[level-membership-for-anesthesiology-category]
9 Pharmacology of Anesthetic Drugs
An enormous body of literature has been accumulated describing the protean effects of the different anesthetic agents on the heart and the pulmonary and systemic regional vascular beds. The effects on the heart, especially, have spawned innumerable publications. More recently, this has been because of the great interest in anesthesia-induced preconditioning (APC). However, even before the initial description of APC, the literature detailing the influence of anesthetic agents on the myocardium was prodigious and not always consistent.1–3 This likely reflected not only the challenges inherent in quantitating the direct effects of volatile agents on the myocyte/myocardium, but the presence of several potential confounding variables including effects on coronary blood flow (CBF), the systemic vasculature, and the baroreceptor reflex arc. This chapter divides the discussion of the effects of volatile agents, fixed agents, and narcotics on the cardiovascular system (CVS) into those that involve acute and delayed effects. Under acute effects, the influence of anesthetic agents is described on: (1) myocardial function, (2) electrophysiology, (3) coronary vasoregulation, (4) systemic and pulmonary vasoregulation, and (5) the baroreceptor reflex. Within delayed effects, the focus is on APC.
Volatile agents

Acute Effects
Myocardial Function
The influence of volatile anesthetics on contractile function has been investigated extensively in several animal species and in humans using various in vitro and in vivo models.4–11 In general, it is now widely agreed that volatile agents cause dose-dependent depression of contractile function (Box 9-1). Moreover, different volatile agents are not identical in this regard and the preponderance of information indicates that halothane and enflurane exert equal but more potent myocardial depression than do isoflurane, desflurane, or sevoflurane. This reflects, in part, reflex sympathetic activation with the latter agents. It is also widely accepted that in the setting of preexisting myocardial depression, volatile agents have a greater effect than in normal myocardium.12,13 Early studies indicating that volatile agents may not have a deleterious effect on function in the setting of acute myocardial infarction (AMI) likely reflected the fact that the limited infarction did not compromise overall myocardial function.14,15 At the cellular level, volatile anesthetics exert their negative inotropic effects, mainly by modulating sarcolemmal (SL) L-type Ca++ channels, the sarcoplasmic reticulum (SR), and the contractile proteins. L-type Ca++ currents are decreased and, secondarily, SR Ca++ release is depressed (Figures 9-1 and 9-2).16 Moreover, the contractile response to lower Ca++ levels is further attenuated in the presence of volatile agents in that the response is decreased by volatile agents at any given Ca++ level; that is, volatile agents also decrease Ca++ sensitivity (Figure 9-3).16 However, the mechanisms whereby anesthetic agents modify ion channels are not completely understood. Ion channels usually are studied in ex vivo circumstances in which, by definition, multiple modulating influences of the specific channel under study may be altered. Moreover, these studies frequently are undertaken in nonhuman tissue. Well-recognized species differences make extrapolation to humans difficult.17 Nitrous oxide causes direct mild myocardial depression but also causes sympathetic activation.18
BOX 9-1. Volatile Anesthetic Agents
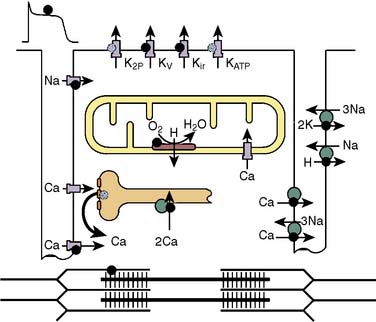
It is well recognized that even in the setting of normal systolic function, diastolic dysfunction occurs with increasing frequency in the elderly and is an important cause of congestive heart failure (CHF).19–25 Diastolic dysfunction and its more severe clinical counterpart, diastolic heart failure, have protean causative factors and can be mechanistically complex23 (Table 9-1). However, the mechanisms underlying these conditions can be categorized into those involving alterations in myocardial relaxation (e.g., SR Ca++ handling, phospholamban), those related to intrinsic properties of myocardial tissue (e.g., myocyte cytoskeletal elements), and those that are extramyocardial (e.g., loading conditions). Indices of diastolic function were not readily and reliably measured noninvasively in the past; hence the relatively more recent recognition and description of diastolic dysfunction and diastolic heart failure compared with perturbations in systolic function. This likely also explains the relative paucity of literature detailing the modulating effects of volatile agents on diastolic function. There is reasonable agreement in the literature that volatile agents prolong isovolumic relaxation and do so in a dose-dependent manner.21,22,26–30 The effects of volatile agents on chamber stiffness are more controversial; for example, halothane has been reported to both decrease compliance and have no effect on myocardial stiffness.21,22,26,28–31 The effect of nitrous oxide on diastolic function has not been investigated in a manner that critically rules out confounding variables. At a molecular level, alterations in relaxation likely reflect modulation of Ca++ currents, including SR Ca++ reuptake mechanisms. Paradoxically, in the setting of reperfusion injury and Ca++ overload, the volatile agent sevoflurane improves indices of diastolic relaxation and attenuates myoplasmic Ca++ overload.32
| Abnormalities of myocardial relaxation |
| Ischemia |
| Hypertrophy |
| Hypertension |
| Valvular heart disease |
| Abnormalities of myocardial compliance |
| Aging |
| Fibrosis |
| Hypertrophy |
| Diabetes mellitus |
| Metabolic syndrome |
| Infiltrative disorders—amyloidosis |
| Cardiomyopathies |
| Constrictive pericarditis |
Cardiac Electrophysiology
The molecular mechanisms underlying this effect of volatile anesthetics are poorly understood. Anesthetic agent–induced modulation of ion channels is important mechanistically in excitation-contraction coupling (vide supra), in preconditioning (vide infra), and in modulating automaticity and arrhythmia generation17 (Table 9-2). Although the effects of any particular volatile agent on a specific cardiac ion channel may have been characterized, this does not allow a ready extrapolation into clinical situations. This partly reflects those issues already discussed (species differences, ex vivo studies) but also the recognition that it is impossible to predict the arrhythmogenic effect that might ensue after modulation with a particular volatile agent. This should be one of the lessons garnered from the experience with the antiarrhythmic drugs such as encainide and flecainide.33 Moreover, even in the clinical setting, not all volatile agents have the same effect.34

Coronary Vasoregulation
Animal studies indicate that halothane has little direct effect on the coronary vasculature.35–37 Likewise, clinical studies investigating the effect of halothane indicate that it has either minimal or mild coronary vasodilator effects.38–41 The effect of isoflurane on coronary vessels was controversial and dominated much of the literature in this area in the 1980s and early 1990s. The current assessments of the effects of isoflurane have been succinctly detailed by Tanaka et al.42 Several reports have indicated that it caused direct coronary arteriolar vasodilatation in vessels of 100 μm or less, and that isoflurane could cause “coronary steal” in patients with “steal-prone” coronary anatomy; that is, in patients with significant coronary stenosis in a vessel subserving a region of ischemic myocardium, when, presumably, vessels were maximally dilated because of local metabolic autoregulation, and in whom isoflurane-induced vasodilatation in adjacent vessels resulted in diversion of coronary flow away from the ischemic region.43,44 Several animal and human studies in which potential confounding variables were controlled indicated clearly that isoflurane did not cause coronary steal.45–51 Studies of sevoflurane and desflurane showed similar results and are consistent with a mild direct coronary vasodilator effect of these agents.52,53
Ultimately, CBF (in the setting of normal systemic hemodynamics) is controlled by coronary vascular smooth muscle tone, which can be modulated directly (endothelium-independent) or indirectly via the endothelium (endothelium-dependent). Teleologically, it can be predicted that in vital organs, control of blood flow is predominantly local, acting through either endothelium-dependent or -independent mechanisms. Thus, volatile agents have the capacity to modulate mechanisms underlying vascular tone: (1) Halothane and isoflurane have been shown to attenuate endothelial-dependent tone (by receptor-dependent and receptor-dependent plus -independent mechanisms, respectively) in coronary microvessels54; (2) several volatile agents cause coronary vasodilation via K+ATP-channel–dependent mechanisms54–57; and (3) sevoflurane induced K+– and Ca++-channel–mediated increases in coronary collateral blood flow.58 The effects in vivo are likely to be modest because local control mechanisms are likely to predominate.
Systemic Regional and Pulmonary Vascular Effects
Reversible inhibition of endothelium-dependent relaxation in aortic and femoral vessels was first demonstrated for halothane and also has been demonstrated for enflurane, isoflurane, and sevoflurane in both capacitance and resistance vessels.54,59–63 However, these observations mask the differential effects of volatile agents on underlying endothelium-dependent mechanisms. Halothane and enflurane decrease agonist (bradykinin) and ATP-induced Ca++ increases in bovine endothelial cells, whereas isoflurane does not.64 In contrast, isoflurane does attenuate histamine-induced Ca++ influx into human endothelial cells.65 Alterations in endothelium-dependent mechanisms by volatile agents are not confined to attenuation of agonist-dependent and -independent activation of endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) release but also may extend to other mechanisms. For example, the effects of sevoflurane on endothelial cell function may be partially because of sevoflurane-induced changes in endothelin-1 (ET-1) production and in the redox milieu of the endothelial cells (i.e., increased superoxide anion production).66
The effect of volatile agents on vascular smooth muscle mechanisms is equally complex and varies among agents. In endothelial cell–denuded aortic rings, halothane decreases both SL Ca++ influx via voltage-dependent calcium channels and SR Ca++ release, but sevoflurane does not.67 Sevoflurane also inhibits angiotensin II–induced vascular smooth muscle contraction in aortic rings.68 In mesenteric vessels, sevoflurane accentuates endothelium-dependent mechanisms and attenuates endothelium-independent mechanisms in the presence of norepinephrine.69 Studies of the influence of volatile agents on vascular smooth muscle Ca++ currents indicate that both halothane and enflurane stimulate SR release and reuptake from the caffeine-sensitive pool. In contrast, halothane, enflurane, and isoflurane all increase calcium-induced calcium release (CICR) mechanisms, but sevoflurane decreases CICR mechanisms.70 Finally, volatile agents also have been demonstrated to modulate Ca++ sensitivity. In mesenteric vessels, halothane relaxation is largely mediated by Ca++ and myosin light-chain desensitizing mechanisms.71
The pulmonary circulation has unique features that must be taken into account when interpreting studies of this vascular bed. In addition to those issues that also apply to systemic vascular beds (vessel size, etc.), the pulmonary vasculature is a low-resistance bed (requiring preconstriction to access vasoactive effects), is not rectilinear (thus, changes in flow per se can change certain parameters used to calculate resistance), is contained within the chest (and thus subject to extravascular pressures, which are not atmospheric and change during the respiratory cycle), and exhibits the unique vascular phenomenon of hypoxia-induced vasoconstriction. It is clear that volatile agents modulate not only the baseline pulmonary vasculature but also multiple vasoactive mechanisms that control pulmonary vascular tone. Moreover, the effect of volatile agents is agent specific. For example, halothane causes flow-independent pulmonary vasoconstriction.72 In contrast, the hypoxic pulmonary vasoconstrictor response does not appear to be altered by at least two currently used volatile agents: sevoflurane and desflurane.73 The pulmonary vascular endothelial response appears to be impaired by the volatile agents halothane and isoflurane.74,75 Finally, pulmonary vascular smooth muscle regulatory mechanisms also can be modified by volatile agents. Halothane, enflurane, and isoflurane all attenuate pulmonary vasodilatation induced by K+ATP channel activation.76,77 Although the effects of the different volatile agents on K+ATP-channel activation are similar, β-adrenergic receptor–induced pulmonary vasodilatation is differently modulated. Halothane and isoflurane potentiate the vasodilatory response, but enflurane has no effect.78
Baroreceptor Reflex
All volatile agents attenuate the baroreceptor reflex. Baroreceptor reflex inhibition by halothane and enflurane is more potent than that observed with isoflurane, desflurane, or sevoflurane, each of which has a similar effect.79,80 Each component of the baroreceptor reflex arc (afferent nerve activity, central processing, efferent nerve activity) is inhibited by volatile agents. Inhibition of afferent nerve traffic results, in part, from baroreceptor sensitization,81,82 whereas attenuation of efferent activity is due, in part, to ganglionic inhibition as manifest by differential preganglionic and postganglionic nerve activity.81–83

Delayed Effects
Reversible Myocardial Ischemia
Prolonged ischemia results in irreversible myocardial damage and necrosis (Box 9-2). Shorter durations of myocardial ischemia can, depending on the duration and sequence of ischemic insults, lead to either preconditioning or myocardial stunning (Figure 9-4).84 Stunning, first described in 1975, occurs after brief ischemia and is characterized by myocardial dysfunction in the setting of normal restored blood flow and by an absence of myocardial necrosis.85 Ischemic preconditioning (IPC) was first described by Murry et al86 in 1986 and is characterized by an attenuation in infarct size after sustained ischemia, if this period of sustained ischemia is preceded by a period of brief ischemia (Figure 9-5). Moreover, this effect is independent of collateral flow. Thus, short periods of ischemia followed by reperfusion can lead to either stunning or preconditioning with a reduction in infarct size (Figure 9-6).84
BOX 9-2 Volatile Agents and Myocardial Ischemia
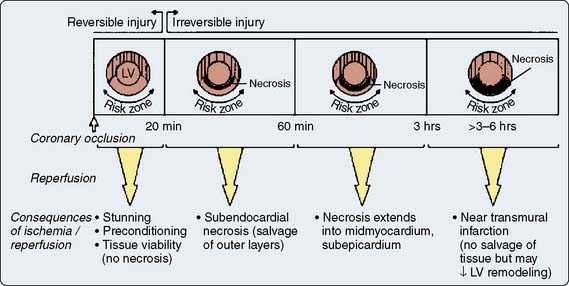
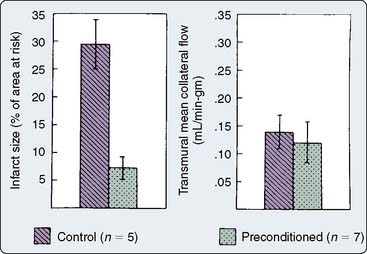
Figure 9-5 Infarct size and collateral blood flow in the 40-minute study.
(From Warltier DC, al-Wathiqui MH, Kampine JP, et al: Recovery of contractile function of stunned myocardium in chronically instrumented dogs is enhanced by halothane or isoflurane, Anesthesiology 69:552, 1988.)
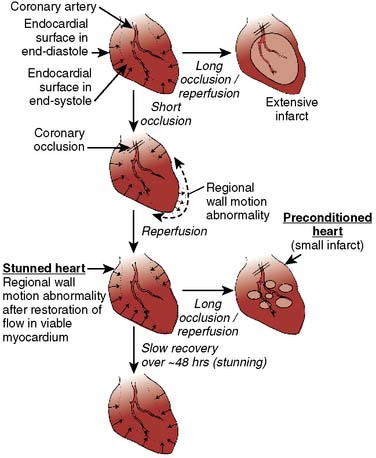
Figure 9-6 Schematic of stunning and preconditioning.
(From Kloner RA, Jennings RB: Consequences of brief ischemia: stunning, preconditioning, and their clinical implications, Part I, Circulation 104:2981, 2001.)
As discussed previously, work in the 1970s indicated that volatile anesthetic agents attenuated ST-segment elevations in the setting of short-duration ischemia and limited infarct size and lactate production after prolonged ischemia.87,88 Moreover, these effects seemed to be independent of the main determinants of myocardial oxygen supply and demand, and suggested that the volatile agents may be exerting a beneficial effect at the level of the myocyte. On resolution of the isoflurane “coronary steal” controversy, the first description of the salutary effects of volatile agents on the consequences of brief ischemia was made in 1988. Warltier and coworkers89 described the beneficial effects of halothane and isoflurane in facilitating the recovery of contractile function in stunned myocardium (Figure 9-7). However, it was almost a decade later before the effects of volatile agents on preconditioning were outlined2,3 and the term APC was used1 (Figure 9-8).
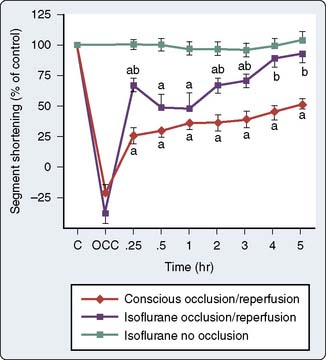
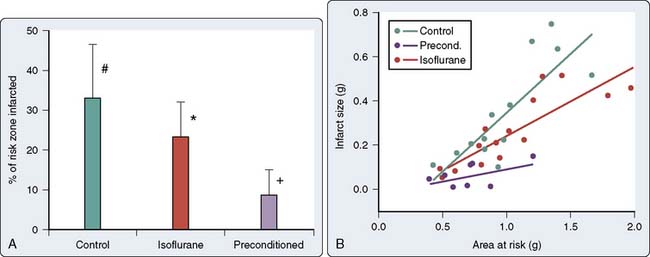
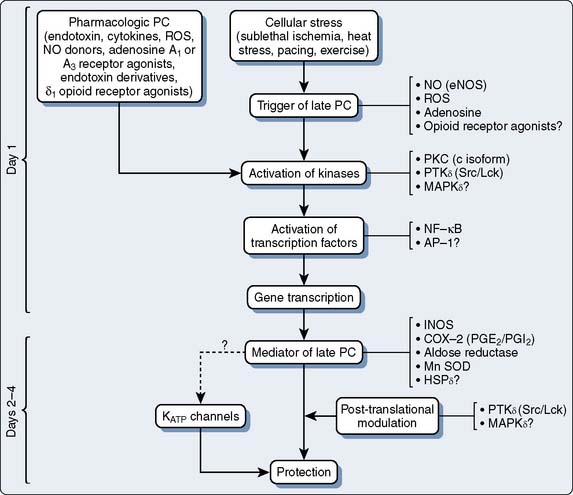
The phenomenon of and the mechanisms underlying IPC are the focus of extensive investigation. IPC has the following characteristics: (1) results in two periods (termed windows) of protection—the first (termed early or classic) occurs at 1 to 3 hours, and the second (termed late or delayed) occurs 24 to 96 hours after the preconditioning stimulus; (2) occurs also in noncardiac tissue, such as brain and kidney; (3) is ubiquitous across species; (4) is most pronounced in larger species with lower metabolism and slower heart rates (HRs); (5) seems to be important clinically because angina within the 24-hour period preceding an AMI is associated with an improved outcome (Figure 9-9)90; and (6) is mediated by multiple endogenous signaling pathways91 (Figure 9-10).92 As might be predicted from the time frame of delayed IPC, it is mediated, at least in part, by transcriptional and posttranslational mechanisms91 (Figure 9-11). Finally, and of the utmost importance, preconditioning can be triggered by events other than ischemia (cellular stress of various forms, pharmacologic agonists, anesthetic agents; see Figure 9-11).91 Moreover, the benefits of IPC are not necessarily confined to and may not include limitation of infarct size, and depend on the specific trigger for IPC, the species under study, and classic versus delayed IPC. For example, rapid pacing affords protection against arrhythmias but not against infarct evolution. In contrast, cytokine-induced IPC limits infarct size but has no effect on arrhythmias.91 Different triggers of IPC modulating different end points suggest that, although there are fundamental mechanisms common to various triggers of IPC, there also exist mechanistic differences across triggers. Thus, APC may not be identical to IPC mechanistically.
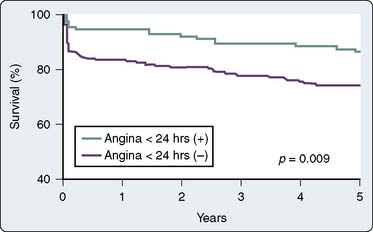
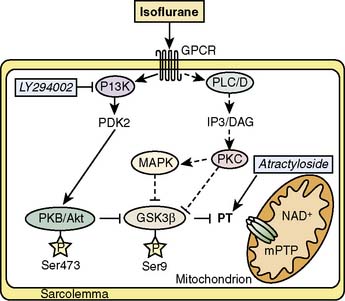
Figure 9-10 Investigated signaling pathways (full lines).
(From Feng J, Lucchinetti E, Ahuja P, et al: Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3β, Anesthesiology 103:987–995, 2005.)
Anesthetic Agents: Pre- and Post-Conditioning
This is an area of intense investigation as reflected by two issues of Anesthesiology being devoted predominantly to the subject.93,94 After the initial description of APC,1–3 subsequent investigations have indicated that volatile agents can elicit delayed (late), as well as classic (early), preconditioning.95,96 Moreover, APC is dose dependent,97–99 exhibits synergy with ischemia in affording protection,100,101 and perhaps not surprisingly, in view of differential uptake and distribution of volatile agents, has been demonstrated to require different time intervals between exposure and the maintenance of a subsequent benefit that is agent dependent42 (see Chapters 6 and 7).
The contributions of both SL and mitochondrial K+ATP channels in IPC have been extensively investigated, and it is now widely agreed that mitochondrial K+ATP channels play a critical role in this process. Volatile agents that exhibit APC activate mitochondrial K+ATP channels, and this effect is blocked by specific mitochondrial K+ATP channel antagonists. However, the precise relative contributions of SL versus mitochondrial K+ATP channel activation to APC remain to be elucidated (Figure 9-12).42 The original descriptions of APC indicated that volatile agents can trigger preconditioning without concurrent ischemia during the “triggering” period1–3 (see Figure 9-8). However, studies of mitochondrial activation (via mitochondrial K+ATP channels) indicate that volatile agents on their own do not activate mitochondria but do potentiate the effects of direct mitochondrial K+ATP channel openers99 (Figure 9-13). These apparent inconsistencies are likely explained by the presence of multiple parallel and redundant pathways activated during APC (and IPC)96 (see Figure 9-12). For example, it is now well established that the adenosine A-1 and δ1 opioid G-coupled receptors can trigger IPC. Moreover, pharmacologic blockade of these receptors attenuates the positive effects of volatile agents.98,102 Protein kinase C (PKC) and the nuclear signaling pathway, mitogen-activated protein kinase (MAPK), are important signaling pathways in preconditioning, and volatile agents have been shown to modulate at least PKC translocation.103 Oxidant stress is a central feature of reperfusion and, depending on the specific moiety, the enzymatic source and, most importantly, the oxidant stress load may trigger preconditioning on the one hand or mediate reperfusion injury on the other. Both indirect and direct evidence indicate that volatile agents can increase oxidant stress to levels that trigger preconditioning.104–106
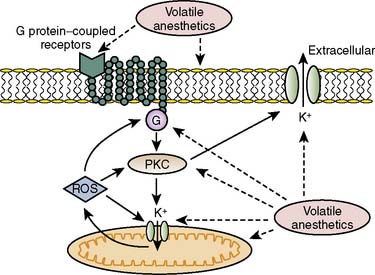
Activation of eNOS also has been shown to play a role, as has depolarization of the mitochondrial internal membrane.107 This may prevent the opening of the mitochondrial permeability transition pore (MPTP) and inhibit Na+-H+ exchange, attenuating Ca+2 overload and cell edema.107 Inhibition of mitochondrial permeability by APC has been suggested to decrease myocyte death, and PKC also has been thought to play a role in IPC-induced delay of MPTP opening.108,109 For the first time, a recent study demonstrates that isoflurane has been shown to activate PKC-dependent signaling pathways resulting in the delay of MPTP opening,110 suggesting a possible mechanism for isoflurane in APC. With respect to postconditioning, Ge et al111 demonstrated that NO may, in fact, act as both a trigger and a mediator for isoflurane-induced cardiac protection in mouse hearts. This implies that an eNOS-dependent mechanism prevents the opening of the MPT pore, although other pathways including glycogen synthase kinase-3β also have been implicated (Figure 9-14).92,111
It is clear that mitochondrial activation attenuates ischemia-induced oxidant stress, favorably modulates mitochondrial energetics, decreases cytochrome c egress into the cytoplasm, and attenuates mitochondrial and cytoplasmic Ca++ overload. Mitochondrial cytochrome c release is one of the important mechanisms underlying caspase activation, and thus the apoptotic process112 (Figure 9-15). Whether by Ca++-mediated or by apoptotic mechanisms, or both, volatile agents clearly attenuate cell death in models of APC101 (Figure 9-16). Although the mechanisms underlying mitochondrial activation have been aggressively studied, they remain incompletely understood. Finally, these salutary effects of volatile agents seem to have a clinical correlate101 (Figure 9-17).
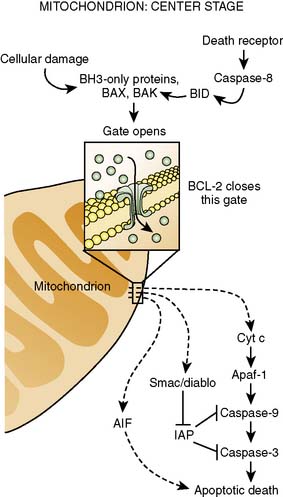
Figure 9-15 Center stage in apoptosis.
(Reprinted from Finkel E: The mitochondrion: is it central to apoptosis? Science 292(5517):624–626, 2001. Illustration: C. Slayden, Copyright 2001 AAAS.)
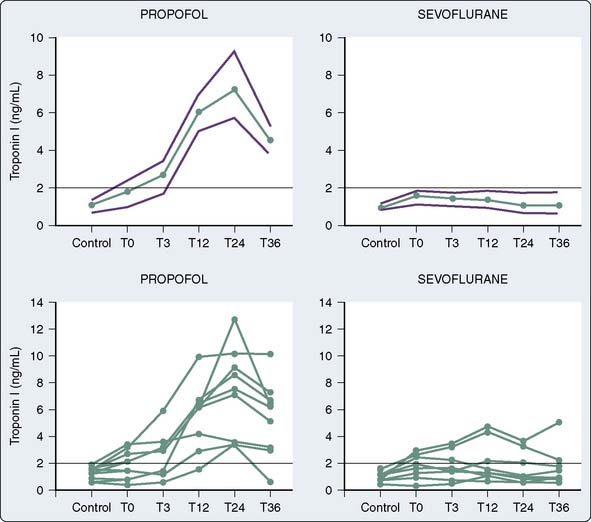
The use of volatile anesthetics also can alter outcomes after cardiac surgery. A meta-analysis by Landoni et al113 demonstrated a significant reduction in postoperative myocardial infarction after cardiac surgery, as well as significant advantages with respect to postoperative cardiac troponin release, inotrope requirements, time to extubation, intensive care unit stay, hospital stay, and survival. Furthermore, another meta-analysis by Bignami et al114 demonstrated that the use of volatile anesthetics may, in fact, have a beneficial role with respect to mortality after cardiac surgery. The duration of the volatile anesthetic exposure seemed to have some impact—the longer the exposure, the greater the effect. De Hert et al115 demonstrated the cardioprotective effects of volatile anesthetics if used throughout the surgical procedure rather than only before and after cardiopulmonary bypass (CPB).
Intravenous induction agents
Unlike the inhalation anesthetic agents that augment IPC, there is no good evidence that the intravenous hypnotic agents demonstrate these protective effects. There is, however, emerging evidence that propofol, the mainstay of induction agents, may enhance antioxidant activity in the heart and thus may prevent lipid peroxidation after ischemia/reperfusion, offering a potential protective effect on the heart.116

Acute cardiac effects
Myocardial Contractility
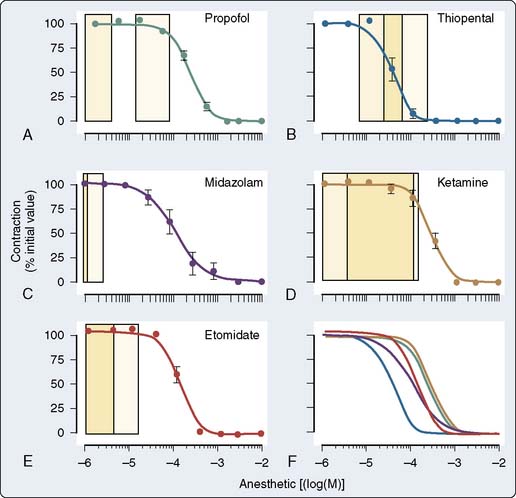
The effect of the agents may be species dependent, thus further confounding the literature regarding mechanism. For instance, van Klarenbosch et al117 demonstrated that in contrast with rat, propofol directly depresses myocardial contractility in isolated muscle preparations from guinea pig, probably by decreasing trans-SL Ca++ influx. However, there was little influence of propofol on Ca++ handling by the SR or on the contractile proteins in rat. In one of the few human studies using isolated atrial muscle tissue (Figure 9-18), no inhibition of myocardial contractility was found in the clinical concentration ranges of propofol, midazolam, and etomidate. In contrast, thiopental showed strong negative inotropic properties, whereas ketamine showed slight negative inotropic properties (Figure 9-19). Thus, negative inotropic effects may explain, in part, the cardiovascular depression on induction of anesthesia with thiopental but not with propofol, midazolam, and etomidate. Improvement of hemodynamics after induction of anesthesia with ketamine cannot, therefore, be explained by intrinsic cardiac stimulation but is a function of sympathoexcitation.118
The effect of drugs such as propofol also may be affected by the underlying myocardial pathology.119,120 For instance, Sprung et al120 determined the direct effects of propofol on the contractility of human nonfailing atrial and failing atrial and ventricular muscles obtained from the failing human hearts of transplant patients or from nonfailing hearts of patients undergoing coronary artery bypass graft surgery (CABG). They concluded that propofol exerts a direct negative inotropic effect in nonfailing and failing human myocardium, but only at concentrations larger than typical clinical concentrations. Negative inotropic effects are reversible with β-adrenergic stimulation, suggesting that propofol does not alter the contractile reserve but may shift the dose responsiveness to adrenergic stimulation. The negative inotropic effect of propofol is at least partially mediated by decreased Ca++ uptake into the SR; however, the net effect of propofol on contractility is insignificant at clinical concentrations because of a simultaneous increase in the sensitivity of the myofilaments to activator Ca++.120
Molecular Mechanisms: Adrenergic Signaling, Ca++ Influx, and Ca++ Sensitivity
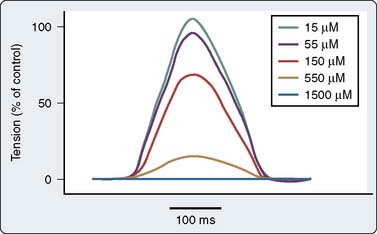
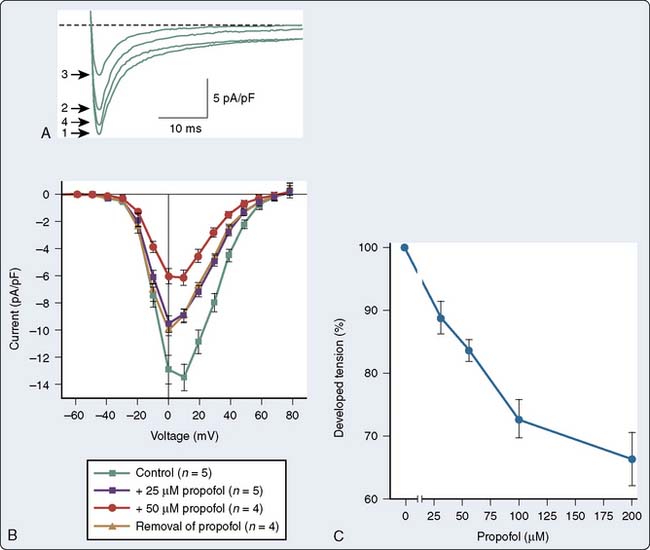
There are a number of suggested molecular mechanisms by which a drug such as propofol may alter cardiac contractility. Propofol may inhibit cardiac L-type calcium current by interacting with the dihydropyridine-binding site121 (Figure 9-20A), with resultant alteration in developed tension (see Figure 9-20B). Furthermore, as mentioned earlier, propofol may alter adrenergic signaling in cardiac myocytes. Experiments in membranes and cardiac preparations isolated from rat heart demonstrate that relatively high concentrations of propofol (25 to 200 μmol/L) are required to antagonize β-adrenoceptor binding and tissue responsiveness.122 Kurokawa et al123 observed that clinically relevant concentrations of propofol attenuated β-adrenergic signal transduction in cardiac myocytes via inhibition of cyclic adenosine monophosphate (cAMP) production (Figure 9-21). The inhibitory site of action of propofol appears to be upstream of adenylyl cyclase and involves activation of PKCα.
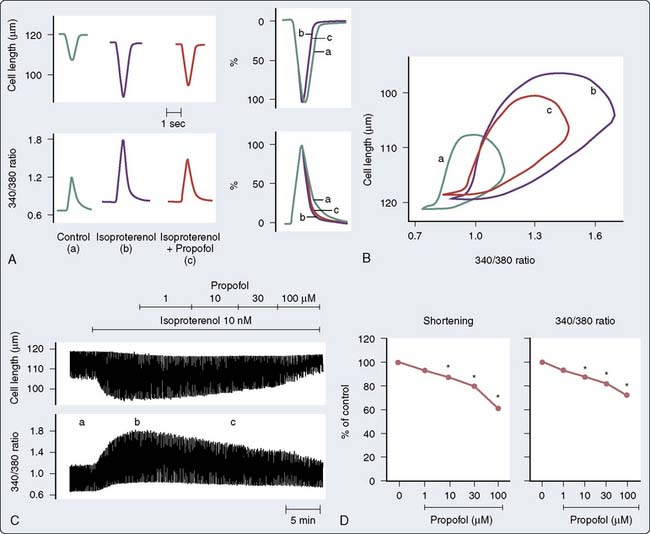
Although propofol may decrease contractile response to adrenergic stimulation, there is emerging evidence that it may enhance myofilament sensitivity to Ca++. Propofol caused a leftward shift in the extracellular Ca++-shortening relationship, suggesting that propofol increases the sensitivity of myofibrillar actomyosin ATPase to Ca++ (i.e., increases myofilament Ca++ sensitivity; Figure 9-22). This is mediated, at least in part, by increasing pH via PKCc-dependent activation of Na+-H+ exchange124 or by a PKC-dependent pathway involving the phosphorylation of MLC2.124
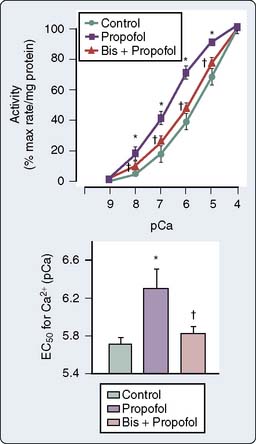
Integrated Cardiovascular Responses
The use of combined conductance-manometric catheters, which allows the simultaneous measurement of pressure and volume in the ventricle, has enabled the precise determination of the effects of anesthetic agents on integrated cardiovascular responses. These parameters include load-independent measures of contractility (slope of the end-systolic pressure-volume relation [ESPVR], Ees), as well as indices of ventricular-vascular coupling (ratio of arterial elastance to ventricular elastance [Ea/Ees]; see Chapters 5 and 14). In a study, the effects of propofol and pentobarbital on integrated cardiovascular function were assessed in pigs both at baseline and after an acute increase in ventricular afterload.125 At baseline, Ees was lower during pentobarbital versus propofol anesthesia, suggesting a greater negative inotropic effect of barbiturates versus propofol (Figure 9-23). On the other hand, the responses to ventricular afterload induced by aortic banding were maintained in the pentobarbital-anesthetized animals, whereas the responses were markedly attenuated in the propofol-anesthetized pigs, suggesting an attenuation of the baroreflex responses with propofol. Furthermore, a decrease in arterial pressure with propofol is consistent with this drug acting as a vasodilator.
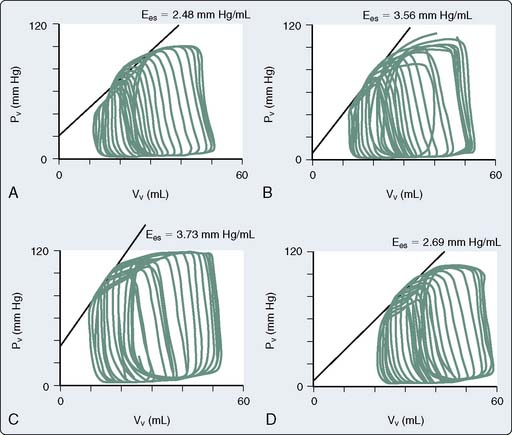
One of the questions with regard to intravenous induction agents is: What represents a clinically relevant dose? As would be predicted, with regard to the myocardial depressant effects, the coronary concentrations of propofol have been shown to be the major contributor to the cardiac depression caused by propofol, but were a less significant contributor to the hypotension caused by this drug.126
The effects of propofol on cardiac contractility are manifest not only on ventricular but also atrial function.127 Propofol depresses contractile function of left atrial myocardium and reduces the active left atrial contribution to left ventricular filling in vivo. Compensatory decreases in chamber stiffness, however, contribute to relative maintenance of left atrial reservoir function during the administration of propofol.
Oxidative Stress
Oxidative stress remains an important pathophysiologic mechanism for cellular injury in critically ill patients and represents an imbalance between the production of these radicals and the enzymatic defense system that removes them (Box 9-3). This has potential therapeutic implications because these agents are used routinely for sedation in the intensive care unit, in which disease processes associated with increased oxidative stress are treated.
BOX 9-3 Intravenous Induction Agents
Animal data also suggest that propofol decreases postischemic myocardial mechanical dysfunction, infarct size, and histologic evidence of injury128–132 (Figure 9-24). On further analysis, it becomes clear that propofol has a chemical structure similar to that of phenol-based free radical scavengers, such as vitamin E, and may therefore act as a free radical scavenger.133,134 Studies by Tsuchiya et al135,136 demonstrated in vitro the potential for both propofol and midazolam to act as free radical scavengers at near-therapeutic doses. Propofol also impairs the activity of neutrophils by inhibiting the oxidative burst and supports a potential role for propofol in modulating injury at the critical phase of reperfusion by reducing free radicals, Ca++ influx, and neutrophil activity.137 Furthermore, microsomes prepared from animals anesthetized with propofol demonstrate a significantly increased resistance to lipid peroxidation.138 The evidence does not support propofol as an APC-inducing agent because protection was observed when the heart was treated with propofol solely during reperfusion rather than before or during the ischemic insult,131 although the addition of glibenclamide, a K+ATP channel blocker, does not abolish the protection afforded by propofol.132 There is little evidence for other intravenous induction agents having protective effects on the heart. In fact, ketamine may block IPC139–141 by deactivating SL K+ATP channels.142
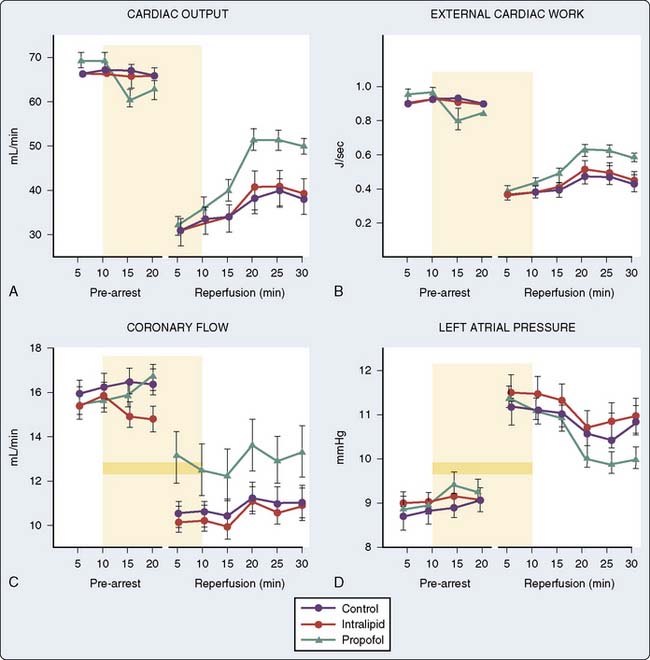

Vasculature
Systemic Vasoregulation
It is now well established that propofol decreases SVR in humans. This was demonstrated in a patient with an artificial heart in whom the CO remained fixed.143 As discussed later, the effect is predominantly mediated by alterations in sympathetic tone; however, in isolated arteries, propofol decreases vascular tone and agonist-induced contraction. The mechanism by which propofol mediates these effects has been attributed, in part, to inhibition of Ca++ influx through voltage- or receptor-gated Ca++ channels, as well as inhibition of Ca++ release from intracellular Ca++ stores regulated by the ryanodine receptor.144–147 Much of the experimental data obtained in isolated studies have focused on conduit arteries (rat aorta); however, some properties of the resistance arteries differ from the rat bioassay. Modulation of vasoconstriction by vascular smooth muscle may be mediated by (1) an alteration in endothelium-independent vasodilation145 or (2) an alteration in the sensitivity of the myofilaments to Ca++, primarily mediated by the rho activation, and thereby the activation of Rho kinase. In an elegant study examining the effects of propofol on resistance arteries, Imura et al148 examined the effect of propofol on simultaneous measurements of force and in mesenteric resistance arteries. The authors concluded that propofol attenuates norepinephrine-induced contraction through an inhibition of Ca++ release, as well as Ca++ influx through L-type Ca++ channels, thus partially explaining the effects of propofol on vascular adrenergic signaling (Figure 9-25). Propofol also may modulate vascular tone by interfering with other signaling pathways involved in vasoregulation, such as ET-1.148,149 In addition, propofol also may attenuate the myogenic tone/response in pressure-flow autoregulation.150
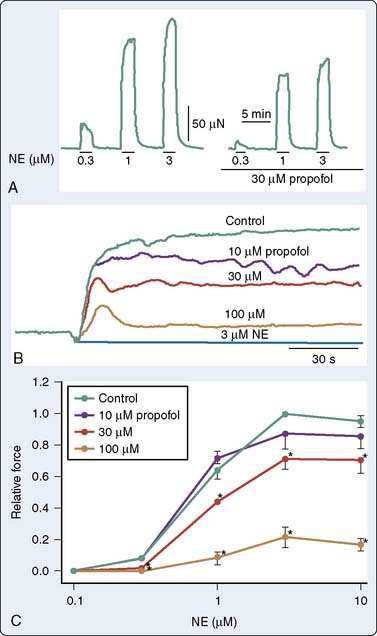
Pulmonary Vasoregulation
The effects of induction agents on pulmonary vasoregulation may have important implications for the management of patients whose primary pathologies involve the pulmonary circulation when they present for cardiothoracic surgery (primary pulmonary hypertension for lung transplantation and chronic thromboembolic disease for pulmonary endarterectomy). In addition, the effects may be of importance in patients with right ventricular failure.151 Furthermore, the effect of these agents in modulating hypoxic pulmonary vasoconstriction may have an effect on intraoperative A-a (alveolar-arterial) gradients, particularly during one-lung ventilation. Murray and colleagues152,153 have systematically studied the effects of anesthetic agents on pulmonary vasoregulation. Specifically, they have demonstrated that propofol attenuates endothelium-dependent vasodilatation152 via a mechanism that involves NO and endothelium-dependent hyperpolarizing factor.153 With regard to the vascular smooth muscle, the effect appears somewhat different in the pulmonary circulation. Rather than attenuate vasoconstriction and thereby decrease tone, propofol appears to increase the sensitivity of the contractile myofilaments to Ca++,154 and thereby potentiates the effect of catecholamines on pulmonary artery smooth muscle cells155 (Figure 9-26).
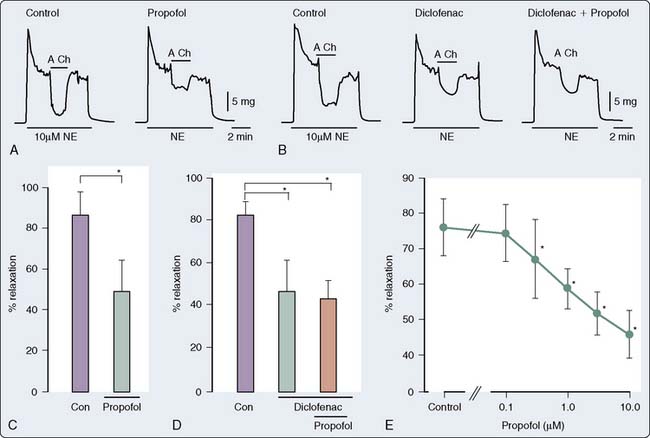
Endothelial Function
Propofol modulates the function of the endothelium, thus altering the underlying tone of the vessels. The data as to the direction and mechanism underlying this effect are widely divergent and are dependent on the vascular bed, species, and experimental conditions. Early studies suggested that propofol-mediated vasodilatation was a function of stimulation of NO and vasodilator prostanoids from the endothelium.140 Other studies have demonstrated an inhibitory effect of propofol and ketamine, but not of midazolam, on endothelium-dependent relaxation.156 Moreover, the suppressive effect of ketamine on endothelium-dependent relaxation appears to be mediated by suppression of NO formation, whereas that of propofol may be mediated, at least partly, by suppression of NO function. In a rabbit mesenteric resistance artery preparation, Yamashita et al157 demonstrated that propofol inhibits prostacyclin-mediated endothelium-dependent vasodilatation by inhibition of vascular hyperpolarization (Figure 9-27). This inhibition of hyperpolarization, and thereby inhibition of relaxation, appears to be mediated by the blockade of ATP-sensitive K+ channels. The clinical implications of these findings are unclear; however, it may be predicted, based on the data, that the effects of propofol on the vasculature may be significantly different in patients in whom endothelial dysfunction, such as hypertension and atherosclerosis, is a significant factor.
With respect to vasorelaxation, Gursoy et al158 described the dose-dependent vasorelaxation produced by intravenous anesthetic agents on human radial artery grafts. Thiopental and ketamine were found to be more potent than etomidate and propofol with respect to relaxant properties. These observations may have implications for the perioperative management of coronary artery graft vasospasm.158
Sympathetic and Parasympathetic Nervous System
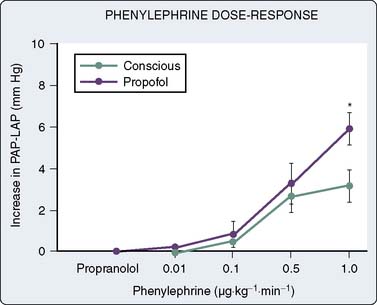
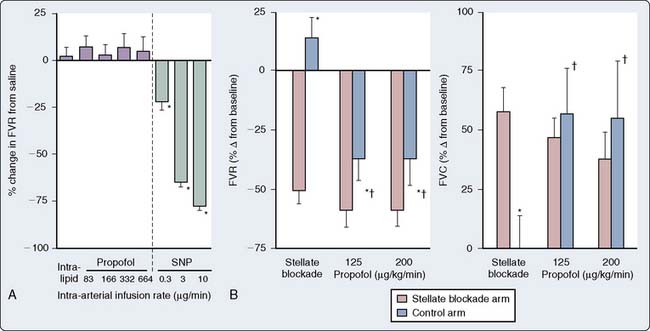
In human in vivo studies, it appears that the cumulative effects of propofol on peripheral arterial venous capacitance are mediated primarily, but not solely, by its effects on the sympathetic nervous system (SNS). In a well-designed study in which the effect of local infusion of propofol into the brachial artery was compared with systemic intravenous administration with induction of anesthesia, it was demonstrated that direct brachial infusion had little effect on resistance and capacitance, whereas the effect of intravenous administration was similar to the effect observed with sympathectomy induced by stellate ganglion block (Figure 9-28). Thus, the peripheral vascular effects of propofol appear to be primarily mediated by reduced sympathetic vasoconstrictor nerve activity.159 In another elegant study, Sellgren et al160 measured the effect of propofol on the SNS using percutaneous recordings of muscle sympathetic nerve activity. The authors demonstrated a profound decrease in SNS activity with a reciprocal increase in blood flow (measured by laser Doppler) with propofol-induced anesthesia. Furthermore, sympathetic baroreflex sensitivities were also depressed by propofol, highlighting the profound effect of this agent on central sympathetic modulation of integrated cardiovascular function. The precise locations of the central modulation by propofol have been investigated by Yang et al,161,162 who demonstrated that propofol principally inhibits the vasomotor mechanism in the dorsomedial and ventrolateral medulla to effect its hypotensive actions. The implications of these effects are significant in that this sympathoinhibition is amplified in patients in whom SNS activity is high. Thus, caution is indicated in the administration of these agents to patients with shock, CHF, or other pathophysiologic circumstances in which the SNS is paramount in maintaining arterial and venous tone.
Remodeling and Cell Proliferation
Emerging literature suggests that the effects of intravenous anesthetic agents may not be solely mediated by direct modulation of vascular tone through alteration of the contractile state of the vascular smooth muscle. New studies suggest that intravenous anesthetic agents may alter vascular smooth muscle proliferation, as well as modulate pathways that are important in angiogenesis. For example, Shiga et al163 demonstrated the effect of ketamine (but not propofol) on inhibiting vascular smooth muscle proliferation through a PKC-dependent pathway. On the other hand, midazolam, but not ketamine, was demonstrated to release vascular endothelial growth factor, a growth factor important in angiogenesis and cellular proliferation from vascular smooth muscle cells.164 The clinical importance of these findings remains unstudied and unclear. However, they serve to emphasize that the administration of these agents (and others) by the anesthesiologist may have effects that last well after the drug has disappeared from the core.
Individual agents

Thiopental
General Characteristics
Thiopental has survived the test of time as an intravenous anesthetic drug (Box 9-4). Since Lundy introduced it in 1934, thiopental has become the most widely used induction agent because of the rapid hypnotic effect (one arm-to-brain circulation time), highly predictable effect, lack of vascular irritation, and general overall safety.165 The induction dose of thiopental is less for older than for younger healthy patients.166 Pharmacokinetic analyses167–169 confirm the findings from the early classic studies of Brodie and Mark170 relating the awakening from thiopental to rapid redistribution. Thiopental has a distribution half-life (t1/2α) of 2.5 to 8.5 minutes, and the total body clearance varies, according to sampling times and techniques, from 0.15 to 0.26 L/kg/hr.159–161,163,164 The elimination half-life (t1/2β) varies from 5 to 12 hours.168,169,171,172 Barbiturates173 and drugs such as propofol174 have increased volumes of distribution (Vd) when used during CPB. Young (< 13 years old) patients seem to have a greater total clearance and quicker plasma thiopental clearance than do adults, which theoretically might result in earlier awakening, especially after multiple doses.175 Because of the affinity of fat for this drug, its relatively large Vd, and its low hepatic clearance, thiopental can accumulate in tissues, especially if given in large doses over a prolonged period.
BOX 9-4 Intravenous Anesthetics
Cardiovascular Effects
The hemodynamic changes produced by thiopental have been studied in healthy patients166,176–182 and in patients with cardiac disease (Table 9-3).183–188 The principal effect is a decrease in contractility,180,181,189 which results from reduced availability of calcium to the myofibrils.190 There is also an increase in HR.166,177,179–181,186–189 The cardiac index (CI) is unchanged177,185–188 or reduced,176,178,181 and the mean aortic pressure (MAP) is maintained177,187,188,191 or slightly reduced.178,179,185–187 In the dose range studied, no relation between plasma thiopental and hemodynamic effect has been found.166 Early hemodynamic investigations demonstrated that thiopental (100 to 400 mg) significantly decreased CO (24%) and systemic BP (10%), presumably by reducing venous return, because of an increase in venous capacitance.178,192
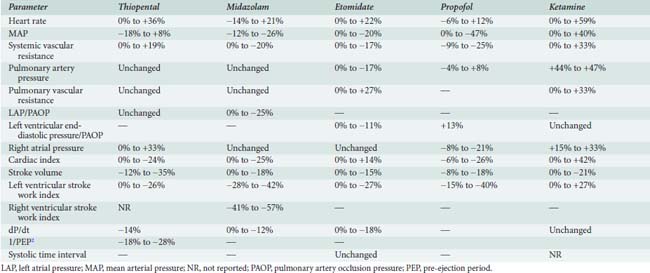
Mechanisms for the decrease in CO include (1) direct negative inotropic action; (2) decreased ventricular filling, resulting from increased venous capacitance; and (3) transiently decreased sympathetic outflow from the central nervous system (CNS). The increase in HR (10% to 36%) that accompanies thiopental administration probably results from the baroreceptor-mediated sympathetic reflex stimulation of the heart. Thiopental produces dose-related negative inotropic effects that appear to result from a decrease in calcium influx into the cells with a resultant diminished amount of calcium at sarcolemma sites.193,194 Patients who had compensated heart disease and received 4 mg/kg thiopental had a greater (18%) BP decline than did other patients without heart disease. The increase in HR (11% to 36%) encountered in patients with CAD, anesthetized with thiopental (1 to 4 mg/kg), is potentially deleterious because of the obligatory increase in myocardial oxygen consumption (Mvo2).
Despite the well-known potential for cardiovascular depression when thiopental is given rapidly in large doses, this drug has minimal hemodynamic effects in healthy patients and in those who have heart disease when it is given slowly or by infusion. Significant reductions in cardiovascular parameters occur in patients who have impaired ventricular function. When thiopental is given to patients with hypovolemia, there is a significant reduction in CO (69%), as well as a large decrease in BP, which indicate that patients without adequate compensatory mechanisms may have serious hemodynamic depression with a thiopental induction.195 Clearly, thiopental produces greater changes in BP and HR than does midazolam when used for induction of American Society of Anesthesiologists (ASA) Class III and IV patients.
Uses in Cardiac Anesthesia
A possible additional use for thiopental infusion is cerebral protection during CPB in patients undergoing selected cardiac operations.196 However, the cerebral protective effect of thiopental during CPB has been challenged by Zaidan et al,197 who demonstrated no differences in outcome between thiopental and control patients undergoing hypothermic CPB for CABG. Although the administration of a barbiturate during CPB may result in myocardial depression, necessitating additional inotropic support, Ito et al’s198 study suggested beneficial effects of a thiopental infusion during CPB in maintaining peripheral perfusion, which allowed more uniform warming, decreased base deficit, and decreased requirements for postoperative pressor support.

Midazolam
General Characteristics
Midazolam (Versed; Figure 9-29), a water-soluble benzodiazepine, was synthesized in the United States in 1975, in contrast with most new anesthetic drugs, which are synthesized and first tested in European countries. It is unique among benzodiazepines because of its rapid onset, short duration of action, and relatively rapid plasma clearance.199 Although controversial, the dose for induction of general anesthesia is between 0.05 and 0.2 mg/kg, and depends on the premedication and speed of injection.200–203
The pharmacokinetic variables of midazolam reveal that it is cleared significantly more rapidly than are diazepam and lorazepam. The rapid redistribution of midazolam, as well as high liver clearance, accounts for its relatively short hypnotic and hemodynamic effects. The t1/2β is about 2 hours, which is at least 10-fold less than for diazepam.204–208
Cardiovascular Effects
The hemodynamic effects of midazolam have been investigated in healthy subjects,179,209,210 in ASA Class III patients,211 and in patients who have ischemic212–219 and valvular211 heart disease (VHD). Table 9-3 summarizes the hemodynamic changes after induction of anesthesia with midazolam. In general, there are only small hemodynamic changes after the intravenous administration of midazolam (0.2 mg/kg) in premedicated patients who have coronary artery disease (CAD).215,217 Changes of potential importance include a decrease in MAP of 20% (from 102 to 81 mm Hg) and an increase in HR of 15% (from 55 to 64 beats/min).215 The CI is maintained.215,217 Filling pressures are either unchanged or decreased in patients who have normal ventricular function,215,217 but are significantly decreased in patients who have an increased pulmonary capillary wedge pressure (PCWP; ≥ 18 mm Hg).216 There seems to be little effect of differences in doses on hemodynamics: 0.2,217 0.25,214 and 0.3 mg/kg219 all produce similar effects. Sedation with midazolam (0.05 mg/kg) in patients undergoing cardiac catheterization is devoid of any hemodynamic effect.212 Marty et al showed that induction with 0.2 mg/kg produced a 24% reduction in CBF and a 26% reduction in Mvo2 in patients with CAD.213 As in patients with ischemic heart disease, the induction of anesthesia in patients with VHD is associated with minimal changes in CI, HR, and MAP after midazolam.212 When intubation follows anesthesia induction with midazolam, significant increases in HR and BP occur, because midazolam is not an analgesic.179,217–219 Adjuvant analgesic drugs are required to block the response to noxious stimuli.
There is a suggestion that midazolam affects the capacitance vessels more than diazepam does, at least during CPB, when decreases in venous reservoir volume of the pump are greater with midazolam than with diazepam. In addition, diazepam decreases SVR more than midazolam during CPB.220
Midazolam (0.15 mg/kg) and ketamine (1.5 mg/kg) have proved to be a safe and useful combination for a rapid-sequence induction for emergency surgery.182 This combination was superior to thiopental alone because it caused less cardiovascular depression, more amnesia, and less postoperative somnolence. If midazolam is given to patients who have received fentanyl, significant hypotension may occur, as seen with diazepam and fentanyl.221 However, midazolam routinely is combined with fentanyl for induction and maintenance of general anesthesia during cardiac surgery without adverse hemodynamic sequelae.222,223

Etomidate
General Characteristics
Etomidate is a carboxylated imidazole derivative synthesized by Godefroi et al224 in 1965. In animal experiments, it was found that etomidate has a safety margin four times greater than the safety margin for thiopental.225 The recommended induction dose of 0.3 mg/kg has pronounced hypnotic effects. Etomidate is moderately lipid soluble226 and has a rapid onset (10 to 12 seconds) and a brief duration of action.227–229 It is hydrolyzed primarily in the liver and in the blood as well.230
The administration of etomidate in a buffered solution was accompanied by a significant incidence of a burning sensation (about 40%) and myoclonic movements (about 40% to 50%).229 The myoclonic movements were not associated with an epileptiform pattern on the electroencephalogram.226
Reports have shown that etomidate infusion and single injections directly suppress adrenocortical function, which, in turn, interferes with the normal stress response.231–233 Blockade of 11-β-hydroxylation mediated by the imidazole radical of etomidate results in decreased biosynthesis of cortisol and aldosterone.234 The clinical significance of etomidate-induced adrenal suppression remains undetermined.
Cardiovascular Effects
In comparative studies with other anesthetic drugs, etomidate is usually described as the drug that changes hemodynamic variables the least.235–241 Studies in noncardiac patients237,240,242 and those who have heart disease187,235,238,239,243,244 document the remarkable hemodynamic stability after administration of etomidate (see Table 9-3). In healthy subjects or patients who have compensated ischemic heart disease, HR, pulmonary artery pressure (PAP), PCWP, left ventricular end-diastolic pressure, right atrial pressure (RAP), CI, SVR, pulmonary vascular resistance (PVR), dP/dt, and systolic time intervals (STIs) are not significantly changed after doses of 0.15 to 0.30 mg/kg.187,235,236,242,244 In comparison with other anesthetics, etomidate produces the least change in the balance of myocardial oxygen demand and supply. Systemic BP remains unchanged in most series235,236,238–241 but may be decreased 10% to 19%239,243,245 in patients who have VHD. Ammon et al235 found modest dose-related decreases in MAP and left ventricular stroke work index (LVSWI). Doses of 0.3, 0.45, and 0.6 mg/kg caused greater decreases in mean BP and LVSWI but had no effect on HR, PAP, PCWP, CVP, CI, SV, or SVR. Therefore, although there was a small dose-related effect on hemodynamics, the remarkable fact is that there is hemodynamic stability despite the twofold increase in dose. Dose-related changes have been demonstrated in the dog and were attributed to three possible causes: (1) decreased CNS sympathetic stimulation, (2) autoregulation secondary to decreased regional O2 consumption, and (3) decreased SV secondary to reduced venous return.246
A dose-dependent direct negative inotropic effect of etomidate was demonstrated in dogs, although at equianesthetic doses, it was half as pronounced as that of thiopental.193 To determine the effects of anesthetic agents on myocardial contractility is difficult in vivo because of concomitant changes in HR, preload, and afterload. In contrast, the effects may be evaluated in vitro, although this does not accurately represent what is occurring in the myocardium as a whole. Riou et al247 studied the effect of etomidate on intrinsic myocardial contractility, using left ventricular papillary muscle and an electromagnetic lever system. Etomidate induced a slightly positive inotropic effect, as manifested by increased maximum shortening velocity. It appears, however, that propylene glycol, the solvent in which etomidate is available, may result in SR dysfunction with a slight negative inotropic effect in some clinical conditions.
Etomidate (0.3 mg/kg intravenously), used to induce general anesthesia in patients with AMI undergoing percutaneous coronary angioplasty, did not alter HR, MAP, and rate-pressure product, demonstrating the remarkable hemodynamic stability of this agent.248 However, the presence of VHD may influence the hemodynamic responses to etomidate. Whereas most patients can maintain their BP, patients with both aortic and mitral VHD had significant decreases of 17% to 19% in systolic and diastolic BP,239,243 as well as decreases of 11% and 17% in PAP and PCWP, respectively.243 CI in patients who had VHD and received 0.3 mg/kg either remained unchanged186,239 or decreased 13%.243 There was no difference in response to etomidate between patients who had aortic valve disease and those who had mitral valve disease.243
Wauquier et al249 anticipated the widespread clinical use of etomidate in their investigation, in which they compared the effects of etomidate and thiopental in hypovolemic dogs. In a hemorrhagic shock model, dogs were bled to an MAP of 40 to 45 mm Hg and then given either etomidate (1 mg/kg) or thiopental (10 mg/kg). There was significantly more hemodynamic depression in the thiopental group and increased survival in the etomidate group. Whether this is true in humans is not known, but certainly clinical evidence suggests that etomidate is useful in patients with hypovolemia.

Ketamine
General Characteristics
Ketamine is a phencyclidine derivative whose anesthetic actions differ so markedly from barbiturates and other CNS depressants that Corssen and Domino250 labeled its effect dissociative anesthesia. The properties of ketamine and its use in anesthesia have been completely reviewed.191 Although ketamine produces rapid hypnosis and profound analgesia, respiratory and cardiovascular functions are not depressed as much as with most other induction agents. Disturbing psychotomimetic activity (described as vivid dreams, hallucinations, or emergence phenomena) remains a problem. Interestingly, preliminary data suggest the possibility of a protective effect against postoperative cognitive dysfunction in patients undergoing cardiac surgery, and a recent study conducted by Hudetz et al suggests that ketamine attenuates the postoperative delirium noted in cardiac surgical patients after surgery.251
Cardiovascular Effects
The hemodynamic effects of ketamine have been examined in noncardiac patients,239,252–257 critically ill patients,258 geriatric patients,259 and patients who have a variety of heart diseases.254,260–270 Table 9-3 contains the range of hemodynamic responses to ketamine. One unique feature of ketamine is stimulation of the CVS. The most prominent hemodynamic changes are significant increases in HR, CI, SVR, PAP, and systemic artery pressure. These circulatory changes cause an increase in Mvo2 with an apparently appropriate increase in CBF.260,269 Although global increases in Mvo2 occur, there is some evidence that the increased work may be borne primarily by the right ventricle, because of significantly greater increases in PVR than SVR271; however, both ventricles certainly demonstrate increased work. The hemodynamic changes observed with ketamine are not dose related in the relatively small dose ranges examined; there is no significant difference between changes after administration of 0.5 and 1.5 mg/kg intravenously.272 It is interesting that a second dose of ketamine produces hemodynamic effects opposite to those of the first.268 Thus, the cardiovascular stimulation seen after ketamine induction of anesthesia (2 mg/kg) in a patient who has VHD is not observed with the second administration, which is accompanied instead by decreases in the BP, PCWP, and CI.
Ketamine produces similar hemodynamic changes in healthy patients and in patients who have ischemic heart disease.256 In patients who have increased PAP (as with mitral valvular disease), ketamine appears to cause a more pronounced increase in PVR than in SVR. The presence of marked tachycardia after administration of ketamine and pancuronium also can complicate the induction of anesthesia in patients who have CAD or VHD with atrial fibrillation.273 In a recent study of patients undergoing elective coronary artery bypass grafting, the use of S-(+)-ketamine did not lead to increased cardiac troponin T levels after surgery when used in combination with propofol.274
The mechanism responsible for ketamine’s stimulation of the circulatory system remains enigmatic. The direct effects of ketamine on the myocardium remain controversial. Riou et al275 demonstrated that ketamine has a dual opposing action on the myocardium: (1) a positive inotropic effect, probably secondary to increased Ca++ influx; and (2) an impairment of SR function. This impairment is significant only at supratherapeutic ketamine concentrations or in cardiomyopathic myocardium,276 and overcomes this positive inotropic effect only under these circumstances. Myocardial depression has been demonstrated in isolated rabbit hearts,277 intact dogs, and isolated dog heart preparations.278,279 Although the precise site of cardiovascular stimulation is still unknown, Ivankovich and colleagues280 showed that small doses of ketamine injected directly into the CNS result in immediate hemodynamic stimulation. Ketamine also causes the sympathoneuronal release of norepinephrine, which can be measured in venous blood.260,272,281 Blockade of this effect is possible with barbiturates, benzodiazepines,272,280–282 and droperidol.261 Animal work supports the hypothesis that the primary hemodynamic effect of ketamine is central and not peripheral.283–290 The role of ketamine’s cocaine-like neuronal inhibition of norepinephrine reuptake has yet to be defined in its overall influence on the CVS.291,292 It also is unknown whether ketamine exerts the same effect centrally, preventing reuptake of norepinephrine in the brain.
One of the most common and successful approaches to blocking ketamine-induced hypertension and tachycardia is the prior administration of benzodiazepines. Diazepam, flunitrazepam, and midazolam all successfully attenuate the hemodynamic effects of ketamine.182,262,265,282,293–295 For example, in a study involving 16 patients with VHD, ketamine (2 mg/kg) did not produce significant hemodynamic changes when preceded by diazepam (0.4 mg/kg).262 Indeed, HR, MAP, and rate-pressure product were unchanged; however, there was a slight but significant decrease in CI.262 Hatano and associates294 reported their experience with 200 cardiac surgical patients in whom the administration of diazepam (0.3 to 0.5 mg/kg) and then a ketamine infusion (0.7 mg/kg/hr) provided a stable hemodynamic course during induction, intubation, and incision. In fact, the combination of diazepam and ketamine rivals the high-dose fentanyl technique with regard to hemodynamic stability. No patient had hallucinations, although 2% had dreams and 1% had recall of events in the operating room.294 Levanen et al296 suggested that premedication with 2.5 μg/kg intramuscular dexmedetomidine before ketamine-based anesthesia is as effective as midazolam in blocking the hemodynamic effects of ketamine and was more effective in reducing adverse CNS effects. Because of the propensity of dexmedetomidine to produce bradycardia, concomitant use of an anticholinergic agent was suggested.
Studies have demonstrated the safety and efficacy of induction with ketamine (2 mg/kg) in hemodynamically unstable patients who required emergency operations.258,297,298 Most of these patients were hypovolemic because of trauma or massive hemorrhage. Ketamine induction was accompanied in the majority of patients by the maintenance of BP and, presumably, of CO as well.258,297 In patients who have an accumulation of pericardial fluid, with or without constrictive pericarditis, induction with ketamine (2 mg/kg) maintains CI and increases BP, SVR, and RAP.299,300 The HR in this group of patients was unchanged by ketamine, probably because cardiac tamponade already produced a compensatory tachycardia.

Propofol
Propofol is the most recent intravenous anesthetic to be introduced into clinical practice. It is an alkylphenol with hypnotic properties. The pharmacokinetics of propofol have been evaluated by numerous investigators and have been described by both two-compartment301,302 and three-compartment303,304 models.
Cardiovascular Effects
The hemodynamic effects of propofol have been investigated in healthy ASA Class I and II patients,305 elderly patients,306,307 patients with CAD and good left ventricular function,308,309 and patients with impaired left ventricular function (see Table 9-3). Numerous studies also have compared the cardiovascular effects of propofol with the most commonly used induction drugs, including the thiobarbiturates and etomidate.310–314 Comparison of the findings between investigators is, however, difficult because of the variations in the anesthetic techniques used, doses of drugs administered, and techniques used for measuring data. It is clear that with propofol, systolic arterial pressure declines 15% to 40% after intravenous induction with 2 mg/kg and maintenance infusion with 100 μg/kg/min. Similar changes are seen in both diastolic arterial pressure and MAP.
The effect of propofol on HR is variable. The majority of studies have demonstrated significant reductions in SVR (9% to 30%), CI, SV, and LVSWI after propofol. Although controversial, the evidence points to a dose-dependent decrease in myocardial contractility. Bendel et al,315 in a double-blind, randomized, controlled trial, compared the effects of propofol and etomidate in patients undergoing elective aortic valve surgery for aortic stenosis. They concluded that propofol was twice as likely to result in hypotension during the induction of patients with severe aortic stenosis compared with etomidate.315 Finally, it has been suggested that propofol increases triglyceride levels.316–318 Oztekin et al319 conducted a study evaluating the effect of propofol and midazolam on lipid levels early in the postoperative period in patients undergoing CABG surgery. Serum triglyceride levels and very-low-density lipoproteins were significantly increased in patients receiving intraoperative propofol infusions 4 hours after surgery. It remains to be seen what effect this increase has on clinical outcomes and postoperative course.319
Uses
In a study of the cerebral physiologic effects of propofol during CPB, Newman et al320 demonstrated that when given during nonpulsatile CPB, propofol produced statistically significant reductions in cerebral blood flow and cerebral metabolic rate in a coupled manner without adverse effects on cerebral arteriovenous oxygen content difference or jugular bulb venous saturation. The coupled reductions in cerebral blood flow and cerebral metabolic rate suggest the potential for propofol to reduce cerebral exposure to emboli during CPB.
The effect of propofol on hypoxic pulmonary vasoconstriction was minimal in thoracic surgical patients undergoing one-lung ventilation. In comparison with isoflurane, maintenance of anesthesia with propofol resulted in lower CI and right ventricular ejection fraction, but avoided the threefold increase in shunt fraction observed with isoflurane on commencement of one-lung ventilation.321

Dexmedetomidine
Cardiovascular Effects
The cardiovascular effects of dexmedetomidine are dose related. Furst and Weinger322 demonstrated that an increase in systemic BP was associated with the pretreatment of rats with high-dose dexmedetomidine, and that dexmedetomidine had little effect on arterial blood gases in spontaneously ventilating rats, consistent with minimal respiratory depression. Canine studies with medetomidine in doses of 30 μg/kg intravenously or 80 μg/kg intramuscularly showed decreases in HR and CO with increased SVR after drug administration. The intravenous administration of 5 to 10 μg/kg medetomidine to anesthetized, autonomically blocked dogs suggested that the decline in CO was not mediated by decreased contractility but rather by the effects of increased vascular resistance and decreased HR. At high doses, the increase in SVR is most likely due to the activation of peripheral postsynaptic α2-adrenoreceptors in vascular smooth muscle.
In human studies, ASA Class I women who received low-dose premedication with 0.5 μg/kg dexmedetomidine demonstrated modest decreases in BP and HR. Intramuscular dexmedetomidine in a dose of 2.5 μg/kg administered 45 minutes before induction of a ketamine/N2O/oxygen anesthetic resulted in effective attenuation of the cardiostimulatory effects of ketamine but also resulted in increased intraoperative and postoperative bradycardia.296 The use of perioperative intravenous infusions of low-dose dexmedetomidine in vascular patients at risk for CAD produced lower preoperative HR and systolic BP, and less postoperative tachycardia, but also resulted in a greater intraoperative requirement for pharmacologic intervention to support BP and HR. The precise cause of this effect is unknown, but it is possibly due to the attenuation of sympathetic outflow from the CNS.
Uses
Clinical studies have suggested that α2-adrenergic agonists can safely reduce anesthetic requirements and improve hemodynamic stability. These agents may enhance sedation and analgesia without producing respiratory depression or prolonging the recovery period. Barletta et al323 compared postoperative opioid requirements in patients undergoing cardiac surgery who received intraoperative propofol versus dexmedetomidine. Although dexmedetomidine resulted in lower opioid use compared with propofol, it did not translate to a shorter duration of mechanical ventilation but did result in significantly greater sedation-related costs.323 Levanen et al296 suggested that dexmedetomidine may be an effective alternative to benzodiazepines in attenuating the postanesthetic delirium effects of ketamine. Because α2-adrenergic agonists potentially inhibit opiate-induced rigidity, it is clear that they may be of use as adjuvants with high-dose opioid anesthetics for cardiac surgery. In contrast with other anesthetic adjuvants, such as the benzodiazepines, the α2-adrenoreceptor agonist dexmedetomidine does not further compromise cardiovascular or respiratory status in the presence of high-dose opioids. The use of dexmedetomidine as a sedative adjunct in the management of patients after surgery in the intensive care unit is becoming increasingly popular.324 In general, the concept whereby the type and amount of agent used intraoperatively can influence the postoperative course, specifically the neuropsychologic events, is emerging as an important paradigm.325 The management of patients after surgery is covered extensively in Chapters 33, 35, 36, and 37. In summary, pharmacologic evidence suggests dexmedetomidine may be useful as an adjuvant in cardiac anesthesia.
Opioids in cardiac anesthesia

Terminology and Classification
Modification of the morphine molecule, although simultaneously preserving the basic five-ring structure, will result in semisynthetic compounds that also exhibit analgesic effects (e.g., hydromorphone, heroin). Progressive removal of these rings results in synthetic opioids. As long as the common core or T shape is retained stereochemically and is shared by these synthetic derivatives, opioid properties will be retained. The piperidine ring forms the crossbar and the hydroxyl phenyl group forms the vertical axis of the T shape (see Figure 9-29).

Opioid Receptors
The postulation of opioid receptor existence was stated in the pioneering work of Beckett and Casy,326 which, in 1965, allowed Portoghese327 to theorize the existence of separate opioid receptors by correlating analgesic activity to the chemical structure of many opioid compounds (Box 9-5). The idea of multiple opioid receptors is an accepted concept, and a number of subtypes for each class of opioid receptors have been identified. Through biochemical and pharmacologic methods, the μ, δ, and κ receptors have been characterized.328–330 Pharmacologically, it is well-known that δ-opioid receptors consist of two subtypes: δ1 and δ2.331–334 Table 9-4 lists the opioid receptors and their associated agonists and antagonists.
BOX 9-5. Opioids
TABLE 9-4 List of Opioid-Receptor Agonist and Antagonist Drugs for the μ-, κ-, and δ-Opioid Receptors
| Opioid Receptor | Agonist | Antagonist |
|---|---|---|
| μ | Morphine | Naloxone |
| Fentanyl | Naltrexone | |
| Levorphanol | β-Funaltrexamine* | |
| Methadone | Nalbuphine† | |
| κ | U-50,488H | Nor-BNI |
| EKC | MR-2266 | |
| U-62,066E (spiradoline) | Naloxone | |
| U-69,593 | ||
| δ | (−)TAN-67 | Naltrindole |
| SIOM‡ | BNTX | |
| SNC 80§ | Naltriben |
BNTX, 7-benzylidene naltrexone; Nor-BNI, nor-binaltorphimine.
* β-FNA is an irreversible opioid receptor antagonist.
† Nalbuphine is an opioid receptor antagonist with partial agonist properties.
§ (+)-4-[((αR)-α(2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-methoxy-benzyl]-N, N-diethylbenzamide chloride.
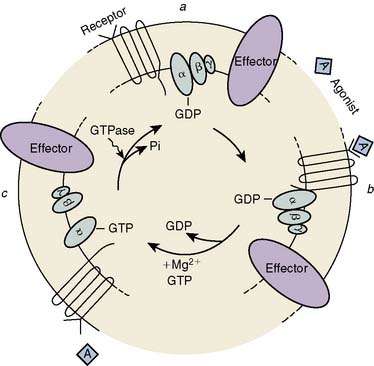
The μ-, κ-, and δ-opioid receptors have all been cloned, and experimental data indicate that they belong to the family of G-protein–coupled receptors.335–338 Three steps have been identified in the opioid-induced transmembrane signaling process: (1) recognition by the receptor of the extracellular opioid agonist, (2) signal transduction mediated by G protein, and (3) altered production of an intracellular second messenger (Figure 9-30). The opioid receptors preferentially couple to a pertussis toxin–sensitive G protein (Gi/Go) to influence one or more of three second-messenger pathways: cytoplasmic-free Ca++ [Ca++]i, the phosphatidylinositol-[Ca++]i system, and the cyclic nucleotide cAMP. The actions of opioids are primarily inhibitory. Opioids close N-type, voltage-operated Ca++ channels and open Ca++-dependent inwardly rectifying K+ channels. This results in hyperpolarization and a reduction in neuronal excitability.339 κ receptors may only act on Ca++ channels.340 Evidence has been reported that P/Q-type Ca++ channels may be inhibited by μ-, but not δ-, receptor opioids.341 These effects may be mediated via a direct coupling between G protein and the ion channels or indirectly via changes in intracellular Ca++. Both Ca++ fluxes and the consequences of these fluxes, such as calmodulin activation, are altered by opioids. Ca++ ions are essential in nociception, and Ca++-channel blockers potentiate opioid analgesia342,343 and reduce fentanyl requirements during cardiac surgery.344 Opioid-induced inhibition of adenylyl cyclase, and the resultant decrease in the concentration of cAMP, may be responsible for modulation of neurotransmitter release (e.g., substance P).
Opioids also have excitatory effects that involve both disinhibition of interneurons and direct excitation of neurons themselves. Nanomolar concentrations, acting via G proteins, stimulate adenylyl cyclase activity in certain neurons.345 Another important stimulatory effect is a transient increase in cytoplasmic-free Ca++ secondary to Ca++ influx via L-type Ca++ channel opening, as well as mobilization of Ca++ from inositol triphosphate–sensitive intracellular stores.346 Mu-agonists also may stimulate Ca++ entry into neurons via G-protein–coupled activation of phospholipase C to increase inositol-1,4,5-triphosphate formation, secondary to Ca++ influx via L-type Ca++ channel opening.346–348
Opioid receptors involved in regulating the CVS have been localized centrally to the cardiovascular and respiratory centers of the hypothalamus and brainstem, and peripherally to cardiac myocytes, blood vessels, nerve terminals, and the adrenal medulla. It generally is accepted that opioid receptors are differentially distributed between atria and ventricles. The highest specific receptor density for binding of κ-agonists is in the right atrium and least in the left ventricle.349,350 As with the κ-opioid receptor, the distribution of the δ-opioid receptor favors atrial tissue and the right side of the heart more than the left.351 The data confirming the presence of the μ-opioid receptor subtype in the heart are less conclusive, and most conclude that this receptor subtype is not present in cardiac tissue.349,350,352,353 Whether these differences in receptor location are important cannot be determined, as it has not been shown whether these receptors are differentially located in cardiac muscle or cardiac nerves, or are expressed on immunomodulatory cells within the heart. Based on these studies, the ability of opioid agonists to produce cardioprotection is most likely the effect of δ- and κ-receptor stimulation, and the current studies appear to support a role for the δ-opioid receptor as the primary receptor responsible for IPC.

Endogenous Opioids in the Heart
Myocardial cells are capable of the synthesis, storage, and release of peptides, such as opioid receptor peptides.354 Opioid-receptor peptides may be either secreted from nerves that innervate the heart or produced in myocardial tissue. Regardless of the manner of production, these peptides are devoid of activity until they undergo enzymatic proteolysis of the precursor by convertases into one (or more) active peptide products. These large stores of endogenous opioid peptide (EOP) precursors, which reside in the myocardial tissue, include proenkephalin, proendorphin, and prodynorphin.
The EOP system consists of the peptides endorphin, dynorphin, and enkephalin, and their associated μ-, δ-, and κ-opioid receptors (Table 9-5). These opioid peptides and receptors are widely distributed in the body, and all have complex actions. In the heart, κ- and δ-receptor agonists have been shown to inhibit ventricular contractility without altering atrial function.351 A study of the actions of peptides on both electrical and mechanical properties of the isolated rat heart has shown that δ- and κ-opioid receptor agonists can directly depress cardiac function.355 The rate of vagal firing has been shown to be regulated by opioid peptides.356 Vagal bradycardia is inhibited by the administration of the intrinsic cardiac opioid heptapeptide MERF (met-enkephalin-arg-phe), presumably by the activation of δ-opioid receptors on prejunctional cardiac vagal nerves or parasympathetic ganglia, reducing acetylcholine release.357 Therefore, EOPs can mediate direct and indirect actions in various regions of the heart. Thus, in addition to complex differences in the general tissue distribution of opioid receptors, cardiac opioid peptide function is complicated by the fact that receptor expression is modulated by both physiologic states and disease.
TABLE 9-5 Endogenous Opioid Peptide Precursors and Some of Their Active Opioid Peptide Products Involved in Cardiovascular Regulation and Function
| Precursor | Opioid Peptide | Receptor* |
|---|---|---|
| Proenkephalin† | [Met]Enkephalin | δ > μ >>> κ |
| [Leu]Enkephalin | ||
| POMC‡ | β-Endorphin | μ ≈︀ δ >> δ |
| Prodynorphin§ | Dynorphin A | κ >>> μ > δ |
| Dynorphin A(1–8) | κ >> μ > δ | |
| Dynorphin B | κ >>> μ > δ | |
| Pronociception | Nociception | ORL1 |
ORL1, opoid receptor-like-1; POMC, proopiomelanocortin.
* The efficacy of the opioid peptide to its receptor is only qualitatively described in this table for the μ-, κ-, and δ-opioid receptor subtypes found in the central nervous system.534
† An additional opioid peptide product from this precursor that exhibits relevant cardiovascular actions in MERF (Zhang et al, 1996).
‡ An additional POMC peptide cleavage product is adrenocorticotropic hormone, which is converted to α-melanocyte–stimulating hormone-related peptides in cardiac tissue (Millington et al, 1999).
§ Additional active peptides include leumorphin, α-neoendorphin, and β-neoendorphin.534
A number of investigators have demonstrated that certain opioid peptides are released during stressful situations into the peripheral circulation.358–361 These peptides can result in the modulation of the ANS.362 In the heart, opioid peptides (leu- and met-enkephalins) have been shown to increase with age,363–365 as well as disease.366–369 EOPs are involved in the modulation of hypertension and other cardiovascular conditions such as CHF and appear to be involved in arrhythmogenesis.370 Myocardial ischemia/reperfusion has been shown to induce synthesis and release of opioid peptides.369,371–376 In fact, several studies in humans have demonstrated that levels of circulating β-endorphins are greater in patients with acute myocardial ischemia or those undergoing angioplasty.371,374,375,377 Although κ-receptor agonists have no effect on cardiovascular indices in healthy humans,378 activation of δ and κ receptors during CHF decreases myocardial mechanical performance and alters regional blood flow distribution.372 The mechanism for the negative inotropic effects is thought to be an increase of intracellular free Ca++ by increasing the mobilization of calcium from intracellular stores subsequent to increased production of inositol-1,4,5-triphosphate. The increase in Ca++ may manifest in cardiac arrhythmias, whereas depletion of Ca++ from intracellular stores is responsible for a reduction in contractility.372,379
In patients with acute heart failure, the concentrations of EOPs are increased,380 whereas the concentrations are decreased in patients with chronic heart failure. This has been interpreted as exhaustion of the opioid system.381 Many studies suggest that EOPs mediate depression of myocardial function in CHF states.359,380,382,383 Clinically, increased levels of EOPS (β-endorphin, met-enkephalin, and dynorphin) have been found in CHF patients, and these may correlate with severity. Naloxone administration to these CHF patients increased BP and HR, suggesting a homeostatic regulatory role for EOPs in CHF. However, not all clinical studies suggest that inhibition of opioid peptides is of benefit to patients with acute and chronic heart failure.381 Oldroyd et al384 found that plasma levels of β-endorphins were normal in patients with acute and chronic heart failure, and did not correlate with the severity of heart failure observed in their study.384 They also found that naloxone administration did not alter cardiopulmonary exercise in these patients, and suggest that EOP inhibition is not likely to have any therapeutic potential.

Cardiac Effects of Opioids
At clinically relevant doses, the cardiovascular actions of narcotic analgesics are limited. The role that endogenously or exogenously administered opioids play in the regulation of the CVS is difficult to interpret because the physiologic effects they impart depend on pharmacologic variables such as dose, site, and route of administration, as well as receptor specificity and species. The actions opioids exhibit are mediated both by opioid receptors located centrally in specific areas of the brain and nuclei that regulate the control of cardiovascular function and peripherally by tissue-associated opioid receptors. The opioids, in general, exhibit a variety of complex pharmacologic actions on the CVS359 (Figure 9-31).
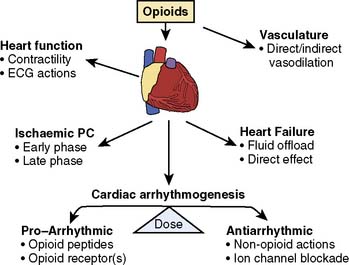
Most of the hemodynamic effects of opioids in humans can be related to their influence on the sympathetic outflow from the CNS. Current evidence shows that sympathetic overactivity favors the genesis of life-threatening ventricular tachyarrhythmias, and its control has protective effects during acute myocardial ischemia.385 Moreover, an imbalance of the ANS, characterized by increased sympathetic activity and reduced vagal activity, results in myocardial electrical instability and promotes the occurrence of ischemic events. The pharmacologic modulation of the sympathetic activity by centrally or peripherally acting drugs elicits cardioprotective effects. Opioid-receptor agonists such as fentanyl are known to exhibit significant central sympathoinhibitory effects.386
Fentanyl and sufentanil enhance the calcium current that occurs during the plateau phase (phase 2) of the cardiac action potential and depress the outward potassium current responsible for terminal repolarization,387 resulting in a significant prolongation of the duration of the action potential. Blair et al388 suggested that the cardiac electrophysiologic effects of fentanyl and sufentanil represented a direct membrane effect resembling that produced by Class III antiarrhythmic drugs. In patients, large doses of opioids prolong the QT interval of the electrocardiogram.387 This may explain the reported antiarrhythmic properties of opioids, particularly in the presence of myocardial ischemia.284 Fentanyl, 60 μg/kg, and sufentanil, 10 μg/kg, significantly increased the ventricular fibrillation threshold in dogs after coronary artery occlusion.389
Isolated heart or heart-muscle studies have demonstrated dose-related inotropic effects for morphine, meperidine, fentanyl, and alfentanil.390–393 However, these effects occurred at concentrations one hundred to several thousand times those found clinically. In canine hearts, the direct intracoronary injection of fentanyl in concentrations up to 240 ng/mL produced no changes in myocardial mechanical functions.394
Morphine produced dose-related decreases in the contractility of atria obtained from nonfailing and failing human hearts, but the concentration–response curve was significantly shifted to the right in preparations from failing hearts395 (Figure 9-32). The negative inotropic effects induced by morphine in both failing and nonfailing preparations were not antagonized by naloxone, indicating that opioid receptors do not play a part in this cardiac effect of morphine. One explanation could be an interaction with β-adrenoceptors, unrelated to the binding of opioids to opioid receptors. Opioids inhibit β-adrenoceptor–sensitive adenylyl cyclase.396
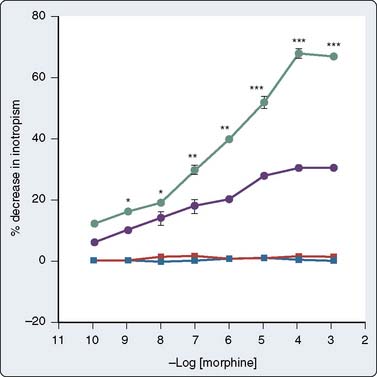
Hypotension can occur after even small doses of morphine and is primarily related to decreases in SVR. The most important mechanism responsible for these changes is probably histamine release. The amount of histamine release is reduced by slow administration (< 10 mg/min). Pretreatment with a histamine H1 or H2 antagonist does not block these reactions, but they are significantly attenuated by combined H1 and H2 antagonist pretreatment.397 Neither morphine nor fentanyl, in clinically relevant concentrations, blocks α-adrenergic receptors in isolated vascular tissue studies.398,399 Opioids also may have a direct action on vascular smooth muscle, independent of histamine release. In the isolated hind limbs of dogs anesthetized with halothane, high doses of alfentanil (500 μg/kg), fentanyl (50 μg/kg), and sufentanil (6 μg/kg) caused significant decreases in SVR of 48%, 48%, and 44%, respectively. Neither pretreatment with naloxone nor denervation changed the responses, and it was concluded that the three opioids produced vasodilation by a direct action on vascular smooth muscle.400 Although fentanyl-induced relaxation in the rat aorta may be mediated by α-adrenergic receptors, this effect occurs only at concentrations several hundred times greater than those encountered clinically.401
The effects of κ-agonists on BP have been examined402 and been shown to be dose, species, and route dependent.402,403 U-50,488H, for example, displays a markedly different cardiovascular profile when injected intravenously as compared with injected directly into the CNS. In anesthetized dogs, κ-agonists produce dose-related decreases in BP, HR, peak systolic pressure, and cardiac contractility when administered intravenously. The cardiovascular responses to both opioid agonists were abolished by previous administration of naloxone.404 In addition to causing a reduction in BP, κ-opioid agonist dose-dependently reduced the HR in anesthetized rats.402 This is suggestive of an effect on either the reflex mechanisms that regulate the HR during hypotension or direct action on the electrical or mechanical properties responsible for normal cardiac contractility. κ-Agonists cause slight depressant action on the HR and BP at low doses that is not inhibited by naloxone, suggesting a lack of involvement of opioid receptors and a possible direct effect on cardiac muscle.403 In anesthetized rats, the cardiovascular responses to κ-opioid agonists in the presence of opioid antagonists were not changed.403,405,406 It was concluded from these studies and others407 that because opioid receptor antagonists did not block these responses, they were not mediated by opioid receptors.
Opioid-receptor agonists and antagonists have been shown to block ion channels that constitute the genesis of the action potential in neurons. Voltage-gated Na+ channels are responsible for the initiation of membrane depolarization and the conduction of action potentials in electrically excitable cells, resulting in contraction of the heart or transmission of electrical impulses in nerves. K+ channels are responsible for repolarization of the cell membrane and cessation of action potentials in excitable cells. Morphine and naloxone have been shown to block the propagation of action potentials in many nerve and cardiac muscle preparations by directly inhibiting voltage-dependent Na+ and K+ currents. In addition to the opioid-receptor–independent–mediated actions of U-50,488H and related κ-agonists on BP and HR (i.e., actions not blocked by opioid-receptor antagonist), these drugs produce changes in the electrocardiogram indicative of a drug interaction with cardiac ion channels.402,408 Table 9-6 summarizes the action of several κ-opioid agonists and their actions on BP, HR, and the PR, QRS, RSh, and Q-T intervals on rat electrocardiograms. The electrocardiographic changes, including PR interval prolongation, QRS widening, and increased amplitude of the RSh, are indicative of cardiac Na+ channel blockade. The widening of the Q-T interval, an index of cardiac repolarization, is suggestive of K+-channel blockade.
TABLE 9-6 D25 Drug Doses of Arylacetamide κ-Opioid Agonists in Intact Rats: Effects on Heart Rate, Blood Pressure, and Electrocardiogram Measures
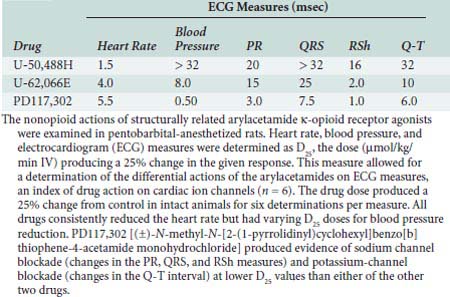
The results of studies with opioid drugs, such as sufentanil387 and morphine,409 corroborate the suggestion of interaction of κ-opioids with cardiac K+ channels. However, the opioid receptor–independent nature of the ion-channel blockade may not be isolated to Na+ and K+ channels and also may include L-type Ca+ channels found in cardiac muscle. The use of Ca++ fluorescent techniques for measurement of cardiac myocyte contractility suggested that the negative inotropic actions for κ-opioid agonists also may be because of inhibition of L-type Ca++ currents.410,411 While investigating the possible cardiotoxic effects produced by opioids in human poisoning, Wu et al412 found that opioids, like meperidine and dextropropoxyphene, exert negative inotropic actions in cardiac muscle by blockade of Ca++ currents in myocytes in the presence of naloxone.

Ischemic Preconditioning
Myocardial IPC is a phenomenon that occurs in cardiac muscle in which brief periods of ischemia (usually < 5 minutes) render the muscle tolerant to tissue damage that occurs during a subsequent period of ischemia, after an interlude of perfusion86 (Figure 9-33). Such a phenomenon has been shown to occur in many species and is known to be mediated by a well-defined intracellular cascade.413 The intercellular mediator or IPC-induction trigger appears to be diverse and has been shown to involve opioids and other substances. Thus, the nature of IPC may not be consistent between the species examined, despite a common end result.
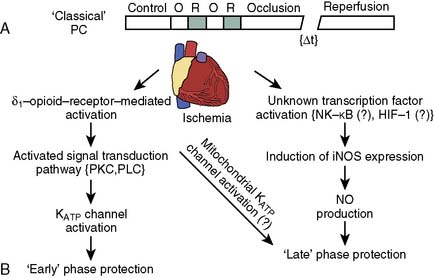
In vivo studies show that IPC can reduce the size of an infarct resulting from prolonged ischemia.414 There also is a reduction in damage to myocardial intracellular structure, a decrease in the dysfunction of the cardiac contractile machinery, and a direct reduction in arrhythmias associated with IPC.414,415 The ability of IPC to limit myocardial damage occurs chronologically in two distinct phases. The first (or early) phase provides a window of protection to the heart muscle that occurs soon after IPC and declines with time during the first 3 hours of reperfusion. The second, late (or delayed) phase of IPC provides a second window of protection to heart muscle that emerges after 24 hours of reperfusion and may last up to 72 hours416–418 (see Chapters 3, 6, and 18).
Importance of Opioid Receptors in Early Preconditioning
The involvement of opioids in IPC resulted from the recognition of their value at increasing survival time and tissue preservation before surgical transplantation, and their possible role in enhancing tolerance to hypoxia.419,420 The first evidence to demonstrate a role for opioids in early IPC was published by Schultz et al421 in the intact blood-perfused rat heart. These investigators demonstrated that the nonselective opioid receptor antagonist naloxone completely antagonized the ability of IPC to reduce infarct size whether administered before the IPC stimulus or after the IPC stimulus just before the index ischemia. These results suggested that endogenous opioids serve as both a trigger and end-effector of IPC in rat hearts (Figure 9-34). Chien and Van Winkle422 found similar results in the rabbit heart with the use of the active enantiomer, (−)naloxone. Furthermore, Schulz et al423 determined the role of endogenous opioids in mediating IPC and myocardial hibernation in pig hearts, and observed that naloxone blocked IPC but not the effects of short-term hibernation. These data clearly suggest that an opioid receptor is mediating the effect of endogenous opioids to elicit IPC in the rat, rabbit, and pig.
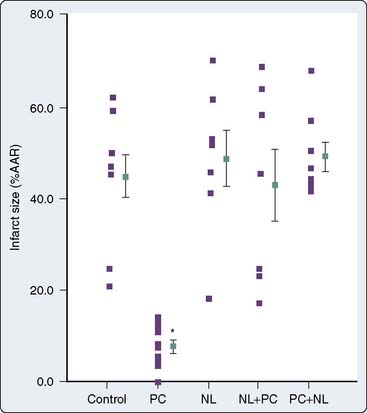
Takashi et al424 performed a study in isolated adult rabbit cardiomyocytes to determine which EOPs are responsible for the cardioprotective effect observed during and after IPC. They found that metenkephalin, leuenkephalin, and met-enkephalin-ang-phe (MEAP) produced a reduction in the incidence of cell death, suggesting that the enkephalins are the most likely candidates that serve as triggers and distal effectors of IPC in the rabbit heart. Huang et al425,426 studied the role of δ-opioid-receptor activation in reducing myocardial infarct size in human and rat hearts. Their studies indicated that endogenous adrenergic-receptor activation and downstream signaling were important mediators in attenuating infarct size.425,426
Cardioprotective Effects of Exogenous Opioid Agonists
In 1996, Schultz and colleagues427 were the first to demonstrate that an opioid could attenuate ischemia/reperfusion damage in the heart. Morphine, at the dose of 300 μg/kg, was given before left anterior descending coronary artery occlusion for 30 minutes in rats in vivo. Infarct area/area at risk was diminished from 54% to 12% by this treatment. The infarct-reducing effect of morphine has been shown in hearts in situ, isolated hearts, and cardiomyocytes.424,428,429 Morphine also improved post-ischemic contractility.430 It is now well accepted that morphine provides protection against ischemia/reperfusion injury. Furthermore, Gross et al431 reported a significant reduction in infarct development in rats after administration of morphine or a selective δ-receptor ligand at reperfusion. The authors reported that the effects are mediated via a glycogen synthase kinase β and the phosphatidylinositol-3 kinase pathway.431 There is also some evidence to suggest that remifentanil, when added to a standard anesthetic regimen, may reduce myocardial damage after coronary artery bypass surgery (Figure 9-35).432
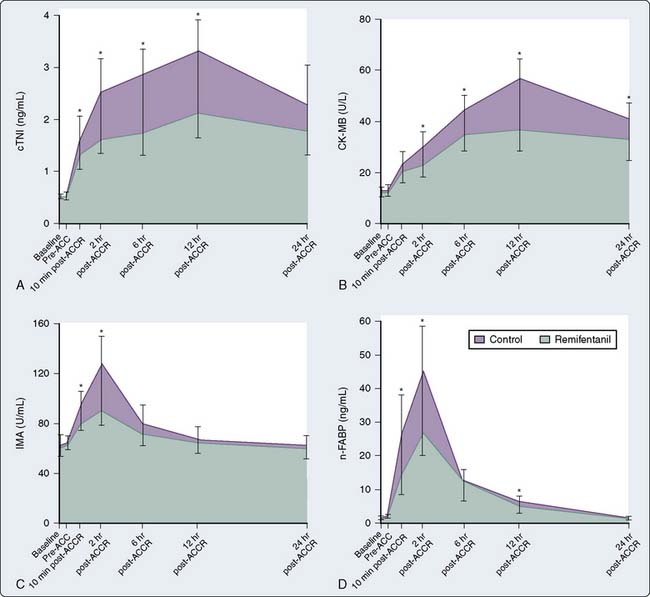
Fentanyl has been studied in a limited fashion and has had mixed results as far as its ability to protect the myocardium.430,433,434 This may be because of differences in species studied, fentanyl concentrations, or both. Pentazocine and buprenorphine improved postischemic contractility in rabbits in vitro.430 Overall, the effects of opioids other than morphine have not been sufficiently investigated to allow conclusions to be drawn.
Schultz et al435 also demonstrated that the effect of IPC to reduce infarct size was mediated by the δ1-receptor but not the δ2, μ, or κ receptor, because the effects of IPC were blunted by the selective δ1-receptor antagonist BNTX (7-benzylidenenaltrexone) but not the δ2-antagonist naltriben. They also showed that cardioprotection was not induced by the administration of the selective μ-agonist DAMGO (D-Ala2, N-Me-Phe, (4) glycerol5-enkephaline) and that IPC was not attenuated by the μ-antagonist B-FNA (beta-funaltrexamine).435 In that same study, they excluded the involvement of the κ receptor because a κ-selective antagonist could not reverse the effects of IPC to reduce infarct size. These data suggest that the δ1-opioid receptor appears to be the primary opioid receptor involved in IPC in the intact rat heart.
Although the exogenous activation of the δ-opioid receptor subtype by highly specific agonists before ischemia has been shown to reduce infarct size in a number of species, including rats,435 rabbits,436 and swine,437 the role of the κ-opioid receptors in preconditioning has been a subject of much controversy. Cao et al438,439 demonstrated that the cardioprotective effects caused by κ-opioid receptor stimulation were abolished with a calcium-activated potassium-channel (KCa) blocker. This is consistent with previous reports that the κ-receptor protective effects are mediated via a KCa-channel pathway as seen in IPC.438,439 It has also been reported that preischemic administration of selective κ-agonists will reduce infarct size and ischemia-induced arrhythmias in the isolated rat heart. Conversely, specific activation of the κ-opioid receptor before ischemia also has been shown to increase infarct size419 and arrhythmias440 and induce an “antipreconditioning”-like state in rats. It has been proposed that the κ-opioid receptor agonists exert a biphasic effect on the myocardium, producing proarrhythmic and antiarrhythmic effects in the rat.385 Therefore, it is unclear whether selective or nonselective activation of the κ-opioid-receptor subtype is beneficial during preconditioning, and although such conflicting information exists for the rat, the role of opioid receptor subtypes in IPC and pharmacologic preconditioning in other species is even more limited. Further studies are needed to address the role of the κ receptor in arrhythmias.
Signaling Pathways Involved in Opioid-Induced Cardioprotection
Opioid-induced cardioprotection and IPC appear to share a common pathway, in that the δ-opioid receptor and the mitochondrial K+ATP channel appear to be involved in the beneficial effects observed. Additional studies show that the cardioprotective effect of IPC and δ1-opioid-receptor activation were both mediated via a GI-protein–coupled receptor and may involve NO.435,441,442 G-protein receptors have an established role in the attenuation of ischemic-reperfusion injury mainly by the activation of κ and δ receptors.443,444 The mechanisms underlying this effect involve apoptotic pathways and restriction of internucleosomal DNA fragmentation.445 To further address potential signaling pathways involved in opioid-induced protection, it was observed that morphine produced a cardioprotective effect in isolated rabbit hearts that was blocked by pretreatment with a nonselective PKC inhibitor. Jang et al441 demonstrated the activation of δ-opioid receptors and their role in postconditioning. This effect occurs via modulation of the MPTP and signaling via a NO and PKG-mediated pathway (Figure 9-36). In summary, the beneficial effects were eliminated by a Gi-protein inhibitor, a PKC inhibitor,429,433,446,447 and a selective mitochondrial K+ATP channel blocker.428,433,446,448–451 Furthermore, studies also have demonstrated the role of iNOS as an upstream mediator of COX-2. In iNOS gene knockout mice, this morphine-mediated cardioprotection is attenuated and some reports suggest iNOS and COX-2 are required only during the mediation phase and not the trigger phase.452,453 Figure 9-37 is a schematic summary of the major pathways thought to be involved in acute opioid-induced cardioprotection.
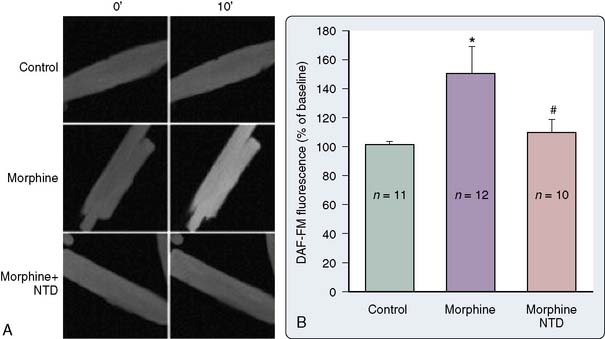
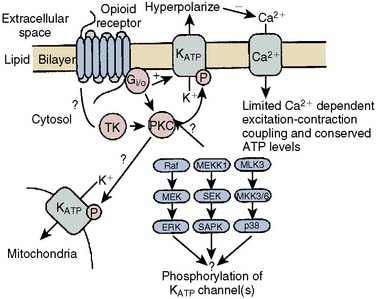
Role of Opioids in Delayed Preconditioning
It appears that opioid receptors are involved in delayed cardioprotection via activation of the δ- and κ-opioid receptors. Fryer et al449 demonstrated that TAN-67, a δ1-opioid agonist, also could induce cardioprotection during the “second window” of IPC. They found no protective effect to reduce infarct size 12 hours after administration of the selective δ1-opioid agonist; however, it produced a marked cardioprotective effect at 24 to 48 hours after drug administration, which disappeared at 72 hours (Figure 9-38). The cardioprotective effects were blocked by pretreatment with a selective δ1-antagonist, a nonselective K+ATP-channel antagonist, and a mitochondrial-selective K+ATP-channel blocker. These results suggest that δ1-opioid receptor activation 24 to 48 hours before an ischemic insult results in a delayed cardioprotective effect that appears to be mediated by the mitochondrial K+ATP channel. Wu and colleagues454 demonstrated that κ-opioid-receptor–induced cardioprotection occurred via two phases: the first window occurred about 1 hour after receptor activation, and the second developed 16 to 20 hours after administration in isolated ventricular myocytes.
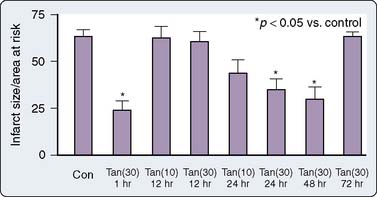
Opioids and Cardioprotection in Humans
Although the animal and cell work reviewed imply a cardioprotective effect of opioid receptor activation, it is important to demonstrate that a similar system exists in humans if these basic studies can be extrapolated to the clinical world. Tomai et al455 have demonstrated that naloxone could abolish the reduction in ST-segment elevation normally observed during a second balloon inflation during coronary angioplasty. In addition, they demonstrated that in naloxone-treated patients, the severity of cardiac pain and time to onset at the end of the second balloon inflation were similar to that of the first inflation, whereas in the placebo-treated patients, the severity of cardiac pain during the second inflation was reduced and the time to onset of pain was lengthened versus the first inflation. This suggests a preconditioning-like effect in humans undergoing coronary angioplasty that could be attenuated by the opioid antagonist naloxone. Similarly, Xenopoulos and coworkers456 have shown that intracoronary morphine (15 μg/kg) mimics IPC, as assessed by changes in ST-segment shifts in humans undergoing percutaneous transluminal coronary angioplasty. Finally, Bell et al448 also demonstrated that δ-opioid-receptor stimulation mimics IPC in human atrial trabeculae via K+ATP channel activation. These results are encouraging and may suggest a possible clinical use for opioids in the therapy of acute or chronic myocardial ischemia.448
Cardioplegia and hypothermia provide considerable myocardial protection against the induced ischemia of cardiac surgery. However, in certain high-risk subgroups and, to some extent, in all patients, current methods of cardioprotection are still suboptimal, and there continues to be poor myocardial tolerance to ischemia. This myocardial ischemia often is evidenced by perioperative ventricular dysfunction, myocardial stunning, and poor functional recovery after an ischemic episode, which may lead to poor surgical outcome (see Chapters 28 and 32). Postsurgical myocardial ischemic changes include hypothermia, intracellular acidosis, hypoxia, depletion of energy stores, and cellular volume shifts, all of which adversely affect myocardial contractility. Hibernating animals demonstrate similar cellular and molecular cardiac changes during hibernation that closely parallel those seen in hypothermic cardioplegic arrest. However, these changes are well tolerated in the myocardium of the hibernating mammals for months at a time, whereas the duration of induced ischemia tolerated surgically is limited. This process is induced by a hibernation-induction-trigger molecule, which has been shown to have an opioid basis.457 It has been shown that opioid peptides can induce mammalian hibernation and may provide protection against the adverse effects of hypothermic myocardial ischemia, providing potential therapeutic applications during CPB and protection of organs for heart transplantation.
In animal studies, Bolling et al458,459 showed the δ-opioid-receptor agonist DADLE protected hearts that were subjected to 18 hours of cold storage at 4°C or 2 hours of global ischemia, respectively, in the presence of a standard cardioplegic solution. Another study460 provided evidence that pentazocine, a δ-opioid agonist, enhanced the myocardial protection of standard cardioplegia at temperatures ranging from 0°C to 34°C. Subsequently, Kevelaitis et al450 showed that stimulation of δ-opioid receptors improved recovery of cold-stored rat hearts to a state similar to IPC. These investigators showed that this opioid-induced cardioprotection is mediated through K+ATP-channel activation.450
Although the initial success of cardiopulmonary resuscitation (CPR) is, on average, 39% (13% to 59%), a majority of victims die within 72 hours, primarily because of heart failure, recurrent ventricular fibrillation, or both. CPR, therefore, yields a functional survival rate of only 1.4% to 5%.461–463 Myocardial function is substantially impaired after successful resuscitation from cardiac arrest. This has led to the investigation of the use of opioid-receptor agonists to improve functional outcome. Tang and colleagues464 demonstrated that pharmacologic activation of δ-opioid receptors significantly reduced Mvo2 during the global myocardial ischemia of cardiac arrest. A follow-up study in rats demonstrated that the nonselective δ-opioid-receptor agonist pentazocine strikingly reduced the severity of postresuscitation myocardial dysfunction and increased the duration of postresuscitation survival.464

Opioids in Cardiac Anesthesia
A technique of anesthesia for cardiac surgery involving high doses of morphine was developed in the late 1960s and early 1970s. This was based on Lowenstein et al’s465 observation that patients requiring mechanical ventilation after surgery for end-stage VHD tolerated large doses of morphine for sedation without discernible circulatory effects. When they attempted to administer equivalent doses of morphine as the anesthetic for patients undergoing cardiac surgery, they discovered serious disadvantages including inadequate anesthesia, even at doses of 8 to 11 mg/kg, episodes of hypotension related to histamine release, and increased intraoperative and postoperative blood and fluid requirements. Attempts to overcome these problems by combining lower doses of morphine with a variety of supplements (such as N2O, halothane, or diazepam) proved unsatisfactory, resulting in significant myocardial depression, with decreases in CO and hypotension.466 However, recently, Murphy et al467 demonstrated that morphine, compared with fentanyl, resulted in better myocardial function after CABG surgery. Specifically, with regard to the protective effects mediated by IPC, it appears that morphine may have some benefit over fentanyl.467 The use of morphine, however, has to be weighed against the multitude of other deleterious effects during the management of cardiac surgical patients.
Because of the above problems associated with the use of morphine, several other opioids were investigated in an attempt to find a suitable alternative. The use of fentanyl in cardiac anesthesia was first reported by Stanley and Webster in 1978.468 Since then there have been extensive investigations of fentanyl, as well as sufentanil and alfentanil, in cardiac surgery. The fentanyl group of opioids has proved to be the most reliable and effective for producing anesthesia for patients with valvular disorders and CABG (see Chapters 18 and 19).
A major advantage of fentanyl and its analogs for patients undergoing cardiac surgery is their lack of cardiovascular depression. This is of particular importance during the induction of anesthesia, when episodes of hypotension can be critical. Cardiovascular stability may be less evident during surgery; in particular, the period of sternotomy, pericardiectomy, and aortic root dissection may be associated with significant hypertension and tachycardia. During and after sternotomy, arterial hypertension increases in SVR and decreases in CO frequently occur.469,470 The variability in the hemodynamic responses to surgical stimulation, even with similar doses of fentanyl, is probably a reflection of differences in the patient populations studied by different authors. One factor is the influence of β-blocking agents. In patients anesthetized with fentanyl undergoing CABG, 86% of those not taking β-blockers became hypertensive during sternal spread versus only 33% of those who were taking β-blockers.471
There may be differences among the opioids with regard to hemodynamic stability during surgery. One study concluded that both fentanyl and sufentanil provide similar hemodynamic stability during induction, whereas alfentanil causes hemodynamic instability and myocardial ischemia.472 Alfentanil also may be less effective in suppressing reflex sympathetic and hemodynamic responses to stimuli than fentanyl or sufentanil.473 In patients undergoing valvular surgery, all three opioids provided satisfactory anesthesia.474 However, controversy still surrounds the best choice of anesthetic, at least for CABG. Two studies involving more than 2000 patients anesthetized with inhalation agents, fentanyl or sufentanil, came to the conclusion that the choice of anesthetic did not significantly influence the outcome after CABG, although the type of anesthetic continues to remain a topic of controversy with respect to postoperative outcomes.113–115,475,476
The degree of myocardial impairment also will influence the response. Critically ill patients or patients with significant myocardial dysfunction appear to require lower doses of opioid for anesthesia. This may reflect altered pharmacokinetics in those patients. A decrease in liver blood flow consequent to decreased CO and CHF reduces plasma clearance. Thus, patients with poor left ventricular function may develop greater plasma and brain concentrations for a given loading dose or infusion rate than patients with good left ventricular function. In addition, patients with depressed myocardial function may lack the ability to respond to surgical stress by increasing CO in the face of progressive increases in SVR.477
An infusion of alfentanil (125-μg/kg bolus followed by 0.5 mg/kg/hr) has been compared with fentanyl, 100 μg/kg, or sufentanil, 20 μg/kg, by bolus injection, as the sole anesthetic for patients undergoing valvular surgery.474 No differences in hemodynamic effects were found in the study, and it was concluded that all three opioids can provide satisfactory anesthesia for valve replacement surgery (see Chapter 19).
Sufentanil appears to offer more stable anesthesia with less hemodynamic disturbance than fentanyl,478 and it has been used successfully for cardiac transplantation.479,480 In patients undergoing mitral or aortic valve surgery, sufentanil (total dose, 9.0 ± 0.4 μg/kg) resulted in less need for supplements and vasodilators than fentanyl, 113 ± 11 μg/kg, but sufentanil produced more hypotension during induction.481 Howie et al482 compared a fentanyl/isoflurane/propofol regimen with remifentanil/isoflurane/propofol for fast-track anesthesia. Significantly more patients in the fentanyl regimen experienced hypertension during skin incision and maximum sternal spread compared with patients in the remifentanil regimen. There was no difference between groups in time to extubation, discharge from the intensive care unit, electrocardiographic changes, catecholamine levels, or cardiac enzymes. The remifentanil-based anesthetic (bolus followed by continuous infusion) resulted in less need for anesthetic interventions compared with the fentanyl regimen.482
Samuelson et al483 compared hemodynamic and stress responses in patients with CAD anesthetized with either sufentanil-oxygen or enflurane-nitrous oxide and oxygen. Both techniques were satisfactory and resulted in stable hemodynamics, but considerable “fine tuning” was required when enflurane was administered. The postoperative hemodynamic effects were compared in patients who received sufentanil, 25 μg/kg, or fentanyl, 100 μg/kg, for anesthesia for CABG.484 Patients who received sufentanil had a more stable course, with higher CO, lower SVR, and a lower incidence of hypertension. The two groups had similar values for time to awakening, response to verbal commands, and extubation.
Collard et al485 compared the intraoperative hemodynamic profiles and recovery characteristics of propofol-alfentanil versus a fentanyl-midazolam anesthetic in elective coronary artery surgery. Cardiovascular parameters and time to extubation were recorded. Throughout surgery, hemodynamic profiles were comparable between groups except after intubation, when the MAP was significantly lower in the propofol-alfentanil group. This group also required less inotropic support, and extubation was performed earlier in this group.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
9 Pharmacology of Anesthetic Drugs
An enormous body of literature has been accumulated describing the protean effects of the different anesthetic agents on the heart and the pulmonary and systemic regional vascular beds. The effects on the heart, especially, have spawned innumerable publications. More recently, this has been because of the great interest in anesthesia-induced preconditioning (APC). However, even before the initial description of APC, the literature detailing the influence of anesthetic agents on the myocardium was prodigious and not always consistent.1–3 This likely reflected not only the challenges inherent in quantitating the direct effects of volatile agents on the myocyte/myocardium, but the presence of several potential confounding variables including effects on coronary blood flow (CBF), the systemic vasculature, and the baroreceptor reflex arc. This chapter divides the discussion of the effects of volatile agents, fixed agents, and narcotics on the cardiovascular system (CVS) into those that involve acute and delayed effects. Under acute effects, the influence of anesthetic agents is described on: (1) myocardial function, (2) electrophysiology, (3) coronary vasoregulation, (4) systemic and pulmonary vasoregulation, and (5) the baroreceptor reflex. Within delayed effects, the focus is on APC.
Volatile agents
Acute Effects
Myocardial Function
The influence of volatile anesthetics on contractile function has been investigated extensively in several animal species and in humans using various in vitro and in vivo models.4–11 In general, it is now widely agreed that volatile agents cause dose-dependent depression of contractile function (Box 9-1). Moreover, different volatile agents are not identical in this regard and the preponderance of information indicates that halothane and enflurane exert equal but more potent myocardial depression than do isoflurane, desflurane, or sevoflurane. This reflects, in part, reflex sympathetic activation with the latter agents. It is also widely accepted that in the setting of preexisting myocardial depression, volatile agents have a greater effect than in normal myocardium.12,13 Early studies indicating that volatile agents may not have a deleterious effect on function in the setting of acute myocardial infarction (AMI) likely reflected the fact that the limited infarction did not compromise overall myocardial function.14,15 At the cellular level, volatile anesthetics exert their negative inotropic effects, mainly by modulating sarcolemmal (SL) L-type Ca++ channels, the sarcoplasmic reticulum (SR), and the contractile proteins. L-type Ca++ currents are decreased and, secondarily, SR Ca++ release is depressed (Figures 9-1 and 9-2).16 Moreover, the contractile response to lower Ca++ levels is further attenuated in the presence of volatile agents in that the response is decreased by volatile agents at any given Ca++ level; that is, volatile agents also decrease Ca++ sensitivity (Figure 9-3).16 However, the mechanisms whereby anesthetic agents modify ion channels are not completely understood. Ion channels usually are studied in ex vivo circumstances in which, by definition, multiple modulating influences of the specific channel under study may be altered. Moreover, these studies frequently are undertaken in nonhuman tissue. Well-recognized species differences make extrapolation to humans difficult.17 Nitrous oxide causes direct mild myocardial depression but also causes sympathetic activation.18
BOX 9-1. Volatile Anesthetic Agents
It is well recognized that even in the setting of normal systolic function, diastolic dysfunction occurs with increasing frequency in the elderly and is an important cause of congestive heart failure (CHF).19–25 Diastolic dysfunction and its more severe clinical counterpart, diastolic heart failure, have protean causative factors and can be mechanistically complex23 (Table 9-1). However, the mechanisms underlying these conditions can be categorized into those involving alterations in myocardial relaxation (e.g., SR Ca++ handling, phospholamban), those related to intrinsic properties of myocardial tissue (e.g., myocyte cytoskeletal elements), and those that are extramyocardial (e.g., loading conditions). Indices of diastolic function were not readily and reliably measured noninvasively in the past; hence the relatively more recent recognition and description of diastolic dysfunction and diastolic heart failure compared with perturbations in systolic function. This likely also explains the relative paucity of literature detailing the modulating effects of volatile agents on diastolic function. There is reasonable agreement in the literature that volatile agents prolong isovolumic relaxation and do so in a dose-dependent manner.21,22,26–30 The effects of volatile agents on chamber stiffness are more controversial; for example, halothane has been reported to both decrease compliance and have no effect on myocardial stiffness.21,22,26,28–31 The effect of nitrous oxide on diastolic function has not been investigated in a manner that critically rules out confounding variables. At a molecular level, alterations in relaxation likely reflect modulation of Ca++ currents, including SR Ca++ reuptake mechanisms. Paradoxically, in the setting of reperfusion injury and Ca++ overload, the volatile agent sevoflurane improves indices of diastolic relaxation and attenuates myoplasmic Ca++ overload.32
| Abnormalities of myocardial relaxation |
| Ischemia |
| Hypertrophy |
| Hypertension |
| Valvular heart disease |
| Abnormalities of myocardial compliance |
| Aging |
| Fibrosis |
| Hypertrophy |
| Diabetes mellitus |
| Metabolic syndrome |
| Infiltrative disorders—amyloidosis |
| Cardiomyopathies |
| Constrictive pericarditis |
Cardiac Electrophysiology
The molecular mechanisms underlying this effect of volatile anesthetics are poorly understood. Anesthetic agent–induced modulation of ion channels is important mechanistically in excitation-contraction coupling (vide supra), in preconditioning (vide infra), and in modulating automaticity and arrhythmia generation17 (Table 9-2). Although the effects of any particular volatile agent on a specific cardiac ion channel may have been characterized, this does not allow a ready extrapolation into clinical situations. This partly reflects those issues already discussed (species differences, ex vivo studies) but also the recognition that it is impossible to predict the arrhythmogenic effect that might ensue after modulation with a particular volatile agent. This should be one of the lessons garnered from the experience with the antiarrhythmic drugs such as encainide and flecainide.33 Moreover, even in the clinical setting, not all volatile agents have the same effect.34
Coronary Vasoregulation
Animal studies indicate that halothane has little direct effect on the coronary vasculature.35–37 Likewise, clinical studies investigating the effect of halothane indicate that it has either minimal or mild coronary vasodilator effects.38–41 The effect of isoflurane on coronary vessels was controversial and dominated much of the literature in this area in the 1980s and early 1990s. The current assessments of the effects of isoflurane have been succinctly detailed by Tanaka et al.42 Several reports have indicated that it caused direct coronary arteriolar vasodilatation in vessels of 100 μm or less, and that isoflurane could cause “coronary steal” in patients with “steal-prone” coronary anatomy; that is, in patients with significant coronary stenosis in a vessel subserving a region of ischemic myocardium, when, presumably, vessels were maximally dilated because of local metabolic autoregulation, and in whom isoflurane-induced vasodilatation in adjacent vessels resulted in diversion of coronary flow away from the ischemic region.43,44 Several animal and human studies in which potential confounding variables were controlled indicated clearly that isoflurane did not cause coronary steal.45–51 Studies of sevoflurane and desflurane showed similar results and are consistent with a mild direct coronary vasodilator effect of these agents.52,53
Ultimately, CBF (in the setting of normal systemic hemodynamics) is controlled by coronary vascular smooth muscle tone, which can be modulated directly (endothelium-independent) or indirectly via the endothelium (endothelium-dependent). Teleologically, it can be predicted that in vital organs, control of blood flow is predominantly local, acting through either endothelium-dependent or -independent mechanisms. Thus, volatile agents have the capacity to modulate mechanisms underlying vascular tone: (1) Halothane and isoflurane have been shown to attenuate endothelial-dependent tone (by receptor-dependent and receptor-dependent plus -independent mechanisms, respectively) in coronary microvessels54; (2) several volatile agents cause coronary vasodilation via K+ATP-channel–dependent mechanisms54–57; and (3) sevoflurane induced K+– and Ca++-channel–mediated increases in coronary collateral blood flow.58 The effects in vivo are likely to be modest because local control mechanisms are likely to predominate.
Systemic Regional and Pulmonary Vascular Effects
Reversible inhibition of endothelium-dependent relaxation in aortic and femoral vessels was first demonstrated for halothane and also has been demonstrated for enflurane, isoflurane, and sevoflurane in both capacitance and resistance vessels.54,59–63 However, these observations mask the differential effects of volatile agents on underlying endothelium-dependent mechanisms. Halothane and enflurane decrease agonist (bradykinin) and ATP-induced Ca++ increases in bovine endothelial cells, whereas isoflurane does not.64 In contrast, isoflurane does attenuate histamine-induced Ca++ influx into human endothelial cells.65 Alterations in endothelium-dependent mechanisms by volatile agents are not confined to attenuation of agonist-dependent and -independent activation of endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) release but also may extend to other mechanisms. For example, the effects of sevoflurane on endothelial cell function may be partially because of sevoflurane-induced changes in endothelin-1 (ET-1) production and in the redox milieu of the endothelial cells (i.e., increased superoxide anion production).66
The effect of volatile agents on vascular smooth muscle mechanisms is equally complex and varies among agents. In endothelial cell–denuded aortic rings, halothane decreases both SL Ca++ influx via voltage-dependent calcium channels and SR Ca++ release, but sevoflurane does not.67 Sevoflurane also inhibits angiotensin II–induced vascular smooth muscle contraction in aortic rings.68 In mesenteric vessels, sevoflurane accentuates endothelium-dependent mechanisms and attenuates endothelium-independent mechanisms in the presence of norepinephrine.69 Studies of the influence of volatile agents on vascular smooth muscle Ca++ currents indicate that both halothane and enflurane stimulate SR release and reuptake from the caffeine-sensitive pool. In contrast, halothane, enflurane, and isoflurane all increase calcium-induced calcium release (CICR) mechanisms, but sevoflurane decreases CICR mechanisms.70 Finally, volatile agents also have been demonstrated to modulate Ca++ sensitivity. In mesenteric vessels, halothane relaxation is largely mediated by Ca++ and myosin light-chain desensitizing mechanisms.71
The pulmonary circulation has unique features that must be taken into account when interpreting studies of this vascular bed. In addition to those issues that also apply to systemic vascular beds (vessel size, etc.), the pulmonary vasculature is a low-resistance bed (requiring preconstriction to access vasoactive effects), is not rectilinear (thus, changes in flow per se can change certain parameters used to calculate resistance), is contained within the chest (and thus subject to extravascular pressures, which are not atmospheric and change during the respiratory cycle), and exhibits the unique vascular phenomenon of hypoxia-induced vasoconstriction. It is clear that volatile agents modulate not only the baseline pulmonary vasculature but also multiple vasoactive mechanisms that control pulmonary vascular tone. Moreover, the effect of volatile agents is agent specific. For example, halothane causes flow-independent pulmonary vasoconstriction.72 In contrast, the hypoxic pulmonary vasoconstrictor response does not appear to be altered by at least two currently used volatile agents: sevoflurane and desflurane.73 The pulmonary vascular endothelial response appears to be impaired by the volatile agents halothane and isoflurane.74,75 Finally, pulmonary vascular smooth muscle regulatory mechanisms also can be modified by volatile agents. Halothane, enflurane, and isoflurane all attenuate pulmonary vasodilatation induced by K+ATP channel activation.76,77 Although the effects of the different volatile agents on K+ATP-channel activation are similar, β-adrenergic receptor–induced pulmonary vasodilatation is differently modulated. Halothane and isoflurane potentiate the vasodilatory response, but enflurane has no effect.78
Baroreceptor Reflex
All volatile agents attenuate the baroreceptor reflex. Baroreceptor reflex inhibition by halothane and enflurane is more potent than that observed with isoflurane, desflurane, or sevoflurane, each of which has a similar effect.79,80 Each component of the baroreceptor reflex arc (afferent nerve activity, central processing, efferent nerve activity) is inhibited by volatile agents. Inhibition of afferent nerve traffic results, in part, from baroreceptor sensitization,81,82 whereas attenuation of efferent activity is due, in part, to ganglionic inhibition as manifest by differential preganglionic and postganglionic nerve activity.81–83
Delayed Effects
Reversible Myocardial Ischemia
Prolonged ischemia results in irreversible myocardial damage and necrosis (Box 9-2). Shorter durations of myocardial ischemia can, depending on the duration and sequence of ischemic insults, lead to either preconditioning or myocardial stunning (Figure 9-4).84 Stunning, first described in 1975, occurs after brief ischemia and is characterized by myocardial dysfunction in the setting of normal restored blood flow and by an absence of myocardial necrosis.85 Ischemic preconditioning (IPC) was first described by Murry et al86 in 1986 and is characterized by an attenuation in infarct size after sustained ischemia, if this period of sustained ischemia is preceded by a period of brief ischemia (Figure 9-5). Moreover, this effect is independent of collateral flow. Thus, short periods of ischemia followed by reperfusion can lead to either stunning or preconditioning with a reduction in infarct size (Figure 9-6).84
BOX 9-2 Volatile Agents and Myocardial Ischemia
Figure 9-5 Infarct size and collateral blood flow in the 40-minute study.
(From Warltier DC, al-Wathiqui MH, Kampine JP, et al: Recovery of contractile function of stunned myocardium in chronically instrumented dogs is enhanced by halothane or isoflurane, Anesthesiology 69:552, 1988.)
Figure 9-6 Schematic of stunning and preconditioning.
(From Kloner RA, Jennings RB: Consequences of brief ischemia: stunning, preconditioning, and their clinical implications, Part I, Circulation 104:2981, 2001.)
As discussed previously, work in the 1970s indicated that volatile anesthetic agents attenuated ST-segment elevations in the setting of short-duration ischemia and limited infarct size and lactate production after prolonged ischemia.87,88 Moreover, these effects seemed to be independent of the main determinants of myocardial oxygen supply and demand, and suggested that the volatile agents may be exerting a beneficial effect at the level of the myocyte. On resolution of the isoflurane “coronary steal” controversy, the first description of the salutary effects of volatile agents on the consequences of brief ischemia was made in 1988. Warltier and coworkers89 described the beneficial effects of halothane and isoflurane in facilitating the recovery of contractile function in stunned myocardium (Figure 9-7). However, it was almost a decade later before the effects of volatile agents on preconditioning were outlined2,3 and the term APC was used1 (Figure 9-8).
The phenomenon of and the mechanisms underlying IPC are the focus of extensive investigation. IPC has the following characteristics: (1) results in two periods (termed windows) of protection—the first (termed early or classic) occurs at 1 to 3 hours, and the second (termed late or delayed) occurs 24 to 96 hours after the preconditioning stimulus; (2) occurs also in noncardiac tissue, such as brain and kidney; (3) is ubiquitous across species; (4) is most pronounced in larger species with lower metabolism and slower heart rates (HRs); (5) seems to be important clinically because angina within the 24-hour period preceding an AMI is associated with an improved outcome (Figure 9-9)90; and (6) is mediated by multiple endogenous signaling pathways91 (Figure 9-10).92 As might be predicted from the time frame of delayed IPC, it is mediated, at least in part, by transcriptional and posttranslational mechanisms91 (Figure 9-11). Finally, and of the utmost importance, preconditioning can be triggered by events other than ischemia (cellular stress of various forms, pharmacologic agonists, anesthetic agents; see Figure 9-11).91 Moreover, the benefits of IPC are not necessarily confined to and may not include limitation of infarct size, and depend on the specific trigger for IPC, the species under study, and classic versus delayed IPC. For example, rapid pacing affords protection against arrhythmias but not against infarct evolution. In contrast, cytokine-induced IPC limits infarct size but has no effect on arrhythmias.91 Different triggers of IPC modulating different end points suggest that, although there are fundamental mechanisms common to various triggers of IPC, there also exist mechanistic differences across triggers. Thus, APC may not be identical to IPC mechanistically.
Figure 9-10 Investigated signaling pathways (full lines).
(From Feng J, Lucchinetti E, Ahuja P, et al: Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3β, Anesthesiology 103:987–995, 2005.)
Anesthetic Agents: Pre- and Post-Conditioning
This is an area of intense investigation as reflected by two issues of Anesthesiology being devoted predominantly to the subject.93,94 After the initial description of APC,1–3 subsequent investigations have indicated that volatile agents can elicit delayed (late), as well as classic (early), preconditioning.95,96 Moreover, APC is dose dependent,97–99 exhibits synergy with ischemia in affording protection,100,101 and perhaps not surprisingly, in view of differential uptake and distribution of volatile agents, has been demonstrated to require different time intervals between exposure and the maintenance of a subsequent benefit that is agent dependent42 (see Chapters 6 and 7).
The contributions of both SL and mitochondrial K+ATP channels in IPC have been extensively investigated, and it is now widely agreed that mitochondrial K+ATP channels play a critical role in this process. Volatile agents that exhibit APC activate mitochondrial K+ATP channels, and this effect is blocked by specific mitochondrial K+ATP channel antagonists. However, the precise relative contributions of SL versus mitochondrial K+ATP channel activation to APC remain to be elucidated (Figure 9-12).42 The original descriptions of APC indicated that volatile agents can trigger preconditioning without concurrent ischemia during the “triggering” period1–3 (see Figure 9-8). However, studies of mitochondrial activation (via mitochondrial K+ATP channels) indicate that volatile agents on their own do not activate mitochondria but do potentiate the effects of direct mitochondrial K+ATP channel openers99 (Figure 9-13). These apparent inconsistencies are likely explained by the presence of multiple parallel and redundant pathways activated during APC (and IPC)96 (see Figure 9-12). For example, it is now well established that the adenosine A-1 and δ1 opioid G-coupled receptors can trigger IPC. Moreover, pharmacologic blockade of these receptors attenuates the positive effects of volatile agents.98,102 Protein kinase C (PKC) and the nuclear signaling pathway, mitogen-activated protein kinase (MAPK), are important signaling pathways in preconditioning, and volatile agents have been shown to modulate at least PKC translocation.103 Oxidant stress is a central feature of reperfusion and, depending on the specific moiety, the enzymatic source and, most importantly, the oxidant stress load may trigger preconditioning on the one hand or mediate reperfusion injury on the other. Both indirect and direct evidence indicate that volatile agents can increase oxidant stress to levels that trigger preconditioning.104–106
Activation of eNOS also has been shown to play a role, as has depolarization of the mitochondrial internal membrane.107 This may prevent the opening of the mitochondrial permeability transition pore (MPTP) and inhibit Na+-H+ exchange, attenuating Ca+2 overload and cell edema.107 Inhibition of mitochondrial permeability by APC has been suggested to decrease myocyte death, and PKC also has been thought to play a role in IPC-induced delay of MPTP opening.108,109 For the first time, a recent study demonstrates that isoflurane has been shown to activate PKC-dependent signaling pathways resulting in the delay of MPTP opening,110 suggesting a possible mechanism for isoflurane in APC. With respect to postconditioning, Ge et al111 demonstrated that NO may, in fact, act as both a trigger and a mediator for isoflurane-induced cardiac protection in mouse hearts. This implies that an eNOS-dependent mechanism prevents the opening of the MPT pore, although other pathways including glycogen synthase kinase-3β also have been implicated (Figure 9-14).92,111
It is clear that mitochondrial activation attenuates ischemia-induced oxidant stress, favorably modulates mitochondrial energetics, decreases cytochrome c egress into the cytoplasm, and attenuates mitochondrial and cytoplasmic Ca++ overload. Mitochondrial cytochrome c release is one of the important mechanisms underlying caspase activation, and thus the apoptotic process112 (Figure 9-15). Whether by Ca++-mediated or by apoptotic mechanisms, or both, volatile agents clearly attenuate cell death in models of APC101 (Figure 9-16). Although the mechanisms underlying mitochondrial activation have been aggressively studied, they remain incompletely understood. Finally, these salutary effects of volatile agents seem to have a clinical correlate101 (Figure 9-17).
Figure 9-15 Center stage in apoptosis.
(Reprinted from Finkel E: The mitochondrion: is it central to apoptosis? Science 292(5517):624–626, 2001. Illustration: C. Slayden, Copyright 2001 AAAS.)
The use of volatile anesthetics also can alter outcomes after cardiac surgery. A meta-analysis by Landoni et al113 demonstrated a significant reduction in postoperative myocardial infarction after cardiac surgery, as well as significant advantages with respect to postoperative cardiac troponin release, inotrope requirements, time to extubation, intensive care unit stay, hospital stay, and survival. Furthermore, another meta-analysis by Bignami et al114 demonstrated that the use of volatile anesthetics may, in fact, have a beneficial role with respect to mortality after cardiac surgery. The duration of the volatile anesthetic exposure seemed to have some impact—the longer the exposure, the greater the effect. De Hert et al115 demonstrated the cardioprotective effects of volatile anesthetics if used throughout the surgical procedure rather than only before and after cardiopulmonary bypass (CPB).
Intravenous induction agents
Unlike the inhalation anesthetic agents that augment IPC, there is no good evidence that the intravenous hypnotic agents demonstrate these protective effects. There is, however, emerging evidence that propofol, the mainstay of induction agents, may enhance antioxidant activity in the heart and thus may prevent lipid peroxidation after ischemia/reperfusion, offering a potential protective effect on the heart.116
Acute cardiac effects
Myocardial Contractility
The effect of the agents may be species dependent, thus further confounding the literature regarding mechanism. For instance, van Klarenbosch et al117 demonstrated that in contrast with rat, propofol directly depresses myocardial contractility in isolated muscle preparations from guinea pig, probably by decreasing trans-SL Ca++ influx. However, there was little influence of propofol on Ca++ handling by the SR or on the contractile proteins in rat. In one of the few human studies using isolated atrial muscle tissue (Figure 9-18), no inhibition of myocardial contractility was found in the clinical concentration ranges of propofol, midazolam, and etomidate. In contrast, thiopental showed strong negative inotropic properties, whereas ketamine showed slight negative inotropic properties (Figure 9-19). Thus, negative inotropic effects may explain, in part, the cardiovascular depression on induction of anesthesia with thiopental but not with propofol, midazolam, and etomidate. Improvement of hemodynamics after induction of anesthesia with ketamine cannot, therefore, be explained by intrinsic cardiac stimulation but is a function of sympathoexcitation.118
The effect of drugs such as propofol also may be affected by the underlying myocardial pathology.119,120 For instance, Sprung et al120 determined the direct effects of propofol on the contractility of human nonfailing atrial and failing atrial and ventricular muscles obtained from the failing human hearts of transplant patients or from nonfailing hearts of patients undergoing coronary artery bypass graft surgery (CABG). They concluded that propofol exerts a direct negative inotropic effect in nonfailing and failing human myocardium, but only at concentrations larger than typical clinical concentrations. Negative inotropic effects are reversible with β-adrenergic stimulation, suggesting that propofol does not alter the contractile reserve but may shift the dose responsiveness to adrenergic stimulation. The negative inotropic effect of propofol is at least partially mediated by decreased Ca++ uptake into the SR; however, the net effect of propofol on contractility is insignificant at clinical concentrations because of a simultaneous increase in the sensitivity of the myofilaments to activator Ca++.120
Molecular Mechanisms: Adrenergic Signaling, Ca++ Influx, and Ca++ Sensitivity
There are a number of suggested molecular mechanisms by which a drug such as propofol may alter cardiac contractility. Propofol may inhibit cardiac L-type calcium current by interacting with the dihydropyridine-binding site121 (Figure 9-20A), with resultant alteration in developed tension (see Figure 9-20B). Furthermore, as mentioned earlier, propofol may alter adrenergic signaling in cardiac myocytes. Experiments in membranes and cardiac preparations isolated from rat heart demonstrate that relatively high concentrations of propofol (25 to 200 μmol/L) are required to antagonize β-adrenoceptor binding and tissue responsiveness.122 Kurokawa et al123 observed that clinically relevant concentrations of propofol attenuated β-adrenergic signal transduction in cardiac myocytes via inhibition of cyclic adenosine monophosphate (cAMP) production (Figure 9-21). The inhibitory site of action of propofol appears to be upstream of adenylyl cyclase and involves activation of PKCα.
