[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 28
Pheochromocytoma
1. What is a pheochromocytoma?
A pheochromocytoma is an adrenal medullary tumor composed of chromaffin cells and capable of secreting biogenic amines and peptides, including epinephrine, norepinephrine, and dopamine. These tumors arise from neural crest–derived cells, which also give rise to portions of the central nervous system and the sympathetic (paraganglion) system. Because of this common origin, neoplasms of the sympathetic ganglia, such as neuroblastomas, paragangliomas, and ganglioneuromas, may produce similar amines and peptides.
2. How common are pheochromocytomas?
Pheochromocytomas are relatively rare. Data from the Mayo Clinic indicate that pheochromocytomas occur in 2 to 8 per million people per year; autopsy data from the same institution reflect an incidence of 0.3% (3/1000 autopsies), indicating that many pheochromocytomas go undetected during life. The incidence of pheochromocytoma from other countries, such as Japan, is lower, 0.4 cases per million people per year.
3. Where are pheochromocytomas located?
Nearly 90% of tumors arise within the adrenal glands, whereas approximately 10% are extra-adrenal and therefore classified as paragangliomas. Sporadic, solitary pheochromocytomas are located more commonly in the right adrenal gland, but familial forms (10% of all pheochromocytomas) are bilateral and multicentric. Bilateral adrenal tumors raise the possibility of multiple endocrine neoplasia 2A or 2B (MEN-2A or MEN-2B) syndromes (see Chapter 51).
4. Where are paragangliomas found?
Paragangliomas occur most commonly within the abdomen but also have been described along the entire sympathetic paraganglia chain from the base of the brain to the testicles. The common locations for paragangliomas are the organ of Zuckerkandl, the aortic bifurcation, and the bladder wall; the mediastinum, heart, carotid arteries, and glomus jugulare bodies are less frequent.
5. Can pheochromocytomas metastasize?
Yes. Demonstration of a metastatic focus in tissue normally devoid of chromaffin cells is the only accepted indication that a pheochromocytoma is malignant. Metastasis occurs in 3% to 14% of cases. The most common sites of metastases are regional lymph nodes, liver, bone, lung, and muscle.
6. What is the rule of 10s for pheochromocytomas?
7. What are the common clinical features of a pheochromocytoma?
The signs and symptoms of a pheochromocytoma are variable. The classic triad, consisting of sudden severe headaches, diaphoresis, and palpitations, carries a high degree of specificity (94%) and sensitivity (91%) for pheochromocytoma in a hypertensive population. The absence of all three symptoms reliably excludes the condition. Hypertension occurs in 90% to 95% of cases and is paroxysmal in 25% to 50% of these (Fig. 28-1). Orthostatic hypotension occurs in 40% of cases because of hypovolemia and impaired arterial and venous constriction responses. Tremor, pallor, and anxiety may also be accompanying signs, whereas flushing is uncommon.
8. What are some of the nonclassic manifestations of pheochromocytomas?
Signs and symptoms of other endocrine disorders may dominate the presentation of a pheochromocytoma. Tumors may elaborate corticotropin (ACTH) with resultant manifestations of Cushing’s syndrome and hypokalemic alkalosis. Vasoactive intestinal peptide (VIP) may be produced, resulting in severe diarrhea and hypokalemia. Hyperglycemia, resulting from catecholamine-associated antagonism of insulin release, and hypercalcemia, resulting from adrenergic stimulation of the parathyroid glands or elaboration of parathyroid hormone–related peptide (PTHrP), have also been encountered. Lactic acidosis may occur as a result of catecholamine-associated decrements in tissue oxygen delivery.
9. Discuss the cardiovascular manifestations of pheochromocytomas.
Cardiovascular manifestations of pheochromocytomas include arrhythmias and catecholamine-induced congestive cardiomyopathy. Atrial and ventricular fibrillations commonly result from precipitous release of catecholamines during surgery or from therapy with tricyclic antidepressants, phenothiazines, metoclopramide, and naloxone. Although cardiogenic pulmonary edema may result from cardiomyopathy, noncardiogenic pulmonary edema may also occur as a result of transient pulmonary vasoconstriction and increased capillary permeability.
10. Describe the intracerebral symptoms related to pheochromocytoma.
Seizures, altered mental status, and cerebral infarctions may occur as a result of intracerebral hemorrhage or embolization.
11. What do pheochromocytomas elaborate?
Most pheochromocytomas secrete norepinephrine. Tumors that produce epinephrine are more commonly intra-adrenal, because the extra-adrenal sympathetic ganglia do not contain phenylethanolamine-N-methyltransferase (PNMT), which converts norepinephrine to epinephrine. Dopamine is most commonly associated with malignant tumors.
12. Why is the blood pressure response among patients with pheochromocytomas so variable?
 Pheochromocytomas elaborate different biogenic amines. Epinephrine, a beta-adrenergic stimulatory vasodilator that causes hypotension, is secreted by some intra-adrenal tumors, whereas norepinephrine, an alpha-adrenergic stimulatory vasoconstrictor that causes hypertension, is produced by most intra-adrenal and all extra-adrenal tumors.
Pheochromocytomas elaborate different biogenic amines. Epinephrine, a beta-adrenergic stimulatory vasodilator that causes hypotension, is secreted by some intra-adrenal tumors, whereas norepinephrine, an alpha-adrenergic stimulatory vasoconstrictor that causes hypertension, is produced by most intra-adrenal and all extra-adrenal tumors.
 Tumor size indirectly correlates with plasma catecholamine concentrations. Large tumors (> 50 g) manifest slow turnover rates and release catecholamine degradation products, whereas small tumors (< 50 g) with rapid turnover rates elaborate active catecholamines.
Tumor size indirectly correlates with plasma catecholamine concentrations. Large tumors (> 50 g) manifest slow turnover rates and release catecholamine degradation products, whereas small tumors (< 50 g) with rapid turnover rates elaborate active catecholamines.

13. How is a pheochromocytoma diagnosed?
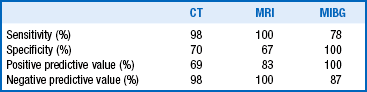
The diagnosis of pheochromocytoma depends on the demonstration of excessive plasma or urine catecholamine levels or urine degradation products. The plasma free metanephrine (PMN) test is probably the best screening method; specificity exceeds that of urinary metanephrine measurement (89% vs. 69%, respectively). Normal PMN values usually reliably exclude pheochromocytoma. Fourfold elevations of PMN values are associated with nearly 100% probability of a pheochromocytoma. The PMN specimen should be drawn with the patient supine for 15 minutes following an overnight fast.
14. How is pheochromocytoma differentiated from essential hypertension?
Confirmation of elevated PMNs involves measurement of urinary metanephrine, normetanephrine (NMN), vanillylmandelic acid (VMA), and free catecholamines produced in a 24-hour period. The ability of such measurements to differentiate pheochromocytomas from essential hypertension varies among institutions: for VMA, sensitivity is 28% to 56% and specificity is 98%; for MN and NMN, sensitivity is 67% to 91% and specificity is 100%; and for free catecholamines, sensitivity is 100% and specificity is 98%. Renal failure may falsely elevate values. Many groups advocate 24-hour urinary levels of metanephrine and catecholamines as good screening tests. Yield is improved when urine is collected after a paroxysmal episode of symptoms.
15. What conditions may alter the diagnostic tests discussed earlier?
Older assays for VMA were sensitive to dietary vanillin and phenolic acids, requiring patients to restrict their intake of such substances. High-pressure liquid chromatography assays have eliminated most false-positive results due to diet and drugs that alter the metabolism of catecholamines.
16. Which drugs alter the metabolism of catecholamines?
 Drugs that reduce plasma and urine concentrations: alpha-2-receptor agonists, calcium channel blockers (long-term therapy), angiotensin-converting enzyme inhibitors, bromocriptine
Drugs that reduce plasma and urine concentrations: alpha-2-receptor agonists, calcium channel blockers (long-term therapy), angiotensin-converting enzyme inhibitors, bromocriptine
 Drugs that decrease VMA and increase catecholamines and MN: methyldopa, monoamine oxidase inhibitors
Drugs that decrease VMA and increase catecholamines and MN: methyldopa, monoamine oxidase inhibitors

 Drugs that produce variable changes in any test: phenothiazines, tricyclic antidepressants, levodopa
Drugs that produce variable changes in any test: phenothiazines, tricyclic antidepressants, levodopa
17. What other medications may interfere with test results?
 Methylglucamine in radiocontrast agents (decreases metanephrine)
Methylglucamine in radiocontrast agents (decreases metanephrine)



18. List two other conditions that may interfere with test results.
 Stimulation of endogenous catecholamines: physiologic stress (ischemia, exercise), drug withdrawal (alcohol, clonidine), vasodilator therapy (nitroglycerin, acute calcium channel blocker administration)
Stimulation of endogenous catecholamines: physiologic stress (ischemia, exercise), drug withdrawal (alcohol, clonidine), vasodilator therapy (nitroglycerin, acute calcium channel blocker administration)
 Administration of exogenous catecholamines: appetite suppressants, decongestants
Administration of exogenous catecholamines: appetite suppressants, decongestants
19. What other biochemical tests are available?
Cases in which screening tests are equivocal may warrant a clonidine suppression test. This test employs a centrally acting alpha-2-receptor agonist that, in patients without a pheochromocytoma, suppresses neurogenically mediated release of catecholamines through the sympathetic nervous system. Blood samples to assess plasma catecholamines (norepinephrine and epinephrine) are drawn through an indwelling venous catheter; clonidine, 0.3 mg, is administered orally; plasma catecholamines are sampled again at 1, 2, and 3 hours. Plasma catecholamine values decrease to less than 500 pg/mL in patients with essential hypertension but exceed this level in patients with pheochromocytomas.
20. Compare computed tomography and magnetic resonance imaging for localization of pheochromocytomas.
The majority of tumors are larger than 3 cm, rendering them detectable by computed tomography (CT) or magnetic resonance imaging (MRI). CT, with special attention to the adrenal glands and pelvis, is advocated as the initial localizing procedure (97% are intra-abdominal). CT is the most cost-effective means of localization. Many authorities also recommend MRI as an adjunctive localizing modality. Advantages of MRI include the lack of radiation exposure and a characteristic hyperintensity on T2-weighted image. The hyperintense image allows definition of tumor size, differentiation from vascular structures, and identification of unsuspected metastases. Also see Chapter 29 for a discussion of adrenal imaging.
 KEY POINTS 1: PHEOCHROMOCYTOMA
KEY POINTS 1: PHEOCHROMOCYTOMA
1. Episodic headache, diaphoresis, and palpitations in a hypertensive patient suggest pheochromocytoma.
2. Ten percent of pheochromocytomas are bilateral, 10% extra-adrenal, 10% familial, and 10% malignant.
3. The best screening assay for pheochromocytoma is measurement of plasma free metanephrines.
4. Confirmation of the diagnosis of pheochromocytoma is elevated 24-hour urine levels of metanephrines and catecholamines.
5. Tumor localization is accomplished with computed tomography (most cost-effective) or magnetic resonance imaging (T2-weighted phase).
6. Therapy is surgical resection after administration of alpha-adrenergic blockade followed by beta-adrenergic blockade.
21. What other modalities are useful for localization of pheochromocytomas?
Scintigraphic localization with meta-iodobenzylguanidine I 123 (MIBG) may also reveal unsuspected metastases. MIBG is actively concentrated by sympathomedullary tissue and is subject to interference by drugs that block reuptake of catecholamines (tricyclic antidepressants, guanethidine, labetalol).
22. Summarize the performance criteria of each localizing procedure.
23. How are pheochromocytomas treated?
24. Why is preoperative preparation with alpha-adrenergic blockade recommended?
Alpha-adrenergic blockade reduces the incidence of intraoperative hypertensive crisis and postoperative hypotension. The most commonly used agent is phenoxybenzamine, a long-acting, noncompetitive antagonist (10-20 mg 2-3 times/day, advanced to 80-100 mg/day), or prazosin, a short-acting competitive antagonist (1 mg t.i.d., advanced to 5 mg t.i.d.). Therapy may be limited by hypotension, tachycardia, and dizziness. Goals of therapy include blood pressure less than 160/90 mm Hg, an electrocardiogram (ECG) free of ST- and T-wave changes over 2 weeks before surgery, and no more than one premature ventricular contraction within 15 minutes. Opinions about the duration of preparation vary between 7 and 28 days before surgery.
25. Discuss the role of beta-blockers and other agents in the preoperative period.
Beta-adrenergic blockade to control tachycardia is added only after alpha-adrenergic blockade has been instituted to prevent unopposed alpha stimulation. Other agents used in the preoperative period include labetalol and calcium channel blockers. Intraoperative hypertension associated with tumor manipulation may be controlled with either phentolamine or nitroprusside. Postoperative hypotension may be minimized by preoperative volume expansion with crystalloid.
26. How are malignant pheochromocytomas treated?
Although evidence of malignancy may be discovered at the time of surgery, metastases from slow-growing pheochromocytomas may remain inapparent for several years. Therapy is rarely curative, because the tumors respond poorly to radiation therapy and chemotherapy; treatment is therefore palliative. Surgical debulking is the therapy of choice, followed by use of alpha-methyltyrosine. This drug is a “false” catecholamine precursor that inhibits tyrosine hydroxylase (the rate-limiting enzyme in catecholamine synthesis) and reduces excessive production of catecholamines.
27. Discuss the role of chemotherapy and MIBG ablation.
Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine may slow tumor growth, as may ablation with MIBG I 131. Unfortunately, neither of these therapeutic measures has resulted in cure. Also see Chapter 29 for further discussion.
28. What is the prognosis for malignant pheochromocytoma?
The prognosis is not dismal. Cases of 20-year survival have been reported, and the 5-year survival rate with malignant pheochromocytomas is 44%.






Eisenhofer, G, Editorial. biochemical diagnosis of pheochromocytoma—is it time to switch to plasma free metanephrines. J Clin Endocrinol Metab 2003;88:550–552.
Jossart, GH, Burpee, SE, Gagner, M. Surgery of the adrenal glands. Endocrinol Metab Clin North Am. 2000;29:57–68.
Krane, NK, Clinically unsuspected pheochromocytomas. experience at Henry Ford Hospital and a review of the literature. Arch Intern Med 1986;146:54–57.
Kudva, YC, Sawka, AM, Young, WF, The laboratory diagnosis of adrenal pheochromocytoma. the Mayo Clinic experience. J Clin Endocrinol Metab 2003;88:4533–4539.
Lenders, JW, Pacak, K, Walther, MM, et al, Biochemical diagnosis of pheochromocytoma. which test is best. JAMA 2002;287:1427–1434.
Pacak, K. 2007 Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069–4079.
Prys-Roberts, C. Phaeochromocytoma—recent progress in management. Br J Anaesth. 2000;85:44–57.
Sawka, AM, Jaeschke, R, Singh, RJ, et al, A comparison of biochemical tests for pheochromocytoma. measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003;88:553–558.
Schwartz, GL, Screening for adrenal-endocrine hypertension. overview of accuracy and cost-effectiveness. Endocrinol Metabol N Am 2011;40:279–294.
Wittles, RM, Kaplan, EL, Roizen, MF. Sensitivity of diagnostic and localization tests for pheochromocytoma in clinical practice. Arch Intern Med. 2000;160:2521–2524.
Xekouki, P, Stratakis, CA. Pheochromocytoma. Translational Endocrinology Metabolism. 2011;2:77–127.
Zuber, SM, Kantorovich, V, Pacak, K, Hypertension in pheochromocytoma. characteristics and treatment. Endocrinol Metab N Am 2011;40:295–311.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 28
Pheochromocytoma
1. What is a pheochromocytoma?
A pheochromocytoma is an adrenal medullary tumor composed of chromaffin cells and capable of secreting biogenic amines and peptides, including epinephrine, norepinephrine, and dopamine. These tumors arise from neural crest–derived cells, which also give rise to portions of the central nervous system and the sympathetic (paraganglion) system. Because of this common origin, neoplasms of the sympathetic ganglia, such as neuroblastomas, paragangliomas, and ganglioneuromas, may produce similar amines and peptides.
2. How common are pheochromocytomas?
Pheochromocytomas are relatively rare. Data from the Mayo Clinic indicate that pheochromocytomas occur in 2 to 8 per million people per year; autopsy data from the same institution reflect an incidence of 0.3% (3/1000 autopsies), indicating that many pheochromocytomas go undetected during life. The incidence of pheochromocytoma from other countries, such as Japan, is lower, 0.4 cases per million people per year.
3. Where are pheochromocytomas located?
Nearly 90% of tumors arise within the adrenal glands, whereas approximately 10% are extra-adrenal and therefore classified as paragangliomas. Sporadic, solitary pheochromocytomas are located more commonly in the right adrenal gland, but familial forms (10% of all pheochromocytomas) are bilateral and multicentric. Bilateral adrenal tumors raise the possibility of multiple endocrine neoplasia 2A or 2B (MEN-2A or MEN-2B) syndromes (see Chapter 51).
4. Where are paragangliomas found?
Paragangliomas occur most commonly within the abdomen but also have been described along the entire sympathetic paraganglia chain from the base of the brain to the testicles. The common locations for paragangliomas are the organ of Zuckerkandl, the aortic bifurcation, and the bladder wall; the mediastinum, heart, carotid arteries, and glomus jugulare bodies are less frequent.
5. Can pheochromocytomas metastasize?
Yes. Demonstration of a metastatic focus in tissue normally devoid of chromaffin cells is the only accepted indication that a pheochromocytoma is malignant. Metastasis occurs in 3% to 14% of cases. The most common sites of metastases are regional lymph nodes, liver, bone, lung, and muscle.
6. What is the rule of 10s for pheochromocytomas?
7. What are the common clinical features of a pheochromocytoma?
The signs and symptoms of a pheochromocytoma are variable. The classic triad, consisting of sudden severe headaches, diaphoresis, and palpitations, carries a high degree of specificity (94%) and sensitivity (91%) for pheochromocytoma in a hypertensive population. The absence of all three symptoms reliably excludes the condition. Hypertension occurs in 90% to 95% of cases and is paroxysmal in 25% to 50% of these (Fig. 28-1). Orthostatic hypotension occurs in 40% of cases because of hypovolemia and impaired arterial and venous constriction responses. Tremor, pallor, and anxiety may also be accompanying signs, whereas flushing is uncommon.
8. What are some of the nonclassic manifestations of pheochromocytomas?
Signs and symptoms of other endocrine disorders may dominate the presentation of a pheochromocytoma. Tumors may elaborate corticotropin (ACTH) with resultant manifestations of Cushing’s syndrome and hypokalemic alkalosis. Vasoactive intestinal peptide (VIP) may be produced, resulting in severe diarrhea and hypokalemia. Hyperglycemia, resulting from catecholamine-associated antagonism of insulin release, and hypercalcemia, resulting from adrenergic stimulation of the parathyroid glands or elaboration of parathyroid hormone–related peptide (PTHrP), have also been encountered. Lactic acidosis may occur as a result of catecholamine-associated decrements in tissue oxygen delivery.
9. Discuss the cardiovascular manifestations of pheochromocytomas.
Cardiovascular manifestations of pheochromocytomas include arrhythmias and catecholamine-induced congestive cardiomyopathy. Atrial and ventricular fibrillations commonly result from precipitous release of catecholamines during surgery or from therapy with tricyclic antidepressants, phenothiazines, metoclopramide, and naloxone. Although cardiogenic pulmonary edema may result from cardiomyopathy, noncardiogenic pulmonary edema may also occur as a result of transient pulmonary vasoconstriction and increased capillary permeability.
10. Describe the intracerebral symptoms related to pheochromocytoma.
Seizures, altered mental status, and cerebral infarctions may occur as a result of intracerebral hemorrhage or embolization.
11. What do pheochromocytomas elaborate?
Most pheochromocytomas secrete norepinephrine. Tumors that produce epinephrine are more commonly intra-adrenal, because the extra-adrenal sympathetic ganglia do not contain phenylethanolamine-N-methyltransferase (PNMT), which converts norepinephrine to epinephrine. Dopamine is most commonly associated with malignant tumors.
12. Why is the blood pressure response among patients with pheochromocytomas so variable?
Pheochromocytomas elaborate different biogenic amines. Epinephrine, a beta-adrenergic stimulatory vasodilator that causes hypotension, is secreted by some intra-adrenal tumors, whereas norepinephrine, an alpha-adrenergic stimulatory vasoconstrictor that causes hypertension, is produced by most intra-adrenal and all extra-adrenal tumors.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
