermediate mesoderm in the notch between the primitive urogenital ridge and the dorsal mesentery.19 These cells proliferate and form cords migrating medially and laterally to the cranial end of the mesonephros (primitive transitional renal structures).90,96 Neural crest cells form a medial mass on the primitive cortex. These cells are surrounded by the cortex cells and differentiate into the adrenal medulla. Later, another wave of cells surrounds the initial cortical cells to form the definitive cortex.19,135 By 8 weeks, the cortex is organized into the fetal zone, with the definitive zone appearing a week later.19,90,96 The fetal zone increases in size from 10 to 15 weeks and dominates from 16 to 20 weeks, being similar in size to the fetal kidneys.19 By 28 to 30 weeks, a transitional zone that is initially similar to the fetal zone appears between the fetal and definitive zones. During the second and third trimesters, the adrenal glands continue to enlarge, with hypertrophy of the fetal zone and hyperplasia of the definitive zone.19,90 Adrenal growth and zonation are stimulated by fetal ACTH, IGF-II, fibroblast growth factor, and epidermal growth factor.19
By midgestation, the fetal zone occupies 85% of the adrenal volume and is highly vascularized.127,128 The adrenals double in size between 20 and 30 weeks and double again between 30 and 40 weeks as steroidogenic activity increases.19 At term, the adrenal glands weigh 3 to 4 g and, relative to adult proportions, are 20 to 30 times larger.19,90,96,127 After birth, the adrenal undergoes extensive remodeling, with a decrease in size and disappearance of the fetal zone.
Adrenal function
As noted above, the fetal adrenal gland initially consists of two zones: the fetal zone (analogous to the adult zona reticularis) and the definitive zone (becomes the postnatal adrenal neocortex producing aldosterone—see Chapter 11). Later the transitional zone, which gives rise to the glucocorticoid-producing zona fasciculata, develops between the fetal and definitive zones. The fetal zone contains large lipid-containing steroidogenic cells, which are the major source of dehydroepiandrosterone (DHEA) and its sulfate (DHEA-S), which are precursors for production of estrone and estradiol-17β in the placenta. DHEA-S is also the precursor for 16-hydroxydehydroepiandrosterone, which is needed for estriol production by the placenta (see Chapter 3). Cholesterol is the precursor for these steroid hormones and the fetus uses both endogenous cholesterol synthesized from low-density lipoproteins and cholesterol transferred across the placenta.57,127
The placenta does not produce these precursors, so is dependent upon the fetal adrenal (primary source) or mother. In fetuses with decreased adrenal function, such as anencephalic fetuses, estriol production by the placenta remains low. Enzymes to synthesize DHEA-S are present by 6 to 8 weeks.90,123 High levels of 17α-hydroxylase (17-OH or CYP17) and probably 21-hydroxylase (21-OH or CYP21) are seen early in gestation (Figure 19-8). 17-OH and 21-OH are cytochrome P-450 enzymes not expressed by the placenta.164 Deficiency of 21-OH is the most common cause of congenital adrenal hyperplasia (see p. 651); a lack of 21-OH can result in virilization of the female external genitalia. Because the genital tract differentiates at 7 to 10 weeks (see Chapter 1), this enzyme must be present early in gestation.19 Levels of 3β-hydroxysteroid dehydrogenase (involved in cortisol production) are low in the fetus.19 The large size of the adrenal glands allows the fetus to secrete larger amounts of adrenal androgens daily, predominantly DHEA-S, than do adults. The fetus supplies 90% of the DHEA-S to the placenta after 16-hydroxylation by the fetal liver (see Figure 19-7).127,128
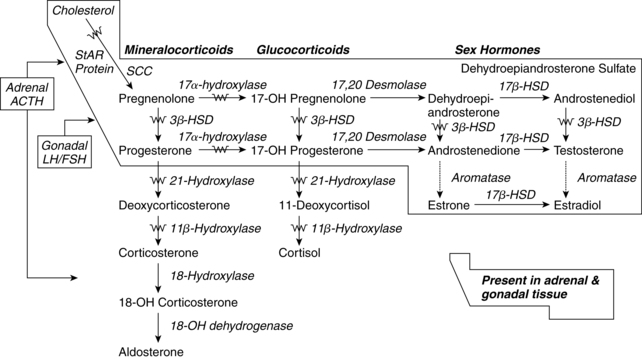
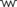 represents one of the potential areas of deficient enzymatic activity causing congenital adrenal hyperplasia; StAR represents steroidogenic auto regulatory protein; SCC represents cholesterol side chain cleavage enzyme; 3β-HSD represents 3β-hydroxysteroid dehydrogenase; 17β-HSD represents 17β-hydroxysteroid dehydrogenase.
represents one of the potential areas of deficient enzymatic activity causing congenital adrenal hyperplasia; StAR represents steroidogenic auto regulatory protein; SCC represents cholesterol side chain cleavage enzyme; 3β-HSD represents 3β-hydroxysteroid dehydrogenase; 17β-HSD represents 17β-hydroxysteroid dehydrogenase. Cortisol is produced as early as 8 to 12 weeks but in small amounts and primarily from progesterone.19,90,123 Production of cortisol from cholesterol, the usual precursor in later life, requires 3β hydroxysteroid dehydrogenase (3β HSD) which is not available in early gestation.162 3β HSD is not expressed before 23 weeks’ gestation, with limited adrenal capacity for synthesis until after 30 weeks.97,123 Cortisol secretion is present by 20 to 24 weeks’ gestation.29 Fetal cortisol levels gradually increase to term. Cortisol is essential for fetal maturation of the lungs, gut, liver, and central nervous system.103 Cortisol is also found in amniotic fluid. During labor, fetal cortisol levels double.96 Most fetal tissues and especially the placenta contain enzymes such as 11β hydroxysteroid dehydrogenase type 2 (11β HSD2) that convert cortisol to inactive cortisone. This serves as a protective mechanism against elevated cortisol in the fetus.
Maternal cortisol enters the placenta. However, since about 85% is deactivated by 11β HSD2, little reaches the fetus.96,162 Maternal cortisol that does reach the fetus has a negative feedback effect on the fetal HPA axis, suppressing fetal cortisol production. Placental CRH stimulates pituitary ACTH production that in turn stimulates the fetal adrenal to produce cortisol, DHEA, and DHEA-S.162 In the third trimester, placental 11β HSD2 activity increases with increased conversion of both fetal and maternal cortisol to cortisone. This reduction in active cortisol reaching the fetal hypothalamus and pituitary further reduces negative feedback to the HPA axis that would normally reduce CRH, ACTH, and cortisol levels. This increases adrenal DHEA-S synthesis. DHEA-S production increases estrogen production, which in turn stimulates increased 11β HSD2.29,103,127,162 CRH plays a major role in regulating fetal adrenal steroidogenesis in late gestation and in labor onset (see Chapter 4).127 Placental CRH stimulates the fetal HPA axis and is itself stimulated by fetal cortisol (whereas hypothalamic CRH is inhibited by high cortisol levels). Seven to ten weeks prior to birth fetal adrenal cortisol production increases with a decrease in the conversion of cortisol to inactive cortisone, mediated by an increase in 11β HSD activity (stimulated by estrogens).29 Factors influencing fetal adrenal function are summarized in Table 19-4.
Table 19-4
Factors Affecting Fetal Adrenal Function
| FACTOR | EFFECT |
| Maternal cortisol passively transferred to the fetus suppresses the fetal HPA axis |
From Watterberg, K.L. (2004). Adrenocortical function and dysfunction in the fetus and neonate. Semin Neonatol, 9, 14.
Growth of the adrenal glands is probably not dependent on ACTH, because the anencephalic fetus with low ACTH levels develops adrenal glands, but ACTH is required for adrenal function.19,128 ACTH may exert its effect via local GFs and other mediators such as fibroblast GF, epidermal GF, transforming growth factor-α (TGF-α), IGF-I, IGF-II, transforming growth factor-β (TGF-β), activin, and inhibin.19,54,90 ACTH, estrogens, and IGF-II stimulate growth of the definitive zone and activation of 3β HSD and P450 17α-hydroxylase, which mediate the increase in fetal cortisol production in late gestation.29 Fisher summarizes the significance of this increase in fetal cortisol in late gestation in anticipation of transition to extrauterine life: “The increase in fetal circulating cortisol concentration during the final weeks of gestation is associated with a variety of physiologic responses. These include (a) increased conversion of T4 to T3 by stimulation of hepatic MDI-I and inhibition of MDI-III leading to an increase in circulating T3 and decrease in rT3 levels; (b) increased epinephrine secretion by sympathetic chromaffin tissue (including adrenal medulla) because of augmented phenylethanolamine transferase activity (which converts norepinephrine to epinephrine); (c) augmented synthesis and secretion of surfactant and maturation of surfactant composition; (d) stimulation of maturation of hepatic gluconeogenic enzyme activities; (e) increased β-adrenergic receptor density in a variety of tissues, including lung, heart, and brown adipose tissue; and (f) induction and maturation of a variety of gut enzymes for nutrient absorption and maturation of gut transport processes, motility, and structure.”29,p.390
Intrauterine stress may activate adrenal cortisol secretion (see Maternal Stress Responses and Fetal Endocrine Programming). This often occurs at the cost of decreased DHEA-S production and thus lowered estriol production. The fetus may be exposed to elevated cortisol passively (due to increased maternal cortisol secondary to maternal stress or lowered placental 11β HSD2 activity) or actively secondary to proinflammatory cytokine secretion.162 Passive exposure tends to suppress the fetal HPA axis and may increase the risk of low birth weight, prematurity, and other complications as well as alter growth and development and increase risks of adult-onset disorders; active exposure tends to enhance cortisol production after birth.21,162,166 The fetus also responds to stress after 18 to 19 weeks by increasing β-endorphin (BE) levels.126,143
Thyroid gland
Anatomic development
The thyroid gland develops during the first 12 weeks of gestation. The thyroid begins as an epithelial thickening at the base of the tongue around 17 days.131 As the primordial gland migrates down the trachea, it becomes bilobular with a small median isthmus and begins to form follicles.63 The thyroid initially descends in front of the pharyngeal gut. Later the thyroid descends in front of the hyoid bone and larynx to reach its final position in front of the trachea by 45 to 50 days of gestation.131 Thyroid gland volume further increases eight- to 10-fold between 30 and 42 weeks’ gestation with increasing iodine, thyroglobulin, iodothyronine, and thyroid hormone reserves.29
Thyroid function
TRH synthesis begins in the hypothalamus by 6 to 8 weeks and is found in fetal serum by 9 weeks.90,100,129,138 TRH is also synthesized by the placenta, fetal pancreas, and gut. Fetal serum TRH levels are high during the first and second trimesters due to these extrahypothalamic sources; hypothalamic TRH production matures by 35 to 40 weeks along with maturation of the hypothalamic-pituitary portal system.29,103 TSH can be detected in fetal serum by 10 to 12 weeks.131 Levels are low until 18 weeks, then increase to 28 weeks, and then plateau and decrease to term.43,90,131
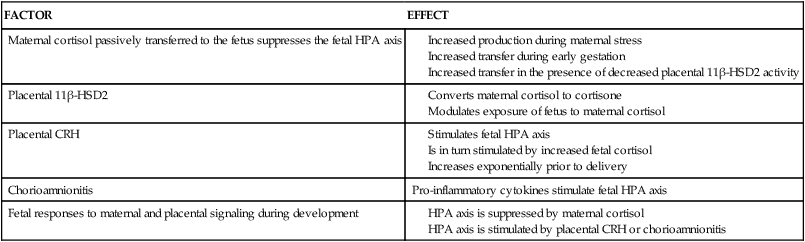
By 10 to 12 weeks, the thyroid gland begins to accumulate and concentrate iodine and has begun to synthesize and secrete iodothyronines. T4 can be detected in fetal serum by 12 weeks, increasing to 2 μg/dL by 20 weeks and 10 μg/dL by term.30,63,90,100,119,129,131,156 Fetal thyroid function remains at basal levels until midgestation, even though the capacity to secrete these hormones as well as TSH and TRH develops earlier.28,43,90 As a result, significant amounts of fetal thyroid hormone are not produced before 18 to 20 weeks’ gestation.95,99 TSH, T4, and free T4 increase from 15 to 42 weeks.168 At term, T4 levels in cord blood are 10% to 20% lower than maternal values; most fetal T4 is bound to TTR and albumin.131 Free T4 concentrations in fetal fluids reflect the interaction between T4-binding proteins and maternal T4, which maintains sufficient free T4 for fetal needs but prevents toxic levels.95 Thyroxine-binding globulin (TBG) can be detected by 12 weeks and reaches term values by midgestation.131 Concentrations of iodine increase after 13 to 15 weeks with peak concentrations at 20 to 24 weeks.72 Changes in fetal thyroid hormones are summarized in Figure 19-9.
Serum T3 levels remain low until 30 weeks and then increase slightly, but never approach maternal values.28,100,156 At term, fetal T3 and free T3 levels are 30% to 50% of maternal values.131 T3 levels remain low because the fetus is unable to convert T4 to T3 peripherally due to incomplete enzyme systems. The increase in T3 after 30 weeks is associated with maturation of these enzymes.28 Type II and type III monodeiodinases (MDI; see Box 19-3 on page 630) are present by midgestation; MDI-I (which converts T4 to the highly active T3) matures later and levels are low during gestation.29,156 As a result, a greater proportion of T4 is converted to the inactive rT3 in the fetus than in the adult.28,98 Iodothyronines are inactivated by sulfation as well, which also helps to regulate the amount of active thyroid hormone; these analogues are also metabolized to the inactive rT3S.168 Increased production of sulfated analogues of thyroid hormones are seen in the fetus.29 In the hypothyroid fetus, MDI-II increases and MDI-I is suppressed so T4 is shunted to the central nervous system (CNS), where it is converted to T3 by MDI-II.29 The control by these monodeiodinases of the levels of active thyroid hormones at different stages of development is an important protective mechanism because too much T3 could damage cells. Organs such as the brain, which are dependent on T3 for normal development, have higher levels of enzymes needed to convert T4 to T3.88,98,99
Serum rT3 levels rise early in the third trimester and then progressively decrease to term as MDI activity increases.90,131,156 As noted earlier, the elevated serum rT3 levels in the fetus are a result of increased MDI-III activity (see also Box 19-3 on page 630).28 MDI-III is the predominant deiodinase in the fetus and placenta with levels 10 to 15 times greater than in adults.28,29 This increases conversion of T4 to the biologically inactive rT3 (rather than to biologically active T3) and T3 to the inactive T2; this may be a way the fetus counteracts high T4 levels and maintains metabolic homeostasis.28,29 Thyroid maturation in the late fetal and early neonatal period is related to progressive increases in hypothalamic TRH secretion, TRH responsiveness by the pituitary (by 26 to 28 weeks, the fetus responds to TRH in a manner similar to that of adults), sensitivity of the thyroid to TSH, and maturation of feedback mechanisms.28,29 Fetal thyroid function is summarized in Table 19-5.
Table 19-5
Characteristics of Fetal Thyroid Function
Placental transfer of iodine and maternal T4
Extrahypothalamic (placenta, pancreas, gut) production of thyrotropin-releasing hormone by the placenta and selected fetal tissue
Progressive maturation of T4 production and T4 negative feedback control of thyroid-stimulating hormone (thyrotropin) secretion
Predominant metabolism of T4 to inactive reverse T3 and sulfated analogues
Local monodeiodinase type II mediated T3 production from T4 in selected fetal tissues including brain
Developmentally programmed maturation of thyroid hormone nuclear receptors in individual fetal tissues
Developmentally programmed maturation of thyroid hormone gene transcription in individual fetal tissues
T3, Triiodothyronine; T4, thyroxine.
Adapted from Fisher, D.A. (1999). Hypothyroxinemia in premature infants: Is thyroxine treatment necessary? Thyroid, 9, 715.
Role of maternal thyroid hormones
Thyroid hormones are critical for brain development, neuronogenesis, neuron migration, axon and dendrite formation, and organization. Thyroid hormone receptors develop early in the fetal brain, appearing by at least 9 weeks’ postmenstrual age, increasing 10-fold by 18 weeks; 25% to 30% of the receptors are occupied by T3 during this time.95 Levels of T3 in the cerebral cortex reach adult values by midgestation.108 Thyroid hormones are much higher in the brain than fetal serum at each stage of development due to activity of MDI-II and brain MDI-III.95
Maternal thyroid hormones are transferred to the fetus in the first trimester, beginning soon after conception and increase rapidly over the next weeks to biologically relevant values that correlate with maternal values.88,94,95,100 Even during the second and third trimesters when the fetal thyroid is producing hormone, much of the T4 needed for development and neuroprotection is of maternal origin.88,94 This has implications for the infant born prematurely, especially very low–birth weight (VLBW) infants, who lose this protection by their early birth.95 Maternal hypothyroidism is particularly damaging to the fetal brain in the first half of pregnancy when fetal T4 production is low.94,129 T3 generation from T4 by MDI-II increases in the cerebral cortex until midgestation, reaching values similar to those in the adult brain. Cerebral T3 levels are dependent on local production in the brain, via MDI-II that converts T4 to T3, and are thus not affected by circulating T3 levels, but by circulating T4. Therefore if the mother is hypothyroxinemic with low T4 levels, the fetal brain will have a deficiency of T3 for normal development, even if circulating T3 is normal or increased.94 Maternal iodine deficiency results in altered thyroid function and inadequate thyroid hormone for development; maternal iodine excess can also block thyroid hormone synthesis and secretion.129
Neonatal physiology
Transitional events
Catecholamines and cortisol surge at birth. Catecholamines, including epinephrine, norepinephrine, and dopamine, increase to levels 20 times greater than those of adults.29 This increase is greater with vaginal delivery than with cesarean delivery. Levels decrease after the first few hours. The catecholamine surge promotes extrauterine cardiorespiratory adaptation (see Chapters 9 and 10) and is accompanied by a surge in other hormones, including renin, angiotensin II, arginine vasopressin (AVP; see Chapter 11), adrenocorticotropic hormone (ACTH), β-endorphins (BEs), and cortisol (see next section). BEs are increased with fetal distress.16 Cord blood and maternal BE levels are not correlated.126 Thyroid hormones are involved in the regulation of thermogenesis, cardiac contractility, and metabolic function during the transition to extrauterine life.11,122
Hypothalamic-pituitary-adrenal axis
Remodeling of the adrenal glands after birth occurs via apoptosis of the fetal zone, remodeling of the fetal zone cells, and development of the other zones.19,90,164 Apoptosis of the fetal zone is mediated by activin and transforming growth factor-β (TGF-β). The size of the adrenals decreases 25% in the first 4 days after birth.164 Adequate pituitary function is seen in both term and preterm infants.86
At birth, the infant’s free cortisol levels are about one third or less than maternal levels, possibly related to higher levels of cortisol-binding globulin (CBG) in the mother. A cortisol surge begins in late gestation, increasing during labor and transition to extrauterine life.29 This surge is blunted in VLBW infants.29 Cortisol levels double during labor, increase further in the first 1 to 2 hours after birth, and then gradually fall during the first week.96 There is an inverse relationship between cortisol levels and gestational age. ACTH and cortisol levels are higher in infants delivered vaginally than in those delivered by cesarean birth.91,134 ACTH and BE levels are elevated in cord blood and decrease significantly by 24 hours and reach adult levels by 4 to 5 days in healthy infants.75,83,126 After birth the high placental corticotropin releasing hormone (CRH) levels fall. This may lead to a refractory period over the first few days where newborn CRH production by the hypothalamus is transiently suppressed, accompanied by a refractory pituitary gland response. These changes result in a decreased ability to increase ACTH to stimulate the adrenal gland. This transition is tolerated well in healthy infants, but in immature or ill infants may lead to a relative adrenal insufficiency and cardiovascular compromise (see Adrenal Cortex Insufficiency).27,105,123
BEs are endogenous opioid-like substances that are derived, along with ACTH and other substances, from a large precursor molecule (pro-opiomelanocortin). BEs increase markedly with birth in both term and preterm infants. The BE increase at birth is greater with perinatal hypoxic-ischemic events or other stress such as forceps delivery and is reported to be inversely related to PO2.16,126,149 Other endogenous opioids, such as the enkephalins, are also increased at birth and further increased with hypoxia, acidosis and ischemia.
Both term and preterm infants in the neonatal intensive care unit (NICU) have higher BE levels than healthy term infants. Preterm infants (especially preterm males) have higher BE levels than both healthy term and ill term infants.75 Elevated BE levels are seen in stressed preterm infants with apnea, but not in nonstressed infants.83 Release of BE is a component of neonatal respiratory control and is triggered by hypoxia.83 BE levels are also higher in infants with respiratory distress.75
The administration of antenatal corticosteroids (see Chapter 10) leads to a transient suppression of the pituitary and adrenal glands. Long-term effects on hypothalamic-pituitary-adrenal (HPA) function have not been well studied.87 Even with this suppression, healthy, preterm infants respond to stress with increased cortisol.103 The ill infant, however, is less able to respond to stress and is more vulnerable to stress-related morbidity (see sections on Neonatal Stress and Adrenal Cortex Insufficiency).
Growth hormone (GH) decreases rapidly after birth as free fatty acids, which block GH release, increase with nonshivering thermogenesis.42 GH levels are higher in preterm infants than in term infants. Somatostatin levels in preterm infants are higher during the second week but decrease by the third to fourth weeks. Levels are even higher in infants with respiratory distress syndrome.55
Prolactin increases in the late fetal and early neonatal periods. Levels are higher in preterm infants than in term infants due to increased sensitivity to thyroid-stimulating hormone (TSH) and estrogens, pituitary insensitivity to dopaminergic inhibition, and reduced renal excretion by the immature kidney (with its lower glomerular filtration rate).149 Prolactin may have a role in lung maturation and fluid homeostasis with transition. Dopamine levels are twice those of adults and function as a prolactin release–inhibiting factor.90
Hypothalamic-pituitary-thyroid axis
The newborn is in a state of physiologic hyperthyroidism because of marked changes in thyroid function at birth (Table 19-6 and Figure 19-9).121,131 Thyroid-releasing hormone (TRH) and TSH levels increase rapidly after birth, followed by increases in T4, free T4, T3, and free T3.124 Exposure of the newborn to the cooler extrauterine environment is believed to stimulate skin thermal receptors, TRH release by the hypothalamus, and TSH release by the pituitary gland.29 The TSH surge is also stimulated by cutting of the umbilical cord and the catecholamine surge at birth.29,121 The increase in TSH (and T4) with cord cutting is limited in extremely low–birth weigh (ELBW) infants.29 TSH increases from 9 to 10 μU/mL (IU/L) at birth to peak at 60 to 70 mU/L within the first hour after birth.29 TSH rapidly decreases to 50% of peak values by 2 hours and to 20% by 24 hours, followed by a progressive decrease over the next 2 to 3 days to levels similar to or lower than cord blood values (see Figure 19-9).28,29,74,90 During the first week, periodic oscillations in serum TSH have been reported, possibly reflecting establishment of a new equilibrium in the negative feedback exerted by thyroid hormone on the anterior pituitary.135 Although TSH generally falls to adult ranges within 3 to 10 days, TSH may remain somewhat elevated for several months.129,131
Table 19-6
Changes in Thyroid Function Associated with Fetal Transition to Extrauterine Life at Term
Neonatal TSH surge
Increased T4 and T3 production
Stimulation of brown adipose tissue thermogenesis
Increased T4 to T3 conversion
Permanently increased serum T3 level
Decreased production of inactive thyroid hormone analogues (rT3, T4S, rT3S, T2S)
Decreased extrahypothalamic thyrotropin-releasing hormone production
Decreased TSH concentration
Resetting of hypothalamic-pituitary free T4 set point for control of TSH secretion
T3, Triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone (thyrotropin).
From Fisher, D.A. (1999). Hypothyroxinemia in premature infants: Is thyroxine treatment necessary? Thyroid, 9, 715.
The TSH surge leads to a rapid threefold to sixfold increase in T4, and twofold increase in free T4.29,168 T3 and rT3 levels are correlated with gestational age and birth weight.129 T4 levels peak at 24 to 48 hours after birth (see Figure 19-9), and then slowly decrease over the next few weeks.29,100,129,131,156 Newborn T4 levels are usually 10% to 20% less than maternal values.131
Cord blood T3 values average 45 to 50 ng/dL (0.69 to 0.77 pmol/L) (30% to 50% of maternal values). The newborn quickly changes from a state of T3 deficiency to T3 excess. T3 levels increase rapidly with birth with a sixfold increase in T3 secretion and eightfold increase in free T3.156 T3 peaks at 24 to 48 hours at 260 to 300 ng/mL (4 to 4.62 pmol/L) (values exceeding those in adults) in most infants and then gradually fall.28,29,129,131 The increase in T3 is due to the TSH surge and increased MDI-I activity (see Box 19-3 on page 630), with increased conversion of T4 to T3 and removal of the placenta (with its high levels of MDI-III that convert T3 to inactive forms).29,156 The increase in T3 is also related to cutting the umbilical cord, which increases liver blood flow and conversion of T3 to T4.29 Serum T3 and T4 levels gradually fall to values seen in infancy with the first few weeks after birth.129
Levels of rT3 remain high for 3 to 5 days and then gradually decrease to adult values by 1 to 2 months.131 Thyroxine-binding globulin (TBG) tends to be high because of transplacental passage of estrogens that stimulate its production. TBG-binding capacity in the infant is higher than in the pregnant woman and about 1.5 times that of the adult.131 Radioactive iodine uptake by the thyroid is higher in the newborn because of increased avidity of the neonatal thyroid for iodine. Iodine concentrations in the thyroid gland at birth are correlated with gestational age with increased 24-hour 123I uptake for the first month and a greater concentration of 123I in infants than adults.131
The TSH surge is also seen in preterm and SGA infants, although it may be blunted.29,168 Thyroid hormones in preterm infants follow patterns similar to those in term infants but may take longer to reach stable values.43,129,131,175 Thyroid hormone levels are inversely related to gestational age.29,99,129 Free T4 and T3 levels are lower and rT3 levels higher in preterm infants than in term infants for the first weeks due to persistence of fetal levels of MDI enzymes (see p. 630) and higher sulfated iodothyronine analogues.29,100,125,156,168 TSH levels and response are generally similar to those in term infants, but TSH may be in the normal adult range.100 In ELBW infants, TSH response to low T4 levels is limited.29 Serum T4 and T3 levels may decrease to below birth levels in VLBW preterm infants during the first week to levels two to three times lower than in term infants.124,129,156 In addition, some ill and VLBW preterm infants develop a characteristic syndrome of transient hyperthyroxinemia with normal or low TSH and low free T4 levels without other evidence of hypothyroidism (see Transient Alterations in Thyroid Function in Preterm Infants).29,99,124,129,156,168 T4 levels are higher in infants of mothers who received a course of antenatal glucosteroids.85 The preterm infant may take 3 to 8 weeks to reach term levels of these hormones.156
Clinical implications for neonatal care
Endocrine adaptation at birth is influenced by gestational age, stresses of delivery, postnatal disease states, and hypoxic-ischemic events.149 Normal endocrine function is critical for normal growth and development both before and after birth. For example, biochemical maturation of the lungs and surfactant production (see Chapter 10) are dependent on the hypothalamic-pituitary-adrenal (HPA) axis and its hormones, including corticotropin-releasing hormone (CRH) and cortisol, as well as thyroid hormones. Thyroid hormones are needed for lung development and surfactant production, bone growth (including calcium and vitamin D homeostasis), thermogenesis, and central nervous system (CNS) maturation.11 Within the CNS, these hormones are critical for differentiation of neural stem cells, cerebral neuronogenesis, and regulation of expression of genes involved in neuronal migration, brain dendritic arborization, synaptogenesis, neuronal migration, axon development, and growth (see Chapter 15).7,11,47,71,144 Effects on bone growth and CNS development continue into early childhood. An intact HPA axis is necessary for the infant to respond appropriately to stress and to prevent maladaptation.
Neonatal stress
Although definitions and depictions of the stress response may vary, all or most propose an integrated biologic response pattern during or after stressor exposure. Activation of endocrine and neurotransmitter systems represents the major mechanisms upon which other aspects of this response are built. Responses to stressors are normally of short duration (acute stress) and are aimed at survival followed by a return to a state of dynamic homeostasis.118,132 Stress responses that go on for extended periods (chronic stress) can produce long-term changes in stress response systems, leading to decreases in adaptive capacity and increased risk for physical and psychological disorders.171
The NICU has the potential for significant stress during a period of critical brain development. There are many sources of stress in the NICU from both internal (e.g., pain, distress, pathologic processes) and external (e.g., physical environment, caregiver interventions, medical or surgical procedures, multiple simultaneous modes of stimulation) demands. Stress and pain (see Chapter 15) interact. Preterm infants may be simultaneously exposed to pain stimuli and other potentially noxious sensations (e.g., handling, light, sound, and temperature changes) that cause further activity in nociceptive pathways and systemic stress responses. An infant’s stress tolerance may be reached or exceeded repeatedly, contributing to short- and long-term morbidity. Infants in the NICU experience both acute and chronic stress. Many handling procedures in small and sick infants lead to stress, which if not controlled, may cause cumulative harm.6
Baseline responses to stress are set early in life and can influence later responses and adaptation.13,33,41,109,159 Stress activates the HPA axis with release of catecholamines, CRH, adrenocorticotropin (ACTH), β-endorphins (BEs), and cortisol. Stress responses affect many systems: cardiovascular (increased blood pressure and heart rate), gastrointestinal (increased gut motility and splanchnic vasoconstriction), renal (sodium and water retention), and respiratory (increased oxygen consumption, decreased tidal volume, and functional residual capacity or ventilation-perfusion mismatch). Stress can alter coagulation, immune function, and cytokine production. Stress has profound metabolic consequences; metabolic stability may be more difficult to maintain in the stressed neonate, especially in immature infants.6 Stress can lead to hyperglycemia or hypoglycemia. Stress increases counterregulatory hormones (e.g., glucagon, ACTH, catecholamines, cortisol) that decrease insulin secretion. Glucagon, fat, and protein are converted to glucose, which—in the face of insulin resistance from the counterregulatory hormones—results in hyperglycemia. In immature infants, this response and the infant’s nutrient stores may become exhausted, so over time the infant becomes hypoglycemic.6
Term infants, whether healthy or ill, have an intact HPA axis and can identify and respond to stress.117 Healthy preterm infants older than 28 weeks’ gestational age respond to stress with increased cortisol secretion, although at lower levels than in term infants.117 Healthy term infants have relatively low basal cortisol levels that rapidly increase with stress.50 Salivary cortisol has been used to examine responses of both term and preterm infants to stressful or soothing events.46,91,137,146,147
However, in preterm infants who are ill or younger than 28 weeks’ gestational age, the HPA is suppressed during the first few weeks after birth. These VLBW infants seem unable to “recognize” stress and to respond by increasing cortisol secretion.50 Preterm infants—especially those who are ill or weigh less than 1000 g—have lower basal cortisol levels and are less able to increase cortisol production with stress. In these infants there is an increase in precursors but not in cortisol, suggesting immature activity of enzymes to convert these precursors to cortisol. In very immature infants, this may reflect the persistence of fetal protective responses against higher maternal cortisol levels.52
Exposure of VLBW infants to significant early pain and stress has been reported to alter later cortisol response patterns with a “resetting” of the basal arousal system postulated.46 Early stress in the fetus and neonate may produce permanent changes in neural pathways, increasing the risk of later disorders such as adult psychopathology and hypertension (see Maternal Stress Responses and Fetal Endocrine Programming).6,13,41,136,159 Maternal stress in the third trimester is associated with increased ACTH and cortisol and increased placental CRH. Because CRH is a primary factor in labor onset, this may lead to preterm labor and decrease the sensitivity of the fetal-neonatal anterior pituitary to CRH, with permanent elevations in cortisol and BEs.136
Adrenal cortex insufficiency
A transient adrenal cortex insufficiency is seen in some ELBW infants. These infants have poor or limited ability to respond to shock and develop hypotension unresponsive to fluid and pressor management.26,104,162 In these infants the adrenal cortex may be unable to produce enough cortisol to maintain blood pressure (cortisol is involved in blood pressure regulation).103 This insufficiency is related to an immature HPA axis with an inadequate pituitary response to CRH stimulation and decreased cortisol production due to immature synthesis of 11β hydroxylase and limited 3β HSD and other enzymes.27,105,123 Some ELBW infants were reported to have an increase in ACTH and cortisol following CRH administration and increased cortisol after ACTH administration, which are expected responses. However, other ELBW infants in this study, especially those who were ill, had very low serum cortisol levels that remained low even under stimulation.50 Others have found that only one third to one half of sick preterm infants were able to achieve a normal response to ACTH and CRH stimuation.51,105,162 Decreased responses to ACTH stimulation have also been reported in small-for-gestational-age (SGA) infants.12
Acutely ill preterm infants may have lower cortisol values than healthy preterm infants with low cortisol associated with hypotension, inflammation, bronchopulmonary dysplasia (BPD), and patent ductus arteriosus and an increase in mortality.8,27,139,162 A multicenter study of prophylactic hydrocortisone to prevent BPD did not improve survival without BPD, although infants who were treated after chorioamnionitis had decreased mortality.163 However, the study was stopped early due to an increased risk of gastrointestinal perforation in the hydrocortisone-treated group, especially with simultaneous use of hydrocortisone and indomethacin or ibuprofen.27,163 Term infants following severe perinatal stress may also demonstrate a transient relative adrenal insufficiency with low circulating levels of cortisol.27,105
Thyroid function and thermoregulation
Thyroid function and neonatal temperature regulation are interrelated (see Chapter 20). T3 and T4 increase basal metabolic rate and heat production, whereas T4 and norepinephrine stimulate metabolism of brown adipose tissue. Occlusion of the umbilical cord activates brown adipose tissue catabolism via a catecholamine surge and withdrawal of placental prostaglandins and adenosine.28,29 T4 enhances the effects of catecholamines to increase oxygen consumption (and metabolic rate) and increases brown adipose tissue lipolysis. Within 6 hours of birth, the term neonate can respond to cold stress by increasing his or her metabolic rate 100%; by 6 to 9 days, this increase may be 170%. The preterm infant responds similarly to cold stress, although at a slower rate and lower percentage increase (∼40%).42
Transient alterations in thyroid function in preterm infants
In healthy preterm infants, cord blood T4 levels range from 5.5 to 6 mcg/dL (72 to 77 nmol/L), increasing to 7 to 9 mcg/dL (90 to 116 nmol/L) by 21 to 28 days and reaching values similar to those of term infants by 4 to 6 weeks or perhaps sooner.17 As a result, preterm infants are more likely to have below normal values on thyroid screening tests compared with term infants.131,141,156 Thyroid hormone values after birth are correlated with gestational age. For example, infants of 25 to 27 weeks’ gestation have free T4 levels that are two to three times lower than those of term infants.29 Maturation of thyroid function in VLBW infants is illustrated in Figure 19-10.
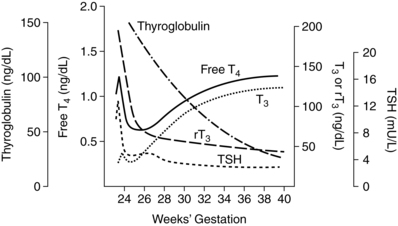
Early loss of placental transfer of maternal T4, which at that point in gestation may account for 30% of fetal T4, along with iodine deficiency from inadequate stores and intake, may alter thyroid homeostasis in preterm infants, especially ELBW infants.7,29,168 As a result some preterm infants develop transient thyroid alterations in the early weeks after birth as T4 and T3 levels fall in the first week to values lower than at birth. Other factors in the etiology of these alterations include decreased liver thyroxine-binding globulin (TBG) production, immaturity of the hypothalamic-pituitary-thyroid axis, inadequate nutrition, decreased MDI-I, decreased brown adipose tissue, and immature tissue thyroid systems (Table 19-7).29,129 Thyroid function tends to return to normal as the infant matures or recovers from the underlying illness, with achievement of stable serum T4 values by 6 to 7 weeks.131 Three transient patterns of altered thyroid function have been described in preterm infants: physiologic hypothyroxinemia, transient primary hypothyroidism, and transient secondary/tertiary hypothyroidism.29,63 These are more likely to occur in VLBW infants who weigh less than 1000 g and infants who are ill or stressed. Ill preterm, such as those with respiratory distress syndrome, patent ductus arteriosus, necrotizing enterocolitis, and prolonged oxygen dependence, may also develop nonthyroidal illness syndrome.29,168 This disorder is similar to the euthyroid sick syndrome seen in severely ill adults and children.140
Table 19-7
Thyroid System Immaturities in the Premature Infant
Loss of maternal T4
Limited postnatal thermogenesis
Hypothalamic-pituitary immaturity
Limited thyroid gland reserve
Persistent fetal thyroid hormone metabolism
Predisposition to nonthyroidal illness syndrome
From Fisher, D.A. (2008). Thyroid system immaturities in very low birth weight premature infants. Semin Perinatol, 32, 391.
Physiologic hypothyroxinemia of prematurity is seen in many preterm infants younger than 35 weeks’ gestational age and referred to as transient hypothyroxinemia of prematurity (THOP).29,168 There is a lack of consensus regarding what values define THOP.168,169 These infants have low T4 levels with low to normal TBG and thyroid-stimulating hormone (TSH) levels.29,129,168 This is a transient phenomenon that usually resolves within 2 to 3 weeks, although it may last for 2 to 3 months.29,63 Thus most preterm infants are hypothyroxinemic. The frequency increases with decreasing gestational age. The mechanism and pattern of this disorder vary with gestational age.29
In infants born at greater than 30 to 32 weeks’ gestational age, T4 and free T4 increase transiently after birth in conjunction with the TSH surge (which is similar to the pattern seen with term infants) and then decrease. This is followed by a gradual increase in TBG, T4, free T4, and T3, reaching term values by 38 to 42 weeks.29 These infants probably have immaturity of the thyroid’s ability to respond to TSH and ability to convert T4 to T3.29
In VLBW infants younger than 30 weeks’ gestational age, there is a limited TSH surge and increase in T4, followed by a decrease in T4 and free T4 to a nadir at 1 to 2 weeks.29,156 These infants usually achieve cord blood levels of serum T4 by 3 to 4 weeks.29 The greatest fall is seen in the lowest gestation infants.156 As T4 decreases, TSH rises, but often remains below 20 μU/L (the cutoff value for hypothyroidism).156 The etiology of the hypothyroxinemia in these infants is not completely understood but probably due to immaturity of the HPT axis, interruption of transfer of maternal thyroid hormones and iodine across the placenta, iodine deficiency, immaturity of MDI enzymes, decreased TBG and probably an inability to increase iodine uptake and T4 production with extrauterine adaptation.7,29,63 Because of immaturity, these infants also have inefficient production of thyroglobulin and a decreased ability to convert T4 to T3 due to low levels of liver MDI-I activity (see Box 19-3 on page 630).29
Transient primary hypothyroidism is less common.29 These infants have low T4 and free T4, with increased TSH levels during the first 2 to 3 weeks after birth.63 Thyroglobulin and iodine stores are low, with increased T4 utilization and demand. The frequency of transient secondary/tertiary hypothyroidism is unknown but may occur in up to 10% of VLBW infants. These infants have low T4 and free T4 levels without changes in TSH during the first 1 to 2 weeks after birth.29 There may be some benefit to treating these infants in that low thyroid hormone may contribute to later neurologic problems, although it is unclear if supplementation improve outcomes.155,156
The decision of whether to treat healthy or ill preterm infants with THOP is controversial.29,63,66,131,155 Exogenous TRH significantly increases serum T4 and TSH levels. However, most studies of use of T4 for thyroid hormone replacement do not demonstrate significant differences in the age at which serum T4 values normalize or in infant growth parameters.130,155,160 Some observational evidence suggests that THOP is associated with adverse developmental outcomes, although trials with thyroid hormone replacement have had mixed results.22,29,155,157,168,169 Osborn and Hunt concluded that prophylactic thyroid hormone administration did not reduce neonatal mortality or morbidity or improve neurodevelopmental outcomes, and there was insufficient evidence regarding the effect of thyroid hormone replacement on neonatal mortality, morbidity, and improvement in neurodevelopmental outcomes.110,111
Neonatal hyperthyroidism
Neonatal hyperthyroidism is rare and is usually a transient disorder associated with transplacental passage of maternal thyroid-stimulating immunoglobulins (TSIs) in women with Graves’ disease (see Chapter 13). This may occur even in euthyroid women after thyroid removal or 131I thyroid ablation, because TSIs are still present in the woman’s system. TSI levels correlate positively with development of neonatal hyperthyroidism; thus maternal screening can identify the infant at risk. An increased fetal heart rate (greater than 160 beats/min) after 22 weeks is of concern.72 The mother may be treated with antithyroid drugs, if she is not already being treated, to maintain the fetal heart rate around 140. The incidence of neonatal effects is 0.6% to 9.5%.72 Although this disorder is usually transient, increased mortality has been reported, usually as a result of respiratory obstruction or a difficult delivery due to the enlarged thyroid, or high-output cardiac failure secondary to tachycardia (heart rates greater than 200 beats/min).72 If the disorder is recognized prior to birth, infants have been treated by giving medications such as propylthiouracil (PTU) to the mother.120,154
Clinical findings such as low birth weight, irritability, hunger, tachycardia, diarrhea, sweating, and arrhythmias may be present at birth. T4 and free T4 levels are increased, but they are also increased in the normal newborn. If the mother is on antithyroid medication, manifestations may be delayed for 2 to 10 days.43 These infants have an advanced bone age and occasionally craniosynostosis. Clinical effects, although transient, last for 1 to 5 months (generally 2 to 3 months) or up to 10 months in some infants.43
Neonatal hypothyroidism
The most common cause of congenital hypothyroidism in North America is thyroid dysgenesis, with an incidence of 1 in 3500 to 4000 live births.76,131 The cause is often unclear. In some infants, the hypothyroidism is due to a single gene defect or familial autoimmune factors. In the majority (85%) of infants, the cause is a nonfamilial embryogenic defect.67 This may be due to mutations in gene coding transcription factors that are needed for thyroid or pituitary gland morphogenesis and differentiation.63,67 Females are affected twice as often as males. Hypothyroidism and cretinism secondary to endemic goiter are rare in North America. Infants with intrauterine hypothyroidism may not experience significant impairment of somatic or brain growth before birth, because maternal T4 crosses the placenta and is catabolized to T3 for normal fetal brain development.28
Most infants appear normal at birth, with clinical signs apparent in fewer than 5% to 30%. Common findings such as a large fontanel, hypotonia, macroglossia, and umbilical hernia may not develop for several weeks or months.43,74,131 By the time these findings are apparent, significant neurologic damage has already occurred, because inadequate thyroid hormone during fetal life and early infancy alters CNS development. The exact mechanism of injury is not well understood, but the degree of neurologic abnormality correlates with the duration and severity of the hypothyroidism.43 Even with relatively early diagnosis and treatment after birth, these individuals may have subtle, selective neurologic impairments.133
Neonatal screening for hypothyroidism
Diagnosis and treatment of congenital hypothyroidism before 1 to 3 months of age are associated with an increased likelihood of normal mental development. Therefore routine screening of newborns for hypothyroidism has been implemented in all U.S. states and Canadian provinces as well as in many other countries.131 Screening involves evaluating T4 or TSH levels. Infants with congenital hypothyroidism have decreased T4 levels because of the thyroidal dysgenesis and elevated TSH levels. TSH levels are elevated because the low T4 level does not provide the usual negative feedback inhibition of the anterior pituitary.28,64,67 TSH screening is more useful to detect subclinical and primary hypothyroidism; T4 screening is most useful in detecting central hypothalamic-pituitary hypothyroidism and for infants with a delayed TSH increase.12,72 Use of TSH as the initial screen has increased in recent years due to increased sensitivity with this test.76,129 Specimens taken immediately after birth (usually within the first 24 hours) are avoided because of the usual TSH surge. Infants screened before 48 hours of age have a higher rate of intermediate results.131 Preterm infants are at greater risk of false-positive tests due to their HPT immaturity.64,141 Infants with a positive screen are further evaluated and those with a positive diagnosis are treated with thyroid hormone replacement therapy to normalize serum T4 levels.
Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) is the most common cause of ambiguous genitalia and adrenal insufficiency.127 CAH comprises a group of autosomal recessive genetic disorders, characterized by enzymatic defects in adrenal cortex hormone synthesis (see Figure 2-10), with a frequency of 1 in 15,000 live births in North America.102 These infants have a defect in cortisol synthesis. Without adequate cortisol negative feedback, ACTH levels are elevated, especially in the last half of gestation. The excess ACTH stimulates the adrenal glands to overproduce precursors for cortisol and leads to adrenal hyperplasia.96,102,131,153
CAH is due to a complete or partial block in one of the enzymes involved in synthesis of adrenal cortex hormones (see Figure 19-8). Symptoms vary from mild to severe depending on the enzyme defect. Depending on the enzyme involved and the severity of the enzyme block, some female infants may have ambiguous genitalia due to virilization. Because some of the same enzymes are found in the gonads as the adrenals, both tissues are affected.96 Prenatal therapy with maternal dexamethasone may prevent or reduce virilization of female genitalia if therapy is begun by 6 to 7 weeks’ gestation.68,97,106,154 This glucocorticoid crosses the placenta, suppressing fetal ACTH secretion (see Chapter 7).68,97 In infants with partial enzyme defects, results may not be apparent until later in childhood or adolescence.
Table 19-8 summarizes the effects of various enzyme blocks that can lead to CAH. The most common block (90% to 95%) is in the cytochrome P-450–type enzyme 21-hydroxylase (21-OH) located on the short arm of chromosome 6, in which multiple types of mutations and clinical phenotypes are seen.96,102,127 The 21-OH mutations block conversion of progesterone to precursors needed for production of aldosterone and cortisol (see Figure 19-8), leading to lower levels of these hormones. The excess precursors are instead converted to androgens that therefore increase. CAH is one of the disorders evaluated in newborn screening. Screening is done by measuring plasma levels of 17-hydroxy-progesterone, which is elevated in CAH. High rates of false positives to true positives have been reported, especially in stressed preterm and low–birth weight infants.12 Treatment involves glucocorticoid therapy to suppress ACTH. Aldosterone may be needed if the infant has a salt-wasting form of the disorder (see Table 19-8).
Table 19-8
Enzyme Deficiencies in Congenital Adrenal Hyperplasia
| DEFICIENT ENZYME | EFFECTS ON HORMONES | EFFECT IN THE NEWBORN |
| 21-Hydroxylase | Inability to synthesize glucocorticoids and aldosterone; increased androgen production in response to increased adrenocorticotropin | Ambiguous genitalia (females), hyponatremia, hyperkalemia, weight loss, acidosis, shock |
| 11-Hydroxylase | Inability to synthesize glucocorticoids; suppression of renin activity | Ambiguous genitalia (females), hypertension, hypernatremia, hypokalemia, alkalosis |
| 3β-Hydrosteroid dehydrogenase | Decreased androgen production; decreased glucocorticoid production | Incomplete genital development (males), mild virilization (females), hypotension, hypoglycemia, hyponatremia, hyperkalemia, acidosis, shock |
| 17α-Hydroxylase | Impaired glucocorticoid and androgen production | Ambiguous genitalia (males), hypernatremia, hypokalemia, hypertension |
| Lipoid adrenal hyperplasia | Decreased glucocorticoid, mineralocorticoid, and androgen production | Ambiguous genitalia (males), hyponatremia, hyperkalemia, hypoglycemia, acidosis, shock |
From Witt, C.L. (1999). Adrenal insufficiency in the term and preterm neonate. Neonatal Netw, 18, 25.
Maturational changes during infancy and childhood
HPA maturation continues during infancy and childhood, followed by marked changes at puberty. Involution of the adrenal gland is most prominent in the first month after birth and is complete during the first year of life. By 1 month the adrenal is 50% of the size at birth.164 Dehydroepiandrosterone sulfate (DHEA-S) rapidly decreases after birth to low levels and then rises again beginning at 5 to 6 years of age, peaking in early adulthood, and then gradually decreasing.51 Circadian rhythmicity of cortisol secretion is seen by 8 to 12 weeks after birth in term infants.60
Within the adrenal gland, the fetal zone involutes after birth and has disappeared by 6 months. The definitive zone (possibly with the transitional zone) develops into the zona glomerulosa and zona fasciculata by 2 years. The final zone, the zona reticularis, develops later becoming fully differentiated by mid-adolescence.123
Adrenarche is maturation of the adrenal cortex, with increased production of DHEA, DHEA-S, and other androgens, and development of the zona reticularis. DHEA rises to a maximum around 25 years of age and then declines gradually thereafter.51 Adrenarche begins in the prepuberty period around 6 to 8 years of age and precedes gonadarche. Steroid hormones increase progressively and are associated with a transient increase in linear growth and bone maturation.112 Adrenarche increases production of androgens and estrogens needed for puberty changes and occurs several years before the onset of puberty. The exact trigger for this change is unknown. It may be related to a decrease in levels of 3β-hydroxysteroid dehydrogenase (3β HSD) in the zona reticularis and may be influenced by insulin-like growth factor-I.37,51 Gonadal maturation and changes in the hypothalamic-pituitary-adrenal (HPA) axis with puberty are discussed in Chapter 2.
HPT function gradually changes during infancy and childhood. Thyroid hormones are critical for continued CNS maturation and bone growth. The critical period for thyroid hormone influences on the CNS continues to 6 to 8 months after birth. It has been estimated that infants with significant hypothyroidism lose 3 to 5 IQ points per month if untreated during this period.28 The ability of the infant to convert T4 to T3 matures over the first month. Twenty-four-hour 123I uptake by the thyroid at 1 month is similar to adult values, but concentrations per gram of thyroid tissue are greater since the thyroid gland at this age is only one tenth that of an adult.131 During childhood, TSH levels tend to remain higher than adult levels. The T4 turnover rate is higher in infants and children, accounting for the increased requirement of children for thyroid hormone per unit weight.131 TBG levels gradually fall over the first few years. Thyroid hormones fall to high adult values by 4 to 6 weeks, but they do not reach mean adult values until mid-puberty due to higher TBS levels in children.131
Summary
Maturation of the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-thyroid (HPT) axes and neuroendocrine function in the fetus and infant is essential for development of the CNS, lungs, bones, and other systems. These functions undergo significant changes during the transition to extrauterine life. In addition, thyroid function is interrelated with thermoregulation and changes dramatically at birth. Newborns normally have a relative hyperthyroidism, but immaturity or illness can lead to transient hypothyroidism. Screening for congenital hypothyroidism and congenital adrenal hyperplasia is a part of newborn metabolic screening. Use of this screening is essential for early identification and reduction of mortality and morbidity in these infants. All neonates should be monitored for stress, and protective interventions should be initiated. Preterm infants—especially those who are ill or younger than 28 weeks’ gestational age—are particularly vulnerable to the adverse consequences of stress. Table 19-9 summarizes clinical recommendations related to neonatal pituitary, adrenal, and thyroid function.
Table 19-9
Know the usual changes in pituitary and adrenal function in the fetus and during the neonatal period (pp. 649-643, 645-646, Table 19-4, and Figure 19-7).
Know the usual changes in thyroid function during the neonatal period in term infants (pp. 645-647, Figure 19-9, Table 19-6).
Know the usual changes in thyroid function during the neonatal period in preterm infants (pp. 646-647 and Table 19-7).
Recognize the normal parameters related to thyroid function in term and preterm neonates (pp. 646-647, 648-658).
Recognize and monitor for transient alterations in thyroid function in the preterm infant (pp. 649-650).
Recognize the signs of and monitor for transient hyperthyroidism in infants of mothers with Graves’ disease (p. 650).
Recognize the clinical signs of hypothyroidism in infants (p. 650).
Screen newborns for congenital hypothyroidism and congenital adrenal hyperplasia per state newborn screening protocol (pp. 650-651).
Monitor growth and neurologic status in infants with congenital hypothyroidism (p. 650).
Monitor for signs of stress and pain in neonates (pp. 647-648 and Chapter 15).
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
Pituitary, adrenal, and thyroid function
Pituitary, adrenal, and thyroid function and the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-thyroid (HPT) axes are critical for normal function and adaptation during pregnancy, growth, and development of the fetus, and adaptation of the newborn to the extrauterine environment. HPA axis function in the mother and fetus are closely interrelated with placental function. The hormones of the HPA and HPT axes (see Figure 2-7) are necessary for many body functions, for development of the central nervous system (CNS) and other growth processes, and for reproductive function. Disorders of these systems are associated with infertility, alterations in normal changes at puberty, and complications of pregnancy. Concentrations of HPA and HPT axis hormones are altered in pregnant women and neonates. In the neonate marked changes in adrenal and thyroid function occur with birth. This chapter examines changes in the glands and hormones of the HPA and HPT axes during pregnancy; development of neuroendocrine function in the fetus and neonate; and implications for the mother, fetus, and neonate.
Maternal physiologic adaptations
Antepartum period
Hypothalamic-pituitary-adrenal axis
Marked changes occur in the hypothalamic-pituitary-adrenal (HPA) axis during pregnancy, resulting in a state of increased HPA function.77,113 (See Figure 2-7 for an illustration of the HPA axis.) These changes are mediated primarily by placental hormones, including placental ACTH, GH, and CRH. Maternal hypothalamic-pituitary function is discussed further in Chapter 2 in conjunction with the hypothalamic-pituitary-ovarian axis. Changes in the hypothalamus and pituitary during pregnancy are summarized in Box 19-1 on page 628.
Anterior pituitary function.
The anterior pituitary is composed of six cell types, each of which produces different hormones. These types of cells and their major hormones include lactotroph (prolactin), corticotroph (pro-opiomelanocortin [POMC] and its derivatives including ACTH, β-endorphin, and β-lipotropin), somatotroph (GH), gonadotroph (follicle-stimulating hormone [FSH] and luteinizing hormone [LH]), and cells producing thyroid-stimulating hormone (TSH) (thyrotropin). The anterior pituitary gland increases in size and weight (from an average of 660 mg in the nonpregnant woman to 760 mg or greater during pregnancy) due to an estrogen-induced increase in the lactotroph cells.32,35,86,93,97,151 The anterior pituitary also develops a more convex, dome-shaped surface, which may cause it to bulge upward in some women, compressing the optic chiasma.32 For these women, this can result in a transient hemianopia. Changes in pituitary function during pregnancy are summarized in Box 19-1 on page 628.
In the nonpregnant woman, the prolactin-producing lactotroph cells make up approximately 20% of the anterior pituitary; this increases to around 60% during pregnancy.151 Prolactin isoforms increase during pregnancy, with the nonglycosylated forms exceeding the N-linked glycosylated form that is most common in nonpregnant women.32,151 The nonglycosylated form may be more bioactive and function to prepare the breast for lactation (see Chapter 5). Prolactin increases 10-fold during pregnancy to peak at delivery at 140 ng/mL (6068 pmol/L).32,35,74,97 Most of the increase in maternal prolactin is from the maternal anterior pituitary, although prolactin is also produced by the maternal decidua. Decidual prolactin is found primarily in amniotic fluid; little enters the maternal circulation. Prolactin levels in the amniotic fluid peak in the second trimester at 6000 ng/mL (260,886 pmol/L).32,35,151
Corticotroph cells do not change in size during pregnancy.151 However, ACTH secretion and plasma ACTH levels increase progressively, peaking during the intrapartum period (Figure 19-1), from approximately 10 pg/mL (2.2 pmol/L) in nonpregnant women to 50 pg/mL (11 pmol/L) at term.32,35,97,151 ACTH secretion is stimulated by CRH and, in turn, stimulates release of cortisol by the adrenal gland. Changes in ACTH parallel the increase in free and total cortisol (see Figure 19-1).35,86 During pregnancy, the adrenal gland is more responsive to ACTH with a blunted HPA axis response to exogenous glucocorticoids.77 There is a two- to fourfold increase in ACTH from the first to third trimesters, in spite of the increased bound and free plasma cortisol.97 The increase in ACTH in the face of increased cortisol suggests a change in the set point for cortisol release that alters the ACTH-cortisol feedback loop.44,97 ACTH maintains its diurnal variation during pregnancy, although this cycling may be blunted.35,43,77,86,97 Placental ACTH increases in the second and third trimesters and it is unclear how much of increased maternal serum ACTH comes from the maternal anterior pituitary versus the placenta.93 The effects of ACTH on the adrenal gland and changes in cortisol during pregnancy are described further under Adrenal Function.
The increased ACTH secretion during pregnancy is thought to be due primarily to increased placental (and some decidual and fetal membrane) CRH, rather than maternal hypothalamic CRH (Figure 19-2, B).32,35,77,86,151,159 Other factors that may contribute to this increase are the decreased pituitary gland sensitivity to cortisol feedback, enhanced pituitary responsiveness to corticotropin-releasing factors such as vasopressin, and CRH.77,159 Maternal serum CRH increases markedly beginning by 8 to 10 weeks and rises from prepregnant values of 10 to 100 pg/mL (47.6 to 476 pmol/L) to 300 to 1000 pg/mL (1429 to 4762 pmol/L) by the third trimester.86,151,159 CRH-binding protein (CRH-BP) decreases the bioactivity of CRH throughout most of gestation. CRH-BP levels are similar to nonpregnant levels until the third trimester and then fall by two thirds during the last 6 weeks in preparation for birth. CRH has multiple roles in establishing and maintaining pregnancy and is produced by placental and uterine tissues in addition to the hypothalamus.159 CRH plays an important role in the onset of parturition; increased CRH prior to term plays a role in preterm labor onset (see Chapter 4).159
Somatotroph and gonadotroph cells of the anterior pituitary decrease during pregnancy.6 Hypothalamic gonadotropin-releasing hormone (GnRH) is suppressed in pregnancy by the elevated CRH, β-endorphins, and cortisol, with a blunted response of the pituitary to GnRH and low LH and FSH levels by 6 to 7 weeks.159 By midpregnancy, LH and FSH levels are undetectable.32,35,97,151 Gonadotropin function is described in Chapter 2. β-endorphins during pregnancy and the intrapartum period are described in Chapter 15.
Pituitary GH (GH-N) decreases after the first trimester as placental GH (GH-V) increases.35,93,97 GH-V is a GH variant that stimulates bone growth and regulates maternal insulin-like growth factor-I (IGF-I), which in turn alters maternal metabolism, stimulating gluconeogenesis and lipolysis.93 GH-V is secreted continuously, as opposed to the pulsatile secretion characteristic of GH-N.5 GH-N is the main maternal GH until 15 to 20 weeks; then it decreases and becomes undetectable by term. GH-V increases progressively from 15 to 20 weeks until term.97 GH-V stimulates IGF-I, with negative feedback suppression of maternal GH-N.5,32
Posterior pituitary function.
The major posterior pituitary hormones are arginine vasopressin (AVP) and oxytocin. Posterior pituitary function changes are associated with osmoregulatory changes and parturition. AVP levels are within normal ranges during pregnancy; however, the threshold at which AVP is secreted is reset so that AVP is secreted at a lower plasma osmolality during pregnancy (see Chapter 11) with a decline in plasma osmolality by 5 to 10 mOsm/kg below the nonpregnant mean of 285 mOsm/kg.97,98 AVP also modulates ACTH release.43 Oxytocin levels progressively increase during pregnancy, with further increase at term (see Chapter 4) and with lactation (see Chapter 5).35,97
Adrenal function.
Pregnancy is characterized by a transient hypercortisolism that begins by at least 12 weeks (see Figure 19-1).59,86 Both total serum cortisol (the primary glucocorticoid) and free cortisol increase, with total cortisol peaking at levels three- to eightfold times higher by term.97,127 Salivary cortisol increases twofold by 25 weeks and then plateaus to term.21 The pregnant woman does not demonstrate signs of hypercortisolism because the free cortisol fraction is still within normal range.21 Urinary cortisol increases at least 180% by term.97 The diurnal secretion of cortisol—with higher levels in the morning versus evening—is blunted but maintained.77,86,97 Physiologic responses to stress are maintained during pregnancy, but appear to be blunted.21,159
The increase in plasma cortisol parallels the increase in ACTH (see Figure 19-1). The increase in total cortisol is primarily due to an estrogen-stimulated increase in cortisol-binding globulin (CBG) that increases to two- to threefold during pregnancy.97 Increased CBG reduces liver catabolism and clearance of cortisol, yielding a twofold increase in cortisol half-life.86 The increase in free cortisol is also due in part to displacement of cortisol from CBG by progesterone.35
The adrenal gland becomes hypertrophic as the zona fasciculata (site of glucocorticoid production) widens, with no changes in the size of the zona glomerulosa or reticularis (see Box 19-2 on page 630).35 Levels of aldosterone (the primary mineralocorticoid) also are markedly increased in pregnancy (see Chapter 11). Synthesis of androgens and dehydroepiandrosterone sulfate (DHEA-S) by the zona reticularis also increases, but less so than the glucocorticoids and mineralocorticoids. An increase in sex hormone binding globulin during pregnancy increases total and protein bound testosterone levels. Levels of free testosterone are low to normal to 28 weeks and then increase along with levels of androstenedione.97 Maternal serum DHEA-S levels remain low due to placental uptake and metabolism.44
Hypothalamic-pituitary-thyroid axis
Marked changes are also seen in the HPT axis during pregnancy. These changes occur primarily in the first half of gestation so that the woman achieves a new steady state in HPT function by midgestation that is maintained until delivery.62,71 The net result of changes in thyroid function during pregnancy is to increase the availability of thyroid hormones by 40% to 100%.142 Morreale de Escobar et al. summarized the major changes in thyroid function during pregnancy: (1) Increased estrogen leads to a two- to threefold increase in thyroid-binding globulin production by the liver. This decreases levels of free thyroid hormone and stimulates the hypothalamic-pituitary-thyroid (HPT) axis; (2) increased human chorionic gonadotropin (hCG), which has a structure similar to thyroid-stimulating hormone (TSH), stimulates increased T3 and T4. This leads to negative feedback to the pituitary gland and a decrease in TSH (especially during weeks 8 to 14 when hCG is peaking); (3) peripheral metabolism of thyroid hormones increases in the second and third trimesters due to increased production of type II and III monodeiodinases (see Box 19-3 below) by the placenta.95
Adaptations during pregnancy related to thyroid physiology mimic hyperthyroidism. The pregnant woman can be described as being in a state of euthyroid hyperthyroxinemia, however, since thyroid function per se does not change during pregnancy.95 Thyroid hormone changes are important in supporting the altered carbohydrate, protein, and lipid metabolism of pregnancy and changes in basal metabolic rate (see Chapter 16).79 The factors primarily responsible for changes in HPT axis function during pregnancy are the elevated hCG and thyroid-binding globulin (TBG) levels and the increased urinary iodide excretion that lowers maternal plasma iodine.40,43 Box 19-3 on page 630, Box 19-4 below and Figure 19-3 review thyroid hormone production and regulation. Figure 19-4 illustrates regulation of maternal thyroid hormone function during pregnancy.
Thyroid hormones are transported in the blood bound to binding proteins, such as TBG, albumin, and transthyretin (TTR; formerly called thyroxine-binding prealbumin). In the nonpregnant individual, about two thirds of the T4 is bound to TBG, increasing to 75% or greater during pregnancy.34,43 Under the influence of estrogen, hepatic synthesis and sialylation of TBG increase twofold to threefold beginning within a few weeks after fertilization and plateau from midgestation to delivery.34,71,99,100,144 Increased sialylation increases the half-life of TBG. The ability of TBG to bind thyroxine doubles during this period; TTR, also influenced by estrogen, decreases.43 These changes increase serum TBG levels, decrease the percent of T4 bound to TTR and increase total T4 and T3.100 The increased TBG is accompanied by 10% to 15% decrease in free T4 and free T3 if iodine is sufficient; if iodine is inadequate, T4 levels increase.99
A transient increase in free T4 is reported in the first trimester, related to the increase in hCG, with a decrease in the second and third trimesters.71,72 Variations in findings are most often due to differences in the iodine status of the populations studied and measurement techniques.73 Free T3 changes parallel those of free T4.72 Concentrations of free T3 and free T4, although low, remain within normal physiologic limits.72,99 The basis for these changes is thought to be primarily related to the interaction of estrogen, TSH, and thyroid-binding proteins.72
In contrast to changes in free T3 and free T4, total T3 and T4 increase, peaking at 10 to 15 weeks’ gestation and then plateauing at levels 40% to 100% higher than nonpregnant values.142,144 This increase is primarily due to increases in TBG and hCG, and possibly to an increase in the production of MDI-III (see Box 19-3 on page 630) by the placenta.71 This enzyme converts T4 to reverse T3 (rT3), an inactive compound, and T3 to T2, another inactive compound.98 Levels of T3 are elevated because of the increased availability of T4 for deiodination to T3 in peripheral tissue rather than due to increased T3 production per se.
The increased T3 and T4 are also related to increased TSH bioactivity stimulated by hCG, which peaks at about the same time.58 hCG has a mild TSH-like activity that increases secretion of T4.34,100 hCG is a partial inhibitor of the pituitary gland and has β subunits similar to TSH (as well as luteinizing and follicle-stimulating hormone). Thus hCG has thyrotropic activity and can activate TSH receptors.34,100 Serum hCG is positively correlated with free T4 and inversely correlated with TSH levels in early pregnancy.40,73 Elevated T3 and T4 suppress endogenous TSH secretion by the anterior pituitary.98 TSH decreases transiently from 8 to 14 weeks’ gestation at the time of the hCG peak, progressively returning to prepregnancy levels by term.34,100,151 Occasionally some otherwise healthy pregnant women will experience a transient hyperthyroxinemia (generally without clinical signs) associated with higher than usual hCG levels in the first trimester and a greater fall in TSH.43,72,99 The increase in total T3 and T4 is less than the increase in TBG, resulting in a decreased T4/TBG ratio, leading to a state of relative hypothyroxinemia during pregnancy.
Pregnancy is characterized by significant changes in iodide metabolism with increased renal iodide clearance and increased iodine needed to make thyroid hormones.38,71 The increased total T4 and T3 stimulate increased serum protein-bound iodine (PBI).38 Because circulating levels of free thyroid hormone are not significantly altered, increased PBI does not reflect maternal hyperthyroidism. Thyroid iodine uptake increases because of a decrease in the total body iodine pool. This pool is altered because of increased renal iodide loss secondary to the increased renal blood flow and glomerular filtration rate (see Chapter 11) and placental transfer of iodine to the fetus.43,71,99,144
The thyroid compensates for the increased loss of iodine by hyperplasia and increased plasma iodine clearance, reducing plasma iodine levels.88,99 Iodine is stored in the colloid of the thyroid gland follicles (see Figure 19-3). With TSH stimulation, thyroglobulin is catabolized to form T4 and T3.99 Serum thyroglobulin increases in the first trimester but is most marked in later pregnancy. This increase is associated with an increase in thyroid volume, especially in areas of low iodine intake.43,72,99 The degree of hyperplasia is related to the degree of imbalance between iodine needs versus iodine intake and stores (see Iodine Needs during Pregnancy).72 Mild thyroid hyperplasia (10% to 15% increase), due primarily to increased vascularity, is seen in areas such as North America, where diets are generally thought to be iodine sufficient.38,99,100 Therefore moderate to marked thyroid enlargement in these women cannot be considered normal and requires further evaluation. Women who live in iodine-poor areas have an increase in thyroid volume of 15% to 30% during pregnancy.36,99 Goiter is generally not a risk if iodine levels are greater than 0.08 mcg/dL (0.006 μmol/L); in North America, levels average 0.3 mcg/dL (0.023 μmol/L). Goiter is a significant risk in areas with low iodine intake.
Intrapartum period
The HPA and HPT axes undergo further alterations during the intrapartum period. CRH appears to be a trigger in the initiation of labor, and activation of the HPA axis may serve as a “biologic clock” timing the length of gestation.99 Maternal plasma CRH, ACTH, β-endorphin, and cortisol levels increase up to sevenfold with labor onset and during labor (see Chapter 4).20,35,77,86,93,172 ACTH reaches its highest level, increasing from 50 pg/mL (11 pmol/L) at term to 300 pg/mL (66 pmol/L) in labor (see Figure 19-1).32,77,151 Further increases in ACTH and cortisol are seen in women with poor progress in labor.31 Low β-endorphin levels at term have been associated with an increased need for pain medication during labor, although a causal relationship is unclear.20
Levels of total and free T3 increase during labor. This change probably reflects the energy demands of labor on the maternal system. T3 and T4 have similar functions, but T3 is three to five times more active. T3 and T4 increase intracellular enzymes (increased cellular metabolism), the number and activity of mitochondria (to provide energy for cellular enzyme systems), and Na-K adenosine triphosphatase (ATPase) (because of the increased energy use for myometrial contractions).47
Postpartum period
The alterations in the HPA (see Figure 19-2, C) and HPT axes during pregnancy are reversed during the postpartum period. CRH levels drop rapidly with removal of the placenta and placental CRH.151 Maternal ACTH and cortisol levels decrease rapidly in the immediate postpartum period and reach nonpregnant values by 1 to 4 days postpartum.35,77,86 The HPA axis is depressed with a reduction in hypothalamic CRH for 3 to 6 weeks, returning to normal levels by 12 weeks.86 This depression of the HPA axis may play a role in postpartum mood disorders or in exacerbation of autoimmune disorders in the postpartum period.1,62 Although ACTH secretion may be suppressed, total serum cortisol is within normal limits, probably secondary to a mild hypertrophy of the adrenal cortex.86 Hyperplasia of the lactotrophs of the anterior pituitary gland peaks in the first 3 days postpartum. This tissue decreases in size by 1 month in nonlactating and more slowly in lactating women, but never returns to nulliparous size.32,93,97 Prolactin falls at delivery and returns to nonpregnant values by 3 months; in breastfeeding women, prolactin increases after delivery.32,35,151 β-endorphins decrease by 24 hours after birth and are higher in colostrum than in maternal plasma.173 Serum growth hormone levels may remain elevated for several months.32 Postpartum changes in prolactin, FSH, and LH are described in Chapter 5.
After delivery with removal of the placenta and reduction in estrogen, hepatic synthesis of TBG decreases, as does the renal excretion of iodine. As a result, the metabolic alterations in thyroid processes gradually reverse over 4 to 6 weeks, although they may persist for up to 6 to 12 weeks.43,144 Thyroid-releasing hormone (TRH) is a (minor) stimulus of prolactin release and has been used to induce relactation.70 Free T4 may be low and TSH elevated in the first 3 to 4 days, which may confound assessment of thyroid function.65 Transient disorders in thyroid function are seen in some postpartum women (see Postpartum Thyroid Disorders).
Thyroid hormones are secreted in breast milk. Levels are low initially and then rise. Breast milk T4 and T3 have been reported to delay the development of hypothyroidism in some infants with this disorder.131
Clinical implications for the pregnant woman and her fetus
Changes in the hypothalamic-pituitary-adrenal (HPA) and hypothalamic-pituitary-thyroid (HPT) axes are critical for maintenance of pregnancy. In addition, the maternal and fetal HPA axes and the interrelationship between maternal and fetal-placental function are essential for initiation of labor (see Chapter 4). Alterations in the HPA axis with infection or stress may lead to preterm labor. These risks are discussed further in Chapter 4. Thyroid disorders are more common in women and are not uncommon in pregnant women, being the second most common endocrine disorder (after diabetes mellitus) seen during pregnancy. Disorders of the adrenal and pituitary gland, such as prolactinomas, which may increase in size during pregnancy due the stimulating effects of elevated prolactin levels, and Cushing disease, are uncommon.78,93,97,98 Cushing disease tends to be exacerbated during pregnancy with remission postpartum and is more often due to adrenal lesions than excess adrenocorticotropin (ACTH).78 Diagnosis of thyroid dysfunction during pregnancy may be more difficult in that symptoms of thyroid disorders often mimic some of the usual physiologic changes of pregnancy, and radioactive iodine tests cannot be used because of fetal risks. Implications of alterations in thyroid function and changes in laboratory tests during pregnancy are discussed in this section, along with disorders of thyroid function unique to the postpartum period.
Iodine needs during pregnancy
Adequate iodine stores and nutrition are essential for normal function of the maternal and fetal thyroid.23,36,39 Iodine needs increase during pregnancy due to increased renal loss and increased placental—and thus fetal—uptake.30,36,131 Dietary iodine is converted to iodide; 20% of this is absorbed by the thyroid gland, the rest is cleared by the kidney. During pregnancy renal iodide clearance doubles due to the increased renal blood flow and glomerular filtration rate (see Chapter 11).99 The World Health Organization recommends an iodine intake of 250 μg/day during pregnancy and lactation; others recommend 200 or up to 300 μg/day.30,76,88,131,174,175 Iodine needs can be met by iodine in prenatal vitamins and use of iodinated salt.43 Mean urinary iodine excretion, used to assess iodine deficiency, is insufficient if less than 150 μg/L during pregnancy or less than 100 μg/L during lactation.30
Iodine deficiency is lowest in North America, higher in Europe, with Southeast Asia accounting for nearly one fourth of the worldwide population with insufficient iodine uptake.88 Iodine stores are low in developing countries and have been falling in many developed countries, for example, 50% to 65% of women in western and central Europe were found to be iodine deficient (versus 11% of women in the United States).36,175 Recent reports suggest a decline in iodine sufficiency in childbearing women in the United States.15 Women who enter pregnancy in an iodine-deficient state fall even further behind so that iodine deficiency in the first trimester tends to become more severe in later pregnancy.71,73 Iodine deficiency is the most frequent cause worldwide of preventable mental retardation and often the damage is done by the time of birth.88,175 Iodine deficiency is also associated with an increased risk of spontaneous abortion and stillbirths.88,175 Even in iodine-sufficient areas, there has been increasing concern that iodine deficiency may increase the risk of later neurodevelopmental impairment of offspring in women with marked subclinical thyroid dysfunction.72 Various professional groups have differing recommendations regarding whether all pregnant women should be routinely screened to identify subclinical hypothyroidism.1,2,9,14,30,62,167
Thyroid function tests during pregnancy
Changes in TBG and thyroid hormones during pregnancy alter parameters for many tests used to assess thyroid status. These alterations, which vary with trimester, must be considered when evaluating thyroid function in pregnant and postpartum women. Free T3 and T4 assays, rather than total values, are generally preferred because of the increased thyroxine-binding globulin (TBG).30 Glinoer and Spencer indicate that serum TSH assay “is the most sensitive index to reliably detect thyroid function abnormalities” during pregnancy.40 Reference ranges for thyroid function tests during pregnancy have been described in several studies, with variations by trimester of testing, number of fetuses, and testing method.30 Table 19-1 summarizes the changes in thyroid function during pregnancy that lead to alterations in these tests. As noted in the previous section, recommendations for routine thyroid screening during pregnancy vary among different professional groups worldwide.1,2,9,14,30,62,167
Table 19-1
Thyroid Function Tests during Pregnancy
| PHYSIOLOGIC CHANGES | RESULTING CHANGE IN THYROID ACTIVITY |
| ↑ Serum estrogens | ↑ Serum TBG |
| ↑ Serum TBG |
Modified from Brent, G.A. (1997). Maternal thyroid function: Interpretation of thyroid function test in pregnancy. Clin Obstet Gynecol, 40, 3, by Fantz, C.R., et al. (1999). Thyroid function during pregnancy. Clin Chem, 45, 2250.
Antithyroid peroxidase enzymes have been reported in 10% to 14% of women at 14 weeks’ gestation and may increase the risk of gestational thyroid dysfunction and postpartum thyroiditis.72 Most women are euthyroid but some have increased thyroid-stimulating hormone (TSH) or decreased free T4 or both.72
Thyroid function and nausea and vomiting in pregnancy
Nausea and vomiting in pregnancy (NVP) has been linked to alterations in T4, TSH, and human chorionic gonadotropin (hCG) (which has TSH-like activity). hCG stimulates receptors for both TSH (increases T4) and hCG (increases estriol and possibly NVP). NVP severity has been correlated with increased free T4 and decreased TSH, with values returning to normal pregnant ranges as the nausea and vomiting resolve.71 These findings may lead to or be a consequence of emesis during early pregnancy. NVP is discussed further in Chapter 12.
Hyperemesis gravidarum in women without any history or evidence of thyroid dysfunction has also been associated with increased free T4 and decreased TSH. These findings, seen in about 60% of women with hyperemesis, are similar to those of the euthyroid sick syndrome seen with severe illness.73 T4 levels in women with hyperemesis return to usual values in 1 to 4 weeks after resolution with or without treatment with antithyroid drugs.1,43,72,98
The pregnant woman with hyperthyroidism
A transient hyperthyroidism is seen in 10% to 20% of otherwise healthy pregnant women during the first trimester; this may be associated with increased hCG levels, multiple gestation, or NVP.34,36 This transient form of subclinical hyperthyroidism is characterized by normal free T4 and decreased TSH.
Another form of transient clinical hyperthyroidism, characterized by increased free T4 and decreased TSH, is seen less frequently and may be associated with multiple gestation, hyperemesis gravidarum, and gestational trophoblast disorders with markedly increased hCG production.34,62,72 Trophoblast disorders such as a molar pregnancy occasionally cause biochemical and, in some women (5% to 64%), clinical findings of hyperthyroidism because of the high levels of hCG secreted by the trophoblastic mass.100
Chronic hyperthyroidism occurs in 0.05% to 0.2% of pregnant women.99 The diagnosis of hyperthyroidism during pregnancy may be difficult in that many of the signs and symptoms associated with this disorder are often seen normally during pregnancy. Findings common to hyperthyroidism and certain stages of pregnancy include fatigue, heat intolerance, warm skin, emotional lability, insomnia, increased appetite, sweating, breathlessness, ankle edema, palpitations, and increased pulse pressure.30,34,43,72,99,100 Failure to gain weight with a good appetite and persistent tachycardia (greater than 100 beats/min) are most suggestive of hyperthyroidism in pregnancy.43 Increased free T3 and total T4 (greater than 15 mcg/dL [193 nmol/L]) and increased or high-normal RT3U are seen with hyperthyroidism. Alterations in thyroid function tests during pregnancy (see Table 19-1) must be considered when interpreting test results. For example, because RT3U is decreased in pregnancy, values in the nonpregnant range suggest hyperthyroidism. TSH levels less than 0.05 μU/L (mIU/mL) and increased free T4 levels greater than 11.6 mcg/dL (149 pmol/L) are diagnostic.99
Hyperthyroidism in pregnant women is almost always (85% to 95%) due to Graves’ disease.34,72,73,81 Graves’ disease is an autoimmune disorder in which thyroid-stimulating immunoglobulins (TSIs), such as TSH receptor antibody (TRAb), attach to and activate TSH receptors on the thyroid follicular cells. This leads to increased production of thyroid hormones and the clinical finding of hyperthyroidism.
Women with mild hyperthyroidism generally do well during pregnancy because increased serum TBG offsets the increased secretion of thyroid hormones. In women with Graves’ disease, their disorder may be aggravated in the first trimester (due to the increased hCG) and then improve during the third trimester, with remission and occasionally complete resolution.30,165 This improvement is related to suppression of the maternal immune system responses (see Chapter 13) by fetal cytokines with lower TSI levels and decreased thyroid hormone production. Relapse or exacerbation generally occurs postpartum, usually within several weeks of delivery, as immune system alterations and production of TBG return to prepregnancy levels.100,165
Pregnant women with hyperthyroidism usually require a caloric intake that is higher than that generally recommended during pregnancy to compensate for their increased metabolic rate. These women are also at risk for fluid loss and dehydration as a result of the diarrhea and tachycardia that often accompany hyperthyroidism.99 Women with hyperthyroidism, particularly if this disorder is poorly controlled, are at increased risk for placental abruption, miscarriage, preterm labor, fetal growth restriction, congestive heart failure, and thyroid storm.30,34,36,69,72,119 Their infants can develop transient hyperthyroidism from transplacental passage of TSIs or hypothyroidism secondary to the effects of maternal antithyroid drugs.36,119 Hyperthyroidism also alters sex steroid metabolism, sperm motility, and fertility.61,131
Transplacental passage of TSIs, especially TRAbs, leads to a transient neonatal hyperthyroidism in about 1% to 5% of infants of mothers with Graves’ disease (see Chapter 13).1,30,33,116 The risk is present even if the woman is euthyroid in pregnancy or has had thyroid ablation via surgery or radiation before pregnancy because TSIs are still present.82,100 The fetal risk increases after 20 weeks when fetal TSH receptors become responsive to TSH.81 TSH-binding inhibitory immunoglobulins may also cross the placenta, causing a transient neonatal hypothyroidism.131
Hyperthyroidism is treated with antithyroid drugs (thioamides), surgical removal, or thyroid ablation with 131I. The most common thioamides are propylthiouracil (PTU) and methimazole (Tapazole or carbimazole, which is metabolized to methimazole). Thioamides cross the placenta and can block synthesis of thyroid hormones by the fetus.119 The lower hormone levels stimulate increased TSH production, which can lead to goiter and tracheal obstruction. Infants of mothers treated with thioamides may have decreased T4 and increased TSH levels after birth. These values are generally within normal neonatal limits by 4 to 5 days of age. PTU is recommended as the first-line drug during pregnancy.1,34,36,69 Although methimazole (MMI) has the advantage of less frequent dosing and fewer tablets per dose, MMI is associated with a risk of fetal scalp defects (cutis aplasia) and possibly esophageal and choanal atresia.30,36,73,100,152 Pharmacologic effects must be monitored carefully, particularly during the third trimester in women with Graves’ disease, when remission can lead to decreased thyroid hormone production and a transient decrease in required drug dosage by 32 to 36 weeks.99 In some women, the drug can be transiently discontinued in late pregnancy.165
Pregnant women may also develop a transient subacute thyroiditis that often occurs in association with viral infections. The inflammation and destruction of thyroid tissue lead to release of stored thyroid hormone into serum and a transient hyperthyroxinemia. As the disorder resolves, the woman may develop hypothyroidism because the released thyroid hormones are used up before the thyroid gland can produce an adequate new supply. If treatment is initiated, β-blockers such as propranolol are required rather than antithyroid drugs that block thyroid hormone production, because with subacute thyroiditis the thyroid is not making hormones.43,72 Propranolol is generally not indicated for long-term treatment of pregnant women with hyperthyroidism. This drug crosses the placenta and has been associated with fetal growth restriction and impaired responses to anoxia and neonatal hypoglycemia and bradycardia.
The pregnant woman with hypothyroidism
Hypothyroidism in iodine-sufficient pregnant women is usually secondary to autoimmune disorders (after surgical removal or ablation of the thyroid with radioactive 131I for Graves’ disease and idiopathic myxedema) or Hashimoto thyroiditis.30,62 Worldwide, iodine deficiency is the most common cause of hypothyroidism and can lead to cretinism (mental retardation, deafness, other neurologic symptoms in the infant).36,48,62,100 Iodine deficiency leads to hypothyroxinemia, thyroid stimulation from thyroid-releasing hormone (TRH) and TSH feedback loops, and development of goiter.71 Women with untreated hypothyroidism have a high incidence of infertility and spontaneous abortion.30,34,43,73,124
The diagnosis of hypothyroidism during pregnancy may be missed because some of the signs and symptoms associated with this disorder—fatigue, weight gain, muscle cramps, constipation, and amenorrhea—are also seen normally during pregnancy.30,43,99 The increase in TBG in pregnancy may mask the decrease in thyroid hormones; however, levels are usually still low for pregnant norms. In addition, free T4 is low and TSH is elevated.72
Pregnancy in women with primary hypothyroidism or autoimmune thyroiditis may be complicated by an increased risk of fetal loss or prolonged pregnancy, possibly due to compromised placental blood flow or the inability of the thyroid gland to meet the metabolic demands of pregnancy. These women are also at greater risk for preterm delivery, preeclampsia, and cesarean section.30,34,36,62,92,107,148 Weight gain patterns must be carefully monitored in the woman with hypothyroidism. These women may also have difficulty with fatigue and constipation during pregnancy.
Thyroid hormone replacement doses usually need to be increased as a result of the increased TBG and T4 demand.3,49,72,100,145,158 For example, levothyroxine may need to be increased 25% to 40% to maintain normal serum TSH levels.3,34,73 Increased requirements may occur as early as the fifth week of gestation and are influenced by maternal estrogen levels, pregestation TSH levels, maternal volume of distribution, parity, and etiology of the hypothyroidism.3,4,34,80 Postpartum levothyroxine levels usually decrease to prepregnancy levels, but some women continue to have a greater requirement.30,34
Although overt hypothyroidism is associated with neurodevelopmental problems in offspring, the role of subclinical hypothyroidism is less clear.1,48,88,101,170 Subclinical hypothyroidism (elevated TSH but normal free T4) occurs in 2% to 5% of pregnancies and increases the risk of prematurity, abruption, and impaired neurologic outcome in infants.1,30,48,72,88 The risk of later neurologic problems in offspring is thought to be greatest with untreated or subclinical hypothyroidism in the first 10 to 12 weeks of gestation because the fetus is completely dependent on maternal thyroid hormone for brain development during this time.115 However, the role of subclinical hypothyroidism in infants’ later development is still an area of debate, due to inadequate data on the causal relationship between subclinical hypothyroidism and lower IQ levels.30,48,114 Routine screening is recommended for all pregnant women or for all who are high risk for this disorder by some, but not all professional organizations1,2,9,14,62,167
Postpartum thyroid disorders
The postpartum period is associated with transient thyroid disorders. Physiologic alterations and experiences of pregnancy can mask clinical findings of hypothyroidism or hyperthyroidism. As a result, these disorders may first become apparent in the postpartum period. Although less common than these transient disorders, postpartum women are also at increased risk of developing Graves’ disease, especially women older than 35 years of age.10
Postpartum thyroid disorder (PPTD) is a transient disorder seen in 6% to 9% of postpartum women.62 The incidence of PPTD is 33% to 50% in women with thyroid peroxidase antibodies in early pregnancy (versus 0.5% in those without these antibodies).62,72 PPTD is also more prevalent in women with type 1 diabetes.62 Classic PPTD accounts for about 28% of these cases and generally appears in the first 6 months (usually by 6 to 8 weeks after delivery). PPTD (Figure 19-5) is generally characterized by weeks or months (average, 2 to 4 months, but can be up to 6 months) of mild hyperthyroidism, followed by weeks or months (up to a year) of hypothyroidism, and finally a return to normal thyroid function in most women—usually by 12 months postpartum.1,43,62,72,100 Others present with only hypothyroidism or hyperthyroidism.
The exact cause of PPTD is unclear but it appears to be an autoimmune disorder with increased susceptibility in the postpartum woman as her immune system rapidly changes from Th2 to Th1 T-lymphocyte dominance (see Chapter 13).1,72 The initial hyperthyroid phase is characterized by thyroid cell destruction with excessive release of thyroid hormone. PPTD may reflect postpartum exacerbation of a subclinical autoimmune disorder that, with release of pregnancy-induced immunosuppression, leads to rebound of immune components with excessive production of thyroid autoantibodies.43,62
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
