[level-membership-for-pediatrics-category]
Chapter 568 Physiology of the Adrenal Gland
568.1 Histology and Embryology
Several transcription factors are critical for the development of the adrenal glands. The three that are associated with adrenal hypoplasia in humans are steroidogenic factor-1 (SF-1; NR5A1), DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia congenita, X chromosome; NR0B1), and the GLI3 oncogene. Disruption of SF-1, encoded on chromosome 9q33, results in gonadal and often adrenal agenesis, absence of pituitary gonadotropes, and an underdeveloped ventral medial hypothalamus. In-frame deletions and frameshift and missense mutations of this gene are associated with 46,XX ovarian insufficiency and 46,XY gonadal dysgenesis. Mutations in the DAX1 gene, encoded on Xp21, result in adrenal hypoplasia congenita and hypogonadotropic hypogonadism (Chapter 569.1). Mutations in GLI3 on chromosome 7p13 cause Pallister-Hall syndrome, other features of which include hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly. Postnatally, both SF-1 and DAX-1 play important roles in regulating steroidogenesis by modulating transcription of steroidogenic enzymes.
Ferraz-de-Souza B, Achermann JC. Disorders of adrenal development. Endocr Dev. 2008;13:19-32.
Hammer GD, Parker KL, Schimmer BP. Minireview: transcriptional regulation of adrenocortical development. Endocrinology. 2005;146:1018-1024.
Kempna P, Fluck CE. Adrenal gland development and defects. Best Pract Res Clin Endocrinol Metab. 2008;22:77-93.
Lourenco D, Brauner R, Lin L, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med. 2009;360:1200-1210.
568.2 Adrenal Steroid Biosynthesis
Cholesterol is the starting substrate for all steroid biosynthesis ( see Fig. 568-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com). Although adrenal cortex cells can synthesize cholesterol de novo from acetate, circulating plasma lipoproteins provide most of the cholesterol for adrenal cortex hormone formation. Receptors for both low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol are expressed on the surface of adrenocortical cells; the receptor is termed scavenger receptor class B, type I (SR-BI). Patients with familial hypercholesterolemia who lack LDL receptors have unimpaired adrenal steroidogenesis, suggesting that HDL is the more important source of cholesterol. Cholesterol is stored as cholesteryl esters in vesicles and subsequently hydrolyzed by cholesteryl ester hydrolases to liberate free cholesterol for steroid hormone synthesis.
see Fig. 568-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com). Although adrenal cortex cells can synthesize cholesterol de novo from acetate, circulating plasma lipoproteins provide most of the cholesterol for adrenal cortex hormone formation. Receptors for both low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol are expressed on the surface of adrenocortical cells; the receptor is termed scavenger receptor class B, type I (SR-BI). Patients with familial hypercholesterolemia who lack LDL receptors have unimpaired adrenal steroidogenesis, suggesting that HDL is the more important source of cholesterol. Cholesterol is stored as cholesteryl esters in vesicles and subsequently hydrolyzed by cholesteryl ester hydrolases to liberate free cholesterol for steroid hormone synthesis.
Zona Fasciculata
In the endoplasmic reticulum of the zona fasciculata, pregnenolone and progesterone are converted by 17α-hydroxylase (P450c17, CYP17) to 17-hydroxypregenolone and 17-hydroxyprogesterone, respectively. This enzyme is not expressed in the zona glomerulosa, which consequently cannot synthesize 17-hydroxylated steroids. 17-Hydroxypregnenolone is converted to 17-hydroxyprogesterone and 11-deoxycortisol by the same 3β-hydroxysteroid and 21-hydroxylase enzymes, respectively, as are active in the zona glomerulosa. Thus, inherited disorders in these enzymes affect both aldosterone and cortisol synthesis (Chapter 570). Finally, 11-deoxycortisol reenters mitochondria and is converted to cortisol by steroid 11β-hydroxylase (P450c11, CYP11B1). This enzyme is closely related to aldosterone synthase but has low 18-hydroxylase and nonexistent 18-oxidase activity. Thus, under normal circumstances the zona fasciculata cannot synthesize aldosterone.
Fetoplacental Unit
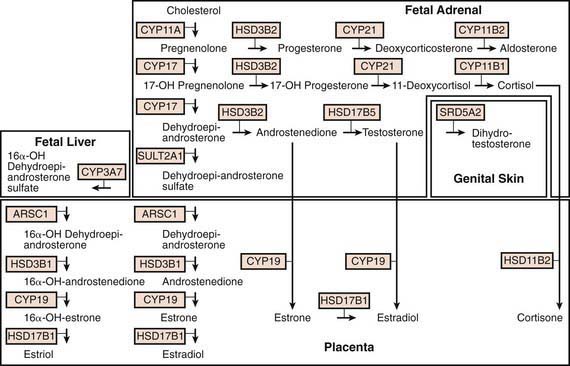
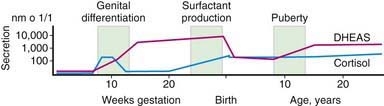
Steroid synthesis in the fetal adrenal varies during gestation (Figs. 568-1 and 568-2). Shortly after the fetal adrenal gland forms (wk 8-10), it efficiently secretes cortisol, which is able to negatively feed back on the fetal pituitary and hypothalamus to suppress ACTH secretion. This is critical time for differentiation of the external genitalia in both sexes (Chapter 570.1); to prevent virilization, the female fetus must not be exposed to high levels of androgens of adrenal origin, and placental aromatase activity must remain low during this time to minimize conversion of testosterone to estradiol in male fetuses, which would interfere with masculinization. After wk 12, HSD3B activity in the fetal adrenal gland decreases and steroid sulfokinase activity increases. Thus, the major steroid products of the midgestation fetal adrenal gland are DHEA and DHEA sulfate (DHEAS) and, by 16α-hydroxylation in the liver, 16α-hydroxy DHEAS. Aromatase activity increases in the placenta at the same time, and steroid sulfatase activity is high as well. Thus, the placenta uses DHEA and DHEAS as substrates for estrone and estradiol and 16α-OH DHEAS as a substrate for estriol. Cortisol activity is low during the 2nd trimester, which might serve to prevent premature secretion of surfactant by the developing fetal lungs; surfactant levels can affect the timing of parturition. As term approaches, fetal cortisol concentration increases as a result of increased cortisol secretion and decreased conversion of cortisol to cortisone by 11β-hydroxysteroid dehydrogenase type 2 (HSD11B2). Low levels of aldosterone are produced in mid gestation, but aldosterone secretory capacity increases near term.
Arlt W, Stewart PM. Adrenal corticosteroid biosynthesis, metabolism, and action. Endocrinol Metab Clin North Am. 2005;34:293-313. viii
Connelly MA. SR-BI-mediated HDL cholesteryl ester delivery in the adrenal gland. Mol Cell Endocrinol. 2009;300:83-88.
Ghayee HK, Auchus RJ. Basic concepts and recent developments in human steroid hormone biosynthesis. Rev Endocr Metab Disord. 2007;8:289-300.
Goto M, Piper Hanley K, Marcos J, et al. In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J Clin Invest. 2006;116:953-960.
Miller WL. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta. 2007;1771:663-676.
Miller WL. Steroidogenic enzymes. Endocr Dev. 2008;13:1-18.
Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25:947-970.
White PC. Ontogeny of adrenal steroid biosynthesis: why girls will be girls. J Clin Invest. 2006;116:872-874.
568.3 Regulation of the Adrenal Cortex
Regulation of Cortisol Secretion
Glucocorticoid secretion is regulated mainly by adrenocorticotropic hormone (corticotropin, ACTH), a 39-amino-acid peptide that is produced in the anterior pituitary. It is synthesized as part of a larger-molecular-weight precursor peptide known as pro-opiomelanocortin (POMC). This precursor peptide is also the source of β-lipotropin (β-LPH). ACTH and β-LPH are cleaved further to yield α- and β-melanocyte-stimulating hormone, corticotropin-like intermediate lobe peptide (CLIP), γ-LPH, β- and γ-endorphin, and enkephalin (Chapter 550).
Corticotropin-releasing hormone (CRH), synthesized by neurons of the parvicellular division of the hypothalamic paraventricular nucleus, is the most important stimulator of ACTH secretion. Arginine vasopressin (AVP) augments CRH action. Neural stimuli from the brain cause the release of CRH and AVP (Chapter 550). AVP and CRH are secreted in the hypophyseal-portal circulation in a pulsatile manner. This pulsatile secretion appears to be responsible for the pulsatile (ultradian) release of ACTH. The circadian rhythm of corticotropin release is probably induced by a corresponding circadian rhythm of hypothalamic CRH secretion, regulated by the suprachiasmatic nucleus with input from other areas of the brain. Cortisol exerts a negative feedback effect on the synthesis and secretion of ACTH, CRH, and AVP. ACTH inhibits its own secretion, a feedback effect mediated at the level of the hypothalamus. Thus the secretion of cortisol is a result of the interaction of the hypothalamus, pituitary, and adrenal glands and other neural stimuli.
Bassett MH, White PC, Rainey WE. The regulation of aldosterone synthase expression. Mol Cell Endocrinol. 2004;217:67-74.
Hammer GD, Parker KL, Schimmer BP. Minireview: transcriptional regulation of adrenocortical development. Endocrinology. 2005;146:1018-1024.
Miller WL. Androgen synthesis in adrenarche. Rev Endocr Metab Disord. 2009;10:3-17.
Nakamura Y, Gang HX, Suzuki T, et al. Adrenal changes associated with adrenarche. Rev Endocr Metab Disord. 2009;10:19-26.
Rainey WE, Nakamura Y. Regulation of the adrenal androgen biosynthesis. J Steroid Biochem Mol Biol. 2008;108:281-286.
568.4 Adrenal Steroid Hormone Actions
Actions of Glucocorticoids
Growth
In excess, glucocorticoids inhibit linear growth and skeletal maturation in children, apparently through direct effects on the epiphyses. However, glucocorticoids are also necessary for normal growth and development. In the fetus and neonate, they accelerate the differentiation and development of various tissues, including the hepatic and gastrointestinal systems, as well as the production of surfactant in the fetal lung. Glucocorticoids are often given to pregnant women at risk for delivery of premature infants in an effort to accelerate these maturational processes (Chapter 90.8).
Actions of Mineralocorticoids
Mineralocorticoids have their most important actions in the distal convoluted tubules and cortical collecting ducts of the kidney, where they induce reabsorption of sodium and secretion of potassium. In the medullary collecting duct, they act in a permissive fashion to allow vasopressin to increase osmotic water flux. Thus, patients with mineralocorticoid deficiency can develop weight loss, hypotension, hyponatremia, and hyperkalemia, whereas patients with mineralocorticoid excess can develop hypertension, hypokalemia, and metabolic alkalosis (Chapters 569–572).
The mineralocorticoid receptor has similar affinities in vitro for cortisol and aldosterone, yet cortisol is a weak mineralocorticoid in vivo. This discrepancy results from the action of HSD11B2, which converts cortisol to cortisone. Cortisone is not a ligand for the receptor, whereas aldosterone is not a substrate for the enzyme. Pharmacologic inhibition or genetic deficiency of this enzyme allows cortisol to occupy renal mineralocorticoid receptors and produce sodium retention and hypertension; the genetic condition is termed apparent mineralocorticoid excess syndrome (Chapter 570.3).
Actions of the Adrenal Androgens
Many actions of adrenal androgens are exerted through their conversion to active androgens or estrogens such as testosterone, dihydrotestosterone, estrone, and estradiol. In men, <2% of the biologically important androgens are derived from adrenal production, whereas in women approximately 50% of androgens are of adrenal origin. The adrenal contribution to circulating estrogen levels is mainly important in pathologic conditions such as feminizing adrenal tumors. Adrenal androgens contribute to the physiologic development of pubic and axillary hair during normal puberty. They also play an important role in the pathophysiology of congenital adrenal hyperplasia, premature adrenarche, adrenal tumors, and Cushing syndrome (Chapters 570 and 571).
Canalis E. Mechanisms of glucocorticoid action in bone. Curr Osteoporos Rep. 2005;3:98-102.
Datson NA, Morsink MC, Meijer OC, et al. Central corticosteroid actions: search for gene targets. Eur J Pharmacol. 2008;583:272-289.
De BK, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol. 2009;23:281-291.
Fuller PJ, Young MJ. Mechanisms of mineralocorticoid action. Hypertension. 2005;46:1227-1235.
Haller J, Mikics E, Makara GB. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Front Neuroendocrinol. 2008;29:273-291.
Reid DM, Devogelaer J-P, Saag K, et al. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. and for the HORIZON investigators. Lancet. 2009;373:1253-1263.
Tait AS, Butts CL, Sternberg EM. The role of glucocorticoids and progestins in inflammatory, autoimmune, and infectious disease. J Leukoc Biol. 2008;84:924-931.
Tasker JG, Di S, Malcher-Lopes R. Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 2006;147:5549-5556.
van der LS, Meijer OC. Pharmacology of glucocorticoids: beyond receptors. Eur J Pharmacol. 2008;585:483-491.
van Raalte DH, Ouwens DM, Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: towards expansion of therapeutic options? Eur J Clin Invest. 2009;39:81-93.
Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70:407-417.
568.5 Adrenal Medulla
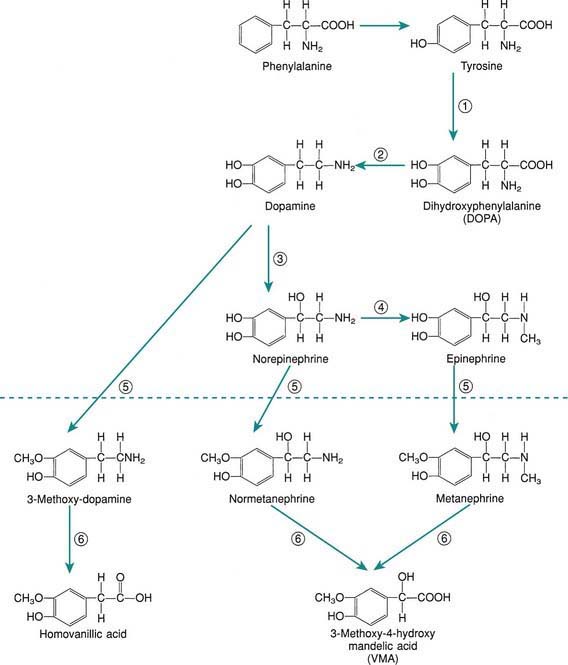
The principal hormones of the adrenal medulla are the physiologically active catecholamines: dopamine, norepinephrine, and epinephrine (see Fig. 568-3 on the Nelson Textbook of Pediatrics  website at www.expertconsult.com). Catecholamine synthesis also occurs in the brain, in sympathetic nerve endings, and in chromaffin tissue outside the adrenal medulla. Metabolites of catecholamines are excreted in the urine, principally 3-methoxy-4-hydroxymandelic acid (VMA), metanephrine, and normetanephrine. Urinary metanephrines and catecholamines are measured to detect pheochromocytomas of the adrenal medulla and sympathetic nervous system (Chapter 574).
website at www.expertconsult.com). Catecholamine synthesis also occurs in the brain, in sympathetic nerve endings, and in chromaffin tissue outside the adrenal medulla. Metabolites of catecholamines are excreted in the urine, principally 3-methoxy-4-hydroxymandelic acid (VMA), metanephrine, and normetanephrine. Urinary metanephrines and catecholamines are measured to detect pheochromocytomas of the adrenal medulla and sympathetic nervous system (Chapter 574).
Adams MS, Bronner-Fraser M. Review: the role of neural crest cells in the endocrine system. Endocr Pathol. 2009;20:92-100.
Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56:331-349.
Goldstein DS, Kopin IJ. Adrenomedullary, adrenocortical, and sympathoneural responses to stressors: a meta-analysis. Endocr Regul. 2008;42:111-119.
Wong DL. Why is the adrenal adrenergic? Endocr Pathol. 2003;14:25-36.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 568 Physiology of the Adrenal Gland
568.1 Histology and Embryology
Several transcription factors are critical for the development of the adrenal glands. The three that are associated with adrenal hypoplasia in humans are steroidogenic factor-1 (SF-1; NR5A1), DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia congenita, X chromosome; NR0B1), and the GLI3 oncogene. Disruption of SF-1, encoded on chromosome 9q33, results in gonadal and often adrenal agenesis, absence of pituitary gonadotropes, and an underdeveloped ventral medial hypothalamus. In-frame deletions and frameshift and missense mutations of this gene are associated with 46,XX ovarian insufficiency and 46,XY gonadal dysgenesis. Mutations in the DAX1 gene, encoded on Xp21, result in adrenal hypoplasia congenita and hypogonadotropic hypogonadism (Chapter 569.1). Mutations in GLI3 on chromosome 7p13 cause Pallister-Hall syndrome, other features of which include hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly. Postnatally, both SF-1 and DAX-1 play important roles in regulating steroidogenesis by modulating transcription of steroidogenic enzymes.
Ferraz-de-Souza B, Achermann JC. Disorders of adrenal development. Endocr Dev. 2008;13:19-32.
Hammer GD, Parker KL, Schimmer BP. Minireview: transcriptional regulation of adrenocortical development. Endocrinology. 2005;146:1018-1024.
Kempna P, Fluck CE. Adrenal gland development and defects. Best Pract Res Clin Endocrinol Metab. 2008;22:77-93.
Lourenco D, Brauner R, Lin L, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med. 2009;360:1200-1210.
568.2 Adrenal Steroid Biosynthesis
Cholesterol is the starting substrate for all steroid biosynthesis (see Fig. 568-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com). Although adrenal cortex cells can synthesize cholesterol de novo from acetate, circulating plasma lipoproteins provide most of the cholesterol for adrenal cortex hormone formation. Receptors for both low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol are expressed on the surface of adrenocortical cells; the receptor is termed scavenger receptor class B, type I (SR-BI). Patients with familial hypercholesterolemia who lack LDL receptors have unimpaired adrenal steroidogenesis, suggesting that HDL is the more important source of cholesterol. Cholesterol is stored as cholesteryl esters in vesicles and subsequently hydrolyzed by cholesteryl ester hydrolases to liberate free cholesterol for steroid hormone synthesis.
Zona Fasciculata
In the endoplasmic reticulum of the zona fasciculata, pregnenolone and progesterone are converted by 17α-hydroxylase (P450c17, CYP17) to 17-hydroxypregenolone and 17-hydroxyprogesterone, respectively. This enzyme is not expressed in the zona glomerulosa, which consequently cannot synthesize 17-hydroxylated steroids. 17-Hydroxypregnenolone is converted to 17-hydroxyprogesterone and 11-deoxycortisol by the same 3β-hydroxysteroid and 21-hydroxylase enzymes, respectively, as are active in the zona glomerulosa. Thus, inherited disorders in these enzymes affect both aldosterone and cortisol synthesis (Chapter 570). Finally, 11-deoxycortisol reenters mitochondria and is converted to cortisol by steroid 11β-hydroxylase (P450c11, CYP11B1). This enzyme is closely related to aldosterone synthase but has low 18-hydroxylase and nonexistent 18-oxidase activity. Thus, under normal circumstances the zona fasciculata cannot synthesize aldosterone.
Fetoplacental Unit
Steroid synthesis in the fetal adrenal varies during gestation (Figs. 568-1 and 568-2). Shortly after the fetal adrenal gland forms (wk 8-10), it efficiently secretes cortisol, which is able to negatively feed back on the fetal pituitary and hypothalamus to suppress ACTH secretion. This is critical time for differentiation of the external genitalia in both sexes (Chapter 570.1); to prevent virilization, the female fetus must not be exposed to high levels of androgens of adrenal origin, and placental aromatase activity must remain low during this time to minimize conversion of testosterone to estradiol in male fetuses, which would interfere with masculinization. After wk 12, HSD3B activity in the fetal adrenal gland decreases and steroid sulfokinase activity increases. Thus, the major steroid products of the midgestation fetal adrenal gland are DHEA and DHEA sulfate (DHEAS) and, by 16α-hydroxylation in the liver, 16α-hydroxy DHEAS. Aromatase activity increases in the placenta at the same time, and steroid sulfatase activity is high as well. Thus, the placenta uses DHEA and DHEAS as substrates for estrone and estradiol and 16α-OH DHEAS as a substrate for estriol. Cortisol activity is low during the 2nd trimester, which might serve to prevent premature secretion of surfactant by the developing fetal lungs; surfactant levels can affect the timing of parturition. As term approaches, fetal cortisol concentration increases as a result of increased cortisol secretion and decreased conversion of cortisol to cortisone by 11β-hydroxysteroid dehydrogenase type 2 (HSD11B2). Low levels of aldosterone are produced in mid gestation, but aldosterone secretory capacity increases near term.
