Chapter 7 Pharmacology of Antithrombotic Drugs
Atherothrombotic disease and atherosclerotic plaque rupture is the leading cause of death worldwide. Its prevalence among adults in the United States is estimated at over 81 million, with costs exceeding $503 billion annually.1 Thrombus and clot formation involved in atherothrombotic disease develop as a complex and dynamic interaction between platelets, blood vessel wall, and coagulation cascades. Increased understanding of the mechanism of these interactions has provided for the development of novel drugs. Antiplatelet drugs have come to the forefront in managing atherothrombotic disease, owing in large part to platelets’ involvement in the initiation and propagation of thrombus. Our understanding of platelet function has expanded from a rudimentary knowledge of aspirin as a cyclooxygenase (COX) inhibitor within the arachidonic acid pathways, to a more complex picture of multiple receptor-modulating agents including thienopyridines, glycoprotein (GP)IIb/IIIa receptor antagonists, von Willebrand factor (vWF)-GPIb/IX, and collagen-GPVI inhibitors. Despite advances with newer inhibitors and combinations, treatment failures persist, necessitating development of new antiplatelet agents.
Platelets, Thrombosis, Coagulation, and Atherothrombotic Vascular Disease
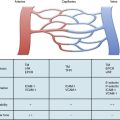
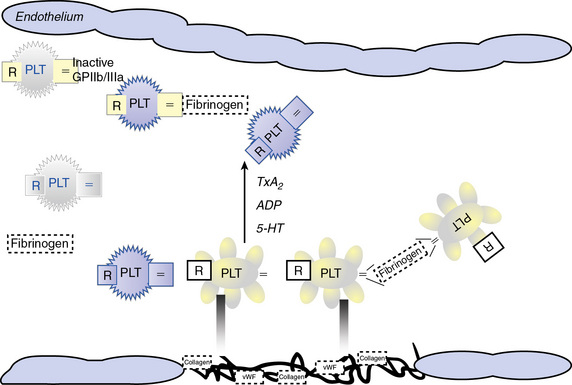
Atherosclerotic plaque rupture and endothelial cell (EC) disruption lead to platelet activation and formation of occlusive thrombi, triggering acute ischemic events in patients with atherothrombotic disease. Platelet activation and aggregation involve multiple signaling molecules and their receptors. Initially, platelets adhere to the subendothelial proteins vWF and collagen), which are exposed at sites of vascular injury. Adenosine diphosphate (ADP), thromboxane A2 (TxA2), serotonin, collagen, and thrombin activate platelets through unique intracellular signaling pathways, resulting in further platelet activation and secretion of mediators, thus further amplifying and sustaining the initial platelet response.2 Adenosine diphosphate, serotonin, and calcium are released by activated platelets via degranulation; thromboxane from arachidonic acid; and thrombin from activated coagulation cascade pathways.3 Activation of platelets occurs through binding of their primary blood-soluble agonists to their respective platelet receptors: ADP binds P2Y1 and P2Y12, thrombin binds to protease- activated receptor 1 (PAR1) and PAR4, and thromboxane binds to the thromboxane/prostanoid (TP) receptor.4 The final common pathway for all autocrine and paracrine activation signals is GPIIb/IIIa activation, which mediates fibrinogen and vWF binding to platelets and contributes to platelet aggregation. Thus, in both physiological hemostasis and pathological states, platelets are recruited to form a platelet-fibrin thrombus.3,4 Various classes of antiplatelet drugs act synergistically through complementary yet independent mechanisms, preventing platelet aggregation and thus acute thrombus formation. Currently available drugs and those under investigation target the thromboxane-induced (aspirin, sulfinpyrazone, indobufen, and triflusal) and ADP-induced (ticlopidine, clopidogrel, prasugrel, ticagrelor, cangrelor and elinogrel) pathways of platelet activation and their final common pathway of GPIIb/IIIa-induced (abciximab, eptifibatide, and tirofiban) platelet aggregation.4,5 The processes of platelet adhesion, activation, and aggregation, along with the targets of platelet-inhibiting drugs, are shown in Figure 7-1. Antiplatelet drugs, in addition to inhibiting acute arterial thrombosis, interfere with the physiological role of platelets in hemostasis. Thus the range of adverse effects, particularly bleeding, is a major factor in evaluating the utility of available and upcoming antiplatelet drugs and their combination regimens. Coagulation cascades are intimately activated through atherosclerotic plaque rupture and platelet activation. Targets of drug therapy to regulate the effects of thrombus formation and propagation are accomplished through oral anticoagulation (warfarin); thrombin inhibitors, both direct and indirect (heparin, low-molecular-weight heparin [LMWH], fondaparinux, hirudins, bivalirudin, argatroban, ximelagatran, dabigatran, etexilate, rivaroxaban, apixaban, DU-176b, LY517717, betrixaban, and YM150); factor IX inhibitors; and factor Xa inhibitors.
Pharmacology of Platelet Inhibitors
Platelets circulate in blood with their activation inhibited by both nitric oxide (NO) and prostaglandin I2 released from ECs.6,7 Activated platelets prevent bleeding by catalyzing the formation of stable blood clot in conjunction with activated coagulation pathways. In the initiation phase of primary hemostasis, platelets roll, adhere, and spread along the exposed collagen matrix of injured blood vessels to form an activated platelet monolayer.8 During the rolling phase, platelet adhesion and tethering is mediated by the platelet GPIb/V/IX receptor complex and vWF, which itself is bound to collagen (see Fig. 7-1). Additional tethering is accomplished between the GPVI and GPIa proteins directly with collagen at sites of vascular injury.6–8 The binding of GPIb/V/IX to vWF has a fast off rate insufficient to mediate stable adhesion, but instead, able to maintain platelets in close contact with the endothelial surface. Platelet activation stimulates high-affinity integrins to form stable adhesion complexes.
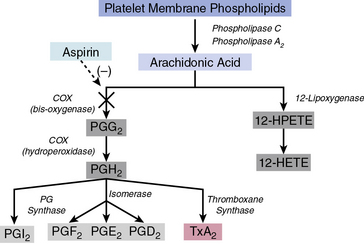
Blood flows with greater velocity in the center of the vessel than near the wall, thereby generating shear forces between adjacent layers of fluid. In conditions of high shear, such as those of small arteries, arterioles, and stenosed arteries, the tethering process is integral in the mechanisms of platelet adhesion. von Willebrand factor binds to collagen within the extracellular matrix (ECM) and to platelet receptors (GPIb/V/IX and GPIIb/IIIa [αIIβ3 integrin]).8 Binding of ECM collagen triggers intracellular signals that shift platelet integrins to a higher-affinity state and induce release of the secondary mediators ADP and TxA2. Both ADP and TxA2, along with thrombin produced from the coagulation cascade, synergistically induce full platelet activation. Upon platelet activation, arachidonic acid is liberated from membrane phospholipids by phospholipase A2 and C, thereby producing TxA2. Aspirin and other agents, such as sulfinpyrazone, indobufen, and triflusal, act to inhibit enzymes within the arachidonic acid cascade, thereby limiting production of TxA2. Adenosine diphosphate binds to P2Y1 and P2Y12 surface platelet receptors, which are targets of clopidogrel, prasugrel, and ticagrelor.
Thrombin is produced at the surface of activated platelets by tissue factor and is responsible for generating fibrin from fibrinogen, which contributes to formation of the hemostatic plug and platelet thrombus growth. Thrombin also directly activates platelets through stimulation of the PAR1.7 Both direct and indirect inhibitors of thrombin inhibit thrombin and affect thrombin activation, respectively. Release of ADP and TxA2 from adherent platelets contributes to recruitment of circulating platelets, thereby inducing a change in platelet shape, increased expression of proinflammatory molecules (P-selectin, CD40 ligand), expression of platelet procoagulant activity, and conversion of the GPIIb/IIIa receptor into an active form, leading to pathological thrombosis.6–8 Activation of GPIIb/IIIa (αIIβ3 integrin) mediates platelet aggregation and spreading on the exposed ECM of the injured vessel wall by means of fibrinogen bridges.8
Fibrinogen bridges activated platelets and contributes to thrombus stabilization.8 Activation of platelets results in a conformational change in the αIIβ3 integrin (IIb/IIIa) receptor, enabling it to bind fibrinogen-enhancing cross-links to adjacent platelets, resulting in aggregation and formation of a platelet plug. Simultaneous activation of the coagulation system results in thrombin generation and fibrin clot formation, which further stabilizes the platelet plug. Abciximab, eptifibatide, and tirofiban inhibit GPIIb/IIIa, thereby inhibiting platelet aggregation.
Cyclooxygenase Inhibitors and the Arachidonic Acid Cascade
Arachidonic acid is liberated from membrane phospholipids by phospholipase A2 and C upon platelet stimulation9 (Fig. 7-2). Prostaglandin (PG) H-synthase catalyzes the conversion of arachidonic acid to PGG2 and PGH2.10 Prostaglandin-synthase possesses two catalytic sites, a COX site, responsible for the formation of PGG2, and a hydroperoxidase site, which reduces the 15-hydroperoxyl group of PGG2 to produce PGH2.11 Subsequent enzymatic catalyzation of PGH2 generates PGs, D2, E2, F2α, I2, and TxA2.11 Aspirin irreversibly binds and inhibits the COX site by acetylating the hydroxyl group of a serine residue at position 529 (Ser 529) without affecting the hydroperoxidase activity of the enzyme, thus inhibiting production of PGG2 and therefore PGH2 circumventing TxA2 production.12
Aspirin
Aspirin produces dose-dependent inhibition of platelet COX activity after a single oral dose. A single dose of 100 mg effectively suppresses biosynthesis of TxA2 within several minutes of administration via acetylation of platelet COX in the presystemic circulation.13 Owing to aspirin’s irreversible inhibition of COX and the inability of platelets to synthesize new proteins, aspirin’s effect is maintained for the lifespan of the platelet (7-10 days). Cyclooxygenase activity returns only as new platelets are generated.
Aspirin reduces acute coronary and cerebrovascular events such as unstable angina, myocardial infarction (MI), sudden cardiac death, and stroke.13 Its utility is enhanced by its modest cost and nominal side effects. Although aspirin effectively reduces platelet secretion and aggregation, it is a relatively weak platelet inhibitor. The inhibitory effects of aspirin are pronounced when using relatively weak platelet agonists, but less so against stronger agonists like thrombin that can induce platelet activation in the absence of TxA2. Importantly, the majority of platelet responses remain unaffected by aspirin treatment. Aspirin does not inhibit shear stress–induced platelet activation and platelet adhesion. In addition to its antiplatelet properties, aspirin also exerts antiinflammatory effects.14 In a meta-analysis of antiplatelet therapy studies in patients with acute coronary syndrome (ACS), administration of several doses of aspirin (from 75 mg/day up to 150 mg/day) significantly decreased the overall risk of nonfatal MI, nonfatal stroke, and death rates.15 Several studies, however, have demonstrated that aspirin monotherapy is inadequate because of high intraindividual variability to aspirin response, as well as increased aspirin resistance, especially observed in patients with diabetes mellitus.16
Nonsteroidal antiinflammatory drugs
There are several nonsteroidal antiinflammatory drugs (NSAIDs) that act as competitive reversible inhibitors of PGH-synthase. Sulfinpyrazone, indobufen, and triflusal are several of the drugs in this class evaluated for their antithrombotic activity in randomized clinical trials. The active sulfide metabolite of sulfinpyrazone administered in the highest dose allowable (200 mg four times a day) inhibits only 60% of platelet COX activity, with results suggesting no significant clinical efficacy.17 The clinical, biochemical, and functional effects of the more effective inhibitor, indobufen, are similar to those of aspirin. An oral dose of indobufen 200 mg twice daily inhibits 95% of platelet TxA2 synthesis.18 Triflusal, a derivative of salicylic acid, is also able to inhibit platelet COX, but only after conversion to a longer-lasting metabolite.19 None of the three are currently approved for use as antiplatelet drugs in the United States.
Adenosine Diphosphate, Purinergic Receptors, and Thienopyridine Inhibitors
Adenosine diphosphate is a key mediator in activating platelet aggregation and thrombus formation. Inhibiting the effects of ADP activity has lead to development of numerous P2Y12 receptor–targeting antiplatelet drugs. Adenosine diphosphate is released from dense granules of activated platelets, providing a soluble positive feedback mediator binding to the receptors P2Y1 and P2Y12. Both P2Y1 and P2Y12 are platelet surface-bound purinoreceptors belonging to the G protein–coupled receptor (GPCR) class, with P2Y1 being coupled to Gq and P2Y12 to Gi. Adenosine diphosphate binding to both P2Y1 and P2Y12 receptors activates distinct intracellular signaling pathways.20 Binding of ADP to the P2Y1 receptor and its Gq protein mobilizes intracellular calcium, triggering a change in the platelet shape and rapid, reversible aggregation.21 Adeosine diphosphate binding through the P2Y12 receptor and its Gi protein results in reduced levels of cyclic adenosine monophosphate (cAMP), resulting in amplification of the platelet response, stabilization of resulting aggregates, and secretion of further mediators from platelet granules.22 Binding of ADP to both P2Y1 and P2Y12 purinoreceptors is necessary for normal ADP-induced aggregation. P2Y12 is considered the major platelet ADP receptor and, since it is more restricted in its expression throughout cell lines, has become an attractive therapeutic target for antithrombotic agents.20
Thienopyridine platelet P2Y12 receptor antagonists
The thienopyridine class of antiplatelets (ticlopidine, clopidogrel, and prasugrel) selectively and irreversibly inhibit the P2Y12 purinoreceptor throughout the life of the platelet. Clopidogrel is the dominant member within the class and provides for modest platelet inhibition, delayed onset of action, and significant interpatient variability, including nonresponsiveness to the drug, which necessitated the search for more potent and stable alternatives.23 Ticlopidine has been eclipsed because of its adverse hematological side effects, including neutropenia and thrombotic thrombocytopenic purpura.24 The opposite, however, is true of third-generation thienopyridines, namely prasugrel.
Oral thienopyridines are prodrugs that require conversion into their active metabolites by hepatic cytochrome P450 (CYP) enzymes (CYP3A4 isozyme). Whereas clopidogrel requires esterase inactivation and a two-step CYP-dependent activation, prasugrel requires only one reaction to yield its active metabolite.25 This difference in metabolism translates into different patient responses and drug interactions. Genetic variations of P450 (CYP) enzymes affect clopidogrel’s active metabolite formation, resulting in lower platelet inhibition and, most importantly, a higher rate of major adverse cardiovascular events. Prasugrel’s pharmacology, however, is not affected by CYP polymorphisms and provides a stable platform for antiplatelet therapy. Prasugrel has a faster onset of action and a tenfold higher potency than clopidogrel.26
Clopidogrel
Clopidogrel is metabolized by CYP P450. Its active metabolite irreversibly binds to the platelet P2Y12 receptor, thus inhibiting the effect of ADP on platelets. As a result, GPIIb/IIIa receptors have decreased activation, thereby resulting in reduced platelet function. The Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) study, a randomized trial that included patients with ischemic stroke, MI, or symptomatic atherosclerotic peripheral artery disease, showed that clopidogrel-treated patients had an 8.7% relative risk reduction for acute MI, stroke, or vascular death, compared to those patients treated with aspirin.27 The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial was the first study establishing the significance of dual antiplatelet therapy (acetylsalicylic acid [ASA] plus clopidogrel) in patients with ACS28 and involved a population of 12,562 patients presenting with non-ST-elevation ACS who were randomized to receive either a combination of ASA (75-325 mg/day) and clopidogrel (300 mg loading dose, followed by 75 mg/day) or ASA and placebo for 3 to 12 months. At 12 months, a lower incidence of MI, stroke, or cardiovascular death was observed in the clopidogrel plus ASA group compared to the placebo group; however, the risk of major bleeding was increased among patients treated with clopidogrel plus ASA.
Similar results were reported by the COMMIT study.29 Patients received either a combination of clopidogrel (75 mg/day) and ASA (162 mg/day) or placebo and ASA for 28 days or until hospital discharge. There was a 9% relative risk reduction in the composite endpoint of stroke, vascular reinfarction, or death and a 7% relative risk reduction for death in the group receiving dual antiplatelet therapy. In the CREDO study, which was the first to evaluate the significance of dual antiplatelet therapy pre- and post-percutaneous coronary intervention (PCI), patients undergoing PCI received either a 300-mg loading dose of clopidogrel 3 to 24 hours before the procedure and then 75 mg/day for 12 months, or 75 mg/day for 30 days after the procedure without a loading dose.30 All patients also received a 325 mg/day dose of ASA during the 12-month follow-up period. A 27% relative risk reduction for death, MI, or stroke was observed in the group receiving long-term dual antiplatelet therapy. Given the established antiplatelet effect of clopidogrel, the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization Management and Avoidance (CHARISMA) study investigated the potential benefit from 28-month dual antiplatelet therapy (75 mg/day clopidogrel and 75-162 mg/day ASA) in patients 45 years of age or older who experienced one of the following conditions: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented symptomatic peripheral artery disease.31 The control group received ASA and placebo. No benefit was observed in the primary endpoint of MI, stroke, or cardiovascular death in the dual antiplatelet therapy group. By contrast, asymptomatic patients of the group with risk factors but without clinical evidence of CAD had a higher risk of bleeding.32 A further analysis in the subgroup of patients with documented atherothrombotic cardiovascular disease showed a significant risk reduction for new MI, stroke, or cardiovascular death in those patients compared with controls.33
Prasugrel
Prasugrel is a P2Y12 inhibitor acting similarly to clopidogrel. Prasugrel is rapidly converted to its active metabolite by the P450 cytochrome and has higher bioavailability than clopidogrel.26 Recently, a 60-mg loading dose of prasugrel achieved high platelet inhibition both in healthy subjects and in patients scheduled for PCI, whereas healthy clopidogrel poor-responders achieved satisfactory platelet inhibition of up to 80% after prasugrel administration.26 The beneficial effect of prasugrel was also established in the TRITON-TIMI 38 study.34 Patients scheduled for PCI were randomized to receive either clopidogrel (300-mg loading dose and 75 mg/day afterwards) or prasugrel (60-mg loading dose and 10 mg/day afterwards). All patients also received ASA, and about half the patients from each group were treated with a GPIIb/IIIa inhibitor as well. The prasugrel group demonstrated a significant risk reduction for the composite primary endpoint of death, nonfatal MI, and nonfatal stroke. In addition, risk was significantly lower in the diabetes mellitus patient subgroup34; however, incidence of major hemorrhagic events was more frequent in the prasugrel group, but even when this parameter was added to the study’s primary endpoints, the net clinical benefit findings still favored prasugrel compared to clopidogrel. Subgroup analysis demonstrated a higher rate of major bleeding in those with body weight of less than 60 kg, history of stroke or transient ischemic attack, and age older than 75 years.34
Non-thienopyridine platelet P2Y12 receptor antagonists
Novel non-thienopyridine platelet P2Y12 receptor antagonists include ticagrelor, cangrelor, and elinogrel, which are direct and reversible P2Y12 antagonists with rapid onsets and short durations of action. Ticagrelor is highly selective and very specific for the P2Y12 receptor, and it exhibits a greater, more consistent inhibition of platelet aggregation than clopidogrel.35 Ticagrelor is administered orally and does not require metabolic activation, providing a rapid onset peaking within 2 to 4 hours of dosing. The metabolism of ticagrelor yields an active molecule (AR-C124910XX) that has similar P2Y12-blocking activity as its parent molecule. Ticagrelor’s plasma half-life is approximately 12 hours, which corresponds to twice-daily dosing and a 1- to 2-day restoration of normal platelet-mediated hemostasis upon discontinuation. These pharmacokinetics are in contrast to clopidogrel and prasugrel, which require discontinuation approximately 5 days before restoration of normal platelet-mediated hemostasis is achieved. Ticagrelor’s potential advantage in plasma half-life also carries the risk of increased thrombotic events if patients miss a dose.36
Multiple trials have demonstrated the benefits of ticagrelor in clinical practice. The Dose Confirmation Study Assessing Antiplatelet Effects of AZD6140 vs. Clopidogrel in Non-ST-Segment Elevation Myocardial Infarction (DISPERSE-2) trial showed no difference in major bleeding or MI, with an increase in minor bleeding at higher doses of ticagrelor in 990 patients with non-ST-segment elevation ACS.37 In the PLATO (the Study of Platelet Inhibition and Patient Outcomes) trial comparing ticagrelor and clopidogrel with respect to their efficacy in preventing cardiovascular events and safety showed ticagrelor significantly reduced the rate of death from vascular causes, MI, or stroke, without an increase in the rate of overall major bleeding compared to clopidogrel in 18,624 patients with ACS.38 Ticagrelor is the first investigational antiplatelet drug to demonstrate a reduction in cardiovascular death when compared to clopidogrel in patients with ACS.
Finally, research has shown ticagrelor to produce platelet inhibition regardless of genotypic variations in the three genes that had been associated with variability to clopidogrel in platelet inhibition.39 All these trials underline the potential for ticagrelor to achieve a rapid and sustained antiplatelet effect that could be reversed and could overcome nonresponsiveness and interpatient variability to clopidogrel, thus addressing the main limitations of clopidogrel therapy.24 Nonetheless, its adverse effects (e.g., dyspnea, bradycardia) and weight-based dosing require further investigation before ticagrelor may advance toward routine use in antiplatelet therapy.38
Role of Integrin Receptors in Platelet Function
Abciximab
Abciximab is a large chimeric monoclonal antibody Fab fragment with high affinity for the GPIIb/IIIa receptor.40 Abciximab is the largest agent with binding sites located on the β-chain of the GPIIb/IIIa receptor which, because of its large size, causes a steric hindrance to ligand access.
The efficacy and safety of abciximab in patients undergoing PCI has been evaluated in several trials, including EPIC, EPILOG, and EPISTENT.41–43 The Intracoronary Stenting and Antithrombotic Regimen–Rapid Early Action for Coronary Treatment (ISAR-REACT) trial demonstrated that the rate of death, MI, and urgent revascularization at 30 days was low and comparable between patients undergoing elective PCI after pretreatment with clopidogrel 600 mg and allocated to abciximab vs. placebo.44 Rates of major bleeding were similar between groups, although abciximab was associated with a significantly higher rate of thrombocytopenia. ISAR-REACT-2 evaluated the same abciximab and clopidogrel treatment regimens in high-risk patients with non-ST-segment elevation ACS undergoing PCI.45 Death, MI, and urgent revascularization at 30 days occurred significantly less frequently with abciximab compared to placebo; however, the treatment benefit of abciximab was confined to patients with elevated troponin levels. Rates of major bleeding were similar between groups. Overall, the findings suggest that in the modern era of interventional cardiology using high clopidogrel dosing regimens, GPIIb/IIIa inhibition should be reserved only for high-risk ACS patients with positive cardiac markers.
Eptifibatide
Eptifibatide is a competitive antagonist of the GPIIb/IIIa receptor. It is a synthetic small-molecule inhibitor that fits directly into the Arg-Gly-Asp binding pocket of the GPIIb/IIIa receptor, directly competing with the binding of ligands such as fibrinogen and vWF.46 Eptifibatide rapidly dissociates from its receptor, is cleared by the kidney largely as active drug, and has a plasma half-life of approximately 1.5 to 2.5 hours. The return of hemostatic platelet function is largely dependent on clearance of the drug from plasma. Cessation of drug infusion restores platelet function and, in patients with normal renal function, normal hemostasis returns within 15 to 30 minutes after drug discontinuation. Unlike abciximab, however, the platelet inhibitory effect of eptifibatide is not significantly influenced by platelet transfusion.46
Eptifibatide has demonstrated efficacy and safety in patients with non-ST-segment elevation ACS or undergoing PCI in a number of randomized clinical trials. Most recently, the Early Glycoprotein IIb/IIIa Inhibition in Non-ST-Segment Elevation Acute Coronary Syndrome (EARLY ACS) trial demonstrated that early administration of eptifibatide vs. provisional eptifibatide after angiography (delayed eptifibatide) resulted in similar 30-day rates of death, MI, urgent revascularization, or thrombotic complications during PCI in patients with non-ST-segment elevation ACS undergoing invasive management.47 Major and minor bleeding rates were significantly higher with early eptifibatide vs. delayed eptifibatide. Overall, these findings do not support the use of upstream compared with ad hoc GPIIb/IIIa inhibition in ACS patients undergoing PCI.
Tirofiban
Tirofiban is a tyrosine-derived nonpeptide inhibitor associated with rapid onset and short duration of action, with a plasma half-life of approximately 2 hours. Tirofiban, like eptifibatide, is a competitive inhibitor of the GPIIb/IIIa receptor that has high specificity but relatively low affinity.46 Tirofiban is excreted by the kidney, predominantly as unchanged drug; it rapidly dissociates from the receptor and has a biological half-life of 1.5 to 2.5 hours. Restoration of hemostasis is best achieved by discontinuing the drug infusion. Efficacy and safety of tirofiban in PCI patients has been investigated in several trials.
Platelet Adhesion
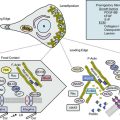
von Willebrand factor is present in plasma, platelets, and vascular subendothelium, and is synthesized and stored by megakaryocytes and ECs.48 von Willebrand factor can be released into the circulation by ECs upon activation by vasopressin (desmopressin/DDAVP [1-deamino-8-D-arginine-vasopressin]) or thrombin, for example.48 von Willebrand factor serves two important functions in the hemostatic response of platelets: initially through platelet/platelet binding (GPIba/vWF A1 domain), and subsequently through collagen/platelet binding (GPIba/vWF A3 domain). The vWF A1 domain specifically serves to assist with platelet aggregation by binding to platelet GPIba in the GPIb/V/IX complex for platelet/platelet adhesion, especially under conditions of high shear stress. von Willebrand factor additionally promotes platelet adhesion by binding to collagen in exposed vascular subendothelium via the vWF A3 domain. The adhesion function of the GPIb/V/IX complex that interacts with vWF resides in the GPIba chain; mutations within this segment may alter its affinity for the vWF A1 domain. 49
Von willebrand factor–GPIP/IX inhibitors
A number of vWF-GPIb/IX inhibitors, including aurin tricarboxylic acid, peptide fragments from vWF A1 domain, and a soluble GPIb-immunoglobulin (Ig)G, have been examined for inhibiting vWF and platelet interactions.50 Glycoprotein-290 is a recombinant protein consisting of a 290 amino acids sequence of the N-terminal of human GPIba, with two gain-of-function mutations that increase the affinity for the vWF A1 domain. The in vivo antithrombotic and antihemostatic effects have been evaluated with good GPIba/vWF inhibition, which could be reversed with a currently approved (DDAVP) treatment regimen. A lower clopidogrel dose combined with GPIba/vWF inhibition could theoretically have improved efficacy with less bleeding risk.
AJvW-2 is a murine monoclonal antibody to human vWF A1 domain that blocks the GPIba/vWF interaction and has demonstrated antithrombotic activity in animal models.51 In order to reduce immunological response, AJvW-2 was humanized and converted from an IgG1 to an IgG4.52 This humanized antibody (AJW200) exhibited similar inhibition of in vitro vWF-mediated platelet activation to AJvW-2. In human volunteer studies, AJW200 demonstrated no clinically significant adverse events or immunogenicity. Ristocetin cofactor (Ri:CoF) assays showed a significant reduction at 1 hour post infusion compared with baseline that lasted for up to 12 hours. The template bleeding time was not significantly prolonged at any time or dose of AJW200. Platelet function as measured by the PFA-100 was reduced by up to 3 to 6 hours at the lower dose and 12 hours at the highest dose administered.
ARC1779 is an aptamer that blocks the GPIba/vWF A1 domain interaction. Aptamers are nucleic acid molecules with high affinity and specificity for a selected target molecule, discovered through in vitro selection on the basis of their ability to fold into unique three-dimensional structures that promote binding to that target.53 ARC1779 is a modified deoxyribonucleic acid/ribonucleic acid (DNA/RNA) oligonucleotide composed of hybrid terminal ends to minimize endonuclease and exonuclease digestion, with nucleotide segments designed to enhance affinity for vWF. ARC1779 has demonstrated efficacy comparable or superior to that of previously published dosing regimens of abciximab with respect to protection from thrombus formation and average time to occlusion. ARC1779 was evaluated in a randomized double-blind placebo-controlled study that demonstrated it was well tolerated, and no bleeding was observed.54 An S-nitroso derivative of a mutated fragment of the A1 domain (S-nitroso-AR545C) was shown to inhibit effectively arterial thrombosis in the carotid artery.55 In unpublished observations, it has also been reported that targeting the vWF A1 domain with the recombinant nanobody ALX-0081, a novel class of antibody therapeutics, resulted in inhibition of vWF-mediated platelet activation, providing novel options for future therapies.
Collagen-GPVI inhibitors
Glycoprotein VI is also expressed on the surface of platelets. Signaling by GPVI is via an immunoreceptor tyrosine activation motif (ITAM) promoting phosphorylation and initiating the syk signaling cascade. Syk activation results in activation of integrin-induced platelet aggregation, release of ADP and thromboxane, and procoagulant activity.56 Rat monoclonal antibody (JAQ1) to mouse GPVI results in inhibition of collagen-induced aggregation.57 In animal models, JAQ1 caused mild thrombocytopenia and resulted in a 34% decrease in platelet counts within 24 hours of treatment, which returned to normal levels within 3 days of treatment. Platelets showed no indication of activation or change in surface protein expression. A single dose of JAQ1 resulted in 14 days of inhibition of ex vivo collagen-induced aggregation. Further evaluation of in vivo response determined that binding of antibody to the platelet GPVI resulted in depletion of platelet GPVI. Collagen-induced adhesion was significantly reduced in these GPVI-depleted platelets. Bleeding times of JAQ1-treated mice were significantly elevated over control mice (330 seconds vs. 158 seconds), but less than that seen following inhibition of GPIIb/IIIa (330 seconds vs. > 600 seconds).
Pharmacology of Antithrombotics and Thrombin Inhibitors
Overview of Coagulation
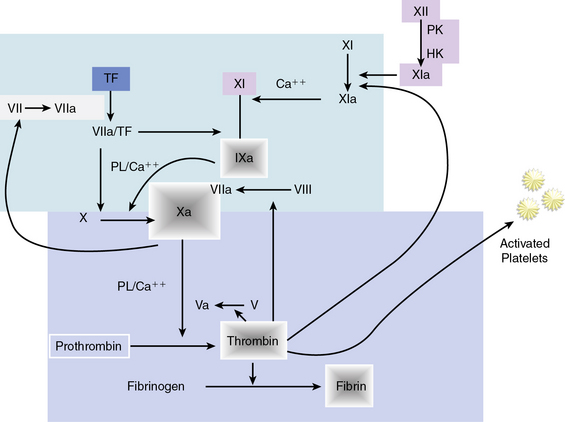
Hemostasis is accomplished by a complex sequence of interactions among platelets, endothelium, and multiple circulating and membrane-bound coagulation factors. As shown in Figure 7-3, the coagulation cascade typically has two intersecting pathways. The intrinsic pathway is initiated with factor XII and involves a cascade of enzymatic reactions that activate factors XI, IX, and VII. In the intrinsic pathway, all factors leading to fibrin clot formation are intrinsic to the circulating plasma, and no surface is required to initiate the process. The extrinsic pathway, however, requires exposure of tissue factor on the surface of the injured vessel wall to initiate the cascade, beginning with factor VII. The two arms of the coagulation cascade merge to a common pathway at factor X, which activates factors II (prothrombin) and I (fibrinogen). The formation of clot is dependent upon the proteolytic conversion of fibrinogen to fibrin.
Pharmacology of Thrombin Inhibitors: Indirect and Direct
Indirect Thrombin Inhibitors
Heparin
Heparin is a sulfated polysaccharide and is isolated from mammalian tissues rich in mast cells, most notably porcine intestinal mucosa. Heparin acts as an anticoagulant by activating antithrombin (antithrombin III), thereby accelerating the rate at which antithrombin inhibits clotting enzymes, particularly thrombin and factor Xa. Heparin activates antithrombin by binding to a unique pentasaccharide sequence to induce a conformational change within antithrombin, rendering it more readily accessible to its target proteases (Fig. 7-4). This conformational change enhances the rate at which antithrombin inhibits factor Xa but has little effect on the rate of thrombin inhibition. To catalyze thrombin inhibition, heparin serves as a template that simultaneously binds antithrombin and thrombin. Formation of this ternary structure allows close apposition, promoting formation of a stable covalent thrombin-antithrombin complex. Heparin additionally causes release of TFPI from the endothelium, a tissue factor–bound factor VIIa inhibitor, further enhancing its anticoagulant effects.
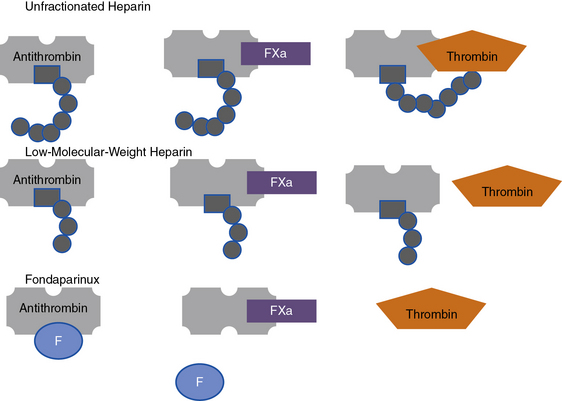
Low-molecular-weight heparin
Like heparin, LMWH exerts its anticoagulant activity by activating antithrombin. Even though LMWH consists of shorter pentasaccharide-containing chains, they retain greater capacity to accelerate factor Xa inhibition by antithrombin (see Fig. 7-4). Consequently, many forms of LMWH catalyze factor Xa inhibition by antithrombin more than thrombin inhibition.
Fondaparinux
Fondaparinux is a synthetic analog of the antithrombin-binding pentasaccharide sequence that differs from LMWH in several ways. As a synthetic analog of the antithrombin-binding pentasaccharide sequence found in heparin and LMWH, fondaparinux binds only to antithrombin and is too short to bridge thrombin to antithrombin (see Fig. 7-4). Consequently, fondaparinux catalyzes factor Xa inhibition by antithrombin and does not enhance the rate of thrombin inhibition. Fondaparinux is licensed for thromboprophylaxis in general surgical and high-risk orthopedic patients and as an alternative to heparin or LMWH for initial treatment of patients with established venous thromboembolism (VTE).
Parenteral Direct Thrombin Inhibitors
Hirudins
Hirudin consists of a single polypeptide chain of 65 amino acids with increased affinity for thrombin. Various recombinant hirudins have since been developed (lepirudin and desirudin). They are bivalent DTIs and bind thrombin with high affinity, forming noncovalent irreversible complexes. Their plasma half-life is approximately 1 to 2 hours, and they distribute widely in the extravascular space. Clearance is primarily through the kidney, so dose adjustment is required in the setting of impaired renal function.58 Monitoring hirudin is challenging, since plasma levels must be determined using enzyme-linked immunosorbent assays (ELISA), which are costly and limited in availability. Hirudins are associated with bleeding, thus requiring monitoring to avoid excessive anticoagulation. Since hirudin prolongs APTT, it is widely used to monitor therapy. Lepirudin is licensed for use in patients with HIT with thrombosis. Major bleeding occurs in 18% to 20% of patients receiving lepirudin for treatment of HIT. Clinical use of commercially available hirudins is complicated by the development of antibodies. Studies have shown that over 40% of lepirudin- treated patients develop IgG antibodies against lepirudin.59 Antilepirudin antibodies typically form 1 to 4 weeks after initiation of treatment and in most cases are not associated with any adverse clinical outcomes. In 10% of lepirudin-treated patients, however, formation of antibodies delay clearance of lepirudin, enhancing its anticoagulant effect.59 Anaphylaxis, which can be fatal, has also been reported in patients receiving intravenous bolus lepirudin for treatment of HIT.60 Lepirudin use in patients with HIT is based on the findings of three prospective, historically controlled cohort studies, HAT-1, HAT-2, and HAT-3. These studies found that compared with historical controls, lepirudin reduced the frequency of the composite endpoint of all-cause mortality, new thrombosis, or limb amputation in patients with HIT.61 Lepirudin was also associated with significantly reduced new thrombosis. Lepirudin additionally has been studied in the setting of non-ST-segment ACS. When compared with heparin, 10,141 patients with ACS lepirudin significantly reduced the incidence of cardiovascular death, refractory angina, or new MI at 7 days from 6.7% to 5.6%, but also significantly increased the rate of major bleeding from 0.7% to 1.2%.62 Lepirudin has not been approved for use in ACS, but such results support the hypothesis that hirudins are superior to heparin in preventing recurrent ischemia; however, they do so within a narrow therapeutic window.
Desirudin is a hirudin used for thromboprophylaxis in patients undergoing elective hip or knee surgery. Desirudin is used for 9 to 12 days, or until the patient is fully ambulatory. Unlike lepirudin, desirudin monitoring is unnecessary. Like lepirudin, bleeding is the most common side effect. When receiving desirudin for thromboprophylaxis, approximately 10% of patients also develop antibodies of the IgG class against hirudin. These antibodies have not been associated with altered plasma concentrations of desirudin, deep vein thrombosis (DVT), pulmonary embolism (PE), allergic reactions, or hemorrhage.63 Desirudin is currently approved in Europe for thromboprophylaxis in patients undergoing elective hip or knee surgery. Approval is based on findings of two multicenter randomized double-blind trials. In the first study, desirudin 15 mg subcutaneously twice daily was compared to UHF, 5000 international units, three times daily in patients undergoing primary elective total hip replacement (THR). Desirudin was associated with a significantly lower rate of all DVT and proximal DVT.64 In the second study, desirudin 15 mg subcutaneously twice daily was compared to enoxaparin 40 mg subcutaneously once daily in patients undergoing elective THR. Duration of treatment was 8 to 12 days, and DVT during the treatment period was verified by mandatory bilateral venography. Desirudin was associated with a significantly lower rate of proximal DVT and overall DVT. There was no significant difference in bleeding, transfusion requirements, or thrombocytopenia between the groups.65
Bivalirudin
Bivalirudin is a hirudin analog with high-affinity binding to thrombin. Once bound, however, thrombin cleaves the Arg-Pro in the N-terminus of bivalirudin, allowing for recovery of thrombin activity and subsequent competing of fibrinogen with the bivalirudin remnant for thrombin.66 Unlike hirudin, only 20% of bivalirudin is excreted via the kidney; the remainder is eliminated by proteolytic enzymatic degradation. Both APTT and activated clotting time (ACT) have been used to monitor bivalirudin. Bivalirudin has been licensed as an alternative to heparin in patients undergoing PCI and for patients with HIT who require PCI. A prospective trial comparing bivalirudin with high-dose heparin in 1261 patients demonstrated a lower rate of death, MI, or repeat revascularization.67 The REPLACE-1 trial compared bivalirudin to heparin in 1056 patients undergoing coronary stenting with GPIIb/IIIa inhibitors. There was a trend toward a reduction in the combined endpoint of death, MI, or revascularization with bivalirudin at 48 hours; there was no difference in major bleeding.68 In REPLACE-2, bivalirudin was as effective as heparin plus GPIIb/IIIa inhibition in reducing death, MI, and revascularization. Bivalirudin was also associated with significant reduction in the incidence of bleeding and thrombocytopenia.69 In the Acute Catheterization and Urgent Intervention Triage Strategy (ACUITY) trial, patients with moderate- to high-risk unstable angina or non-ST-segment elevation myocardial infarction (NSTEMI) undergoing early invasive management70 demonstrated that bivalirudin alone was noninferior to heparin (either UFH or enoxaparin) plus GPIIb/IIIa inhibitors in terms of mortality and the primary endpoint. It was also associated with significantly less bleeding. At 1 year, bivalirudin was still noninferior to heparin in terms of the primary endpoint and mortality rates; however, bivalirudin, either alone or with GPIIb/IIIa inhibitors, continued to be associated with significantly less bleeding. Results were similar in patients triaged to medical management, PCI, or coronary artery bypass surgery. The ACUITY trial showed a strong association between major bleeding in the first 30 days and risk of death over 1 year.70
Argatroban
Argatroban is a potent agent that differs from parenteral DTIs in that it binds reversibly to the active site of both free and clot-bound thrombin.71 Argatroban is metabolized by the liver, and its clearance is reduced in patients with moderate hepatic impairment (Child-Pugh > 6). Thus, significant reductions in argatroban dose are required for individuals with moderate hepatic impairment, and the drug is contraindicated in patients with severe hepatic dysfunction. No dose adjustment is needed in the setting of renal impairment.72 Argatroban increases APTT, PT/INR, thrombin time, ecarin clotting time, and ACT in a dose-dependent fashion.
The major side effect of argatroban is bleeding. Because there is no specific antidote, excessive bleeding can only be managed by stopping the argatroban infusion and providing supportive therapy. In patients with normal hepatic function, the anticoagulant effect of argatroban disappears 2 to 4 hours after stopping the infusion. However, the anticoagulant effects of argatroban may persist for up to 24 hours in patients with hepatic impairment. A major challenge of argatroban is its effect on PT/INR. When overlapped with warfarin, PT/INR is prolonged beyond what would be expected with warfarin alone, making dose adjustment of either drug difficult.73
Two multicenter phase III prospective trials of argatroban in HIT have been completed. When compared with historical controls, patients on argatroban had reduced rates of thrombosis and death due to thrombosis, without an increase in bleeding.74 On the basis of these findings, argatroban has been approved for treatment of thrombosis and for thromboprophylaxis in patients with HIT, including those undergoing PCI with HIT.75 Increasing data supporting the use of argatroban in patients without HIT undergoing PCI has been emerging. A 2007 multicenter prospective pilot study evaluated efficacy and safety of argatroban in combination with the GPIIb/IIIa inhibitors abciximab or eptifibatide in 152 patients. The primary efficacy endpoint (a composite of death, MI, or urgent revascularization at 30 days) occurred in 2.6% of patients, and major bleeding occurred in 1.3% of patients.76 This study also showed that argatroban in combination with GPIIb/IIIa inhibition was an adequate anticoagulant with an acceptable bleeding risk.
Oral Direct Thrombin Inhibitors
Ximelagatran
Because of the significant limitations of warfarin, alternative oral DTIs have undergone significant development and clinical study. Ximelagatran, the orally available prodrug of the univalent DTI melagatran, was the first drug in this class to generate widespread interest.77 Although ximelagatran was shown to be as effective as warfarin in preventing stroke or systemic embolism in the setting of atrial fibrillation,78 there were questions about its safety profile. The open-label SPORTIF III trial compared ximelagatran with warfarin for prevention of stroke and systemic embolism, and although ximelagatran was shown to be noninferior to warfarin in preventing stroke and systemic embolism, serum alanine aminotransferase levels rose to greater than three times the upper limit of normal in 6% of individuals in the ximelagatran group and greater than five times the upper limit of normal in 3.4% of individuals in the ximelagatran group.79 Significant safety concerns regarding increased liver toxicity without a significant offsetting advantage in major bleeding led the U.S. Food and Drug Administration (FDA) to reject the sponsor’s application for ximelagatran in 2004.
Dabigatran etexilate
Like ximelagatran, dabigatran etexilate is a prodrug. Once absorbed, the drug is rapidly converted by esterases to dabigatran, whose levels peak in approximately 1 to 2 hours. Dabigatran is a small-molecule reversible inhibitor that binds to the active site of thrombin. The half-life of dabigatran is approximately 12 hours, and it is primarily eliminated via the kidney.80 The half-life is prolonged in the elderly, reflecting their impaired renal function.81 The pharmacokinetics and pharmacodynamics of dabigatran are not influenced by CYP P450 enzymes and other hepatic oxidoreductases and thus do not interfere with drugs that are metabolized by the P450 enzyme system.80 Dabigatran produces a predictable anticoagulant response. Therefore, routine coagulation monitoring is not necessary. Dabigatran prolongs ecarin clotting time, APTT, and PT/INR in a dose-dependent fashion.81 Because these widely available tests have not been used in the clinical setting for monitoring, target levels are unknown.
The major side effect of dabigatran is hemorrhage. No specific antidote is available. Consequently, bleeding complications must be managed symptomatically. Although not well studied, dialysis or hemoperfusion likely removes this compound from the circulation, and administration of activated coagulation factor complexes such as FEIBA, Autoplex, or rFVIIa may overcome its anticoagulant effect.82 Several clinical trials have demonstrated dabigatran’s antithrombotic effect. The phase II Boehringer Ingelheim Study in Thrombosis II (BISTRO II) trial compared oral dabigatran with enoxaparin as thromboprophylaxis after total hip or total knee replacement.83 Dabigatran was administered at doses of 50 mg, 150 mg, or 225 mg twice daily, or 300 mg once daily for 6 to 10 days. A significant dose-dependent decrease in VTE occurred with increasing doses of dabigatran etexilate. Overall VTE rates were 28.5% in patients receiving 50 mg of dabigatran twice daily, 17.4% in patients receiving 150 mg of dabigatran twice daily, 13.1% in patients receiving 225 mg of dabigatran twice daily, 16.6% in patients receiving 300 mg of dabigatran once daily, and 24% in patients receiving enoxaparin. The risk of serious bleeding with dabigatran increased in a dose-dependent manner as well, but did not reach statistical significance at any dose. Serious bleeding occurred in 0.3% of patients receiving 50 mg of dabigatran twice daily, 4.1% of patients receiving 150 mg of dabigatran twice daily, 4.8% of patients receiving 225 mg of dabigatran twice daily, and 4.7% of patients receiving 300 mg of dabigatran once daily. Serious bleeding occurred in 2.0% of patients receiving enoxaparin.
The RE-NOVATE study demonstrated that dabigatran etexilate was as effective as enoxaparin for preventing VTE after THR, with a similar safety profile.84 This double-blind, noninferiority trial randomized 3494 patients to treatment for 28 to 35 days with dabigatran etexilate 220 mg or 150 mg once daily, or subcutaneous enoxaparin 40 mg once daily. The primary efficacy outcome was the composite of total VTE and all-cause mortality. Both doses of dabigatran etexilate were noninferior to enoxaparin, with the primary efficacy outcome occurring in 6.7% of patients in the enoxaparin group vs. 6.0% of patients in the dabigatran etexilate 220-mg group, and 8.6% of patients in the 150-mg group. There was no significant difference in major bleeding rates with either dose of dabigatran etexilate compared to enoxaparin. There was no difference in the frequency of liver enzyme elevation.
The subsequent REMODEL trial reproduced these results in 2076 patients undergoing total knee replacement (TKR).85 The primary efficacy outcome occurred in 37.7% of the enoxaparin group vs. 36.4% of the dabigatran etexilate 220-mg group and 40.5% of the 150-mg group. Both doses of dabigatran etexilate were thus noninferior to enoxaparin. Incidence of major bleeding did not differ significantly between the three groups (1.3% vs. 1.5% and 1.3%, respectively), and there were no significant differences in liver enzyme elevation.
The RE-MOBILIZE trial was similar in design to the RE-MODEL and, in contrast to REMODEL, failed to show equivalence for a composite endpoint of proximal DVT, distal DVT, PE, and all-cause mortality.84 Warfarin is often used for thromboprophylaxis after knee arthroplasty in centers in North America, but it has not been directly compared to dabigatran in a clinical trial. Dabigatran has undergone study for initial and long-term treatment of patients with established VTE. The Randomised Evaluation of Long-Term Anticoagulant Therapy (RE-LY) trial demonstrated that in 18,133 patients with atrial fibrillation, primary outcome of stroke or embolism was lower in patients on dabigatran as compared to warfarin, as was bleeding.86 Dabigatran has been approved for use in the United States and is in use in Europe.
Oral Factor Xa Inhibitors
Rivaroxaban
Rivaroxaban is in development for prevention and treatment of thromboembolic disorders, including VTE prevention following orthopedic surgery, treatment of DVT and PE, ACS, and stroke prevention in patients with atrial fibrillation. Rivaroxaban is a potent inhibitor of factor Xa and does not inhibit thrombin-induced platelet aggregation, but it attenuates tissue factor–induced platelet aggregation indirectly through inhibition of thrombin generation.87 The antithrombotic efficacy of rivaroxaban has been demonstrated in various animal models of arterial or venous thrombosis across doses that do not prolong bleeding times.87 When combined with ASA or clopidogrel, the antithrombotic potency of rivaroxaban is enhanced.88
In healthy subjects and patients undergoing orthopedic surgery, rivaroxaban displays predictable pharmacokinetics and pharmacodynamics.89 The half-life is between 7.6 and 9.1 hours. Plasma levels of rivaroxaban correlate well with both inhibition of factor Xa activity and prolongation of PT, as assessed in healthy subjects who received multiple doses of rivaroxaban across a wide dose range.90 The pharmacodynamic effects of rivaroxaban (as measured by endogenous thrombin potential) are sustained for 24 hours after single oral doses, thus supporting once-daily dosing.91 Age, gender, and body weight have not been shown to exert clinically significant effects on the pharmacokinetic or pharmacodynamic profiles of rivaroxaban. Rivaroxaban has a dual mode of elimination: one third is excreted unchanged via the kidneys and the remaining two thirds of the drug is metabolized by the liver; there are no major or active circulating metabolites.92 It does not interact with or mobilize PF4 on platelets, and is therefore unlikely to induce the conformational changes in PF4 necessary for cross-reaction with HIT antibodies.
Four dose-finding clinical studies (one phase IIa and three phase IIb) have assessed the potential efficacy and safety of rivaroxaban for thromboprophylaxis in patients undergoing major orthopedic surgery.84,93–95 All four studies assessed rivaroxaban relative to conventional anticoagulants and measured the composite of the incidence of any DVT or objectively confirmed nonfatal PE or all-cause mortality as the primary endpoint and major bleeding as the primary safety endpoint. Results from these studies support the feasibility of daily dosing. The 10 mg daily dose provided the optimal balance between efficacy and safety in the phase II trials and was therefore selected for further study in phase III trials. There has been no evidence of liver toxicity. The RECORD program (Regulation of Coagulation in Major Orthopedic Surgery Reducing the Risk of DVT and PE) was initiated in December 2005 and enrolled more than 12,500 patients worldwide to participate in four multicenter randomized, active-controlled, double-blind studies of rivaroxaban prophylaxis in patients undergoing THR (RECORD1 and RECORD2) and TKR (RECORD3 and RECORD4).96–99 Rivaroxaban was significantly more effective in prevention of VTE after TKR and THR. These three phase III studies demonstrate that a fixed daily unmonitored dose of rivaroxaban provides a safe and effective option for short-term and extended thromboprophylaxis after major orthopedic surgery.
Rivaroxaban was also assessed for treatment of VTE in two randomized phase IIb double-blind, dose-ranging studies of rivaroxaban administered for 12 weeks in patients with acute symptomatic proximal DVT (without PE) vs. parenteral UFH or LMWH and a vitamin K antagonist.100 These two studies of more than 1150 patients suggested that efficacy of rivaroxaban for treatment of proximal DVT was similar to that achieved with standard anticoagulation therapy, with no significant dose-response relationship for the primary efficacy endpoints and low rates of VTE recurrence and bleeding events. Following on from the promising findings in the VTE treatment studies, phase III studies of long-term rivaroxaban for stroke prevention in patients with atrial fibrillation are underway. ROCKET-AF is a randomized double-blind study designed to assess efficacy and safety of rivaroxaban (20 mg daily) relative to dose-adjusted warfarin for stroke prevention in approximately 14,000 patients with atrial fibrillation. A large dose-finding randomized, double-blind, placebo-controlled phase II study is also underway to investigate the efficacy and safety of rivaroxaban alone or in combination with ASA or ASA and thienopyridine, for secondary prevention of fatal and nonfatal cardiovascular events in patients with recent ACS.
Apixaban
Efficacy and safety of apixaban for prevention of VTE in patients undergoing TKR was evaluated in a phase IIb trial (APROPOS).101 In this study, 1217 patients were randomized to receive one of six doses of apixaban (5 mg, 10 mg, or 20 mg, administered once or twice daily), open-label enoxaparin 30 mg twice daily, or warfarin for 10 to 14 days. Apixaban and enoxaparin were initiated 12 to 24 hours after surgery, whereas warfarin was started in the evening of the day of surgery. Rates of VTE and all-cause mortality, the primary efficacy endpoint, were significantly lower in the combined apixaban groups (8.6%) than in either the enoxaparin or warfarin groups (15.6% [P < 0.02] and 26.6% [P < 0.001], respectively). The primary safety endpoint of major bleeding was similar between treatment groups.
A recent double-blind randomized dose-finding trial investigated the efficacy and safety of apixaban in patients with confirmed DVT (proximal DVT or extensive calf DVT). Frequency of the primary efficacy endpoint (composite of symptomatic recurrent VTE and deterioration of the thrombotic burden, as assessed by repeat bilateral compression ultrasound and perfusion lung scan) was similar in the apixaban 5 mg twice daily and 10 mg twice daily groups (6.0% and 5.6%, respectively) and lower in the apixaban 20 mg daily group (2.6%). Early evaluation of results from phase III study of apixaban for prevention of VTE in patients undergoing TKR, however, indicate that the primary endpoint was not met.102
DU-176b
DU-176b is an oral direct factor Xa inhibitor in early clinical development for prophylaxis and treatment of thrombotic disorders. It is a potent inhibitor of factor Xa, with a 10,000-fold higher selectivity for factor Xa than for thrombin. DU-176b dose-dependently prolongs clotting times and decreases thrombin generation and platelet aggregation. A phase I study in 12 healthy adults demonstrated that DU-176b was able to reduce thrombus formation ex vivo in a Badimon chamber. The antithrombotic effects of DU-176b were sustained for up to 5 hours, with maximum inhibition of factor Xa activity occurring 1.5 hours after administration.103 A recent phase II study evaluating the efficacy and safety of DU-176b for prevention of VTE in patients undergoing total knee arthroplasty demonstrated significant dose-dependent reductions in VTE in patients undergoing total knee arthroplasty, with a bleeding incidence similar to placebo.104 In a phase II study of prevention of stroke in atrial fibrillation, patients will receive DU-176b or warfarin for 3 months.
LY517717
LY517717 is an indol-6-yl-carbonyl derivative in development for treatment and prophylaxis of thromboembolic disorders. It is a factor Xa inhibitor with 1000-fold higher selectivity for factor Xa than other serine proteases, and high oral availability.105 In humans, anticoagulant activity of LY517717 peaked within 0.5 to 4 hours of administration, and a terminal half-life of approximately 27 hours was observed, with the gastrointestinal tract as the main elimination route. A phase II double-blind parallel-group, dose-ranging study of LY517717 was undertaken in 511 patients undergoing THR or TKR. LY517717 was investigated relative to enoxaparin.106 The primary efficacy endpoint was a composite of DVT, and for the higher doses of LY517717, incidences of VTE were 19% (100 mg), 19% (125 mg), and 16% (150 mg), compared to 21% for enoxaparin, indicating that LY517717 at these doses was noninferior to enoxaparin according to prespecified criteria. Further development of LY517717 is planned, with phase III trials for prevention of VTE.
Betrixaban
Betrixaban is a potent inhibitor of factor Xa, with a half-life of 19 hours. The antithrombotic activity of betrixaban, demonstrated in different animal models of arterial and venous thrombosis, has been shown to occur at doses that inhibit thrombin generation in human blood. A phase I dose-escalation study in 64 subjects revealed that betrixaban had minimal interactions with food and predictable pharmacokinetics and pharmacodynamics.107 Furthermore, betrixaban undergoes minimal renal excretion because it is predominantly eliminated unchanged in bile. In a phase IIa proof-of-concept study (EXPERT), betrixaban was investigated relative to enoxaparin administered for 10 to 14 days.108 The primary efficacy endpoint was the incidence of VTE (symptomatic DVT or PE or asymptomatic DVT on a mandatory venogram) on days 10 to 14. Rates of VTE were 20% and 15%, respectively, in patients receiving betrixaban, and 10% in patients receiving enoxaparin. Further clinical studies for prevention and treatment of VTE, stroke prevention in atrial fibrillation, and secondary prevention of stroke and MI are planned.
YM150
YM150 is in development for prevention of VTE. YM150 has a major active metabolite, YM-222741, against factor Xa. A randomized open-label, phase IIa dose-escalation trial in 178 patients undergoing THR assessed YM150 for 7 to 10 days after surgery, relative to enoxaparin.109 The primary endpoint was major and/or clinically relevant nonmajor bleeding, and the main efficacy endpoint was the composite of DVT detected by mandatory bilateral venography, confirmed symptomatic DVT, PE, and all-cause mortality. There were no major bleeding events during the study and three clinically relevant nonmajor bleeding events. Venous thromboembolism incidence was dose dependent, ranging from 52% for 3-mg dosing to 19% at 60 mg; incidence of VTE in the enoxaparin group was 39%.110 A phase IIb study of YM150 (5-120 mg daily) for prevention of VTE after THR has been completed recently.110 Incidence of the primary efficacy endpoint (composite of DVT, symptomatic VTE, PE, and death up to day 7 to 10 of treatment) ranged from 31.7% to 13.3% and decreased significantly with increasing doses of YM150 (P < 0.0002). A further phase II study will assess the pharmacokinetics, pharmacodynamics, safety, and tolerability of YM150 in an atrial fibrillation patient population.
Factor IX Inhibitors
Coagulation is a complex process in which circulating soluble proteins, cellular elements, and tissue-based proteins interface to form an insoluble clot at sites of vascular injury. Thrombin generation is maximized on the platelet surface during the propagation phase of clot formation. Activated platelets bind the factor IXa/VIIIa complex. Additional IXa is generated by factor XIa on the platelet surface.111 The factor IXa/VIIIa complex, in physical proximity to factor Va, recruits factor X for activation. The Xa/Va complex on the platelet surface is protected from inhibition by TFPI and AT. Activation of factor X by the factor IXa/VIIIa complex is nearly 50 times more efficient than its activation by the TF/VIIa complex.112 The platelet factor Xa/Va complex then catalyzes thrombin formation, resulting in a stable fibrin-platelet clot.113 A severe bleeding tendency is typically associated with less than 1% factor IX activity. A moderate bleeding risk is incurred among individuals with 1% to 5% FIX activity, and a 5% to 40% factor IX activity causes a relatively modest hemostatic defect. Factor IXa plays a role in angiogenesis, wound healing, vascular repair, and platelet-mediated hemostasis. Factor IXa/VIIIa complex may play a pivotal role in amplifying thrombin generation initiated by the TF-VIIa complexes after vascular injury. Binding of factors IX and IXa to thrombin-activated human platelets is well described. In the presence of factors VIII and X, the affinity of receptors for factor IXa increases fivefold.
Active-site competitive antagonists
The earliest investigation of FIXa inhibitors was based on an active-site competitive antagonist, IXai, a protein without functional anticoagulant activity.114 Intravenous infusion of IXai inhibited thrombosis in animal models of coronary thrombosis and stroke in a dose-dependent fashion and produced less bleeding than UFH.114–118 To date, clinical trials of factor IXai have not been conducted.
Monoclonal antibodies as anticoagulants
Monoclonal antibodies are currently used to treat cancer, autoimmune disease, and allergy. Several antibodies directed against the Gla domain of factor IX have been developed. The 10 C12 clone was an effective anticoagulant, prolonging APTT as well as inhibiting platelet-mediated clotting in vitro.119 It effectively inhibited arterial thrombosis in a rabbit model of carotid artery injury, without increasing blood loss from a standardized cutaneous incision.120 A humanized monoclonal antibody, SB 249417, is a chimeric molecule directed against the human FIX Gla domain. In a rat arterial thrombosis model, the antibody produced significant reductions in thrombus formation, with modest APTT prolongation. In a murine stroke model, SB 249417 reduced infarct volume and was associated with reduced neurological deficits compared to tPA.121 Suppression of FIX activity and APTT prolongation were rapid and dose dependent. A phase I clinical trial with SB 249417 has been completed. Designed as a single-blind randomized placebo-controlled, single intravenous infusion dose-escalating trial, the study was undertaken to establish pharmacokinetic and pharmacodynamic properties. The antibody displayed a dose-dependent effect on clotting times, with a maximal effect at completion of a 50-minute continuous infusion.122 There were no major safety concerns.
RIBONUCLEIC ACID Aptamers as anticoagulants
Aptamers are short oligonucleotides (< 100 bases) selected for their ability to bind a chosen target, typically a protein or small molecule.123 A complex between RNA and the selected target protein (or small molecule) involves a three-dimensional folding of the RNA such that it is complementary with the surface of the target protein. Molecular recognition of a target protein by an aptamer can involve several types of RNA protein interactions, including hydrogen bonding, salt bridges, van der Waals forces, and stacking with aromatic amino acids.124
Aptamer 9.3 t, specific for factor IXa, showed that the aptamer bound factors IX and IXa with high affinity but exhibited minimal affinity for the structurally related proteins, factors VII, X, or XI, or protein C.125 Since factor VIIa binds FIX via the Gla and EGF domains, the aptamer may interact with the EGF domain.126 An RNA antidote (5.2) to the FIXa aptamer has been made and can reverse 9.3 t-induced anticoagulation in human plasma.127 Other advantages of the aptamer/antidote pair include reduced generation of thrombin and inflammatory mediators (interleukin [IL]-1b, IL-6), reduced postoperative hemorrhage, and improved cardiac output.128 The anti-IX aptamer/antidote pair 9.3 t and its antidote 5-2 were subsequently optimized for in vivo stability and manufacturability to generate the REG-1 anticoagulation system. Regado-1A was a subject-blinded dose-escalation placebo-controlled study that randomized 85 healthy volunteers to receive a bolus of drug (FIX aptamer RB006) or placebo, followed 3 hours later by a bolus of antidote (RB007) or placebo.129 Among subjects treated with RB006, APTT and ACT increased rapidly in a dose-dependent fashion, and the observed pharmacodynamic effect was stable over a 3-hour time period. The Regado-1 C study randomized 39 healthy human subjects in a double-blind fashion to either three consecutive weight-adjusted drug/antidote treatment cycles or double placebo. Each treatment cycle consisted of an intravenous bolus of RB006, followed an hour later by an ascending dose of RB007. There was a graded response to varying doses of antidote, showing an ability to titrate anticoagulant response and reversibility. There were no major bleeding or other serious adverse events.130 Potential clinical applications for this injectable factor IXa–specific drug antidote system include percutaneous and surgical coronary revascularization procedures, bridging therapy for elective noncardiac surgery in patients on Coumadin therapy, prophylaxis and treatment of venous and arterial thromboembolic disorders, and maintenance of hemodialysis circuit patency. A subcutaneous formulation that is currently being studied may extend the pharmacodynamic half-life of the drug, minimizing the number of daily injections and enabling home use. A key concern will be the potential for equipment-related thrombosis, a phenomenon that has hindered clinical development of other specific coagulation protease inhibitors.131
TTP889
TTP889 is an orally available small-molecule selective partial antagonist of factor IX/IXa. The FIXIT study group conducted a phase II clinical trial to determine the safety and antithrombotic efficacy of TTP889 in patients at risk for VTE. This multicenter placebo-controlled trial enrolled 261 hip fracture surgery patients,132 and there was no significant difference between treatment groups in the composite primary outcome of venographic or symptomatic DVT or PE at the end of the study period. However, TTP889 had no effect on markers of thrombin generation and fibrin degradation (D-dimer) compared with placebo, despite the use of TTP889 dose levels considered sevenfold higher than that required to prevent venous thrombosis in animal models. This apparent lack of pharmacodynamic effect raises concerns about the appropriateness of the dose of TTP889 selected.
Factor IX-binding proteins
It is known that natural anticoagulants occur from snake venom, including a family of homologous proteins that complex with factor IX (IX-binding proteins [bp]), factor X (X-bp), or both (IX/X-bp). The family includes habu IX-bp and habu IX/X-bp of Trimeresurus flavoviridis, echis IX/X-bp of Echis carinatus leucogaster, and acutus X-bp of Deinagkistrodon acutus.133 The venom of Agkistrodon acutus contains agkisacutacin, a homologous protein that binds both platelet GPIb and coagulation factors IX and X.134 These proteins have structures similar to disulfide-linked heterodimers of C-type lectin-like subunits. In vitro studies with IX-bp from T. flavoviridis showed anticoagulant activity, with prolongation of APTT and interference of FIXa binding to phosphatidyl serine on the plasma membrane.
1 Writing Group MembersLloyd-Jones D., Adams R.J., et al. Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215.
2 Kamath S., Blann A.D., Lip G.Y. Platelet activation: assessment and quantification. Eur Heart J. 2001;22:1561–1571.
3 Quinton T.M., Murugappan S., Kim S., et al. Different G protein-coupled signaling pathways are involved in α granule release from human platelets. J Thromb Haemost. 2004;2:978–984.
4 Hamilton J.R. Protease-activated receptors as targets for antiplatelet therapy. Blood Rev. 2009;23:61–65.
5 Sabatine M.S. Novel antiplatelet strategies in acute coronary syndromes. Cleve Clin J Med. 2009;76:S8–S15.
6 Davì G., Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494.
7 LF B. Thrombin and platelet activation. Chest. 2003;124:18S–25S.
8 Varga-Szabo D.P., Irina, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28(3):403–412.
9 Smith J.B., Dangelmaier C., Mauco G. Measurement of arachidonic acid liberation in thrombin-stimulated human platelets. Use of agents that inhibit both the cyclooxygenase and lipoxygenase enzymes. Biochim Biophys Acta. 1985;835:344–351.
10 Smith W.L. Prostanoid biosynthesis and mechanisms of action. Am J Physiol. 1992;263:F181–F191.
11 Smith W.L., Marnett L.J. Prostaglandin endoperoxide synthase: structure and catalysis. Biochim Biophys Acta. 1991;1083:1–17.
12 Funk C.D., Funk L.B., Kennedy M.E., et al. Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J. 1991;5:2304–2312.
13 Pedersen A.K., FitzGerald G.A. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N Engl J Med. 1984;311:1206–1211.
14 Awtry E.H., Loscalzo J. Aspirin. Circulation. 2000;101:1206–1218.
15 Berge E., Sandercock P. Anticoagulants versus antiplatelet agents for acute ischaemic stroke. Cochrane Database Syst Rev. 2002. CD003242
16 DiChiara J., Bliden K.P., Tantry U.S., et al. The effect of aspirin dosing on platelet function in diabetic and nondiabetic patients: an analysis from the aspirin-induced platelet effect (ASPECT) study. Diabetes. 2007;56:3014–3019.
17 Pedersen A.K., FitzGerald G.A. Cyclooxygenase inhibition, platelet function, and metabolite formation during chronic sulfinpyrazone dosing. Clin Pharmacol Ther. 1985;37:36–42.
18 Rebuzzi A.G., Natale A., Bianchi C., et al. Effects of indobufen on platelet thromboxane B2 production in patients with myocardial infarction. Eur J Clin Pharmacol. 1990;39:99–100.
19 Ramis J., Torrent J., Mis R., et al. Pharmacokinetics of triflusal after single and repeated doses in man. Int J Clin Pharmacol Ther Toxicol. 1990;28:344–349.
20 Marczewski M.M., Postula M., Kosior D. Novel antiplatelet agents in the prevention of cardiovascular complications–focus on ticagrelor. Vasc Health Risk Manag. 2010;6:419–429.
21 Mangin P., Ohlmann P., Eckly A., et al. The P2Y1 receptor plays an essential role in the platelet shape change induced by collagen when TxA2 formation is prevented. J Thromb Haemost. 2004;2:969–977.
22 Storey R.F. Biology and pharmacology of the platelet P2Y12 receptor. Curr Pharm Des. 2006;12:1255–1259.
23 Schomig A. Ticagrelor–is there need for a new player in the antiplatelet-therapy field? N Engl J Med. 2009;361:1108–1111.
24 Patrono C., Coller B., FitzGerald G.A., et al. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:234S–264S.
25 Wallentin L., Varenhorst C., James S., et al. Prasugrel achieves greater and faster P2Y12 receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur Heart J. 2008;29:21–30.
26 Brandt J.T., Payne C.D., Wiviott S.D., et al. A comparison of prasugrel and clopidogrel loading doses on platelet function: magnitude of platelet inhibition is related to active metabolite formation. Am Heart J. 2007;153(66):e9–e16.
27 CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet. 1996;348:1329–1339.
28 Yusuf S., Zhao F., Mehta S.R., et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494–502.
29 Chen Z.M., Jiang L.X., Chen Y.P., et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1607–1621.
30 Steinhubl S.R., Berger P.B., Mann J.T., et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA. 2002;288:2411–2420.
31 Bhatt D.L., Topol E.J. Current role of platelet glycoprotein IIb/IIIa inhibitors in acute coronary syndromes. JAMA. 2000;284:1549–1558.
32 Wang T.H., Bhatt D.L., Fox K.A., et al. An analysis of mortality rates with dual-antiplatelet therapy in the primary prevention population of the CHARISMA trial. Eur Heart J. 2007;28:2200–2207.
33 Bhatt D.L., Flather M.D., Hacke W., et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol. 2007;49:1982–1988.
34 Wiviott S.D., Trenk D., Frelinger A.L., et al. Prasugrel compared with high loading- and maintenance-dose clopidogrel in patients with planned percutaneous coronary intervention: the Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation-Thrombolysis in Myocardial Infarction 44 Trial. Circulation. 2007;116:2923–2932.
35 Tantry U.S., Bliden K.P., Gurbel P.A. Azd6140. Expert Opin Investig Drugs. 2007;16:225–229.
36 Sabatine M.S. Novel antiplatelet strategies in acute coronary syndromes. Cleve Clin J Med. 2009;76(Suppl 1):S8–S15.
37 Cannon C.P., Husted S., Harrington R.A., et al. Safety, tolerability, and initial efficacy of AZD6140, the first reversible oral adenosine diphosphate receptor antagonist, compared with clopidogrel, in patients with non-ST-segment elevation acute coronary syndrome: primary results of the DISPERSE-2 trial. J Am Coll Cardiol. 2007;50:1844–1851.
38 Wallentin L., Becker R.C., Budaj A., et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–1057.
39 Gurbel P.A., Bliden K.P., Butler K., et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120:2577–2585.
40 Schror K., Weber A.A. Comparative pharmacology of GP IIb/IIIa antagonists. J Thromb Thrombolysis. 2003;15:71–80.
41 Moliterno D.J., Califf R.M., Aguirre F.V., et al. Effect of platelet glycoprotein IIb/IIIa integrin blockade on activated clotting time during percutaneous transluminal coronary angioplasty or directional atherectomy (the EPIC trial). Evaluation of c7E3 Fab in the Prevention of Ischemic Complications trial. Am J Cardiol. 1995;75:559–562.
42 Roe M.T., Moliterno D.J. The EPILOG trial. Abciximab prevents ischemic complications during angioplasty. Evaluation in PTCA to improve long-term outcome with abciximab GP IIb/IIIa blockade. Cleve Clin J Med. 1998;65:267–272.
43 Lincoff A.M., Califf R.M., Moliterno D.J., et al. Complementary clinical benefits of coronary-artery stenting and blockade of platelet glycoprotein IIb/IIIa receptors. Evaluation of Platelet IIb/IIIa Inhibition in Stenting Investigators. N Engl J Med. 1999;341:319–327.
44 Kastrati A., Mehilli J., Schuhlen H., et al. A clinical trial of abciximab in elective percutaneous coronary intervention after pretreatment with clopidogrel. N Engl J Med. 2004;350:232–238.
45 Kastrati A., Mehilli J., Neumann F.J., et al. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA. 2006;295:1531–1538.
46 Tcheng J.E. Differences among the parenteral platelet glycoprotein IIb/IIIa inhibitors and implications for treatment. Am J Cardiol. 1999;83:7E–11E.
47 Giugliano R.P., White J.A., Bode C., et al. Early versus delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med. 2009;360:2176–2190.
48 Ruggeri Z.M., Ware J. von Willebrand factor. FASEB J. 1993;7:308–316.
49 Weiss H.J., Sussman I.I., Hoyer L.W. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J Clin Invest. 1977;60:390–404.
50 Hennan J.K., Swillo R.E., Morgan G.A., et al. Pharmacologic inhibition of platelet vWF-GPIb alpha interaction prevents coronary artery thrombosis. Thromb Haemost. 2006;95:469–475.
51 Montalescot G., Philippe F., Ankri A., et al. Early increase of von Willebrand factor predicts adverse outcome in unstable coronary artery disease: beneficial effects of enoxaparin. French Investigators of the ESSENCE Trial. Circulation. 1998;98:294–299.
52 Kageyama S., Yamamoto H., Nakazawa H., et al. Pharmacokinetics and pharmacodynamics of AJW200, a humanized monoclonal antibody to von Willebrand factor, in monkeys. Arterioscler Thromb Vasc Biol. 2002;22:187–192.
53 Brody E.N., Gold L. Aptamers as therapeutic and diagnostic agents. J Biotechnol. 2000;74:5–13.
54 Gilbert J.C., DeFeo-Fraulini T., Hutabarat R.M., et al. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007;116:2678–2686.
55 Gurevitz O., Eldar M., Skutelsky E., et al. S-nitrosoderivative of a recombinant fragment of von Willebrand factor (S-nitroso-AR545C) inhibits thrombus formation in guinea pig carotid artery thrombosis model. Thromb Haemost. 2000;84:912–917.
56 Samaha F.F., Kahn M.L. Novel platelet and vascular roles for immunoreceptor signaling. Arterioscler Thromb Vasc Biol. 2006;26:2588–2593.
57 Nieswandt B., Schulte V., Bergmeier W., et al. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193:459–469.
58 Markwardt F., Nowak G., Sturzebecher J. Clinical pharmacology of recombinant hirudin. Haemostasis. 1991;21(Suppl 1):133–136.
59 Eichler P., Friesen H.J., Lubenow N., et al. Antihirudin antibodies in patients with heparin-induced thrombocytopenia treated with lepirudin: incidence, effects on aPTT, and clinical relevance. Blood. 2000;96:2373–2378.
60 Greinacher A., Lubenow N., Eichler P. Anaphylactic and anaphylactoid reactions associated with lepirudin in patients with heparin-induced thrombocytopenia. Circulation. 2003;108:2062–2065.
61 Lubenow N., Eichler P., Lietz T., et al. Lepirudin in patients with heparin-induced thrombocytopenia – results of the third prospective study (HAT-3) and a combined analysis of HAT-1, HAT-2, and HAT-3. J Thromb Haemost. 2005;3:2428–2436.
62 Organisation to Assess Strategies for Ischemic Syndromes (OASIS-2) Investigators. Effects of recombinant hirudin (lepirudin) compared with heparin on death, myocardial infarction, refractory angina, and revascularisation procedures in patients with acute myocardial ischaemia without ST elevation: a randomised trial. Lancet. 1999;353:429–438.
63 Greinacher A., Eichler P., Albrecht D., et al. Antihirudin antibodies following low-dose subcutaneous treatment with desirudin for thrombosis prophylaxis after hip-replacement surgery: incidence and clinical relevance. Blood. 2003;101:2617–2619.
64 Eriksson B.I., Ekman S., Lindbratt S., et al. Prevention of thromboembolism with use of recombinant hirudin. Results of a double-blind, multicenter trial comparing the efficacy of desirudin (Revasc) with that of unfractionated heparin in patients having a total hip replacement. J Bone Joint Surg Am. 1997;79:326–333.
65 Eriksson B.I., Wille-Jorgensen P., Kalebo P., et al. A comparison of recombinant hirudin with a low-molecular-weight heparin to prevent thromboembolic complications after total hip replacement. N Engl J Med. 1997;337:1329–1335.
66 Bates S.M., Weitz J.I. Direct thrombin inhibitors for treatment of arterial thrombosis: potential differences between bivalirudin and hirudin. Am J Cardiol. 1998;82:12P–18P.
67 Bittl J.A., Chaitman B.R., Feit F., et al. Bivalirudin versus heparin during coronary angioplasty for unstable or postinfarction angina: final report reanalysis of the Bivalirudin Angioplasty Study. Am Heart J. 2001;142:952–959.
68 Lincoff A.M., Bittl J.A., Kleiman N.S., et al. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol. 2004;93:1092–1096.
69 Lincoff A.M., Kleiman N.S., Kereiakes D.J., et al. Long-term efficacy of bivalirudin and provisional glycoprotein IIb/IIIa blockade vs heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary revascularization: REPLACE-2 randomized trial. JAMA. 2004;292:696–703.
70 Stone G.W., Ware J.H., Bertrand M.E., et al. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA. 2007;298:2497–2506.
71 Berry C.N., Girardot C., Lecoffre C., et al. Effects of the synthetic thrombin inhibitor argatroban on fibrin- or clot-incorporated thrombin: comparison with heparin and recombinant Hirudin. Thromb Haemost. 1994;72:381–386.
72 Swan S.K., St Peter J.V., Lambrecht L.J., et al. Comparison of anticoagulant effects and safety of argatroban and heparin in healthy subjects. Pharmacotherapy. 2000;20:756–770.
73 Sheth S.B., DiCicco R.A., Hursting M.J., et al. Interpreting the International Normalized Ratio (INR) in individuals receiving argatroban and warfarin. Thromb Haemost. 2001;85:435–440.
74 Lewis B.E., Wallis D.E., Leya F., et al. Argatroban anticoagulation in patients with heparin-induced thrombocytopenia. Arch Intern Med. 2003;163:1849–1856.
75 Lewis B.E., Matthai W.H.Jr, Cohen M., et al. Argatroban anticoagulation during percutaneous coronary intervention in patients with heparin-induced thrombocytopenia. Catheter Cardiovasc Interv. 2002;57:177–184.
76 Cruz-Gonzalez I., Sanchez-Ledesma M., Baron S.J., et al. Efficacy and safety of argatroban with or without glycoprotein IIb/IIIa inhibitor in patients with heparin-induced thrombocytopenia undergoing percutaneous coronary intervention for acute coronary syndrome. J Thromb Thrombolysis. 2008;25:214–218.
77 Francis C.W., Davidson B.L., Berkowitz S.D., et al. Ximelagatran versus warfarin for the prevention of venous thromboembolism after total knee arthroplasty. A randomized, double-blind trial. Ann Intern Med. 2002;137:648–655.
78 Albers G.W., Diener H.C., Frison L., et al. Ximelagatran vs warfarin for stroke prevention in patients with nonvalvular atrial fibrillation: a randomized trial. JAMA. 2005;293:690–698.
79 Olsson S.B. Stroke prevention with the oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with non-valvular atrial fibrillation (SPORTIF III): randomised controlled trial. Lancet. 2003;362:1691–1698.
80 Blech S., Ebner T., Ludwig-Schwellinger E., et al. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos. 2008;36:386–399.
81 Stangier J., Stahle H., Rathgen K., et al. Pharmacokinetics and pharmacodynamics of the direct oral thrombin inhibitor dabigatran in healthy elderly subjects. Clin Pharmacokinet. 2008;47:47–59.
82 Wienen W., Stassen J.M., Priepke H., et al. Effects of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate, on thrombus formation and bleeding time in rats. Thromb Haemost. 2007;98:333–338.
83 Eriksson B.I., Dahl O.E., Buller H.R., et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost. 2005;3:103–111.
84 Eriksson B.I., Dahl O.E., Rosencher N., et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet. 2007;370:949–956.
85 Eriksson B.I., Dahl O.E., Rosencher N., et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost. 2007;5:2178–2185.
86 Connolly S.J., Ezekowitz M.D., Yusuf S., et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139–1151.
87 Perzborn E., Strassburger J., Wilmen A., et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59–7939–an oral, direct factor Xa inhibitor. J Thromb Haemost. 2005;3:514–521.
88 Gerotziafas G.T., Elalamy I., Depasse F., et al. In vitro inhibition of thrombin generation, after tissue factor pathway activation, by the oral, direct factor Xa inhibitor rivaroxaban. J Thromb Haemost. 2007;5:886–888.
89 Kubitza D., Becka M., Mueck W., et al. Rivaroxaban (BAY 59–7939)–an oral, direct factor Xa inhibitor–has no clinically relevant interaction with naproxen. Br J Clin Pharmacol. 2007;63:469–476.
90 Kubitza D., Becka M., Wensing G., et al. Safety, pharmacodynamics, and pharmacokinetics of BAY 59–7939–an oral, direct factor Xa inhibitor–after multiple dosing in healthy male subjects. Eur J Clin Pharmacol. 2005;61:873–880.
91 Graff J., von Hentig N., Misselwitz F., et al. Effects of the oral, direct factor xa inhibitor rivaroxaban on platelet-induced thrombin generation and prothrombinase activity. J Clin Pharmacol. 2007;47:1398–1407.
92 Weinz C., Schwarz T., Kubitza D., et al. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab Dispos. 2009;37:1056–1064.
93 Eriksson B.I., Dahl O.E., Buller H.R., et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost. 2005;3:103–111.
94 Eriksson B.I., Dahl O.E., Rosencher N., et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost. 2007;5:2178–2185.
95 Ginsberg J.S., Davidson B.L., Comp P.C., et al. Oral thrombin inhibitor dabigatran etexilate vs. North American enoxaparin regimen for prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty. 2009;24:1–9.
96 Eriksson B.I., Borris L.C., Friedman R.J., et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med. 2008;358:2765–2775.
97 Lassen M.R., Ageno W., Borris L.C., et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med. 2008;358:2776–2786.
98 Kakkar A.K., Brenner B., Dahl O.E., et al. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet. 2008;372:31–39.
99 Turpie A.G., Lassen M.R., Davidson B.L., et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial. Lancet. 2009;373:1673–1680.
100 Agnelli G., Gallus A., Goldhaber S.Z., et al. Treatment of proximal deep-vein thrombosis with the oral direct factor Xa inhibitor rivaroxaban (BAY 59–7939): the ODIXa-DVT (Oral Direct Factor Xa Inhibitor BAY 59–7939 in Patients With Acute Symptomatic Deep-Vein Thrombosis) study. Circulation. 2007;116:180–187.
101 Lassen M.R., Davidson B.L., Gallus A., et al. The efficacy and safety of apixaban, an oral, direct factor Xa inhibitor, as thromboprophylaxis in patients following total knee replacement. J Thromb Haemost. 2007;5:2368–2375.
102 Pfizer B-MSa:. Bristol-Myers Squibb and Pfizer provide update on apixaban clinical development program, [press release],. 2008. August 26, 2008
103 Zafar M.U., Vorchheimer D.A., Gaztanaga J., et al. Antithrombotic effects of factor Xa inhibition with DU-176b: phase-I study of an oral, direct factor Xa inhibitor using an ex-vivo flow chamber. Thromb Haemost. 2007;98:883–888.
104 Fuji T., Fujita S., Tachibana S., et al. A dose-ranging study evaluating the oral factor Xa inhibitor edoxaban for the prevention of venous thromboembolism in patients undergoing total knee arthroplasty. J Thromb Haemost. 2010;8:2458–2468.
105 Spyropoulos A.C. Investigational treatments of venous thromboembolism. Expert Opin Investig Drugs. 2007;16:431–440.
106 Agnelli G., Haas S., Ginsberg J.S., et al. A phase II study of the oral factor Xa inhibitor LY517717 for the prevention of venous thromboembolism after hip or knee replacement. J Thromb Haemost. 2007;5:746–753.
107 Turpie A.G. Oral, direct factor Xa inhibitors in development for the prevention and treatment of thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2007;27:1238–1247.
108 Turpie A.G., Bauer K.A., Davidson B.L., et al. A randomized evaluation of betrixaban, an oral factor Xa inhibitor, for prevention of thromboembolic events after total knee replacement (EXPERT). Thromb Haemost. 2009;101:68–76.
109 Eriksson B.I., Turpie A.G., Lassen M.R., et al. Prevention of venous thromboembolism with an oral factor Xa inhibitor, YM150, after total hip arthroplasty. A dose finding study (ONYX-2). J Thromb Haemost. 2010;8:714–721.
110 Eriksson B.I., Turpie A.G., Lassen M.R., et al. A dose escalation study of YM150, an oral direct factor Xa inhibitor, in the prevention of venous thromboembolism in elective primary hip replacement surgery. J Thromb Haemost. 2007;5:1660–1665.
111 Oliver J.A., Monroe D.M., Roberts H.R., et al. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol. 1999;19:170–177.
112 Lawson J.H., Kalafatis M., Stram S., et al. A model for the tissue factor pathway to thrombin. I. An empirical study. J Biol Chem. 1994;269:23357–23366.
113 Bowen D.J. Haemophilia A and haemophilia B: molecular insights. Mol Pathol. 2002;55:1–18.
114 Benedict C.R., Ryan J., Wolitzky B., et al. Active site-blocked factor IXa prevents intravascular thrombus formation in the coronary vasculature without inhibiting extravascular coagulation in a canine thrombosis model. J Clin Invest. 1991;88:1760–1765.
115 Spanier T.B., Oz M.C., Madigan J.D., et al. Selective anticoagulation with active site blocked factor IXa in synthetic patch vascular repair results in decreased blood loss and operative time. ASAIO J. 1997;43:M526–M530.
116 Wong A.G., Gunn A.C., Ku P., et al. Relative efficacy of active site-blocked factors IXa Xa in models of rabbit venous and arterio-venous thrombosis. Thromb Haemost. 1997;77:1143–1147.
117 Spanier T.B., Oz M.C., Minanov O.P., et al. Heparinless cardiopulmonary bypass with active-site blocked factor IXa: a preliminary study on the dog. J Thorac Cardiovasc Surg. 1998;115:1179–1188.
118 Choudhri T.F., Hoh B.L., Prestigiacomo C.J., et al. Targeted inhibition of intrinsic coagulation limits cerebral injury in stroke without increasing intracerebral hemorrhage. J Exp Med. 1999;190:91–99.
119 Suggett S., Kirchhofer D., Hass P., et al. Use of phage display for the generation of human antibodies that neutralize factor IXa function. Blood Coagul Fibrinolysis. 2000;11:27–42.
120 Refino C.J., Jeet S., DeGuzman L., et al. A human antibody that inhibits factor IX/IXa function potently inhibits arterial thrombosis without increasing bleeding. Arterioscler Thromb Vasc Biol. 2002;22:517–522.
121 Toomey J.R., Valocik R.E., Koster P.F., et al. Inhibition of factor IX(a) is protective in a rat model of thromboembolic stroke. Stroke. 2002;33:578–585.
122 Benincosa L.J., Chow F.S., Tobia L.P., et al. Pharmacokinetics and pharmacodynamics of a humanized monoclonal antibody to factor IX in cynomolgus monkeys. J Pharmacol Exp Ther. 2000;292:810–816.
123 Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822.
124 Hermann T., Patel D.J. Adaptive recognition by nucleic acid aptamers. Science. 2000;287:820–825.
125 Rusconi C.P., Scardino E., Layzer J., et al. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature. 2002;419:90–94.
126 Gopinath S.C., Shikamoto Y., Mizuno H., et al. A potent anti-coagulant RNA aptamer inhibits blood coagulation by specifically blocking the extrinsic clotting pathway. Thromb Haemost. 2006;95:767–771.
127 Nimjee S.M., Keys J.R., Pitoc G.A., et al. A novel antidote-controlled anticoagulant reduces thrombin generation and inflammation and improves cardiac function in cardiopulmonary bypass surgery. Mol Ther. 2006;14:408–415.
128 Rusconi C.P., Roberts J.D., Pitoc G.A., et al. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat Biotechnol. 2004;22:1423–1428.
129 Dyke C.K., Steinhubl S.R., Kleiman N.S., et al. First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation. 2006;114:2490–2497.
130 Chan M.Y., Rusconi C.P., Alexander J.H., et al. A randomized, repeat-dose, pharmacodynamic and safety study of an antidote-controlled factor IXa inhibitor. J Thromb Haemost. 2008;6:789–796.
131 Califf R.M. Fondaparinux in ST-segment elevation myocardial infarction: the drug, the strategy, the environment, or all of the above? JAMA. 2006;295:1579–1580.
132 Eriksson B.I., Dahl O.E., Lassen M.R., et al. Partial factor IXa inhibition with TTP889 for prevention of venous thromboembolism: an exploratory study. J Thromb Haemost. 2008;6:457–463.
133 Atoda H., Kaneko H., Mizuno H., et al. Calcium-binding analysis and molecular modeling reveal echis coagulation factor IX/factor X-binding protein has the Ca-binding properties and Ca ion-independent folding of other C-type lectin-like proteins. FEBS Lett. 2002;531:229–234.
134 Li W.F., Chen L., Li X.M., et al. A C-type lectin-like protein from Agkistrodon acutus venom binds to both platelet glycoprotein Ib and coagulation factor IX/factor X. Biochem Biophys Res Commun. 2005;332:904–912.