[level-membership-for-neonatal-and-perinatal-medicine-category]
Pharmacology and pharmacokinetics during the perinatal period
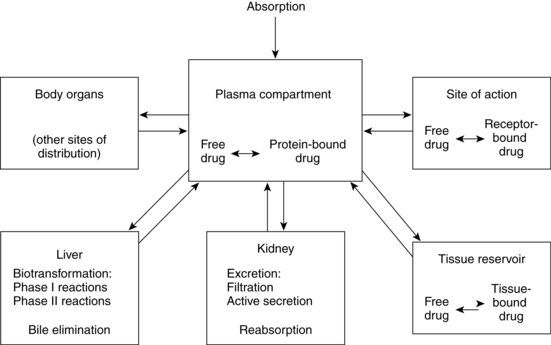
Pharmacokinetics refers to the processes involved in drug absorption, distribution, metabolism, and biotransformation and excretion (Figure 7-1). Site of absorption depends on route of administration (e.g., oral, intramuscular, intravenous, topical, inhaled). Drugs are carried in the blood, either bound to plasma proteins or as free drug. Protein-bound drugs provide a reservoir for future use, whereas free drug is the active component available to interact with the target cell. Biotransformation occurs primarily in the liver with a series of enzymatic reactions to modify and convert the drug into more polar (water-soluble) compounds. Intermediary steps in biotransformation may result in active metabolites. For example, codeine is demethylated to morphine, and in the neonate theophylline is methylated to caffeine.32 Biotransformation also takes place in the lungs, intestinal mucosa, and kidneys. Hepatic biotransformation occurs in two phases. Phase I (nonsynthetic reactions) takes place primarily in the microsomes, although some occurs in the mitochondria and cytosol. Phase I reactions modify the activity of a drug. Phase I enzymes include the cytochrome P450 (CYP450) monooxygenase system. Phase II (synthetic) reactions require adenosine triphosphate (ATP) and, except for glucuronidation, are extramicrosomal.32 Phase II reactions generally inactivate the drugs by converting them to more polar substances. Table 7-1 provides examples of drugs metabolized by various phase I and phase II reactions. Not all drugs undergo hepatic metabolism before elimination; some drugs are excreted directly via the kidneys.
Table 7-1
| REACTION | EXAMPLES OF DRUG SUBSTRATES |
| PHASE I (NONSYNTHETIC REACTIONS) | |
| Oxidation | Phenytoin, phenobarbital, ibuprofen, acetaminophen, morphine, codeine, diazepam, cimetidine |
| Reduction | Prontosil, chloramphenicol, ethanol |
| Hydrolysis | Acetylsalicylic acid, indomethacin |
| PHASE II (SYNTHETIC REACTIONS: CONJUGATIONS) | |
| Glucuronide conjugation | Morphine, acetaminophen |
| Glycine conjugation | Salicylic acid |
| Sulfate conjugation | Acetaminophen, α-methyldopa |
| Glutathione conjugation | Ethacrynic acid |
| Methylation | Dopamine, epinephrine |
| Acetylation | Sulfonamides, clonazepam |
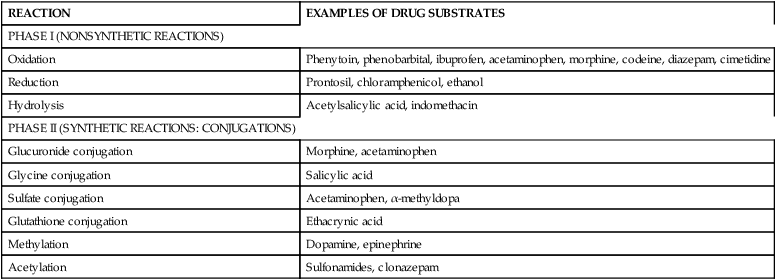
Adapted from Chemtob, S. (2004). Basic pharmacologic principles. In R.A. Polin, W.W. Fox, & S.H. Abman (Eds.). Fetal and neonatal physiology (3rd ed.). Philadelphia: Saunders.
Variations in drug pharmacokinetics and pharmacodynamics occur among individuals at all ages, even with similar doses of a given medication, due to extrinsic (diet, environment, other therapies) and intrinsic (development, genetic constitution, diseases) factors.95 Currently much research focuses on understanding these factors, especially the contributions of individual genetic polymorphisms (see Chapter 1). This has led to new fields of study such as pharmacogenetics and pharmacogenomics. Pharmacogenetics is the study of “genetic variations that give rise to interindividual responses to drugs,”95, p.256 while pharmacogenomics integrates pharmacology and genomics into “the broader application of genome-wide technologies and strategies to identify both disease processes that represent new targets for drug development and factors predictive of therapeutic efficacy and risk of adverse drug reactions.”95, p.256 Genetic polymorphisms are small variations in an individual’s genome that may either be nonfunctional (have no effect on the individual) or modify expression of a protein that may alter the effects of exposures to environmental health hazards or result in individual differences in responses to drugs (changing efficacy, risk of adverse effects, or therapeutic dosing regimens).76,95 Understanding these variations, which is just beginning, can individualize pharmacologic management. During pregnancy, these individual differences may occur in the other, fetus and/or placental, or in any combination of these to influence the expression of specific genes involved in biotransformation, amount and activity of enzymes produced and thus levels of the drug and potential risks.76
The term drug is used generically in this chapter to refer to prescription and nonprescription pharmacologic agents, herbal agents, and vitamins, as well as drugs of abuse. In addition, the principles that govern placental transfer of drugs are also applicable to transfer of chemicals, food additives, and environmental agents. Definitions of common terms used in pharmacokinetics are summarized in Box 7-1 on p. 185.
Pharmacokinetics during pregnancy
Drug use during pregnancy
A survey of drug utilization by 14,000 pregnant women in 22 countries found that 86% of these women took some medication during pregnancy, with an average woman taking 2.9 medications (range, 1 to 15).73 A longitudinal survey from the United States surveyed 578 women throughout pregnancy. These women took a mean of 1.14 prescribed drugs per person, excluding iron and vitamins, with the most frequent being antibiotics, respiratory agents, gastrointestinal (GI) agents, and opioids. Of these women, 59.7% used at least one prescribed drug; 3.1% used more than five. In addition, these women took a mean of 2.95 over-the-counter drugs per person (92.6% of the women studied used at least one; 20.8% used more than five), with the most frequent being analgesics and agents for gastrointestinal or upper respiratory symptoms. Herbal agents were used by 45.2% of study participants, with a mean of 0.77 herbal agents per person; most frequently used were peppermint tea, cranberry juice, aloe, and other herbal teas.49 A retrospective chart review of eight U.S. health maintenance organizations (n = 152,531 women) from 1996 to 2000 found that 59% of the women received drugs other than vitamin or mineral supplements after their first prenatal care visit (39% in the first, 34.4% in the second, and 37.9% in the third trimester). Approximately one third of the women were on prescribed drugs for which there is evidence of some fetal risk (FDA category C, D, or X).8 More recently Mitchell and associates reviewed drug use during pregnancy between 1976 and 2008. They found that (1) average use of prescribed and over the counter drugs increased over these years from 2.5 to 4.2; (2) use of prescribed drugs increased 63% during the first trimester and by 2008, 94% of women took at least one prescribed drug; and (3) <1% of pregnant women took an antidepressant in 1988-90 versus 7.5% in 2006-08.89 Petersen et al. reported a four-fold increase in use of antidepressants during pregnancy from 1992 to 2006.104
Adherence to prescribed drug regimens is a problem during pregnancy, with up to 50% of women either not taking a prescribed drug or stopping before receiving a full course of the drug.27,72,104 This can lead to maternal risks, especially in women with chronic diseases.72,104 Peterson et al. found that antidepressants were more likely to be discontinued by pregnant than nonpregnant women, often leading to increased depression and other complications.104 Problems with adherence stem primarily from concerns about teratogenic effects. Most women overestimate the teratogenic risks of common agents.69 For example, in one study women personally estimated that the risk of having a baby with a birth defect was 25% (which is the risk associated with a potent teratogen such as thalidomide). After counseling, however, they more correctly estimated the risk at 5%.69 The background risk of a birth defect for the general population is 3% to 5%. However, the risk may be higher or lower for an individual woman depending on factors such as her health status, teratogen exposures, and genetic makeup.
Several principles have been identified to guide drug therapy during pregnancy: (1) avoid medications in the first trimester whenever possible; (2) anyone prescribing drugs for a woman of childbearing age must consider a potential pregnancy before prescribing; (3) women receiving long-term drug therapy require counseling before pregnancy of the potential implications and risks; (4) a necessary treatment should not be stopped without good reason; (5) drugs may have altered effects during pregnancy (e.g., the increased metabolism of methicillin or increased renal elimination of digoxin may result in decreased levels, resulting in the need for higher doses); (6) use single action, short-acting medications rather than long-acting or combination drugs; (7) use the lowest effective dose of the safest available medication; (8) use drugs only if the benefits outweigh the risks; (9) use alternate routes of administration if available (i.e., topical or inhaled rather than systemic agents); (10) select drugs that have a history of use during pregnancy without adverse effects, rather than the “latest” drug; and (11) use doses at the low end of the normal dose range while recognizing that for some drugs (e.g., digoxin, phenytoin, lithium), the woman’s usual dose may need to be increased due to increased volume of distribution (Vd) and clearance of these agents during pregnancy.73,109 These principles also apply to all women of childbearing age and to preconceptional and interconceptional use of drugs, in that approximately half of all pregnancies are unplanned and critical developmental processes occur before most women realize they are pregnant.73
Alterations in drug absorption, distribution, metabolism, and excretion during pregnancy
Maternal responses to drugs during pregnancy are influenced by both maternal physiology and the presence of the placental-fetal unit.81 Variations in handling of specific drugs by the pregnant woman may occur due to effects of the normal physiologic changes of pregnancy on drug absorption, distribution, metabolism, and excretion (Table 7-2). The presence of the placental-fetal compartment can also alter maternal kinetics.81 For example, many antibiotics are lipid-soluble and may readily cross the placenta and become sequestered in the fetal compartment and thus be unavailable to the mother. This can lead to lower maternal levels.
Table 7-2
Influence of Pregnancy on Physiologic Aspects of Drug Disposition
| PHARMACOKINETIC PARAMETER | CHANGE IN PREGNANCY |
| ABSORPTION | |
| Intestinal motility | Decreased |
| Ventilation | Increased |
| Cardiac output | Increased |
| Blood flow to skin | Increased |
| DISTRIBUTION | |
| Plasma volume | Increased |
| Total body water | Increased |
| Plasma proteins | Decreased |
| Body fat | Increased |
| METABOLISM | |
| Hepatic metabolism | ± |
| Extrahepatic metabolism | ± |
| EXCRETION | |
| Uterine blood flow | Increased |
| Renal blood flow | Increased |
| Glomerular filtration rate | Increased |
| Ventilation | Increased |
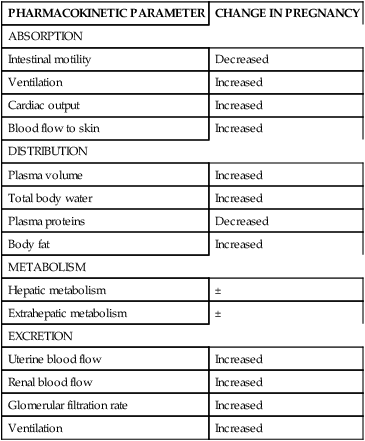
From McClary, J., Blumer, J.L., & Aranda, J.V., et al. (2011). Developmental pharmacology. In R.J. Martin, A.A. Fanaroff, & M.C. Walsh (Eds.). Fanaroff and Martin’s neonatal-perinatal medicine: Diseases of the fetus and infant (9th ed.). Philadelphia: Mosby Elsevier, p. 711.
Evidence regarding effects, effectiveness, and safety of most pharmacologic agents during pregnancy is limited.69,80,87 Pharmacokinetic data on specific drugs during pregnancy is often minimal because pregnant women are excluded from drug trials. As a result, many drugs are not labeled for use in pregnancy, because the effects of the drug on the fetus are unknown.70,131 This is protective for the fetus but may also limit benefits to the pregnant women from these drugs. The net result of changes in drug handling during pregnancy depends on the individual drug, with both increases in levels (with need for a lower dose or less frequent dosing) and decrease in levels (with a need for a higher dose or more frequent dosing) seen.78 In general, both peak and steady-state serum concentrations are lower or unchanged in the pregnant woman.20 Although changes may not be clinically significant the efficacy and toxicity of drugs can be harder to predict, because of the physiologic changes during pregnancy that have the potential to alter pharmacokinetics.20,57
Little analyzed data on pharmacokinetics of drugs in pregnancy and found that across all agents (1) drug oral bioavailability decreased during pregnancy in 47% of studies of different agents; (2) Vd increased in 39%, with no change in 48%; (3) peak plasma levels decreased in 33%, with no change in 57%; (4) steady-state plasma levels decreased in 45%, with no change in 45%; (5) half-life decreased in 41%, with no change in 44%; (6) clearance increased in 55%, with no change in 34%; and (7) protein binding decreased in 86%.78 Therefore during pregnancy, there are considerable variations in the direction of pharmacokinetic changes among different drugs. This increases the complexity for the practitioner in choosing a specific pharmacologic agent and monitoring its effects. Many studies have significant methodologic problems, for example, sample sizes may be small, the composition of the control group may vary (e.g., the group may include nonpregnant women, adult males, or the same subjects postpartum), women of various gestational ages may be grouped together, and dosing methods may vary.36,78 Pregnancy drug registries have been established to collect experiences with specific drugs or groups of drugs (such as antiepileptic agents) during pregnancy and fetal and neonatal outcomes (see Box 7-2). Three types of drug registries have been established: individual academic sites; pharmaceutical drug company sites; and population-based sites (from individual countries or a group of countries).24,128
Drug absorption
Pulmonary, GI, and peripheral blood flow changes during pregnancy can alter absorption of drugs from the lungs, gut, and skin (see Chapters 9, 10, and 12). The increased minute ventilation that occurs in pregnancy can increase the rate of drug uptake across the alveoli.81,86 As a result, the pulmonary system may have a greater role in drug and metabolite excretion during pregnancy.91 Alveolar uptake of inhalation agents is also influenced by changes in cardiac output, which leads to increased pulmonary blood flow and thus increased alveolar uptake. As a result, doses of volatile anesthetics (e.g., halothane, isoflurane, methoxyflurane) may need to be decreased to compensate for these changes in the pregnant women.81,102 Inhaled aerosols such as anti-inflammatory and bronchodilators may have enhanced absorption.106
Absorption of oral medications is influenced by gastric acidity, gastric motility, presence of bile acids or mucus, nausea and vomiting of pregnancy, and intestinal transit time. During pregnancy there is decreased gastric acid (hydrochloric acid [HCl]) secretion, altered intestinal emptying time, increased mucus, and decreased gastric and intestinal motility.36,106 Decreased gastric motility can increase the oral bioavailability of slowly absorbed drugs (such as digoxin) by prolonging transit time, and can decrease peak plasma levels of rapidly absorbed drugs. Factors altering GI function in pregnancy may initially delay, then prolong absorption of oral medications. Peak plasma levels may be lower, and the time to peak levels may be later than in nonpregnant women. These changes tend to peak in the third trimester.68 Changes in pH affect ionization, and absorption of drugs.36,81 For example, decreased gastric pH slows the rate at which weak acids (e.g., aspirin) are absorbed and increases absorption of weak bases (e.g., narcotic analgesics).102 Decreased GI motility may alter initial absorption of oral antibiotics and lead to unpredictable patterns of intestinal absorption. Absorption of hydrophilic drugs may be enhanced due to the longer intestinal transit time (from delayed intestinal emptying and decreased motility).83 Increased cardiac output and peripheral blood flow may increase the rate of absorption of drugs from the stomach and small intestine.78 Iron chelates with some drugs taken concurrently, preventing absorption. However, even with these changes in GI function, the clinical effect for most drugs is not significant. This is because therapeutic windows are usually wide enough for drugs administered orally that these factors do not significantly alter therapeutic effects.91
Cardiovascular and integumentary changes during pregnancy may lead to more rapid absorption of agents delivered via transdermal, intranasal, intravascular, epidural, and subcutaneous routes.102 The increased extracellular water and peripheral blood flow to the skin during pregnancy may enhance absorption and alter distribution of topical agents. For example, topical and vaginal iodine-based preparations are not recommended during pregnancy because of the increased absorption of iodine. Iodine is of concern because it is actively transported across the placenta to the fetus, whose thyroid gland has a high avidity for this substance (see Chapter 19). Increased peripheral perfusion secondary to decreased peripheral vascular resistance and vasodilation may increase absorption of intramuscular drugs.36
Drug distribution
The distribution of drugs within the body depends on many factors, including the amount of body water, which affects Vd; degree to which a drug is bound to plasma proteins or body tissues; and the presence of the fetal-placental unit. For many drugs the Vd is increased during pregnancy because of the increased plasma volume, blood volume, cardiac output, total body water, and body mass.20,36,91 The increased plasma and blood volumes, and thus Vd, may result in decreased drug levels in the central compartment, reduce serum levels of drugs, and necessitate larger loading doses.83,103 Increases in total body water during pregnancy affect primarily water-soluble (polar) drugs that tend to stay in the extracellular space. Initial doses may need to be increased to achieve therapeutic concentrations; however, maternal doses for most drugs are not significantly altered since drug steady state is influenced by bioavailability and clearance as well as Vd.68 Due to changes in the Vd, some pregnant women may notice that their prepregnancy dose of a drug taken regularly, such as digoxin or phenytoin, may not achieve the same therapeutic effect during pregnancy.
Some lipid-soluble drugs (e.g., caffeine, diazepam, thiopental) have a longer half-life in the pregnant woman, whereas polar drugs (e.g., oxazepam, ampicillin) tend to have a shorter half-life.52,130 The Vd for lipophilic drugs is increased during pregnancy because of the accumulation of fat, which may serve as a reservoir for some drugs via tissue binding.36,78 Tissue binding is a mechanism by which a drug is removed from circulation and stored in tissue such as hair, bone, teeth, and adipose tissue. This mechanism can result in storage of significant quantities of a drug, because tissue storage sites may need to be saturated before there is sufficient free drug to be effective at receptor sites. Factors reducing maternal serum drug levels increase the risk of subtherapeutic drug levels. When the drug is discontinued, tissue deposits may give up their stores slowly, resulting in persistent drug effects. Because lipid-soluble drugs are stored in adipose tissue, the increased adipose tissue during pregnancy can lead to a slight decrease in the amount of free lipid-soluble drugs, such as sedatives and hypnotics, and persistence of drug effects (“hangover”) after the drug has been discontinued.
Drugs in plasma are either unbound (free) or bound to plasma proteins, primarily albumin or α1-acid glycoprotein. The reduction in plasma proteins, especially albumin, during pregnancy may increase plasma levels of free (active) drug. Albumin is the major binding protein for acidic drugs such as salicylates, anticonvulsants, nonsteroidal anti-inflammatory agents, and some neutral drugs such as warfarin and diazepam.81 Serum albumin levels fall during pregnancy, increasing the unbound or free drug fraction. Total plasma drug concentration stays the same during pregnancy, but the unbound fraction of albumin-bound drugs is greater; therefore the drug may be cleared faster or transported more rapidly across the placenta.81 Increases in free drug do not necessarily lead to an increase in plasma levels of that drug if the increase in free drug is balanced by more rapid hepatic biotransformation or renal elimination.36,81,102 However, with regard to doses of some drugs that are bound to albumin (such as many antiepileptic drugs), total and free drug concentrations may be altered, and doses may need to be adjusted during pregnancy. For example, phenytoin total plasma concentration is decreased by 55% to 65% and levels of free drug by 18% to 31% during pregnancy; carbamazepine total concentration is reduced 0% to 43% and free drug 0% to 28%; phenobarbital total concentration is reduced by 50% to 55%.102 Thus when monitoring serum concentrations of drugs such as phenytoin during pregnancy, free versus total levels should be evaluated (many laboratories report free levels if specified when ordering).36
Free fatty acids and steroid hormones, both of which are increased in pregnancy, may compete with drugs for albumin binding sites, further increasing levels of unbound drug. The decrease in albumin, coupled with increases in free fatty acids and steroid hormones, are most prominent in late pregnancy. This change can result in unpredictable transient increases in free levels of some drugs, such as phenytoin, valproate acid, carbamazepine, sulfisoxazole, theophylline, and phenobarbital as well as the potential for more rapid elimination increases the risk of subtherapeutic maternal levels.36,46 α1-Acid glycoprotein is the major protein for binding basic drugs such as local anesthetics, most opioids, and β-blockers. α1-Acid glycoprotein is decreased slightly during pregnancy.6,106
Changes in protein binding during pregnancy not only influence availability of free drug in the mother but also the amount of drug available to cross the placenta. This may alter fetal-to-maternal drug ratios, thus further altering placental transfer. Drugs with hepatic blood flow–limited biotransformation (e.g., propranolol, lidocaine) are rapidly eliminated in their first pass through the liver, regardless of whether they are bound or unbound.20,103 Therefore even with the decreased protein binding in pregnancy, hepatic clearance and total drug concentration generally are not significantly altered, although concentrations of free drug, Vd, and half-life may increase. Conversely, with drugs whose hepatic biotransformation is independent of hepatic blood flow (e.g., warfarin, phenytoin), only the unbound fraction is removed by the liver. For these drugs, total drug concentrations may be lower than usual, with a decreased half-life and greater fluctuations in peak and trough levels.
Hepatic drug metabolism
The hepatic changes during pregnancy (see Chapter 12) can alter biotransformation of drugs by the liver and clearance of drugs from the maternal serum. Drugs that are primarily (greater than 70%) metabolized in their first pass through the liver (high extraction ratio) are usually cleared rapidly by the liver, because clearance of these drugs is dependent on hepatic blood flow. Hepatic elimination of drugs with high extraction ratios is generally unchanged in pregnancy. Drugs with low (less than 30%) first-pass hepatic clearance (low extraction ratio) are more dependent on liver enzyme systems. The rate of elimination of these drugs (e.g., theophylline, caffeine, and diazepam) is related to free drug levels and tends to be decreased in pregnancy with increased half-lives. Hepatic elimination of other drugs—including many antibiotics, pancuronium, phenytoin, and acetaminophen—is increased in the pregnant woman.
Some liver enzymatic processes may be slower during pregnancy, delaying drug metabolism and degradation; other processes have increased activity. The cytochrome P450 (CYP450) monooxygenase system is a group of phase I enzymes essential for hepatic metabolism of drugs. The enzymes of this system are indicated by the prefix CYP (cytochrome) followed by a series of numbers and letters, (e.g., CYP1A2, CYP3A4, CYP2D6). Activity of the following are increased during pregnancy: CYP3A4 (50% to 100%), CYP2A6 (54%), CYP2D6 (50%), and CYP2C9 (20%).5,57,72,123 Up regulation of CYP2C9 decreases plasma concentrations and increases clearance of phenytoin, nonsteroidal anti-inflammatory agents and some oral antidiabetic agents.57,68 Changes to CYP3A4 may increase the metabolism of methadone, nifedipine, and indinavir during pregnancy.102,123 However, activity of CYP1A2, which is responsible for hepatic elimination of many drugs, is decreased (33% in the first, 50% in the second, and 65% in the third trimester); activity of CYP2C19 is also decreased (50%).5,6,78,123 CYP1A2 is also involved in clearance of caffeine so decreased activity may lead to slower clearance and enhance effects during pregnancy.72,102
Activity of phase II enzymes is also altered during pregnancy, including some of the uridine 5′-diphosphatase glucuronosyltransferase (UGT) enzymes.5,102,106 For example, UGT1A4 is increased 200% in the first and second trimesters and 300% in the third trimester; UGT2B7 is increased 50% to 200% by the third trimester.6,123 Some extrahepatic enzymes, particularly cholinesterase, are also decreased, which may alter the woman’s response to neuromuscular agents.28,36,131 For example, the decrease in pseudocholinesterase during pregnancy has been enough to impair breakdown of suxamethonium with subsequent prolonged paralysis after anesthesia in some women. The relative increase in hepatic metabolism and blood flow during pregnancy (as a consequence of the increased cardiac output), may increase clearance of polar drugs.36 In general, slowly metabolized drugs that are cleared primarily by the liver tend to be cleared more slowly in pregnancy due to decreased enzymatic activity and the net decrease in liver blood flow. This may also increase the length of time potentially teratogenic intermediary metabolites remain in circulation.
Clearance and metabolism of drugs by the liver during pregnancy may also be influenced by the increases in steroid hormones that stimulate hepatic microsomal enzyme activity. For example, progesterone may induce enzyme activity for phenytoin clearance, whereas progesterone and estradiol alter hepatic elimination of caffeine and theophylline by competitive inhibition of microsomal oxidase.36,81 Estrogens have cholinergic effects and may interfere with clearance of drugs such as rifampin that are excreted into the biliary system.81 Smoking and alcohol may also induce hepatic enzymes.
Renal drug excretion
Many drugs are excreted primarily by the kidneys. Thus the increased glomerular filtration rate (GFR) in pregnancy (see Chapter 11) alters renal excretion of drugs, especially water-soluble drugs such as penicillin G, digoxin, aminoglycosides, lithium, and pancuronium. These drugs are eliminated more rapidly, leading to lower and potentially subtherapeutic blood and tissue levels.36,78,91 These effects are most prominent in late pregnancy.72 In many cases the changes are not clinically significant enough to require alteration in drug dose.36 However, this is not always the case. For example, pregnant women with paroxysmal atrial tachycardia may experience an increased frequency of attacks when taking usual doses of digoxin. These women may require up to a 50% increase in their digoxin dose during pregnancy.81 In addition, shorter dosage intervals are recommended for nifedipine and labetalol due to a threefold increase in clearance in the pregnant woman.81 The half-life of atenolol, digoxin, and peripheral myorelaxants is shorter during pregnancy.102 Because lithium is eliminated entirely by GFR, the pregnant woman may require a higher dose of this drug, especially during the third trimester.88 More rapid clearance and shorter half-lives are also seen with many penicillins, cephalosporins, aminoglycosides, and sulfonamides as a consequence of the changes in renal function during pregnancy.102 Morphine metabolites may be cleared more quickly in late pregnancy and may lead to a lowered analgesic effect.72
Alterations in drug absorption, distribution, metabolism, and excretion during the intrapartum and postpartum periods
Drug handling is further altered during the intrapartum period. GI absorption may decrease to a greater extent than seen in pregnancy. Gastric emptying is reported to decrease during labor (see Chapter 12), although this may be secondary to use of opiate agents such as meperidine and pentazocine. Administration of antacids to decrease the risk of acid aspiration with anesthesia can alter ionization by altering gastric pH, prolonging gastric emptying, or complexing with drugs, reducing their absorption. Cardiac output increases in the intrapartum period further increase the Vd and may transiently alter hepatic and renal blood flow.101 Blood flow to the lower extremities may be reduced, especially if the woman is in a supine position. This may result in a slower and more unpredictable rate of absorption of drugs given intramuscularly in the buttock or thigh.101 Plasma nonesterified fatty acids rise during labor and may compete for albumin binding sites, with an increase in free drug levels.101 Both supine positioning and oxytocin may reduce renal blood flow and delay renal excretion of drugs and drug metabolites during labor. Most analgesics and anesthetics used in labor, especially opioids, tend to be low-molecular-weight, lipid-soluble, weak bases that enhance movement across the placenta to the fetus once they enter maternal circulation. The clinical effects depend on the timing of the dose, half-life, drug physicochemical characteristics, placental blood flow, and maternal and fetal drug metabolism and excretion.79
During the immediate postpartum period, the pattern of pharmacokinetics and drug distribution is similar to that of late pregnancy and remains so until the pregnancy-induced physiologic alterations of each system return to nonpregnant status.42 During this transition, drug handling by the woman may remain altered to some extent for some drugs. Transfer of drugs to nursing infants is discussed later in this chapter in “Drugs and Lactation.”
Transfer of drugs across the placenta
Dancis suggested that in thinking of placental transfer one should “Ask not whether a maternal [substance] crosses the placenta. Ask rather, how, how much, and how fast. Ask also as to fetal need.”35 There are few compounds, endogenous or exogenous, that are unable to cross the placenta in detectable amounts given sufficient time and sensitivity of detection.20,23,35,87,91 Drugs with similar physiodynamic properties can differ in placental transfer.87,105 Plonait and Nau suggest that if no other information is available and a group of drugs is not known to have teratogenic effects, then a comparison of lipid solubility, molecular weight (MW), ionization, and degree of protein binding might assist the clinical selection of an appropriate drug for more efficient transfer (fetal drug therapy) or least efficient transfer (limited fetal exposure).105 For example, drugs that are lipid soluble have an extremely low (50 daltons) or low (51 to 599 daltons) MW, and are nonionized with low protein binding in maternal plasma cross the placenta more rapidly and in higher concentrations than drugs that are water soluble (polar), have a high MW (greater than 600 daltons), and are ionized and highly bound to protein in maternal plasma. Transfer of drugs across the placenta varies with developmental changes in the placental, and thus gestational age.121
Patterns of transfer kinetics
Three major patterns of transfer pharmacokinetics are seen: (1) complete transfer (drugs rapidly equilibrate between mother and fetus, with blood flow being the rate limiting step); (2) exceeding transfer (levels are higher in the fetus than in the mother); and (3) incomplete transfer (levels are higher in the mother than the fetus).99 These patterns are illustrated in Figure 7-2. Examples of drugs with each pattern are listed in Table 7-3.
Table 7-3
Examples of Drugs with Transplacental Kinetics*
| MODEL A OR B | MODEL C | MODEL D | MODEL E | MODEL F |
| Some barbiturates: Thiopental Pentobarbital Secobarbital Antipyrine Promethazine Ritodrine Magnesium sulfate Digoxin |
Some benzodiazepines: Diazepam Lorazepam Desmethyl-diazepam Oxazepam Valproate Salicylate Nalidixic acid Nicotine Urea Some penicillins: Ampicillin Penicillin G Methicillin |
Ascorbate Colistimethate† Furosemide Meperidine Some cephalosporins: Cephalothin Cefazolin Cephapirin Cephalexin Some aminoglycosides: Gentamycin Kanamycin |
Amide-type local anesthetic agents: Lidocaine Bupivacaine Some β-adrenoceptor blockers: Propranolol Sotalol Labetalol Dexamethasone Cimetidine Ranitidine Methadone Some sulfonamides‡ |
Heparin Quaternary ammonium compounds: Tubocurarine Succinylcholine Vecuronium Pancuronium Elementary ions: Chlorthalidone Dicloxacillin Erythromycin Nitrofurantoin |
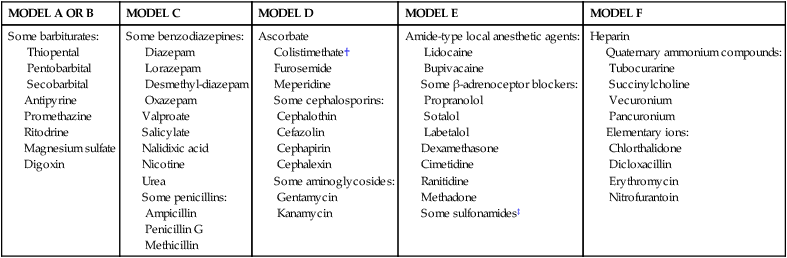
*According to models A through F in Figure 7-2. Differentiation between models A and B, C and D, and E and F is often uncertain because of incomplete data on the initial phase of drug distribution across the placenta. Model B is rarely applied.
†Polypeptide antibiotic, with a molecular weight of 1200 daltons.
Modified from Plonait, S.L. & Nau, H. (2012). Physiochemical and structural properties regulating placental drug transfer. In R.A. Polin, W.W. Fox, & S.H. Abman (Eds.). Fetal and neonatal physiology (4th ed.). Philadelphia: Saunders.
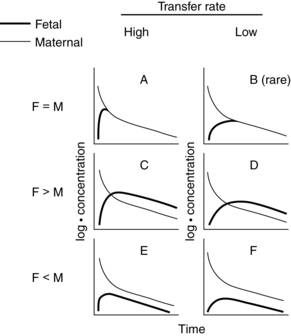
There is a great deal of variability even among drugs with similar pharmacokinetic characteristics. However, in general, complete transfer tends to be seen with drugs that have a high pKa and MW less than 600 daltons, whereas incomplete transfer is seen with drugs that have a high pKa and MW greater than 600 daltons. Strongly ionized drugs generally have incomplete transfer, although exceptions occur. For example, ampicillin and methicillin are strongly ionized but have lipophilic side groups, so they have a complete transfer pattern.98,101 Valproate and salicylates are ionized at the normal (physiologic) pH level but rapidly cross the placenta.81
Factors influencing placental transfer

Placental transfer is influenced by the following: (1) maternal and fetal drug handling; (2) surface area of the placenta; (3) diffusing distance between maternal and fetal blood; (4) physicochemical characteristics of the drug; (5) gradients (drug concentration, electropotential [normally 20 mV], and pH) between mother and fetus; (6) degree of binding of a substance to proteins in maternal and fetal circulations; (7) concentrations of binding proteins and protein binding gradients between mother and fetus; (8) permeability of the placental barrier (primarily the syncytiotrophoblast); (9) rates of maternal and fetal blood flow through the intervillous space and villi; and (10) placental influx and efflux transporters (Table 7-4).58,68,86,98,99,102,126
Table 7-4
Factors Influencing the Distribution of Drugs and Other Substances in the Fetus
| FACTOR | POSSIBLE INFLUENCES |
| Type of drug | Factors that increase transfer include lipid solubility, low molecular weight (less than 500 to 600 daltons for lipid soluble and less than 100 daltons for polar substances), nonioinized, and unbound. |
| Amount of drug | Transfer is increased by greater maternal-to-fetal gradient, especially for drugs transferred by diffusion. |
| Membrane permeability | Diffusion of substances increases with increasing gestation and greater placental efficiency. Some drugs have greater affinity for specific fetal tissues (e.g., tetracycline in teeth, warfarin in bones, phenytoin in the fetal heart [because this is a highly lipid organ in the fetus, whereas in adults the central nervous system has a high lipid content owing to myelin sheaths], streptomycin in the optic nerve). |
| Fetal body water compartment | Drug distribution and dilution change as the total body water compartment decreases with increasing gestation. With an increased volume of distribution, peak volumes of drugs are reduced and excretion may be delayed. |
| Fetal circulation | Maternal and fetal blood flow rates will influence transfer. Upon reaching the fetal circulation, drugs may be shunted by the ductus venosus past the liver (thus missing an opportunity for detoxification), with highest concentrations of these substances in blood going to the heart and upper body. |
| Serum protein binding | Protein binding of a drug in the maternal system limits the amount of free drug available for transfer to the fetus. Binding of drugs to macromolecules in the fetal circulation may increase the maternal-to-fetal transfer by maintaining a concentration gradient from the mother to the fetus. |
| Receptor function | Functional ability of receptors on cell membranes increases with gestation, leading to increased specificity to respond to or exclude certain drugs. |
| Placental enzymes | Enzymes produced by the placenta (e.g., insulinase) may detoxify drugs and reduce transfer to the fetus. |
| Gestational age | Many of the factors identified above are altered with advancing gestation and maturity of the fetus and placenta. The size and efficiency of the placental exchange area increase with increasing gestational age. |
| Fetal pH during labor | The fetal pH is usually 0.1 to 0.15 units below that of the mother. A decrease in the fetal pH may increase the transfer of acidophilic agents from the mother to fetus. Hypoxia alters blood flow and thus drug distribution, metabolism, and excretion. For example, with hypoxia, blood flow to the liver and kidneys may be reduced with preferential flow to the brain. Albumin binding of drugs may also be reduced, resulting in more free drug in the fetal circulation. |
Diffusing distance and surface area.
Increased surface area for exchange, increased concentration gradient, and decreased diffusing distance enhance transfer across the placenta. With increasing gestation and placental growth, efficiency of the placental transfer increases as a result of the increased surface area. As the placenta matures, the distance between maternal and fetal blood decreases due to thinning of the syncytiotrophoblast and connective tissue and increases in the size and number of capillaries in the villi (see Chapter 3). Thus more drug passes to the fetus in the third trimester than in the first trimester. Transfer may be reduced with decreased surface area (small placenta, placental infarcts) or increased diffusing distance (placental edema, infection). Edema with fetal hydrops increases the diffusing distance and decreases transfer so drugs such as digoxin may be less effective in treating arrhythmias in the fetus with hydrops.71
Physiochemical characteristics and concentration gradients.
Physiochemical characteristics that influence movement across the placenta include lipid solubility, MW, degree of ionization, and protein binding. These characteristics can increase, decrease, or deter movement of potentially harmful drugs and other substances from the maternal to the fetal circulation.105 For example, placental transfer may be increased or enhanced if a substance is lipid soluble (e.g., diazepam, lipoproteins, most sedatives) or nonionized (e.g., phenobarbital), has a low MW (e.g., alcohol), or has a lower albumin binding (e.g., ampicillin). A substance will have reduced, slower, or no transport across the placenta if it has a certain charge or molecular configuration (e.g., heparin) or certain size (e.g., heparin, IgM), is altered or bound by enzymes within the placenta (e.g., amines, insulin), or is firmly and highly bound to the maternal red blood cell (e.g., carbon monoxide) or plasma proteins (e.g., dicloxacillin).
Lipid-soluble substances cross the placenta more readily than water-soluble substances, because cell membranes are made primarily of lipids that enhance diffusion. Lipid-soluble substances dissolve in the lipid membranes of the tissues separating maternal and fetal blood, primarily the syncytiotrophoblast. Polar substances cross slowly unless they have low MW and can move through pores. If the syncytiotrophoblast has a high lipid affinity for a drug (e.g., buprenorphine), the drug may be sequestered in the placenta with decreases in both maternal and fetal concentrations.120
The exact MW limit for placental transfer is unknown, although transfer of substances with MW >500 to 600 daltons is limited.105 Insulin (with an MW of more than 6000) does not cross the placenta because of its size and configuration and because of the effects of placental insulinase enzymes.36,99 Substances with higher MW tend to cross more slowly and are influenced by maternal concentrations; for example, if maternal concentrations are high, the substance is more likely to cross. These substances may also cross via active transport if a carrier is present. Immunoglobulin G (IgG) with an MW of 150,000 daltons is transported across the placenta via endocytosis and exocytosis (see Chapters 3 and 13).37 Water-soluble substances with an MW of less than 100 daltons diffuse readily; those with an MW greater than 100 diffuse more slowly. Small lipid-soluble substances (MW less than 500 to 600 daltons) cross rapidly and are blood flow–dependent; those with an MW of more than 500 to 600 daltons cross more slowly and may have incomplete transfer.20,91,120 However, MW is of limited value clinically in that most drugs have MWs less than 600 daltons. Exceptions include thyroid hormones (MW 889), insulin (MW greater than 5000), heparin (MW 20,000 to 40,000), and low-MW heparin (MW 3000 to 5000).52,68,99,102
Maternal-to-fetal concentration or electrochemical gradients also influence transfer. The maternal-fetal pH gradient influences transfer of drugs and can result in “ion trapping,” with higher concentrations of some drugs in the fetus than in the mother. Nonionized substances cross the placenta more efficiently than ionized substances.36 Normally the fetus and amniotic fluid are slightly more acidotic than the mother by 0.1 to 0.15 pH units.99 As a result of this difference, basic drugs may accumulate in the fetus and amniotic fluid with levels higher than in the mother. In the mother, a weak base tends to remain nonionized in her plasma and cross the placenta rapidly. As this weak base comes into contact with the more acidic fetal blood, the drug becomes more ionized, increasing the flow gradient from mother to fetus. The now-ionized drugs cannot return across the placenta to the mother as readily, thus becoming trapped in the fetus.20,81,99,120 For example, most opioids are weak bases that may accumulate in the fetus, which is one reason that effects of these drugs may persist in the neonatal period. The opposite effect is seen with acidic drugs: that is, these drugs are more likely to be ionized in the mother and thus not cross the placenta as readily.20 For nonionized, lipophilic drugs, the rate-limiting step for placental transfer is blood flow; for polar drugs, the rate-limiting step is diffusion.81 Hypoxia and acidosis in the fetus can influence transplacental passage of drugs. For example, local anesthetics are weak bases whose degree of ionization depends on pH. If the fetus becomes acidotic, ionization of these drugs increases with accumulation (“ion trapping”) in the fetus and neonate.47,99,102
Drug metabolites also cross the placenta to the fetus and vice versa. The majority of fetal drug elimination is by transfer back to the mother across the placenta. However, some elimination may occur by fetal metabolism, placental metabolism, or excretion into amniotic fluid.91 This transfer is also based on physicochemical characteristics, primarily ionization and lipid solubility. Most drug metabolites are polar, because polarity enhances biliary and renal elimination. Once polar substances are transferred to the fetus, elimination back across the placenta is slow and may result in accumulation in the fetus or amniotic fluid.91 In addition the fetus may metabolize a drug producing polar metabolites. These metabolites can also become trapped and accumulate in the fetus or be excreted into amniotic fluid and reswallowed by the fetus. Examples of drugs that tend to concentrate in amniotic fluid are the β-lactams, such as penicillin G, dicloxacillin, and amiodarone.36,91
Protein binding.
Drugs that are protein bound, usually to albumin or α1-acid glycoprotein, in the mother or fetus do not cross the placenta. Therefore plasma protein gradients between mother and fetus can also influence transfer. For example, ampicillin has a lower protein binding to albumin, thus increasing the amount of free drug that can cross to the fetus, and resulting in decreased maternal levels. Protein binding of dicloxacillin is 96%. Therefore dicloxacillin would be an appropriate choice to treat maternal infections, but not to treat intrauterine or fetal infection. Another example is propranolol, which is highly protein bound so it crosses much slower than atenolol, which has minimal binding.71
Maternal albumin levels fall during pregnancy and are about 15% to 20% lower than those in the fetus at term; α1-acid glycoprotein levels in the fetus are only about 37% to 40% of maternal levels.91,99,120 Thus at term, albumin (or fetal albumin macromolecule) bound drugs may be more highly bound in the fetus, whereas α1-acid glycoprotein drugs are more highly bound in the mother.91 Binding to proteins in the fetus may increase transfer of the drug by maintaining a concentration gradient across the placenta. Plasma protein binding and thus concentrations of drugs in the mother versus the fetus changes with increasing gestational age for some drugs. The decreased maternal albumin with increasing gestation increases the amount of free drug in the mother available for placental transfer. For example, diazepam binds to albumin. Albumin binding of diazepam is low in the fetus in the first half of gestation. However, in late gestation, as maternal serum albumin levels decrease, more free diazepam is present in maternal serum and, because this is a lipophilic drug, readily crosses the placenta. Thus by term, fetal diazepam levels are greater than maternal levels (Table 7-5).105 This may increase the risk of neonatal hyperbilirubinemia in that diazepam competes with bilirubin for albumin binding (see “Neonatal Pharmacokinetics”).
Table 7-5
| DRUG | PRIMARY BINDING PROTEIN | BOUND IN MOTHER (%) | BOUND IN FETUS (%) | FETUS/MOTHER TOTAL PLASMA CONCENTRATION RATIO |
| Betamethasone | AGP | 60 | 41 | 0.33 |
| Bupivacaine | AGP | 91 | 51 | 0.27 |
| Diazepam | ALB | 97 | 98.5 | 1.6 |
| Lidocaine | AGP | 64 | 24 | 0.66 |
| Mepivacaine | AGP | 55 | 36 | 0.71 |
| N-desmethyl diazepam | ALB | 95 | 97 | 1.7 |
| Phenobarbital | ALB | 41 | 36 | 1.0 |
| Phenytoin | ALB | 87 | 82 | 1.0 |
| Salicylate | ALB | 43 | 54 | 1.2 |
| Valproic acid | ALB | 73 | 88 | 1.7 |
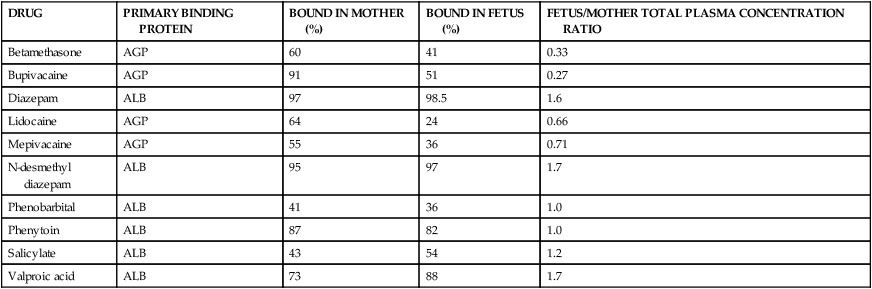
AGP, α1-Acid glycoprotein; ALB, albumin.
From Plonait, S.L. & Nau, H. (2012). Physiochemical and structural properties regulating placental drug transfer. In R.A. Polin, W.W. Fox, & S.H. Abman (Eds.). Fetal and neonatal physiology (4th ed.). Philadelphia: Saunders.
Placental blood flow.
The rate of maternal blood flow to and through the intervillous space and fetal blood flow to and through the villi influence placental transfer, particularly of nonionized, lipophilic substances such as anticonvulsants, alcohol, and opioid derivatives. Contractions decrease transfer so drugs that cross rapidly and are given during a contraction may demonstrate decreased transfer to the fetus. Contractions may also delay clearance of drugs from the fetus.120 In addition to uterine contractions, factors that may alter uteroplacental blood flow include maternal position; anesthesia; nicotine, β-lactams, ototoxics and other drugs; emotional or physical stress; and degenerative changes within the placenta that are seen with hypertension, prolonged pregnancy, diabetes, or renal disease.71
Route of administration.
The route of administration can also affect placental transfer. For example, nalbuphine that is given intramuscularly results in fetal blood levels that are 80% of maternal venous levels; if it is given intravenously, fetal levels are three to six times greater than maternal levels.81,99 Peak drug concentrations in the fetus (and the infant, if delivered after maternal administration) do not necessarily occur at the same time as in the mother. For example, meperidine peaks at 30 to 50 minutes in the mother, but 90 minutes to 3 hours after maternal administration in the fetus as a result of transfer kinetics across the placenta. Therefore meperidine levels are highest in the infant if given to the mother 90 minutes to 3 hours before delivery; on the other hand, fentanyl is higher in the neonate than mother if given shortly before delivery. Therefore it is reasonable to give meperidine if birth is expected within the hour.
Antibiotics are some of the most commonly prescribed drugs in pregnancy. Because antibiotics cross the placenta primarily by simple diffusion, rate-limiting factors include maternal-fetal concentration gradients, MW, and other physicochemical characteristics of the drug, placental surface area, diffusing distance, and the degree to which the drug is bound to maternal plasma proteins (see Table 7-5). A general pattern of antibiotic distribution to the fetus has been described. After an intravenous dose, peak maternal serum concentrations are seen within 15 minutes then fall exponentially. Peak umbilical cord values are seen in 30 to 60 minutes followed by an exponential fall. Peak amniotic fluid levels are seen in 4 to 5 hours. This lag results from slow excretion in fetal urine due to fetal renal immaturity.114
Placental transporters.
Although many drugs cross the placenta by simple diffusion, some drugs use placental influx or efflux transporters. These transporters are located along the apical membrane of the microvillus brush border facing maternal blood in the intervillous space and on the syncytiotrophoblast basal membrane (facing the fetal connective tissue matrix and blood vessels in the villi) or fetal capillary epithelium to facilitate movement of nutrients and other physiologic substances across the placenta.44,58,91,98,102,106,120,126 Some drugs may also be transferred across the placenta by nutrient (influx) transporters, including glucose, carnitine, monoamine, and organic ion carriers.102 Nonphysiologic substances—some drugs, environmental pollutants, and toxins—if structurally similar to nutrients, may compete for nutrient transporters. The drugs that seem to be particularly able to use this method to cross the placenta include amphetamines, cocaine, cannabinoids, and nicotine and other substances from cigarettes.44,129 By successfully competing with nutrients for these transporters, nutrient transfer is reduced and fetal growth and development altered. In addition, cephalosporins, ganciclovir, and corticosteroids cross by facilitated diffusion, probably by using carriers that normally transport dipeptides, hormones, and steroids.98
The placenta also contains a variety of efflux transporters whose role is to remove steroids and other potentially toxic substances and thus protect the fetus.68,71,102,120,129,135 Originally most of the transporters were identified in studies of multidrug resistance in tumors as is reflected in some of their names. Examples of drug efflux transporters in the placenta include members of the APT-binding cassette (ABC) group such as multidrug-resistant gene protein (MRP) 1, commonly called P-glycoprotein (P-gp), MRP-2, MRP-3, and breast cancer resistance protein (BCRP).58,68,102,121,126,129,135 P-gp, MRP-2, and BCRP are located on the apical membrane of the syncytiotrophoblast cells facing maternal blood; MRP-1 and MRP-3 are on the basal or fetal side of the trophoblast (see Figure 7-3).47,68,126 Efflux transporters are also found on the gut, lung, and blood brain barrier and help protect entry of substances into the body and brain. Efflux transporters are under hormonal regulation, for example, BCRP by estradiol, and progesterone and P-gp by glucocorticoids.126 Inflammation and infection may alter regulation of these transporters.89
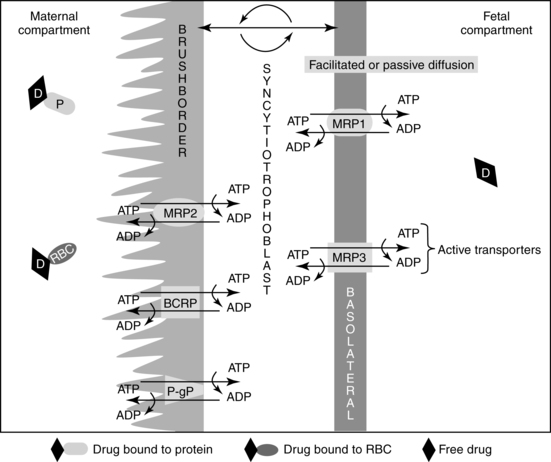
Each transporter appears to have specific substrates for which they provide protection. For example, studies of P-gp suggest that it exports cations, steroids, cytotoxic drugs, some antibiotics, opioids, phenobarbital, erythromycin, phenytoin, verapamil, and other substances that enter the placenta back into maternal blood, thus reducing fetal exposure.20,58,102,135 BCRP is involved in efflux of flavinoids, zidovudine, cimetidine, glyburide, nitrofurantoin, and the food-borne chemical carcinogen PhiP.58,68,102 MRP 1 and 3 prevent entry of organic anions and promote excretion and reduce entry of glutathione/glucuronide metabolites into the fetus.47 Some transporters may have bidirectional flow which can allow xenobiotics to be transported across the placenta to the fetus.68 Thus BCRP and P-gp cannot only decrease the amount of drug reaching the fetus but enhance removal of any drug reaching the fetal circulation.129 A drug that is a P-gp or BCRP substrate would be preferred for treating a maternal disorder with minimal fetal drug exposure, whereas a drug that is not a P-gp or BCRP substrate would be preferred for fetal pharmacotherapy.120
Deficiency of these transporter proteins increases the risk of teratogenesis from fetal drug and chemical exposure.58,135 Gene polymorphisms for the ABC efflux transporters may increase or decrease placental transfer of these substances and thus alter fetal susceptibility to effects of maternal drugs.58
Placental metabolism of drugs
The placenta has many enzyme systems, including various peptidases that can metabolize drugs, thus influencing transfer of specific substances from mother to fetus or fetus to mother.120 Both phase I and phase II drug metabolizing enzymes are present.106,129 Several isoforms of the CYP450 system (phase I reactions) are found in the placenta, although at levels about half of those seen in the liver. The primary CYP450 isoforms seen in the placenta are CYP1A1/1A2, although other isoforms, including CYP2E1, CYP3A4, CYP3A7, and CYP4B1, have also been reported, although with low activity.20,55,91,101,106,112,120 Expression of placental CYP1A1 increases with exposure to polychlorinated biphenyls (PCBs) or maternal cigarette smoking.120 Phase II reactions including UGTs are also present during most of gestation.102,129 UGT has a role in detoxifying endogenous and exogenous substances and in regulating steroid hormones produced in the trophoblast.129 Glutathione S-transferase may help protect the placenta and fetus from oxidative stress and is also involved in placenta hormone metabolism.129 These enzymes may increase with maternal nicotine or alcohol use, although alcohol dehydrogenase (a phase I reactant) is seen in low concentrations and is not inducible.20,106,120,129
Summary
The impact of maternal physiologic alterations on fetal drug transfer can be illustrated by examining pharmacokinetics and distribution of several drug classes, namely hypoglycemic agents, antibiotics, and anticonvulsants. For example glyburide, which has been used as an adjunct to insulin therapy in treating gestational diabetes, has minimal placental transfer because it is >99.8% protein bound with a short elimination half-life, rapid clearance, and rapid transfer from the placenta back to the mother by placental ABC efflux transporters thus minimizing fetal exposure.47,129
Serum levels and the half-life of many antibiotics are reduced during pregnancy, primarily because of a more rapid excretion associated with the increased GFR and expanded plasma volume.96 Most cephalosporins have a shorter half-life in late pregnancy, except for cefoperazone, which is excreted in bile rather than by the kidneys, so it has a similar half-life in pregnant and nonpregnant women.96 Penicillins have decreased serum levels and increased renal clearance.96
The increased GFR augments renal clearance of antibiotics, thus further reducing serum levels. Most antibiotics are lipid soluble and readily cross the placenta. As a result, dosages of some antibiotics may need to be increased or the dosing interval reduced.96,116 With most local infections the reduction in maternal serum concentrations probably does not alter therapeutic efficacy of the antibiotic. Pregnant women with acute infections or who need high serum concentrations may require higher doses than nonpregnant women. In these situations, maternal antibiotic levels must be carefully monitored to ensure that the mother is receiving an adequate therapeutic dose and to avoid maternal or fetal toxicity.
Drugs with lower protein binding such as ampicillin and penicillin are associated with increased levels of free drug that readily crosses into the fetal compartment.96 As a result, fetal and amniotic fluid levels are elevated and maternal serum and plasma levels decreased. The Vd and renal clearance are increased and the half-life of ampicillin in particular is reduced in the pregnant woman.96 Decreased maternal serum and plasma levels of aminoglycosides are also found in some women, increasing the likelihood of subtherapeutic levels. If aminoglycosides are used, drug blood levels (to evaluate the risk of subtherapeutic levels) and maternal renal function (to evaluate the risk of nephrotoxicity) require careful monitoring.96
Higher maternal serum levels and lower fetal and amniotic fluid levels are seen with the use of antibiotics with increased protein binding such as dicloxacillin (96%). As noted earlier, a drug such as dicloxacillin would be appropriate for treating a maternal infection but not effective in treating an intrauterine infection.131 Dicloxacillin and erythromycin (base or stearate) have limited placental transfer, so fetal levels are low. Maternal absorption, serum levels, and tissue levels of erythromycin may be somewhat erratic and unpredictable.96,131 Higher concentrations of antibiotics are found in the fetus and amniotic fluid after bolus administration versus continuous drip infusion to the mother and after multiple versus single doses.
A major factor affecting the pregnant epileptic is the effect of the usual physiologic changes of pregnancy on the metabolism of antiepileptic drugs (AEDs).26,54 As a result of these changes, the apparent plasma clearance of AEDs decreases with subtherapeutic plasma concentrations and increased risk of seizures. However, these changes vary between women and with different AEDs, leading to unpredictability in dosing regimens.103 The pregnant woman on AEDs may need to have the dosages of some medications increased by 30% to 50% above prepregnant levels.4 These changes occur predominantly during the second half of pregnancy and reverse by 6 weeks postpartum. As the physiologic alterations of pregnancy are reversed following delivery, drug levels may fluctuate rapidly, leading to toxicity. Drug levels are monitored regularly during pregnancy to ensure therapeutic dosages, with monitoring continuing after delivery until drug levels have stabilized. Phenytoin, phenobarbital, carbamazepine, valproic acid, primidone, and other AEDs have also been associated with teratogenic effects (see Teratogenesis).24 Valproic acid in particular appears to have a higher risk of major fetal anomalies than carbamazepine and possibly phenytoin and lamotrigine.53 Fetal anticonvulsant drug syndrome is multifactorial in etiology and characterized by various combinations of symptoms including an increased risk of congenital malformations, microcephaly, cognitive dysfunction (especially with valproic acid), and fetal growth restriction.48,53,103,128 In addition, some AEDs, including carbamazepine, valproic acid, and phenytoin, have been associated with an increase in facial dysmorphisms. The risks increase with increasing numbers of AEDs used, so monotherapy is recommended during pregnancy.48,53,103,128 Some agents may also interfere with folic acid increasing the risk of neural tube defects (see Chapter 15).54 The risks of AED therapy must be balanced against the risks to the mother and fetus of increased seizure activity.128
Pharmacokinetics in the fetus
Drug distribution, metabolism, and excretion in the fetus
Distribution, metabolism, and elimination of drugs are altered in the fetus. Factors that influence the effects of drugs in the fetus include individual drug characteristics, dosage, duration of use of the drug, genetic makeup of the mother and fetus (genetic susceptibility), maternal handling and elimination of the drug, timing in relation to stage of embryonic or fetal development, maternal physiology, and placental transfer. Placental transfer is affected by drug physicochemical characteristics (described in the previous section), blood flow, maturation, and metabolic activity. Placental transfer was discussed in the previous section (see “Transfer of Drugs Across the Placenta”).
Fetal drug distribution
Fetal drug distribution is influenced by individual drug characteristics, specificity of individual agents for certain fetal tissues, and receptor function. Some drugs have a greater affinity for a specific fetal tissue that is not necessarily the agent’s expected target tissue. Examples include the high affinity of tetracycline for fetal teeth, warfarin for the fetal bones, and phenytoin for fetal heart tissue. Receptor function of fetal cells increases with increasing gestational age.86 Receptors become more specific in their ability to respond to or exclude certain drugs, whereas in the embryo and young fetus, these receptors are less discriminating in excluding drugs, increasing the likelihood of teratogenic effects. Other factors influencing drug distribution in the fetus are gestational age, degree of serum protein binding, and fetal body water compartment and pH.86,102 Fetal pH is slightly lower than maternal, so weak bases (such as anesthetics, cocaine, meperidine, codeine, hydrocodone, erythromycin, barbiturates, and iodine) become more ionized in the fetus and may accumulate in the relatively more acidotic fetus (“ion trapping”).47,102 This phenomenon is more likely during the intrapartum period when fetal pH is further decreased.
The fetus has lower levels of serum proteins, increasing the amount of free drug. Fetal proteins may bind some drugs with greater affinity than in the mother (salicylates) and others (ampicillin, benzyl penicillin) with less affinity.81 Levels of α1-acid glycoprotein are up to three times lower in the fetus than in the mother with a fetal-to-maternal concentration ratio at term of 0.37.47,91,99,102 The maternal-to-fetal serum concentration ratio of albumin increases to term as fetal albumin concentrations increase and maternal levels decrease (due to hemodilution). Thus this ratio increases from 0.38 at 12 to 15 weeks to 0.66 at 16 to 25 weeks, 0.97 at 26 to 35 weeks and 1.2 after 35 weeks.102 Even as serum binding proteins increase with advancing gestational age, levels of free drug tend to remain high because of the lowered protein binding affinity. The fetus has an increased Vd due to increased total body water. This results in a reduction in peak volume of drugs, but also wider distribution of drugs in the body and delayed excretion. Factors influencing distribution of drugs and other substances within the fetus are summarized in Table 7-4.
Fetal drug metabolism and elimination
Drug metabolites are also excreted by the liver into bile and thus into meconium. Polar metabolites may accumulate in amniotic fluid, reaching levels higher than in either the mother or fetus. Examples of drugs that may accumulate in amniotic fluid are ampicillin, amoxicillin, cefuroxime, and cephalothin.99 Because most drug metabolites are more polar than the original drug, substances that cross the placenta to the fetus may not return across the placenta to the mother as readily. Therefore, these substances can accumulate in fetal tissues and at levels that may be higher than in the mother.36,47,81,102 Metabolites of some drugs that the fetus is exposed to in the third trimester, such as opiates and cocaine may also be found in fetal hair.
Fetal hepatic metabolism.
Immature phase I and II metabolism is present by about 8 weeks’ postconception.36 By the end of the first trimester, the fetal liver can oxidize some substances via the CYP450 system (phase I biotransformation) (see Table 7-1).112 The major fetal isoform is CYP3A7, a form found primarily in the fetal and neonatal liver, although it can also be found in the placenta and endometrium.17,86,102 CYP3A7 accounts for about 50% of the total fetal CYP450 content.55 Levels peak at 20 weeks, then decrease to term, decreasing rapidly after birth.102 CYP3A7 may protect the fetus by detoxifying steroids and retinoic acid derivatives and is involved in estriol biosynthesis.17,66,112 Fetal CYP3A7 has a lower catalytic activity than in adults; levels decrease markedly after birth.99,112 Other isoforms reported in the fetal liver (although in low quantities) include CYP1A1 (involved in metabolism of endogenous toxins) and probably CYP2E1 (found by the second trimester and involved in prenatal alcohol metabolism), CYP1B1 and CYP2D6.31,55,91,102,112 By term, CYP450 enzyme activity is about 30% to 60% that of adults, primarily due to CYP3A7 activity.55,102
Phase II hepatic biotransformation reactions (see Table 7-1) mature at different rates.85,112 Glucuronyl transferase activity is low at term, so substances dependent on this mechanism for elimination (e.g., salicylates or lorazepam) are metabolized much more slowly than in the adult. On the other hand, morphine can be glucuronidated in the first trimester, whereas sulfate conjugation is well developed at birth. Sulfation activity increases with gestational age and activity in infancy eventually exceeds that of adults.76,112
Lipophilic drugs are more dependent on hepatic metabolism for elimination than are polar (water soluble) substances. The reduced hepatic metabolism in the fetus may be an advantage because drug metabolites are polar and thus tend to slowly cross back to the mother. Lipophilic drugs that remain in that form for longer periods without being metabolized are more likely to cross back to the mother for elimination. Phase I metabolites from the CYP450 system generally slowly cross the placenta to the mother for elimination. Phase II metabolites are more polar, an advantage after birth because these are primarily eliminated by biliary excretion or renal elimination. In the fetus these metabolites have decreased placental transfer, so they tend to accumulate in the fetus and amniotic fluid.112
Fetal hepatic metabolism of drugs is limited by a decreased first-pass effect, because some of the blood coming to the fetus from the placenta via the umbilical vein bypasses the liver through the ductus venosus. Biochemically, the left and right lobes of the fetal liver function independently of each other. The more highly oxygenated blood in the umbilical vein coming from the placenta supplies the left lobe, whereas less oxygenated portal vein blood supplies the right lobe. CYP450 activity is higher in the right lobe.112
Thus the fetal liver has the ability to metabolize drugs but at a decreased rate due to lower enzyme activity. Metabolism of drugs by the fetal liver may not be an advantage because the intermediary metabolites in these reactions may be toxic, tend to be more polar, and are cleared more slowly than the original drug, thus prolonging exposure.112 Heroin is a lipophilic drug that readily crosses the placenta, where it is converted by the fetal liver to morphine and other metabolites. These substances are less lipid-soluble than heroin and may be “trapped” on the fetal side of the placenta.36
Fetal renal elimination.
Although polar substances cross the placenta slowly, they are rapidly eliminated by the fetal kidney into amniotic fluid. These drugs may be recycled as the fetus swallows amniotic fluid. Because renal blood flow is low (about 5% of cardiac output), renal elimination in general is not a significant mechanism in the fetus.91
Fetal drug therapy
Planned fetal drug exposure may occur as a method of treating fetal disorders by administration of drugs to the mother. The most common fetal treatment is administration of glucocorticoids to induce surfactant synthesis and reduce the risk of respiratory distress syndrome in preterm infants (see Chapter 10). Not all glucocorticoids are equally effective, because some are metabolized by the maternal-fetal-placental unit and thus rendered inactive. Inactivation of betamethasone and dexamethasone is limited, so these glucocorticoids are more effective in enhancing surfactant synthesis than drugs such as hydrocortisone, which are rapidly metabolized in the placenta.
Glucocorticoids may prevent development of ambiguous genitalia in female fetuses in families with a history of congenital adrenal hyperplasia. If begun prior to onset of adrenal androgen secretion, these drugs suppress excessive adrenal androgen production and may prevent virilization of the external female genitalia.71,97 Glucocorticoids act by crossing the placenta and suppressing ACTH production by the fetal pituitary, which reduces adrenal cortex output. Ideally therapy is begun based on family history soon after conception and at least before 6-7 weeks from conception (∼9th week of pregnancy).16 Maternal adverse effects include Cushing syndrome, hypertension, hyperglycemia, gastric irritation, excess weight gain, and pedal edema.16,97 Drugs may also be given to the mother to treat several specific inborn errors of metabolism.16,71,110
Anti-infective agents are given to the mother as prophylaxis against group B streptococcal infection, to treat congenital syphilis (penicillin) or toxoplasmosis (pyrimethamine and sulfadiazine), listeria (ampicillin usually in combination with an aminoglycoside), or to prevent infection with human immunodeficiency virus (HIV).16,47,71,129 In prophylaxis to prevent HIV transmission, zidovudine with protease inhibitors (such as lamivudine) are not substrates for placental drug efflux transporters. Thus these drugs achieve higher fetal blood concentrations and have greater efficacy than other agents whose transfer across the placenta is reduced by these efflux transporters.47,129 Administration of antibiotics to the mother with the purpose of protecting the fetus is probably reasonably effective for protection of the GI (swallowing of amniotic fluid) and renal (adequate blood flow) systems. The fetal circulation shunts blood away from the lungs, however, which limits delivery of antibiotics to the respiratory tract for protection against congenital pneumonia in the presence of chorioamnionitis. Because amniotic fluid levels of antibiotics are dependent on excretion of the drug in fetal urine, therapeutic levels of antibiotics in amniotic fluid are attained only with a live fetus. This limits the effectiveness of this treatment for chorioamnionitis after intrauterine death.114
Fetal pharmacotherapy has also been used to treat fetal arrhythmias (most often for supraventricular tachycardia, atrial flutter, and fibrillation), hydrops, and congestive heart failure.129 Examples of drugs used include digoxin, procainamine, quinidine, propanolol, flecainide, amiodarone, and sotalol.16,129 Fetal therapy has also been used to treat polyhydramnios (indomethacin), hypothyroidism (intraamniotic thyroxine), hyperthyroidism (propylthiouracil and thiamazole), and severe anemia or thrombocytopenia (blood transfusions).71 Another example of a preventive fetal drug therapy is the use of folic acid for prevention of neural tube defects (see Chapter 15).
Neonatal pharmacokinetics
Absorption, distribution, metabolism, and excretion of drugs are developmentally different in newborns than in adults. Pharmacokinetic changes in the neonate are dependent on both postmenstrual age and postbirth age. Changes in absorption, distribution, metabolism, and elimination of drugs occur at different rates; there may be marked individual differences as well as day-to-day differences. Postbirth age changes are especially marked in the first month. In terms of pharmacokinetics, the neonatal time is the “period of life when most profound and rapid physiologic changes occur.”60
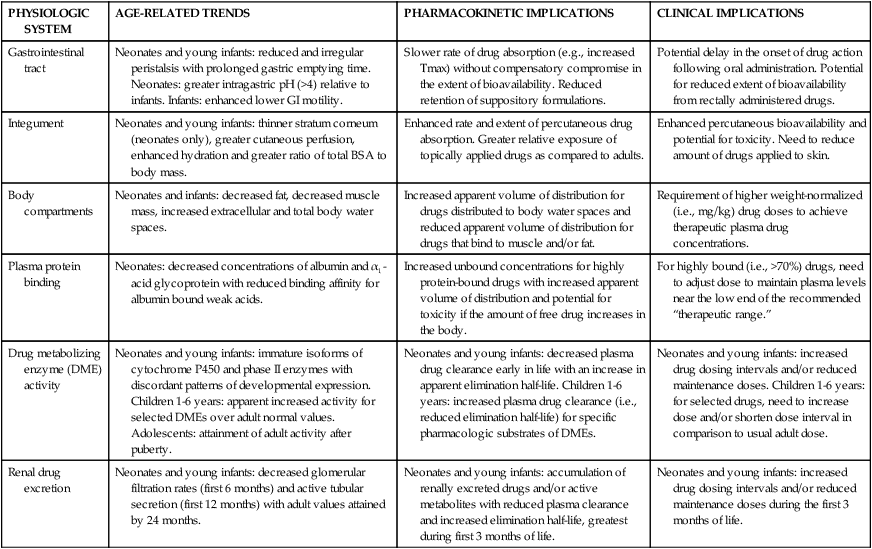
There are relatively few pharmacokinetic studies for most agents used in neonates. The majority of studies involve relatively heterogeneous samples (in terms of age, weight, disease state) of infants being treated with the specific drug rather than randomized studies.60 Alterations in drug absorption, distribution, hepatic metabolism, and renal elimination in the neonate affect pharmacokinetics of drugs used in the neonatal period. Often an individual drug is influenced by multiple pharmacokinetic factors. For example, the prolonged diuretic response to furosemide seen in newborns is due to both an increased plasma half-life (which is eight times greater than in adults and may be up to 24 hours in infants younger than 3 weeks of age), and an increase in volume of distribution (Vd) (four times greater than adults), probably because of decreased protein binding.62 Drug half-lives are almost always longer in the fetus and neonate than in older individuals (Table 7-6). Physiologic differences affecting drug pharmacokinetics in neonates are summarized in Table 7-7.
Table 7-6
Comparative Plasma Half-Lives of Miscellaneous Drugs in Newborns and Adults
| DRUG | PLASMA HALF-LIFE (hours) IN NEWBORNS | PLASMA HALF-LIFE (hours) IN ADULTS |
| Acetaminophen | 3.5 | 2.2 |
| Phenylbutazone | 21-34 | 12-30 |
| Indomethacin | 7.5-51 | 6 |
| Meperidine | 22 | 3.5 |
| Phenytoin | 21 | 11-29 |
| Carbamazepine | 8-28 | 21-36 |
| Phenobarbital | 82-199 | 24-140 |
| Caffeine | 100 | 6 |
| Theophylline | 30 | 6 |
| Chloramphenicol | 14-24 | 2.5 |
| Salicylates | 4.5-11.5 | 2.7 |
| Digoxin | 52 | 31-40 |
Data from Aranda, J.V., et al. (1980). Drug monitoring in the perinatal patient: Uses and abuses. Ther Drug Monit, 2, 39; Morselli, P.L., et al. (1980). Clinical pharmacokinetics in newborns and infants: Age-related differences and therapeutic implications. Clin Pharmacokinet, 5, 485; and Morselli, P.L. (1976). Clinical pharmacokinetics in neonates. Clin Pharmacokinet, 1, 81.
Table 7-7
Summary of Developmental Dependent Changes in Drug Disposition
| PHYSIOLOGIC SYSTEM | AGE-RELATED TRENDS | PHARMACOKINETIC IMPLICATIONS | CLINICAL IMPLICATIONS |
| Gastrointestinal tract | Neonates and young infants: reduced and irregular peristalsis with prolonged gastric emptying time. Neonates: greater intragastric pH (>4) relative to infants. Infants: enhanced lower GI motility. | Slower rate of drug absorption (e.g., increased Tmax) without compensatory compromise in the extent of bioavailability. Reduced retention of suppository formulations. | Potential delay in the onset of drug action following oral administration. Potential for reduced extent of bioavailability from rectally administered drugs. |
| Integument | Neonates and young infants: thinner stratum corneum (neonates only), greater cutaneous perfusion, enhanced hydration and greater ratio of total BSA to body mass. | Enhanced rate and extent of percutaneous drug absorption. Greater relative exposure of topically applied drugs as compared to adults. | Enhanced percutaneous bioavailability and potential for toxicity. Need to reduce amount of drugs applied to skin. |
| Body compartments | Neonates and infants: decreased fat, decreased muscle mass, increased extracellular and total body water spaces. | Increased apparent volume of distribution for drugs distributed to body water spaces and reduced apparent volume of distribution for drugs that bind to muscle and/or fat. | Requirement of higher weight-normalized (i.e., mg/kg) drug doses to achieve therapeutic plasma drug concentrations. |
| Plasma protein binding | Neonates: decreased concentrations of albumin and α1-acid glycoprotein with reduced binding affinity for albumin bound weak acids. | Increased unbound concentrations for highly protein-bound drugs with increased apparent volume of distribution and potential for toxicity if the amount of free drug increases in the body. | For highly bound (i.e., >70%) drugs, need to adjust dose to maintain plasma levels near the low end of the recommended “therapeutic range.” |
| Drug metabolizing enzyme (DME) activity | Neonates and young infants: immature isoforms of cytochrome P450 and phase II enzymes with discordant patterns of developmental expression. Children 1-6 years: apparent increased activity for selected DMEs over adult normal values. Adolescents: attainment of adult activity after puberty. | Neonates and young infants: decreased plasma drug clearance early in life with an increase in apparent elimination half-life. Children 1-6 years: increased plasma drug clearance (i.e., reduced elimination half-life) for specific pharmacologic substrates of DMEs. | Neonates and young infants: increased drug dosing intervals and/or reduced maintenance doses. Children 1-6 years: for selected drugs, need to increase dose and/or shorten dose interval in comparison to usual adult dose. |
| Renal drug excretion | Neonates and young infants: decreased glomerular filtration rates (first 6 months) and active tubular secretion (first 12 months) with adult values attained by 24 months. | Neonates and young infants: accumulation of renally excreted drugs and/or active metabolites with reduced plasma clearance and increased elimination half-life, greatest during first 3 months of life. | Neonates and young infants: increased drug dosing intervals and/or reduced maintenance doses during the first 3 months of life. |
From Rakhmanina, N.Y. & van den Anker, J.N. (2006). Pharmacological research in pediatrics: From neonates to adolescents. Adv Drug Deliv Rev, 58, 10.
Drug absorption
Oral agents
Absorption of oral medications in the neonate is altered by the following: (1) decreased bile salts and pancreatic enzymes; (2) slower gut transit time due to delayed gastric emptying and decreased motility; (3) mucus in the stomach; (4) differences in gastric and duodenal pH; (5) high levels of β-glucuronidase in the duodenum; and (6) intestinal surface area.31,32,66,86,93,106 These alterations are more pronounced in preterm and intrauterine growth–restricted (IUGR) infants.32,93 Oral drug absorption matures by 4 to 5 months of age.31 Decreased bile salts and pancreatic enzymes can result in poorer absorption of lipid-soluble substances such as vitamin K, or drugs that must be hydrolyzed for absorption. Slower gut transit time due to delayed gastric emptying and decreased motility initially delays then prolongs absorption. Mucus in the stomach may also delay absorption. Differences in gastric and duodenal pH may partially or totally inactivate some drugs. Absorption of oral drugs by the neonate is also influenced by the drug’s physicochemical characteristics. Drugs such as phenytoin, acetaminophen, and chloramphenicol are absorbed slowly and erratically, whereas penicillin and ampicillin are absorbed more efficiently than in adults because of the higher gastric pH in the neonate.105 Other drugs such as theophylline, digoxin, and diazepam have oral absorption patterns that are more similar to those in adults.
Gastric and duodenal pH influences drug solubility and ionization. An acid pH increases the absorption of acidic drugs (low pKa) because these drugs tend to stay in a nonionized, lipid-soluble form. A higher pH enhances alkaline drug absorption and slows absorption of acidic drugs. Gastric pH is higher for the first 12 to 24 hours after birth, gradually reaching adult values by 2 years.7 Preterm infants who are less than 32 weeks’ gestational age have decreased gastric acid secretion with a more alkaline pH and thus poorer absorption of weak acids and bases.19,32,93 Gastric emptying is delayed for up to 6 to 8 hours in immature infants and is slower in all infants before 6 to 8 months. This may delay absorption and prolong the time to reach peak drug concentration.19,66 Oral absorption is also influenced by drug efflux transporter expression on the intestinal lining and type of bacterial flora.86,95
The neonate’s duodenum has higher levels of β-glucuronidase, an enzyme that unconjugates drugs (e.g., indomethacin, chloramphenicol) that the liver has metabolized via glucuronyl conjugation.32,66,93 These drugs reenter the blood via the enterohepatic circulation (see Chapter 18) and must be remetabolized by the liver. This increases the half-life and prolongs activity of these drugs.31,93 Intestinal absorption of drugs, gut transit time, and motility depend on perfusion and food intake. Better gastrointestinal (GI) absorption occurs with drugs of low molecular weight (MW) that are nonionized and lipid-soluble.93 An ill infant who is not being fed may have further impairment of gut function and erratic absorption of oral medications. Intestinal surface area is decreased with genetic disorders such as cystic fibrosis and disorders such as short bowel syndrome and necrotizing enterocolitis, thus reducing absorption.19,93
Intramuscular and rectal absorption
The infant’s small muscle mass and alterations in peripheral perfusion influence intramuscular medications. Decreased muscle mass may limit use of intramuscular administration in very low-birth weight (VLBW) infants. Drugs given intramuscularly may have slower, more erratic absorption, depending on the infant’s gestational age and health status. In the first few days after birth, muscle blood flow is decreased, slowing absorption.19 Variations in muscle blood flow last for 2 to 3 weeks after birth.14 Infants with respiratory problems or low cardiac output have decreased peripheral perfusion, leading to even slower and more erratic absorption of drugs given intramuscularly.66 On the other hand, rectal administration may be more effective in neonates than in adults.66
Skin absorption
Percutaneous absorption of substances occurs via the cells of the stratum corneum (transepidermal route) or via the hair follicle–sebaceous gland complex (transappendageal route). The major pathway is most likely transepidermally, with diffusion of a substance through the stratum corneum and epidermis into the dermis and microcirculation. The subepidermal circulation is also readily accessible, enhancing rapid absorption. Preterm infants have greater skin absorption because their skin is thinner than that of the term infant. VLBW infants have minimum to no stratum corneum and higher dermal water content.66,93
Neonates are at increased risk for toxic reactions from absorption of topically applied substances. Skin metabolism is different in neonates, so drugs applied topically may result in the release of metabolites different from those that would occur if the drugs were given by other routes. This increases the risk of toxicity.19,66 Occlusion of the skin (e.g., placement against the mattress) permits more complete absorption, with longer contact enhancing absorption of the substance. In preterm infants, percutaneous absorption occurs even more rapidly and completely as a result of the markedly increased skin permeability. Topical analgesic agents are more easily absorbed, increasing the risk of toxicity (see Chapter 14). Permeability of these agents, such as EMLA cream (lidocaine and prilocaine) is further increased in preterm infants in the first few weeks after birth.93
Neonatal toxicity secondary to the use of topical agents such as hexachlorophene, pentachlorophenol-containing laundry detergents, isopropyl alcohol, and povidone-iodine has been well documented.19,23,28,66 Practices can lead to detrimental effects if not monitored carefully. Topical application of povidone-iodine (Betadine) yields significantly elevated levels of iodine in blood plasma if not removed completely from the skin after completion of invasive procedures (e.g., chest tube insertion, percutaneous line insertion).19 Gentle cleansing of the skin with water reduces this risk.
Drug distribution
The distribution of drugs is influenced by the infant’s large extracellular fluid space, increased total body water, decreased fat, and decreased plasma protein content and binding. The blood-brain barrier is less well developed, which may result in increased amounts of drugs (especially lipid-soluble substances) reaching and being deposited in the brain.95,119 The greater total body water and lower fat content in neonates increases the apparent Vd, especially for water-soluble substances.14,93,116,122
The altered Vd in the neonate may reduce peak volumes of drugs and delay excretion. Because of the increased extracellular water, higher doses per kilogram may be needed in the neonate for water-soluble drugs distributed in extracellular fluid. For example, an increased Vd is seen with drugs such as theophylline, caffeine, phenobarbital, phenytoin, digoxin, and aminoglycosides such as gentamicin.93,116,122 The Vd is further increased with decreasing gestational age, because more immature infants have a greater total body water percentage. VLBW infants may need higher loading doses and a longer dosing interval of some drugs because of their even greater apparent Vd and immature hepatic and renal function. Other factors that alter total body water include those that alter drug distribution by increasing (congestive heart failure, patent ductus arteriosus, syndrome of inappropriate antidiuretic hormone secretion) or decreasing (diuretic use) extracellular water.31,32
Decreased fat in the newborn, especially preterm and IUGR infants, decreases the apparent Vd for lipophilic drugs.31,93 Body fat content increases from 1% to 3% at 28 weeks’ gestation to 15% to 28% by term, thus increasing the deposition and distribution of lipid-soluble drugs with increased gestational age.32,93 In addition, neonatal adipose tissue is more than 50% water (versus approximately 25% in adults), further altering tissue distribution of lipid-soluble drugs.32
APT-binding cassette (ABC) drug efflux transporters (that prevent specific toxins from crossing the blood brain barrier or moving into hepatic, renal tubular or intestinal cells) appear to have decreased activity in the newborn.66,95 This may increase central nervous system permeability to drugs, and alter rates of hepatic and renal clearance.19,66,95,119 Examples of the transporters are P-glycoprotein (P-gp)/multidrug-resistant gene protein (MRP)-1, MRP-2, MRP-3, and breast cancer resistance protein (BCRP).86,95 P-gp increases rapidly in the first 3 to 6 months and reaches adult levels by 2 years.95
Protein binding is lower in the neonate due to decreased concentrations of binding proteins; altered binding affinity, especially in the presence of fetal albumin, which has a poorer binding capacity; and increased levels of nonesterified fatty acids and bilirubin, which compete for protein binding sites.19 The major drug-binding proteins are albumin, which generally binds acidic drugs, and α1-acid glycoprotein, which along with lipoproteins generally binds basic drugs.31,105 Levels of both albumin and α1-acid glycoprotein are low in the neonate, reaching adult levels by about 1 year, resulting in increased free drug levels of substances such as theophylline, digoxin, phenytoin, and many analgesic compounds.7,31,32,93,116
In addition to lower plasma protein levels, the newborn tends to have fewer binding sites and a lower affinity of these sites for drugs such as phenobarbital, phenytoin, penicillin, and theophylline. As a result, the newborn tends to saturate protein binding sites at lower drug concentrations with increased free drug, longer half-lives, and risk of toxicity.66,93 However, increased levels of free drug can also lead to lower drug levels because the free drug may be more rapidly eliminated by the kidneys.32,93 Drugs can also bind to albumin outside the vascular space in sites such as cardiac and skeletal muscle. Newborns tend to have more binding to the myocardium than adults, which can result in an increased volume of distribution for drugs such as digoxin.31
Protein binding of drugs may also be reduced by acidosis, free fatty acids, maternal drugs from placental transfer before birth, and indirect bilirubin. Drugs and bilirubin compete with each other for binding sites. Bilirubin is displaced from albumin by sulfonamides, ceftriaxone, and chloral hydrate, leading to increased free indirect bilirubin levels. However, bilirubin is more likely to displace drugs than to be displaced. Drugs that may be displaced from albumin by bilirubin include ampicillin, penicillin, phenobarbital, and phenytoin, leading to higher free drug concentrations of these agents.93 Other drugs, such as furosemide and indomethacin, if given together, compete against each other for albumin binding sites.93
Hepatic drug metabolism
Neonates and infants have immature organ systems, especially the lungs (see Chapter 10), kidney (see Chapter 11), and liver (see Chapter 12), which are typical routes for drug excretion. The large surface-to-mass ratio and high metabolic rate of the neonate and infant affect drug metabolism.
Liver enzyme systems necessary for metabolism of many drugs are depressed in the newborn. In the mature liver, oxidation and conjugation result in water-soluble metabolites that are readily excreted into bile. In the fetus, depression of these processes may be an advantage, because lipid-soluble metabolites are more readily transferred across the placenta where they can be handled by the maternal system. The liver smooth endoplasmic reticulum (SER) is the location of many hepatic microsomal enzymes. The neonatal liver has little SER, and activities of many microsomal enzymes are reduced or undetectable at birth, altering drug metabolism.85 As a result, drug metabolism may be characterized by slower biotransformation and elimination and increased individual variability.93 The reduced activity of hepatic microsomal enzymes extend the half-life of drugs dependent on them for metabolism and increase the risk of drug intolerance.
Phase I (see Table 7-1) biotransformation reactions are reduced in the fetus and newborn, impairing degradation and increasing half-lives of drugs such as phenytoin, phenobarbital, diazepam, mepivacaine, lidocaine, caffeine, theophylline, phenylbutazone, salicylate, indomethacin, furosemide, and sulfa drugs.32,86
Both microsomal enzyme (e.g., CYP450 system, NADPH-cytochrome reductase) and extramicrosomal enzyme (e.g., deaminases, esterases) activity are decreased in the newborn, with a rapid increase in activity over the first few weeks after birth.32,66 CYP3A7 is predominant at birth, peaking shortly after birth, then decreases to low levels within 1 to 2 weeks.17,55,56,66,102,108 CYP3A4 activity is low at birth but increases 3-fold in the first 3 months, reaching 20% of adult activity by 1 month and around 70% by 1 year.7,56,108 CYP2E1 surges within a few hours after birth.108 Levels of CYP1A1, the isoform responsible for degradation of over half of all drugs in adults, has low activity until 1 to 3 months after birth.17,31,56 By 2 to 3 weeks of age, liver enzyme systems are maturing at varying rates.17 Maturation of some enzyme activity may be rapid; other systems mature more slowly. Activity of CYP1A2 and CYP2B6 are minimal at birth and reach 50% of adult activity around a year.7,56,108 CYPC9 has 20% of adult activity at birth reaching 50% of adult activity by 3 months; CYP2C19 has 30% of adult activity reaching adult activity by 1 year.7,56,108 Alternative pathways of biotransformation are seen for some drugs in the neonate. For example, methylxanthine metabolism is different in neonates versus adults. In the neonate, theophylline is methylated, some is converted to caffeine, and its clearance is 50% of adults; whereas in adults, theophylline is metabolized to 1,3 dimethyluric acid and other metabolites.7,31,93,108
There are marked variations in maturation rates of processes for specific drugs and within individual infants. This increases elimination half-lives and the risk of both underdosing and overdosing.31,32 Maturation is correlated with both postmenstrual age and postbirth age. For example, midazolam and phenytoin are both metabolized by CYP2C9 and CYP2C19. Midazolam clearance increases up to five-fold from 1 to 3 months; low activity of these enzymes in preterm infants increases bioavailability.7,14,108 Phenytoin biotransformation increases and half-life decreases in early infancy with a half-life of approximately 75 hours in preterm infants, decreasing to about 20s by term and 8 hours by 2 weeks post term.7,108
Phase II reactions (see Table 7-1), particularly glucuronidation, are also altered in the newborn. Glucuronic acid conjugation (via uridine 5′-diphosphatase glucuronosyltransferase or UGT enzymes), needed for metabolism of drugs such as morphine, acetaminophen, phenytoin, sulfonamides, chloramphenicol, salicylic acid, and indomethacin, is reduced at birth and does not reach adult levels until 24 to 30 months.31 An example of maturation of the ability to metabolize drug via glucuronidation is seen with morphine. Morphine glucuronidation is found in preterm infants by 24 weeks’ gestation. Morphine metabolism increases with increasing postmenstrual (PMA), due to maturation of UGT isoforms, so that clearance quadruples from 27 to 40 weeks’ PMA, requiring increased dosages for pain management.53,95 Zidovudine is also metabolized by glucuronidation and its plasma clearance in infants <2 weeks of age is half that of infants over 2 weeks.108 Acetaminophen is metabolized by UGT1A6 and UGT1A9. These enzymes are low at birth and reach full activity by 9 to 12 months; bioavailability increases with age due to an increased first pass effect and enzyme maturation.14,108 UGT1A1 is the major enzyme for bilirubin glucuronidation. Variants of this enzyme lead to reduced activity and prolonged neonatal jaundice. Mutations in the gene for this enzyme occur with Criglar-Najjar syndrome.95
Chloramphenicol is eliminated by the kidney after conjugation in the liver. The hepatic glucuronyl transferase system responsible for conjugation of this drug is immature in the neonate (especially in preterm infants), and free (unconjugated) chloramphenicol is eliminated by glomerular filtration, which is reduced in neonates. The result is higher and prolonged peak serum chloramphenicol concentrations. Half-life of chloramphenicol is approximately 20 hours in the neonate and longer in preterm infants, falling to about 10 hours by 21 days (versus 4 to 5 hours in adults).75 This results in increased risk of toxicity (“gray baby syndrome”) with aplastic anemia, shock, and cardiovascular collapse.31,86
Other phase II reactions (sulfate and glycine conjugation) are somewhat decreased but more similar to those in adults.93 Methylation reactions may be more important in infants than in adults for drugs such as theophylline.93 Decreased hydrolysis and increased Vd may prolong the effect of local anesthetic agents. Ligandin (carrier protein Y) needed for hepatocyte uptake of substances including bilirubin and many drugs is low for 5 to 10 days after birth, further altering hepatic clearance of some drugs.
3. The development of individual drug-metabolizing enzymes varies widely among neonates and may be delayed in premature infants. Predicting clearance is difficult, and dosing regimens must be individualized for patients based on the careful observation of the patient’s response and tolerance; monitoring of plasma drug concentration may be useful for selected drugs.”86,p.727
Renal drug excretion
Because of the decreased hepatic metabolism in neonates, the infant is more reliant on renal elimination. However, renal elimination is reduced compared to adults and correlated with both postmenstrual and postbirth ages. Alterations in renal function in neonates can markedly alter the infant’s ability to eliminate drugs and drug metabolites, increasing the half-life and risk of toxic effects and the need for longer dosing intervals. Any pathologic event that alters renal function (e.g., congestive heart failure, respiratory distress syndrome, hypovolemia, ischemia, or hypoxemia) further compromises the ability of the infant to eliminate drugs. In addition, drugs such as indomethacin may decrease glomerular filtration rate (GFR), further impeding excretion.31 Renal processes involved in drug handling are GFR, tubular secretion, and, to a lesser extent, tubular reabsorption.
The GFR is significantly lower in the neonate than in adults (see Chapter 11), although partially compensated by an increase in tubular reabsorption. The reduced GFR delays clearance of drugs that are eliminated by the kidneys, leading to prolonged half-lives and longer dosing intervals.62,127 For example, clearance of indomethacin and many aminoglycosides is 10% to 30% lower than in adults. The GFR is particularly low in the first week after birth, increasing susceptibility to nephrotoxic drugs such as aminoglycosides. Plasma half-lives of many drugs used in the neonate—including penicillin, aminoglycosides, methicillin, furosemide, barbiturates, and digoxin—vary inversely with creatinine clearance (which is a measure of GFR).
Renal maturation progresses at variable rates after birth depending on postmenstrual and post birth ages. The most rapid changes in GFR are seen in the first 7 to 10 days after birth.31 The increased half-life of drugs such as digoxin, vancomycin, and indomethacin correlate with both postmenstrual and postbirth age and may be significantly longer in preterm infants.62,127 Drug levels must be carefully and regularly monitored. Use of drugs such as vancomycin in combination with indomethacin (which decreases GFR) can further decrease vancomycin elimination.93 Renal elimination of drugs is further altered in preterm infants who are younger than 34 weeks’ gestational age. In these infants, GFR increases at birth, but then tends to plateau or increase slowly until about 34 weeks, when nephron maturation is complete (see Chapter 11). As a result, some antibiotics and other drugs are given less frequently to VLBW infants. Serum half-lives of antibiotics change rapidly over the first few weeks after birth as renal function matures. Therefore dosages of these drugs or dosing intervals may need to be increased.66 For example, gentamicin dosing intervals range from 24 to 48 hours in more immature infants to every 8 to 12 hours in the term newborn.31
Elimination of drugs by tubular secretion is reduced in the neonate for the first 6 months as a result of decreased renal blood flow to the peritubular area, shorter tubular length, decreased energy for carriers, and reduced number of carriers to transport drugs from the blood into the tubule.62 This affects drugs such as furosemide and penicillin G, which are therefore more dependent on GFR than tubular secretion for clearance during early infancy. In older children and adults, tubular secretion is the most important mechanism for penicillin excretion (versus GFR in neonates). As a result, penicillin clearance is nearly one fifth that of adults, and penicillin blood levels may persist for three times as long. Urine flow rates can also influence elimination of drugs such as chloramphenicol, phenobarbital, and theophylline by decreasing the concentration gradient due to formation of dilute urine or reducing the time for drug diffusion.62
Summary
The Vd is altered in the neonate because of the increased total body water and increased proportion of extracellular water. As a result, peak volumes of some drugs are reduced and excretion delayed. Immaturity of liver enzyme systems can delay metabolism and clearance of drugs. The reduced GFR seen in neonates increases serum concentrations and prolongs the half-life of many drugs and may result in longer dosing intervals. Doses of some drugs may be higher per weight in neonates than adults due to an increased Vd, reducing plasma levels. Doses may also need to be altered because of increased or decreased sensitivity of neonatal tissue to some drugs. Digoxin doses are relatively higher in the neonate due to increased Vd and decreased sensitivity of the myocardium to both therapeutic and toxic effects.116
Clinical implications for the pregnant women, fetus, and neonate
Teratogenesis
Embryonic and fetal development can be altered by internal events, external events and teratogens, which result in structural and functional defects. Fetal defects can result from malformation, deformation, or disruption. A malformation is an embryonic alteration in morphogenesis due to an intrinsically abnormal developmental process such as may occur with a chromosome or multifactorial disorder (see Chapter 1).64 Deformation defects are the abnormal form, shape, or position of a body part arising from extrinsic mechanical forces (e.g., clubfoot due to fetal restraint or lung hypoplasia with congenital diaphragmatic hernia); a disruption is caused by an external force that alters a previously normal tissue (e.g., teratogenic effects or amputation of a fetal part by an amniotic band).64 Birth defects may be major or minor abnormalities. Minor abnormalities may be normal variations, family traits, or isolated defects, or may provide clues to the presence of internal malformations, genetic disorders, or chromosomal problems. Three or more minor abnormalities are associated with an increased risk for a major birth defect.64 Information about human teratogens can be found in a variety of on-line and print resources (see Box 7-2 on p. 187).23–25,30,86,130
Drugs administered to the mother during pregnancy can affect the fetus in a variety of ways ranging from no effect to major structural or functional deficits. Drugs administered during labor affect the fetus primarily by exaggerating the degree of fetal asphyxia and influencing the rate and quality of infant recovery from birth, adaptation to extrauterine life, and neurobehavioral status. A teratogen is any substance, organism, physical agent, or deficiency state present during gestation that is capable of inducing abnormal postnatal structure or function (biochemical, physiologic, or behavioral) by interfering with normal embryonic and fetal development. Thus many substances can act as teratogens including infectious agents (such as toxoplasmosis, cytomegalovirus, syphilis), ionizing radiation, hyperthermia, pesticides, metal (e.g., mercury and lead), organic solvents, excess or deficient nutrients, effects of maternal chronic disorders (such as diabetes and phenylketonuria), as well as over-the-counter, prescribed, and abused drugs. Some herbal agents are also contraindicated in pregnancy, including those that have the potential to act as abortifacients and/or uterine stimulants (black cohash, chamomile, chasteberry, goldenseal, white horehound, licorice, mistletoe, nettle, pennyroyal, rue, tansy, and yarrow).33
Fewer than 30 drugs are proven teratogens and fewer still are in current clinical use.67,80 Most women exposed to these known teratogens have normal offspring.80 The FDA categories consider both the potential risks of and benefits from the drug and are based largely on preapproval animal studies.100 The category is assigned via structured regulatory process and negotiation with drug sponsors and is not meant to provide estimates of teratogenic risk, although the information is widely used that way in clinical practice.30,40,80,87 There have been efforts in recent years to revise these categories for pregnancy labeling. One proposal is that the revised categories address clinical considerations, summarize risk assessment, and provide data to support this assessment.109 Very few drugs are classified as FDA category A or B or as X (contraindicated during pregnancy). Category X drugs include thalidomide, isotretinoin, acitretin, radioactive iodine, oral contraceptives, and diethylstilbesterol.24 Most drugs are in category C (risk cannot be determined).87 Category D drugs are those for which there are known risks but there may be no safer alternative and the risks outweigh the potential benefits. Examples of drugs in category D include paroxetine, angiotensin converting enzyme inhibitors (captopril, enalapril, and lisinopril), valproic acid, phenytoin, carbamazepine, diazepam, carbimazole, and lithium.24
Lo and Friedman examined 468 drugs approved by the Food and Drug Administration between 1980 and 2000.80 The teratogenic risk was undetermined in 91.2%. Eleven drugs (2.4%) were demonstrated to pose “some” to “high” teratogenic risk, with a mean of 6 years (SD ± 4.1) to determine risk. Only 30 drugs (6.1%) were proven to pose no risk, with a mean of 9 years to recognize apparent safety of these drugs in literature (the longer time was probably due to the fact that studies showing no risk are often not published).80 Lo and Friedman also found poor agreement between TERIS risk rating and the FDA pregnancy categories (A, B, C, D, X) for 163 drugs. TERIS is a teratogenic resource complied via consensus of teratology experts based on published data and does not consider potential risk and benefit to the mother. Recently this group examined 172 drugs approved by the FDA between 2000 and 2008 using the TERIS risk rating system (data were rated by an expert advisory panel).1 They found that 168 (97.7%) of these drugs had “undetermined” teratogenic risk and 126 (73.3%) had no data available regarding safety during pregnancy. The authors also relooked at the 468 drugs from the earlier 1980-2000 study to see if the risk had been revised in the past 10 years.80 Twenty-three drugs (5%) had a change in risk category (some from unknown to risk; some from unknown to no risk). The mean time for drugs classed as undetermined risk to having a more precise risk assignment was 27 years.1
Teratogenesis is often difficult to prove. To detect a defect due to a specific agent, an increase in a specific anomaly or group of anomalies over what would be expected by chance must be documented. Thalidomide was recognized as a teratogen sooner than some other agents because it produced significant malformations, with small doses given once or twice in early pregnancy with a malformation rate of 20% to 30%.70 Documentation of teratogenesis is often difficult to do in a single location, especially with rare anomalies. Koren and colleagues note the following:
“Most congenital malformations occur rarely, and many teratogens, even when known to be associated with an increased risk of a given malformation, do not affect the great majority of exposed fetuses. In fact, very few drugs increase the malformation rate by a factor of more than two (isotretinoin and thalidomide are two such drugs). If, for example, the risk of malformations in a given population is 3%, then at least 220 pregnancies with a specific exposure and a similar number of control pregnancies will be required to show a risk that is increased by a factor of 2.5 with a power of 80%.”70, p. 1133
Other common methodologic issues in establishing teratogenesis, in addition to sample size, are recall bias in retrospective studies, use of nonrandomized observational studies, limitations in voluntary reporting, issues in choosing the right animal model, and the effects of maternal disease.22,37,70,100,131 An example of the difficulty with recall bias occurred with Bendectin in which women whose infants had defects recalled taking this drug, which was commonly used, in early pregnancy. Subsequent studies did not confirm a link between Bendectin and malformations.11,94 Maternal pharmacologic therapy may be administered for an illness that itself leads to a defect or malformations or may produce maternal symptoms requiring treatment with a specific drug; several agents may interact with each other so that the combination is teratogenic rather than either individual drug; or the mother may be taking multiple drugs for different problems.70 Problems with human studies include difficulty studying cause and effect, lack of random sampling, lack of control over dosing and timing, and expense.
Animal studies can also be problematic. Drug studies with animals may involve administration of large doses that exceed the usual therapeutic dose in humans. Selecting the appropriate animal model can be difficult. Drug kinetics and metabolism may differ between different species and between humans and animals.22,70 For example, thalidomide, a potent teratogen in humans, did not produce limb reduction anomalies in the animal models used to study the drug before its release; subsequently, similar anomalies were seen in macaque monkeys. On the other hand, benzodiazepines cause oral clefts in some animal models but not in humans given clinically appropriate doses. Similarly, salicylates cause cardiac defects in some animal species but not humans. Most human teratogens have been discovered by either epidemiology studies or by health care professionals, not animal studies.22 Dicke noted, “We will never be in a position to state that an environmental agent has no teratogenic potential. The most we can say is that an agent poses no measurable risk.”38
Principles of teratogenesis
General principles that govern the action of teratogens include the following:
1. Susceptibility to a teratogenic agent is dependent upon the genotype of the embryo and the manner in which the agent interacts with environmental factors.38,133 The genetic makeup of the developing embryo is the environmental programmer to which the teratogenic agent is introduced. This programming means that the genes and extrinsic factors interact in varying degrees with varied responses in different individuals and species. Because individual sensitivity is dependent upon the biochemical and morphologic makeup of that particular individual, data from animal models and sometimes even from studies on other individuals cannot always be applied. For example, individuals with a gene mutation that reduces levels of an enzyme needed to detoxify anticonvulsant drugs such as phenytoin are at greater risk of having a child with defects.30
2. Susceptibility to teratogenic agents is dependent on the timing of the exposure and the developmental stage of the embryo.38,89,133 A basic precept of biology is that the more immature an organism is, the more susceptible that organism is to change.133 This suggests that there is a critical period where teratogenic events have the greatest impact. The period of greatest susceptibility is during the first trimester, when cell differentiation and organogenesis are occurring. Structural and functional maturation continues after birth, and therefore many systems remain susceptible to alterations in later development. Sometimes the specific period of vulnerability can be identified. For example, the hypoplastic limb defects seen with thalidomide occur only with exposure at 21 to 36 days after fertilization; valproic acid exposure results in a neural tube defect only with exposure between 14 and 27 days.87,117
The most likely period for structural defects to occur is during organogenesis, because exposure of the embryo to a teratogenic agent either during or before a critical stage in development of that organ can lead to anomalies.29,90 The time of greatest susceptibility during development is defined as the time when the highest incidence of defects occurs (usually gross anatomic defects). Other defects, especially neurobehavioral and other functional defects, have peaks in sensitivity at different times during development. These periods of sensitivity vary depending on the timing and duration of the period of cell proliferation (e.g., brain growth and development extend into early childhood and thus are vulnerable for a longer period of time).90,133
Malformations resulting from incomplete morphogenesis within an organ usually originate before organ structure is complete. The exact time at which a specific defect occurs cannot be determined; it can be said only that a defect occurred at some time before a particular point. For example, anencephaly must occur before closure of the anterior neural tube (25 to 26 days); meningomyelocele before closure of the posterior neural tube (27 to 28 days); cleft palate before fusion of the maxillary palatal shelves (10 weeks); cleft lip before closure of the lip (36 days); tracheoesophageal fistula before separation of the foregut into the trachea and primitive esophagus (30 days); ventricular septal defect before closure of the ventricular septum (6 weeks); transposition of the great vessels before development of the aorticopulmonary septum (34 days); diaphragmatic hernia before closure of the pleuroperitoneal canal (6 weeks); omphalocele before or during return of the gut to the abdomen (10 to 10.5 weeks); and imperforate anus before perforation of the anal membrane (8 weeks).
3. Teratogenic agents act in specific ways on cells or tissues to cause pathogenesis.38,133 Unfavorable factors within the environment are able to trigger changes in developing cells that alter their subsequent development. These changes are not specific to the type of causative factor, and initial changes may result in a variety of alterations within the embryo. These early changes may not be discernible because they occur subcellularly at the molecular level. Pathogenesis is the visible sign of cellular damage and may occur from cell necrosis or by secondary interference with cellular interactions—that is, by induction, adhesion, and migration; reduced biosynthesis of macromolecules; or accumulation of foreign materials, fluids, or blood.38,133
4. The final manifestations of abnormal development are death, malformation, growth restriction, and functional disorders.38,133 For the early embryo a teratogenic event will most likely result in death. Once organogenesis begins, teratogenic events lead to malformation in the organs or organ systems. The teratogenic insult might also make the embryo more susceptible to death or general cell necrosis, resulting in a reduced cell mass and slower overall rate of growth. Functional defects may be diagnosed throughout infancy and childhood.
Before conception, damage can occur to the chromosomes of one or both parents, or new gene mutations may arise. Alterations in spermatogenesis; in seminal fluid or sperm transport in the male; or in oogenesis or the environment of the vagina, cervix, or uterus of the female may also alter development.124
The preembryonic stage (conception to 14 days) is a time of little morphologic differentiation in specific organ systems. Exposure to teratogens during this period usually has an all-or-nothing effect; that is, either the damage is so severe that the zygote is aborted or there are no apparent effects.24,87,109 However, this may be the time that syndromes affecting multiple organ systems arise. Teratogens or environmental disturbances may interfere with implantation of the blastocyst or cause death and early abortion. However, most congenital anomalies probably do not arise during this period, possibly due to the lack of cell differentiation, at least in the early part of this period. Many cells within the inner cell mass are not yet programmed to become specific structures. Thus damage to a few cells does not alter development if the preembryo is able to produce sufficient cells to restore the lost volume.133 If a teratogen is potent or the dosage is high, the effect is death or possibly mitotic disjunction during cleavage, with chromosomal alterations that subsequently cause malformation syndromes rather than local defects.64,107 If only a few cells are damaged, development continues, although the genetically programmed schedule may be delayed.90
The period of organogenesis (15 to 60 days after conception) is a period of extreme sensitivity to teratogens; it is the period when many congenital malformations develop. Insults early in this period (15 to 30 days) are likely to result in death if the embryo is damaged. Early events in organ formation are generally most sensitive to external forces, although in some systems (e.g., the sensory organs), critical periods occur during relatively late stages. The more specialized the metabolic requirements of a group of cells, the more sensitive they are to deprivation and damage.90,107,133
From 11 weeks to term, the fetus becomes increasingly resistant to structural damage from toxic agents as the ability to produce major structural deviations is reduced as organ systems become organized. Thus once the definitive form and relationships within a system are established, gross anatomic defects are no longer possible. However, histogenesis continues and the function of organ systems can still be altered.90,133 Defects can occur at the microscopic level, or neurobehavioral or other functional abilities can be altered, resulting in physiologic defects and delayed growth. Insults during fetal life can lead to dysfunction such as brain damage or deafness, prematurity, growth restriction, stillbirth, infant death, or malignancy.
5. Access to the embryo by environmental teratogens depends on the nature of the agent.38,133 There are several routes by which agents reach the embryo or fetus. Agents such as ultrasound, ionizing radiation, and microwaves pass directly through maternal tissue without modification. Chemical agents or their metabolites reach developing tissues indirectly via transmission across the placenta. Whether these agents reach toxic or teratogenic concentrations depends on maternal dosage, rate of absorption, and maternal homeostatic capabilities as well as physical properties of the agent and the placenta. Pathogenic organisms may also reach the fetus by an ascending route via the vaginal canal and cervix.
6. As the dosage increases, manifestations of deviant development increase.38,133 There appears to be a threshold at which embryotoxicity occurs and damage is initiated. When the effect threshold is exceeded, cell damage or death exceeds restoration. For example, diagnostic x-rays with a dose lower than 5 rads (dosage for most common procedures) is not thought to have teratogenic effects. Exposure to 5 to 10 rads is of concern, whereas exposure to more than 10 rads (e.g., with radiation therapy) is associated with significant risk.30 Valproic acid increases the risk of neural tube defects and the risk increases with increased dosage levels.30 Sometimes the specific threshold cannot be determined, as is the case with alcohol. Different types of embryotoxicity exist for different thresholds.
Mechanisms of teratogenesis
Proposed mechanisms of teratogenesis include the following:
1. Gene mutation. Mutation is the basis of heritable developmental defects and is the result of a change in the sequence of nucleotides. If the change appears in the germinal cell line (i.e., oocytes and spermatocytes), it is likely to be heritable. A mutation in a somatic cell will be passed to daughter somatic cells but cannot be transferred to the next generation. Gene mutations can result in biochemical or structural disorders or later development of malignancies (see Chapter 1).133
2. Chromosome breaks and nondisjunction. These alterations lead to excesses, deficiencies, or rearrangements of chromosomes or parts of chromosomes and can be transmitted to offspring. For example, sperm of men exposed to radiation or chemotherapy have increased numbers of chromosomal aberrations for at least 3 to 4 months (chemotherapy) to up to 36 months (ionizing radiation).124
3. Mitotic interference and cell death. Interference with mitosis can result from inhibition of deoxyribonucleic acid (DNA) synthesis, prevention of spindle formation, or failure of chromosome separation. For example, aminopterin and methotrexate inhibit an enzyme needed during the cell cycle (see Chapter 1), leading to cell death during the S (DNA synthesis) phase.30 Viruses and other infectious agents may also interfere with mitosis.
4. Altered nucleic acid integrity or function. These alterations occur secondary to biochemical changes that interfere with nucleic acid replication, transcription, natural base incorporation, or ribonucleic acid (RNA) translation and protein synthesis. Because processes such as protein synthesis are essential for survival of the embryo, interference usually results in death rather than malformation. Diethylstilbestrol (DES) acts on estrogen-responsive tissue by altering RNA, protein, and DNA synthesis with development of columnar epithelium not normally found in the vaginal area. This “foreign” tissue is at risk for later malignant degeneration.30
5. Lack or excess of precursors, substrates, or coenzymes needed for biosynthesis. These deficiencies result in slowed or altered growth and differentiation and occur because of dietary deficiencies, placental transfer failure, maternal absorption failure, or the presence of specific analogues or antagonists. Some agents, such as amphetamines, cocaine, cannabis, and nicotine, may occupy receptors for nutrient, thus decreasing transfer of critical substances and altering fetal growth and development.44 Retinoic acid regulates expression of one of the developmental genes involved in craniofacial and axial skeleton development. Excess levels—as occurs with isotretinoin—leads to malformations in these systems. Coumadin inhibits formation of substances needed for proteins to bind to calcium and can alter bone ossification with craniofacial and other skeletal anomalies.30
6. Altered energy sources. As a result of interference with energy pathways (glucose sources, glycolysis, citric acid cycle, terminal electron transport systems), the energy needs of the rapidly proliferating and synthesizing tissues of the embryo are not met. For example, biotin deficiency—seen with an inborn error of metabolism—decreases activity of a mitochondrial biotin-dependent enzyme.30
7. Enzyme and growth factor inhibitions. Inhibition of critical enzymes interferes with cell functioning, cellular repair, differentiation, and growth. Inadequate folic acid increases the risk of neural tube defects, especially if the deficiency occurs in women with an enzyme defect in folic acid metabolism. Thalidomide may inhibit angiogenesis and thus limb growth by decreasing critical growth factors such as insulin-like growth factor-I and fibroblast growth factor.117 Inhibition of alcohol dehydrogenases, needed to detoxify alcohol, alters retinoic acid metabolism similarly to the action of isotretinoin (which also leads to craniofacial and skeletal defects).30
8. Osmolar imbalance. These imbalances lead to pathogenesis by causing edema that impinges upon the embryo.133
9. Altered membrane characteristics. These changes can result in abnormal membrane permeability and lead to osmolar imbalances and edema.133
10. Altered cell and neuronal migration or CNS organization. These processes are influenced by neurotransmitters, especially monoamines (dopamine, serotonin, and norepinephrine). Inborn errors of metabolism—especially those involved with amino acid metabolism—alter CNS organization. Drugs, such as cocaine, that increase or reduce levels of neurotransmitters may alter migration or later organization of cortical neurons (see Chapter 15).133
Specific proposed teratogenic mechanisms associated with drugs are folate antagonism, oxidative stress, angiotensin-converting enzyme inhibition, angiotensin II receptor antagonism, cyclooygenase-1 and -2 inhibition, 5-hydroxytryptamine-reuptake inhibition, and placental drug transporters.132
Effects of drug exposure in utero
Cigarette smoking
Smoking during pregnancy and its metabolites are “one of the leading preventable causes of adverse maternal and fetal outcomes.”92,p.75 Cigarette smoke contains many chemicals. Nicotine a particularly important in adverse pregnancy outcomes.18,92 Nicotine and cotinine readily cross the placenta with fetal levels often exceeding maternal levels.92 Animal data suggest that one mechanism by which nicotine may influence offspring is by binding to cholinergic receptors, leading to premature onset of cell differentiation and disrupting cell replication.92,115
The effects of smoking include an increased risk of low birth weight and prematurity.11,18,21,30,92 Smoking increases pregnancy and placental complications (e.g., ectopic pregnancy, placental abruption, and miscarriage) and may alter fertility in both female and males.30,92 Nicotine exposure increases the risk of altered cognitive and learning skills, sudden infant death syndrome, and altered respiratory function and increased respiratory infections.92 Chronic exposure to secondhand smoke also reduces fetal growth and increases the risk of low birth weight. The effects of smoking on birth weight and other effects are dose-dependent so that the fewer cigarettes a woman smokes, the less likely her infant will be affected.30
Alcohol
Ethanol is one of the most common abused drugs during pregnancy with specific teratogenic effects of fetal alcohol syndrome seen in 0.5 to 2/1000 births.130 Infants who were exposed to alcohol in utero are at increased risk for a range of alcohol-related damage including fetal alcohol syndrome (FAS) and other forms of alcohol-related damage.43,65,84 These differential effects may be due in part to variations in the metabolism of alcohol in the placenta by CYP2E1 and alcohol dehydrogenases.129 Diagnosis of FAS requires the presence of prenatal or postnatal growth restriction, CNS involvement, and specific craniofacial features. Other forms of alcohol-related damage seen in infants of women with a history of substantial alcohol intake during pregnancy include physical defects (e.g., congenital heart defects) without the characteristic facial features or mental and behavioral abnormalities without defects or facial features.65
Damage from ethanol may be due to chromosomal damage or acetyldehydrate (the placenta deoxidizes ethanol to this substance), which reaches 50% of maternal levels. Acetyldehydrate effects cell membranes and cell migration altering embryonic tissue organization with dysmorphic changes.43 Alcohol interferes with transport of amino acids across the placenta and incorporation into proteins. This may limit the number of fetal cells and lead to fetal growth restriction.43 Decreased placental transfer of linoleic and docosahexanoic acid may also alter fetal growth and development.129
No level of drinking alcohol has been proven safe in that drinking alcohol at any stage of pregnancy can affect the brain and other areas of development.43 Current studies do not support identifying a “safe level” of alcohol consumption by pregnant women. FAS usually occurs in offspring of chronic alcohol abusers, although it has occurred in women who drink less. Mild to moderate alcohol consumption during pregnancy has been associated with a range of later neurobehavioral effects, although the effects of moderate and light drinking are not yet well understood.21
Opiods
The fetus is not immune from developing chemical dependency. Neonatal abstinence syndrome (NAS) refers to particular withdrawal behaviors (CNS hypersensitivity, respiratory distress, autonomic dysfunction, and GI disturbances) observed in neonates exposed to dependency-producing drugs in utero.67 NAS is seen most commonly with maternal heroin or methadone use, but can also occur with drugs such as oxycodone, hydrocodone, and codeine. The timing and severity of withdrawal are based on the type of drug, the mother’s drug dosage, the length of time since the mother’s last dose, the duration of exposure, the neonate’s degree of immaturity, and the neonate’s general health status. Symptoms usually appear within 72 hours after birth but can be seen as late as 2 to 4 weeks of age. The neonate’s withdrawal responses may last from 6 days to 8 weeks.
Methadone has a long elimination half-life, which minimizes fluctuations in maternal serum levels and the risk of drug withdrawal in the fetus, reduces infant morbidity and mortality, and leads to less fetal growth restriction.11 Withdrawal symptoms may be more frequent and severe in infants exposed to methadone, possibly because methadone accumulates in fat tissue and is released slowly. Until the late 1990s, the widespread recommendation was to reduce a pregnant woman’s daily methadone intake to ≤20 mg/day in order to prevent NAS, however, the pregnant woman’s volume of distribution is increased so many women need higher doses to prevent withdrawal.11,34 Buprenorphine is a potent long acting (72 h) synthetic opioid with partial mu-receptor agonist and κappa-receptor antagonist properties that is being used with increased frequency as an alternative to methadone for the treatment of opiate-dependency. A recent multicenter international double–blinded study comparing buprenorphine with methadone treatment (n = 191) reported that neonates from mothers in the buprenorphine group required significantly less morphine, shorter hospital stay, and shorter duration of treatment for NAS. No significant differences were reported between groups in other outcomes or in rates of adverse events.63
The responses to drug withdrawal in the neonate are similar to those in the adult, but because of the nature of neurologic organization, the implications are more severe in neonates. NAS includes both physiologic and behavioral responses. Several scales have been developed to aid observation and measurement of the responses to neonatal abstinence. Interventions are initiated based on the severity of withdrawal as assessed by these scales. Withdrawal may be treated pharmacologically with drugs such as phenobarbital and morphine.67
Cocaine and amphetamines
Cocaine, amphetamine, and methamphetamine are central and peripheral stimulants that produces their effects by interfering with the reuptake of monoamines such as dopamine, norepinephrine, and serotonin at presynaptic adrenergic nerve terminals, thus increasing levels of these neurotransmitters at the neuronal junction (these transporters are inhibited by many antidepressants such as selective serotonin receptor reuptake inhibitors).2,11,45,48,134 The excess monoamines result in prolonged neuronal activation, leading to characteristic neurobehavioral responses (due to binding to central receptors) and cardiovascular and motor effects (due to binding to peripheral receptors located in tissues innervated by the sympathetic nervous system).9 Monoamines have a trophic role in CNS cell proliferation, neural migration, and organization. Thus exposure to elevated levels of monoamines may alter CNS organization, increasing the risk for later alterations in arousal and attention. Serotonin and norepinephrine receptors are found on the maternal facing side of the placental syncytium. Inhibition increases serotonin and norepinephrine in the intervillous space, which may lead to uterine contractions, vasoconstriction, and decreased placental blood flow, thus increasing the risk of preterm labor and fetal growth restriction.11,45,50,61
The actions of cocaine and amphetamines are similar, causing intense sympathetic nervous system activity. Cocaine produces vasoconstriction, tachycardia, and elevation of blood pressure in addition to a sense of excitement and euphoria. Cocaine is lipid-soluble, easily crossing the placenta. In the fetus, it produces increased motor activity and tachycardia; cocaine crosses the fetal blood-brain barrier. Cocaine competes with nutrients for monoamine transporters in the placenta.44 By successfully competing with nutrients for these transporters, cocaine reduces nutrient transfer, which can lead to fetal growth restriction. Levels of cocaine are high in amniotic fluid, resulting in a reservoir that prolongs fetal exposure.
Vasoconstrictive effects with reduction in placental blood flow to the placenta are associated with a high incidence of placental abruption.50 Blood flow to the fetus through the placenta is also reduced. In addition, the umbilical arteries constrict in response to cocaine. The resultant placental ischemia has been attributed as the cause of low birth weight, decreased body length, and smaller head circumference found among infants of cocaine-abusing mothers. Cocaine also increases uterine irritability and results in contractions. Cocaine may have teratogenic effects via damage to developing tissue either by vascular disruption and placental vascular insufficiency or production of reactive oxygen species during biotransformation of cocaine in the liver by P450 enzymes.9 Cocaine may cause cerebral infarcts in the fetus. Cocaine use during pregnancy also poses risks to the mother, including an increased risk of stroke, seizure, and uterine rupture.113
After birth, cocaine does not produce withdrawal behaviors, per se, but its effect appears to be related to an alteration of neurobehavioral organization that probably results from its direct influence on the developing brain. Neonates exposed to cocaine in utero are irritable, tremulous, and difficult to soothe and have rapid respiratory and heart rates; these infants exhibit excessive motor activity, altered sleep-wake patterns, poor feeding and feeding intolerance, and diarrhea.9,12,61 No increase in anomalies has been found in recent studies.77 Later outcomes of prenatal cocaine exposure include catch-up growth by 1-2 years in many but not all studies and an increased risk of behavior, attention, language development, information processing alterations in many studies of 4- to 14-year-olds.61,77 Outcomes are related to cumulative effects of cocaine, environment and possibly alcohol or other drug use.77
Neonatal symptoms from methamphetamine exposure are similar to those with cocaine, with neurobehavioral alterations (decreased arousal, increased stress, altered movements) reported.61,77 Animal and human studies demonstrate an increased risk of adverse pregnancy outcome with isolated anomalies reported.61 These infants are at increased risk of fetal distress, placental abruption, preterm birth, and a 2.5 fold increased risk of fetal growth restriction.61,129 Children usually achieve catch up growth by 2 years, depending on environment.77 While few studies have been done on older children, available studies suggest similar findings to cocaine exposure.77
Drugs and lactation
The majority of breastfeeding women take at least one drug during lactation.51,118 Many pharmacologic agents and environmental pollutants (as well as alcohol, nicotine, and other abused drugs) can be found in human milk, although usually at levels lower than those in the mother. Drugs cross into breast milk across the milk-blood barrier via the following mechanisms: (1) transcellular diffusion of low molecular weight (<100 to 200 daltons), nonionized, lipid soluble substances such as ethanol, which are pulled across by water flow; (2) intercellular movement of large molecules through spaces between alveolar cells; (3) passive diffusion across a concentration gradient; (4) carrier mediated diffusion of polar substances; and (5) active movement via specific drug transporters.15,111 A few drugs—nitrofurantoin, acyclovir, ranitidine, and iodine—are actively transported across the blood-milk barrier into human milk by influx transporters.51
Often, breastfeeding is interrupted or discontinued for maternal therapy even though there are relatively few maternal medications that are not compatible with breastfeeding.39,41 Even if the mother is on drugs with potential risks, careful monitoring and strategies to minimize infants’ exposure can be often used so that the mother can decide to continue breastfeeding if she chooses. In general, serious side effects in infants are uncommon.
The use of any pharmacologic agent in the breastfeeding woman should be carefully evaluated. Most drugs enter breast milk in some quantity, although few drugs are contraindicated during lactation.24 Often little is known about side effects of less commonly used agents. Some drugs should be used with caution and only as absolutely needed in the lactating woman, with careful monitoring of both infant and mother. The nurse and the mother should be aware of potential hazards of any drug, for both mother and infant. There are many reviews of drugs used during breastfeeding and concerns or potential hazards of specific agents3,4,23–25,33,41,51,52,59,87 as well as textbooks and online sites (see Box 7-2 on p. 187).
Drugs taken by a nursing mother reach infants in much smaller amounts through breast milk than drugs from the pregnant woman reach the fetus across the placenta.82,111 Figure 7-4 illustrates transfer of milk from mother to infant. Drugs from the pregnant woman to the fetus move directly from maternal blood to fetal blood, whereas drugs from the nursing mother to infant pass from mother’s blood into milk and then to the infant’s gut. In the infant’s gut, the drug may be destroyed or absorption reduced due to limitations in GI absorption in infants, especially in the early months. In addition, there are more tissue layers between maternal blood and milk than between maternal and fetal blood across the placenta. This increases the diffusing distance and reduces the efficiency of transfer. Calculated doses of maternal drug received by the infant are generally significantly less than the standard therapeutic doses tolerated by infants without toxicity for most drugs.111 Therefore, some drugs that are not recommended during pregnancy may pose little risk during lactation.
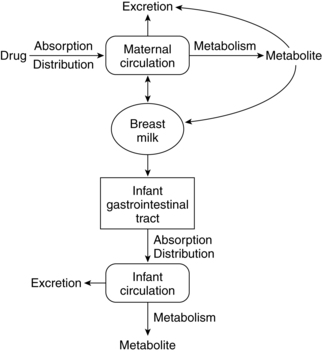
Factors influencing the amount and rapidity of drug excretion into milk include drug dosage and duration; route of administration; drug physicochemical characteristics; blood level in maternal circulation; oral bioavailability; protein binding in maternal circulation; maternal physiology and drug handling; blood-milk barrier (Figure 7-4); and infant physiology, drug handling, and maturity (see Table 7-7). Other infant factors to consider include the age of the infant, drug oral bioavailability, amount of milk intake, and the record of the drug’s use as a therapeutic agent in infants.15,51,75,86,123 Characteristics of drugs most likely to cross into milk are agents that are lipid-soluble, have low molecular weight (MW), have a long half-life in the mother, have low protein binding, are given to the mother in large doses, or are used for chronic conditions or are used long term so that plasma levels are maintained at steady-state conditions.5,51,81,86
Infant drug exposure can be calculated by the milk-to-plasma ratio of a drug, percentage of maternal dose, infant dosage (drug concentration in milk times the volume of milk ingested), and estimated infant serum concentration (using oral bioavailability/clearance in the infants).82 The milk-to-plasma (M/P) ratio is the most common index and ranges from 0.01 to 6.5 (averaging 0.5 to 1 for most drugs used during lactation).10,74 This value may be calculated as a single value, but the M/PAUC (where AUC is the area under the curve), which is based on concentration time curves, provides a better estimate of the ratio over a dosing interval. Highly lipid-soluble and water-soluble substances with MW lower than 200 daltons generally have an M/P ratio near 1. High M/P ratios (greater than 1) are seen with weak bases and actively transported substances; a low M/P ratio (less than 1) is generally found with drugs that are weak acids or highly protein bound substances.75 Drugs with a low (less than 1) M/P ratio and high maternal and infant clearance rates generally have low risk of infant side effects.39 M/P and M/PAUC ratios are available in several resources.10,15,52,74
Most drugs cross by passive or facilitated diffusion from higher to lower concentrations between maternal plasma and milk.51 Some low MW drugs may also cross via membrane pores, while other substances such as immunoglobulins may pass between alveolar cells.111 The blood-milk barrier consists of the maternal capillary epithelium, mammary alveolar cell membrane, intracellular structures (including protein micelles and lipid vesicles), and the apical membrane of the mammary alveolar cell (Figure 7-5). The alveolar cell is a lipid barrier that reduces transfer of water-soluble and ionized drugs.15 Therefore, lipid-soluble drugs cross into milk more readily than water-soluble drugs, although this is somewhat influenced by MW. Concentration of drugs in milk may change with time from birth or during a feeding. For example, drugs such as diazepam and phenobarbital that are highly lipid-soluble may be more concentrated in hindmilk, which has four to five times greater lipid content than foremilk.10,51 Lipid content is higher in mature milk versus colostrum; thus concentrations of lipid-soluble drugs may be reduced initially after birth.82 Efflux transporters such a P-gp may transport some substances out of milk back to maternal plasma.51
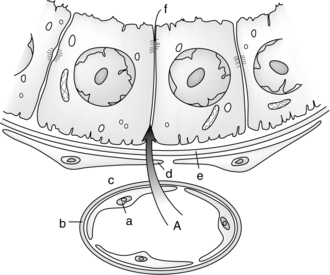
Low MW substances such as ethanol rapidly diffuse across the tissue layers between maternal blood and milk. Water-soluble drugs with an MW of less than 200 daltons can move across the blood-milk barrier via water-filled membrane pores.82 Molecules greater than 200 daltons cannot pass through these pores and are transferred more slowly because they must first dissolve in the outer lipid membrane of the alveolar cell, diffuse across the cell, then dissolve in the apical membrane to reach milk. Substances with MWs larger than 800 daltons, such as insulin and heparin, generally do not cross into milk or do so very minimally.51,75 Larger substances may cross in the first 4 to 10 days after delivery due to larger gaps between alveolar cells during this time.37,87,98,111
The major milk protein (α-lactalbumin) does not significantly bind drugs. One of the main serum drug-binding proteins (α1 acid-glycoprotein) is not present in milk. Therefore the major milk drug-binding proteins are albumin and lactoferrin, which have markedly decreased binding compared to plasma proteins. Drugs that are highly bound in maternal blood (such as warfarin which is 99% bound) are less likely to cross, because plasma levels of free drug are lower. If a drug is at least 85% bound in maternal blood, measureable concentrations of the drug are unlikely to be found in the infant.5 Conversely a drug such as lithium which has no protein binding and a low MW can reach high levels in milk.5 Free drug levels may be increased in the mother in the first 5 to 7 weeks postpartum as the lower albumin levels of pregnancy gradually return to nonpregnant values.82
Milk pH (mean of 7.08 with a range of 6.35 to 7.35) is lower than blood pH.111 Weak acids are highly ionized in maternal plasma and less likely to cross into milk. However, weak bases (e.g., erythromycin, iodides, antihistamines, barbiturates) tend to be transferred and are at equal or higher concentrations in milk than in maternal plasma.82 Colostrum has a mean pH of 7.45, so it is more likely to have higher levels of basic drugs than mature milk.111 Ion trapping may occur in milk. Weak bases are generally nonionized and readily cross into milk. As these drugs encounter the low milk pH, they become ionized. This creates a gradient toward movement of more of the drug into milk and reduces the ability of the drug to return across the blood-milk barrier to maternal blood.15,51,82 Drugs with a pKa greater than 7.2 are most likely to be sequestered in milk.51
Steady-state concentrations of drugs in the infant are influenced by drug oral bioavailability, dose, and infant clearance. Drugs given to the mother via methods other than oral administration often have a low oral bioavailability. Drugs with high oral bioavailability may be sequestered in milk at high levels. Drugs with low oral bioavailability have only minimal milk transfer.51,82 This reduces infant exposure since the infant receives these drugs via milk into the gut. The amount of drugs reaching the nursing infant is also influenced by non–steady-state conditions. Milk is supplied on a continuous basis during sucking, so milk drug concentrations at time of feeding tend to reflect maternal plasma levels at that time. Concentrations of drugs in milk (including most narcotics) are independent of volume of milk.
Drugs may also affect lactation via either central or peripheral mechanisms to increase or reduce milk output.39,51,111 Central mechanisms include increased or decreased oxytocin, prolactin, or CNS dopamine release. Increased dopamine depresses prolactin. Peripheral mechanisms act directly on the breast to alter milk production. Exogenous substances that increase milk production include thyrotropin-releasing hormone, domperidone, metoclopramide, and psychotropic agents such as reserpine, imipramine, and phenothiazine derivatives that interfere with dopamine release.39,51 Nicotine, estrogens, and possibly pseudoephedrine may also depress lactation.51,59
Use of combination oral contraceptives by lactating women is controversial, with many differing opinions. Estrogen-containing oral contraceptives should be avoided as they suppress lactation probably through inhibition of prolactin by estrogen.51 These agents have been associated with decreases in milk production, shorter overall length of time of breastfeeding, and slower infant weight gain, although this is not well documented and the available studies are limited and have significant methodologic limitations.15,51,111,124 Truitt and colleagues conducted a systematic review of randomized controlled studies and concluded that existing trials were “insufficient to establish an effect of hormonal contraception, if any, on milk quality and quantity.”125 The effects on breast milk production are reportedly most prominent in the first 4 to 6 weeks postpartum.39,111 Although progestin only or combination agents have been thought to be safer,39,111 Hale recommends that mothers be advised to avoid progestin agents for 4 to 6 weeks postpartum and that even with use after 6 weeks some mothers report decreased milk synthesis.51
Minimizing infant drug exposure
A drug with a high dose may be safe if given after feeding or before the infant’s longest predictable sleep period. However, a normally safe drug may be problematic if given in a high dose or over a long duration.39 Most data on drug levels in milk are with mature milk, with minimal data on milk from mothers of preterm infants. These infants may be at greater risk for adverse effects from exposure to drugs in breast milk due to their immature hepatic and renal function. In addition, these infants are more likely to absorb large macromolecules across their immature (“open”) gut (see Chapter 13).10 Although the major concern is that the infant will receive a pharmacologically significant concentration of drug, other possible effects are allergic sensitivities and altered gut flora with antibiotics.39
Suggestions for reducing the effects of drugs in breast milk on the infant include: (1) using alternative forms of drugs within the same drug class that pass more poorly into milk, have shorter half-lives, or can be given via alternate routes (topical or inhaled); (2) using single components rather than compound drugs (e.g., a decongestant for allergy rather than a multisymptom drug); and (3) feeding the infant before taking the medication, thus avoiding feeding during peak plasma levels, which tend to occur 1 to 3 hours after a maternal oral dose.13,37,39,52,75,82,87,98,111 Use of a short-acting form of the drug may reduce the risk of accumulation for drugs with concerns at high levels, however use of preparations that can be given at longer intervals (once versus three to four times per day) may be a useful strategy for other agents.13,37,39,52,75,82,87,98,111
If the mother is taking a drug for which there are concerns about the potential for elevated infant plasma levels, drug levels may need to be monitored periodically. Monitor the infant closely (growth pattern, sleep patterns, activity, alertness, behaviors, responsiveness, and general health). Breastfeeding may need to be temporarily discontinued when mothers are taking medication for diagnostic procedures or are taking drugs with a high potential for toxicity that are given once. In such cases, previously expressed milk or formula may be used. The timing for reinstitution of breastfeeding varies with the toxicity of the agent. For mild toxicity, resumption of nursing may be as soon as 1 to 2 maternal half-lives (50% to 75% elimination); for high toxicity, it may be necessary to wait for 4 to 5 maternal half-lives (94% to 97% elimination) or longer.82,111 Rarely, breastfeeding may need to be discontinued if a highly toxic drug is necessary for maternal health such as chemotherapy.
Summary
Drug therapy of the pregnant woman, fetus, neonate, and lactating woman is a complex health challenge. Both pregnant women and neonates have alterations in drug absorption, distribution, metabolism, and elimination that increase the risk of subtherapeutic or toxic levels of individual drugs. An understanding of the principles influencing transfer of drug across the placenta and into breast milk is critical to reducing risk in the fetus and young infant. Drug therapy of the lactating woman generally poses fewer risks to the infants than therapy of the pregnant woman. However, the use of any pharmacologic agent for either the pregnant and breastfeeding woman must be carefully evaluated and drugs used with caution. Neonates present a unique challenge for pharmacologic therapy, not only because of their physiologic immaturity but also because their systems continue to undergo maturation during early infancy, so drug handling can change within a relatively short period of time. Recommendations for clinical practice related to perinatal pharmacology are listed in Table 7-8. Box 7-2 on p. 187 lists print and online resources for information about drugs in pregnancy and lactation.
Table 7-8
Recommendations for Clinical Practice Related to Perinatal Pharmacology
Know the effects of physiologic alterations during pregnancy on pharmacokinetics (pp. 185-189 and Table 7-2).
Recognize the effects of altered gastrointestinal and hepatic function during pregnancy on drug absorption and metabolism (pp. 185-189).
Recognize the effects of altered body water, fat, and plasma proteins on drug distribution and binding during pregnancy (pp. 187-188).
Recognize the effects of altered renal function on drug elimination during pregnancy (p. 189).
Counsel women regarding the side effects of drugs and potential toxicity to mother, fetus, and neonate (pp. 185, 203-208).
Counsel women regarding planned fetal drug therapy (p. 197).
Monitor and evaluate maternal responses to drugs for evidence of subtherapeutic doses (pp. 185-189, 195).
Know or verify the usual doses for medications given during pregnancy (pp. 184-189).
Recognize the effects of maternal physiologic adaptations on the pharmacokinetics potential toxicity to the woman and fetus (pp. 185-189, 195). of antibiotics (p. 195).
Counsel women regarding the side effects of drugs used to treat specific chronic conditions and potential toxicity to the woman and fetus (pp. 185, 203-208).
Counsel women with chronic disorders regarding the impact of their disorder and its pharmacologic treatment during pregnancy and of treatment alternatives (pp. 185-189, 195).
Know the factors that influence distribution of drugs and other substances in the fetus (pp. 195-197 and Table 7-4).
Recognize critical periods of development and principles of teratogenesis (pp. 203-206 and Chapter 3).
Recognize risks associated with fetal exposure to drugs and other environmental agents during pregnancy (pp. 203-208).
Provide appropriate counselling and health teaching to promote optimal fetal development and health (pp. 203-208 and Chapter 3).
Provide counselling and health teaching to reduce exposure of the fetus to adverse environmental influences (pp. 203-208 and Chapter 3).
Counsel women regarding the use of drugs or exposure to other environmental agents during pregnancy (pp. 203-208).
Know the effects of neonatal physiology on pharmacokinetics (pp. 197-202 and Table 7-7).
Recognize the effects of immature gastrointestinal and hepatic function in the neonate on drug absorption and metabolism (pp. 199, 200-202).
Recognize the effects of altered body water, fat, and plasma proteins on drug distribution and binding in the neonate (p. 200).
Recognize the effects of maturation of liver enzyme systems on drug metabolism and risks of side effects in the neonate (pp. 200-202).
Recognize the effects of immature renal function on drug elimination and risks of side effects in the neonate (p. 202).
Know or verify the usual doses for drugs given during the neonatal period (pp. 197-202).
Recognize the side effects of drugs and potential toxicity to the neonate (pp. 197-202).
Monitor and evaluate neonatal responses to drugs, including peak and trough and serum levels (pp. 200-203).
Know the factors that place neonates at risk for toxic responses to topically applied substances (pp. 198-200).
Provide care to reduce the risk of topically applied substances (pp. 198-200).
Counsel women regarding the use of medications during lactation (pp. 208-211).
Recognize the risks associated with use of pharmacologic agents in breastfeeding women (pp. 208-211).
Evaluate the need for use of specific drugs and counsel women regarding side effects (pp. 208-211).
Monitor and evaluate maternal and neonatal responses to drugs taken by the breastfeeding woman (pp. 208-211).
Institute interventions as appropriate to reduce the risks associated with specific drugs (p. 211).
Assess infants for signs of intrauterine drug exposure from maternal substance abuse (pp. 206-208).
Know resources for obtaining information about risks of specific drugs during pregnancy and lactation (Box on p. 187).
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
Pharmacology and pharmacokinetics during the perinatal period
Pharmacokinetics refers to the processes involved in drug absorption, distribution, metabolism, and biotransformation and excretion (Figure 7-1). Site of absorption depends on route of administration (e.g., oral, intramuscular, intravenous, topical, inhaled). Drugs are carried in the blood, either bound to plasma proteins or as free drug. Protein-bound drugs provide a reservoir for future use, whereas free drug is the active component available to interact with the target cell. Biotransformation occurs primarily in the liver with a series of enzymatic reactions to modify and convert the drug into more polar (water-soluble) compounds. Intermediary steps in biotransformation may result in active metabolites. For example, codeine is demethylated to morphine, and in the neonate theophylline is methylated to caffeine.32 Biotransformation also takes place in the lungs, intestinal mucosa, and kidneys. Hepatic biotransformation occurs in two phases. Phase I (nonsynthetic reactions) takes place primarily in the microsomes, although some occurs in the mitochondria and cytosol. Phase I reactions modify the activity of a drug. Phase I enzymes include the cytochrome P450 (CYP450) monooxygenase system. Phase II (synthetic) reactions require adenosine triphosphate (ATP) and, except for glucuronidation, are extramicrosomal.32 Phase II reactions generally inactivate the drugs by converting them to more polar substances. Table 7-1 provides examples of drugs metabolized by various phase I and phase II reactions. Not all drugs undergo hepatic metabolism before elimination; some drugs are excreted directly via the kidneys.
Table 7-1
| REACTION | EXAMPLES OF DRUG SUBSTRATES |
| PHASE I (NONSYNTHETIC REACTIONS) | |
| Oxidation | Phenytoin, phenobarbital, ibuprofen, acetaminophen, morphine, codeine, diazepam, cimetidine |
| Reduction | Prontosil, chloramphenicol, ethanol |
| Hydrolysis | Acetylsalicylic acid, indomethacin |
| PHASE II (SYNTHETIC REACTIONS: CONJUGATIONS) | |
| Glucuronide conjugation | Morphine, acetaminophen |
| Glycine conjugation | Salicylic acid |
| Sulfate conjugation | Acetaminophen, α-methyldopa |
| Glutathione conjugation | Ethacrynic acid |
| Methylation | Dopamine, epinephrine |
| Acetylation | Sulfonamides, clonazepam |
Adapted from Chemtob, S. (2004). Basic pharmacologic principles. In R.A. Polin, W.W. Fox, & S.H. Abman (Eds.). Fetal and neonatal physiology (3rd ed.). Philadelphia: Saunders.
Variations in drug pharmacokinetics and pharmacodynamics occur among individuals at all ages, even with similar doses of a given medication, due to extrinsic (diet, environment, other therapies) and intrinsic (development, genetic constitution, diseases) factors.95 Currently much research focuses on understanding these factors, especially the contributions of individual genetic polymorphisms (see Chapter 1). This has led to new fields of study such as pharmacogenetics and pharmacogenomics. Pharmacogenetics is the study of “genetic variations that give rise to interindividual responses to drugs,”95, p.256 while pharmacogenomics integrates pharmacology and genomics into “the broader application of genome-wide technologies and strategies to identify both disease processes that represent new targets for drug development and factors predictive of therapeutic efficacy and risk of adverse drug reactions.”95, p.256 Genetic polymorphisms are small variations in an individual’s genome that may either be nonfunctional (have no effect on the individual) or modify expression of a protein that may alter the effects of exposures to environmental health hazards or result in individual differences in responses to drugs (changing efficacy, risk of adverse effects, or therapeutic dosing regimens).76,95 Understanding these variations, which is just beginning, can individualize pharmacologic management. During pregnancy, these individual differences may occur in the other, fetus and/or placental, or in any combination of these to influence the expression of specific genes involved in biotransformation, amount and activity of enzymes produced and thus levels of the drug and potential risks.76
The term drug is used generically in this chapter to refer to prescription and nonprescription pharmacologic agents, herbal agents, and vitamins, as well as drugs of abuse. In addition, the principles that govern placental transfer of drugs are also applicable to transfer of chemicals, food additives, and environmental agents. Definitions of common terms used in pharmacokinetics are summarized in Box 7-1 on p. 185.
Pharmacokinetics during pregnancy
Drug use during pregnancy
A survey of drug utilization by 14,000 pregnant women in 22 countries found that 86% of these women took some medication during pregnancy, with an average woman taking 2.9 medications (range, 1 to 15).73 A longitudinal survey from the United States surveyed 578 women throughout pregnancy. These women took a mean of 1.14 prescribed drugs per person, excluding iron and vitamins, with the most frequent being antibiotics, respiratory agents, gastrointestinal (GI) agents, and opioids. Of these women, 59.7% used at least one prescribed drug; 3.1% used more than five. In addition, these women took a mean of 2.95 over-the-counter drugs per person (92.6% of the women studied used at least one; 20.8% used more than five), with the most frequent being analgesics and agents for gastrointestinal or upper respiratory symptoms. Herbal agents were used by 45.2% of study participants, with a mean of 0.77 herbal agents per person; most frequently used were peppermint tea, cranberry juice, aloe, and other herbal teas.49 A retrospective chart review of eight U.S. health maintenance organizations (n = 152,531 women) from 1996 to 2000 found that 59% of the women received drugs other than vitamin or mineral supplements after their first prenatal care visit (39% in the first, 34.4% in the second, and 37.9% in the third trimester). Approximately one third of the women were on prescribed drugs for which there is evidence of some fetal risk (FDA category C, D, or X).8 More recently Mitchell and associates reviewed drug use during pregnancy between 1976 and 2008. They found that (1) average use of prescribed and over the counter drugs increased over these years from 2.5 to 4.2; (2) use of prescribed drugs increased 63% during the first trimester and by 2008, 94% of women took at least one prescribed drug; and (3) <1% of pregnant women took an antidepressant in 1988-90 versus 7.5% in 2006-08.89 Petersen et al. reported a four-fold increase in use of antidepressants during pregnancy from 1992 to 2006.104
Adherence to prescribed drug regimens is a problem during pregnancy, with up to 50% of women either not taking a prescribed drug or stopping before receiving a full course of the drug.27,72,104 This can lead to maternal risks, especially in women with chronic diseases.72,104 Peterson et al. found that antidepressants were more likely to be discontinued by pregnant than nonpregnant women, often leading to increased depression and other complications.104 Problems with adherence stem primarily from concerns about teratogenic effects. Most women overestimate the teratogenic risks of common agents.69 For example, in one study women personally estimated that the risk of having a baby with a birth defect was 25% (which is the risk associated with a potent teratogen such as thalidomide). After counseling, however, they more correctly estimated the risk at 5%.69 The background risk of a birth defect for the general population is 3% to 5%. However, the risk may be higher or lower for an individual woman depending on factors such as her health status, teratogen exposures, and genetic makeup.
Several principles have been identified to guide drug therapy during pregnancy: (1) avoid medications in the first trimester whenever possible; (2) anyone prescribing drugs for a woman of childbearing age must consider a potential pregnancy before prescribing; (3) women receiving long-term drug therapy require counseling before pregnancy of the potential implications and risks; (4) a necessary treatment should not be stopped without good reason; (5) drugs may have altered effects during pregnancy (e.g., the increased metabolism of methicillin or increased renal elimination of digoxin may result in decreased levels, resulting in the need for higher doses); (6) use single action, short-acting medications rather than long-acting or combination drugs; (7) use the lowest effective dose of the safest available medication; (8) use drugs only if the benefits outweigh the risks; (9) use alternate routes of administration if available (i.e., topical or inhaled rather than systemic agents); (10) select drugs that have a history of use during pregnancy without adverse effects, rather than the “latest” drug; and (11) use doses at the low end of the normal dose range while recognizing that for some drugs (e.g., digoxin, phenytoin, lithium), the woman’s usual dose may need to be increased due to increased volume of distribution (Vd) and clearance of these agents during pregnancy.73,109 These principles also apply to all women of childbearing age and to preconceptional and interconceptional use of drugs, in that approximately half of all pregnancies are unplanned and critical developmental processes occur before most women realize they are pregnant.73
Alterations in drug absorption, distribution, metabolism, and excretion during pregnancy
Maternal responses to drugs during pregnancy are influenced by both maternal physiology and the presence of the placental-fetal unit.81 Variations in handling of specific drugs by the pregnant woman may occur due to effects of the normal physiologic changes of pregnancy on drug absorption, distribution, metabolism, and excretion (Table 7-2). The presence of the placental-fetal compartment can also alter maternal kinetics.81 For example, many antibiotics are lipid-soluble and may readily cross the placenta and become sequestered in the fetal compartment and thus be unavailable to the mother. This can lead to lower maternal levels.
Table 7-2
Influence of Pregnancy on Physiologic Aspects of Drug Disposition
| PHARMACOKINETIC PARAMETER | CHANGE IN PREGNANCY |
| ABSORPTION | |
| Intestinal motility | Decreased |
| Ventilation | Increased |
| Cardiac output | Increased |
| Blood flow to skin | Increased |
| DISTRIBUTION | |
| Plasma volume | Increased |
| Total body water | Increased |
| Plasma proteins | Decreased |
| Body fat | Increased |
| METABOLISM | |
| Hepatic metabolism | ± |
| Extrahepatic metabolism | ± |
| EXCRETION | |
| Uterine blood flow | Increased |
| Renal blood flow | Increased |
| Glomerular filtration rate | Increased |
| Ventilation | Increased |
From McClary, J., Blumer, J.L., & Aranda, J.V., et al. (2011). Developmental pharmacology. In R.J. Martin, A.A. Fanaroff, & M.C. Walsh (Eds.). Fanaroff and Martin’s neonatal-perinatal medicine: Diseases of the fetus and infant (9th ed.). Philadelphia: Mosby Elsevier, p. 711.
Evidence regarding effects, effectiveness, and safety of most pharmacologic agents during pregnancy is limited.69,80,87 Pharmacokinetic data on specific drugs during pregnancy is often minimal because pregnant women are excluded from drug trials. As a result, many drugs are not labeled for use in pregnancy, because the effects of the drug on the fetus are unknown.70,131 This is protective for the fetus but may also limit benefits to the pregnant women from these drugs. The net result of changes in drug handling during pregnancy depends on the individual drug, with both increases in levels (with need for a lower dose or less frequent dosing) and decrease in levels (with a need for a higher dose or more frequent dosing) seen.78 In general, both peak and steady-state serum concentrations are lower or unchanged in the pregnant woman.20 Although changes may not be clinically significant the efficacy and toxicity of drugs can be harder to predict, because of the physiologic changes during pregnancy that have the potential to alter pharmacokinetics.20,57
Little analyzed data on pharmacokinetics of drugs in pregnancy and found that across all agents (1) drug oral bioavailability decreased during pregnancy in 47% of studies of different agents; (2) Vd increased in 39%, with no change in 48%; (3) peak plasma levels decreased in 33%, with no change in 57%; (4) steady-state plasma levels decreased in 45%, with no change in 45%; (5) half-life decreased in 41%, with no change in 44%; (6) clearance increased in 55%, with no change in 34%; and (7) protein binding decreased in 86%.78 Therefore during pregnancy, there are considerable variations in the direction of pharmacokinetic changes among different drugs. This increases the complexity for the practitioner in choosing a specific pharmacologic agent and monitoring its effects. Many studies have significant methodologic problems, for example, sample sizes may be small, the composition of the control group may vary (e.g., the group may include nonpregnant women, adult males, or the same subjects postpartum), women of various gestational ages may be grouped together, and dosing methods may vary.36,78 Pregnancy drug registries have been established to collect experiences with specific drugs or groups of drugs (such as antiepileptic agents) during pregnancy and fetal and neonatal outcomes (see Box 7-2). Three types of drug registries have been established: individual academic sites; pharmaceutical drug company sites; and population-based sites (from individual countries or a group of countries).24,128
Drug absorption
Pulmonary, GI, and peripheral blood flow changes during pregnancy can alter absorption of drugs from the lungs, gut, and skin (see Chapters 9, 10, and 12). The increased minute ventilation that occurs in pregnancy can increase the rate of drug uptake across the alveoli.81,86 As a result, the pulmonary system may have a greater role in drug and metabolite excretion during pregnancy.91 Alveolar uptake of inhalation agents is also influenced by changes in cardiac output, which leads to increased pulmonary blood flow and thus increased alveolar uptake. As a result, doses of volatile anesthetics (e.g., halothane, isoflurane, methoxyflurane) may need to be decreased to compensate for these changes in the pregnant women.81,102 Inhaled aerosols such as anti-inflammatory and bronchodilators may have enhanced absorption.106
Absorption of oral medications is influenced by gastric acidity, gastric motility, presence of bile acids or mucus, nausea and vomiting of pregnancy, and intestinal transit time. During pregnancy there is decreased gastric acid (hydrochloric acid [HCl]) secretion, altered intestinal emptying time, increased mucus, and decreased gastric and intestinal motility.36,106 Decreased gastric motility can increase the oral bioavailability of slowly absorbed drugs (such as digoxin) by prolonging transit time, and can decrease peak plasma levels of rapidly absorbed drugs. Factors altering GI function in pregnancy may initially delay, then prolong absorption of oral medications. Peak plasma levels may be lower, and the time to peak levels may be later than in nonpregnant women. These changes tend to peak in the third trimester.68 Changes in pH affect ionization, and absorption of drugs.36,81 For example, decreased gastric pH slows the rate at which weak acids (e.g., aspirin) are absorbed and increases absorption of weak bases (e.g., narcotic analgesics).102 Decreased GI motility may alter initial absorption of oral antibiotics and lead to unpredictable patterns of intestinal absorption. Absorption of hydrophilic drugs may be enhanced due to the longer intestinal transit time (from delayed intestinal emptying and decreased motility).83 Increased cardiac output and peripheral blood flow may increase the rate of absorption of drugs from the stomach and small intestine.78 Iron chelates with some drugs taken concurrently, preventing absorption. However, even with these changes in GI function, the clinical effect for most drugs is not significant. This is because therapeutic windows are usually wide enough for drugs administered orally that these factors do not significantly alter therapeutic effects.91
Cardiovascular and integumentary changes during pregnancy may lead to more rapid absorption of agents delivered via transdermal, intranasal, intravascular, epidural, and subcutaneous routes.102 The increased extracellular water and peripheral blood flow to the skin during pregnancy may enhance absorption and alter distribution of topical agents. For example, topical and vaginal iodine-based preparations are not recommended during pregnancy because of the increased absorption of iodine. Iodine is of concern because it is actively transported across the placenta to the fetus, whose thyroid gland has a high avidity for this substance (see Chapter 19). Increased peripheral perfusion secondary to decreased peripheral vascular resistance and vasodilation may increase absorption of intramuscular drugs.36
Drug distribution
The distribution of drugs within the body depends on many factors, including the amount of body water, which affects Vd; degree to which a drug is bound to plasma proteins or body tissues; and the presence of the fetal-placental unit. For many drugs the Vd is increased during pregnancy because of the increased plasma volume, blood volume, cardiac output, total body water, and body mass.20,36,91 The increased plasma and blood volumes, and thus Vd, may result in decreased drug levels in the central compartment, reduce serum levels of drugs, and necessitate larger loading doses.83,103 Increases in total body water during pregnancy affect primarily water-soluble (polar) drugs that tend to stay in the extracellular space. Initial doses may need to be increased to achieve therapeutic concentrations; however, maternal doses for most drugs are not significantly altered since drug steady state is influenced by bioavailability and clearance as well as Vd.68 Due to changes in the Vd, some pregnant women may notice that their prepregnancy dose of a drug taken regularly, such as digoxin or phenytoin, may not achieve the same therapeutic effect during pregnancy.
Some lipid-soluble drugs (e.g., caffeine, diazepam, thiopental) have a longer half-life in the pregnant woman, whereas polar drugs (e.g., oxazepam, ampicillin) tend to have a shorter half-life.52,130 The Vd for lipophilic drugs is increased during pregnancy because of the accumulation of fat, which may serve as a reservoir for some drugs via tissue binding.36,78 Tissue binding is a mechanism by which a drug is removed from circulation and stored in tissue such as hair, bone, teeth, and adipose tissue. This mechanism can result in storage of significant quantities of a drug, because tissue storage sites may need to be saturated before there is sufficient free drug to be effective at receptor sites. Factors reducing maternal serum drug levels increase the risk of subtherapeutic drug levels. When the drug is discontinued, tissue deposits may give up their stores slowly, resulting in persistent drug effects. Because lipid-soluble drugs are stored in adipose tissue, the increased adipose tissue during pregnancy can lead to a slight decrease in the amount of free lipid-soluble drugs, such as sedatives and hypnotics, and persistence of drug effects (“hangover”) after the drug has been discontinued.
Drugs in plasma are either unbound (free) or bound to plasma proteins, primarily albumin or α1-acid glycoprotein. The reduction in plasma proteins, especially albumin, during pregnancy may increase plasma levels of free (active) drug. Albumin is the major binding protein for acidic drugs such as salicylates, anticonvulsants, nonsteroidal anti-inflammatory agents, and some neutral drugs such as warfarin and diazepam.81 Serum albumin levels fall during pregnancy, increasing the unbound or free drug fraction. Total plasma drug concentration stays the same during pregnancy, but the unbound fraction of albumin-bound drugs is greater; therefore the drug may be cleared faster or transported more rapidly across the placenta.81 Increases in free drug do not necessarily lead to an increase in plasma levels of that drug if the increase in free drug is balanced by more rapid hepatic biotransformation or renal elimination.36,81,102 However, with regard to doses of some drugs that are bound to albumin (such as many antiepileptic drugs), total and free drug concentrations may be altered, and doses may need to be adjusted during pregnancy. For example, phenytoin total plasma concentration is decreased by 55% to 65% and levels of free drug by 18% to 31% during pregnancy; carbamazepine total concentration is reduced 0% to 43% and free drug 0% to 28%; phenobarbital total concentration is reduced by 50% to 55%.102 Thus when monitoring serum concentrations of drugs such as phenytoin during pregnancy, free versus total levels should be evaluated (many laboratories report free levels if specified when ordering).36
Free fatty acids and steroid hormones, both of which are increased in pregnancy, may compete with drugs for albumin binding sites, further increasing levels of unbound drug. The decrease in albumin, coupled with increases in free fatty acids and steroid hormones, are most prominent in late pregnancy. This change can result in unpredictable transient increases in free levels of some drugs, such as phenytoin, valproate acid, carbamazepine, sulfisoxazole, theophylline, and phenobarbital as well as the potential for more rapid elimination increases the risk of subtherapeutic maternal levels.36,46 α1-Acid glycoprotein is the major protein for binding basic drugs such as local anesthetics, most opioids, and β-blockers. α1-Acid glycoprotein is decreased slightly during pregnancy.6,106
Changes in protein binding during pregnancy not only influence availability of free drug in the mother but also the amount of drug available to cross the placenta. This may alter fetal-to-maternal drug ratios, thus further altering placental transfer. Drugs with hepatic blood flow–limited biotransformation (e.g., propranolol, lidocaine) are rapidly eliminated in their first pass through the liver, regardless of whether they are bound or unbound.20,103 Therefore even with the decreased protein binding in pregnancy, hepatic clearance and total drug concentration generally are not significantly altered, although concentrations of free drug, Vd, and half-life may increase. Conversely, with drugs whose hepatic biotransformation is independent of hepatic blood flow (e.g., warfarin, phenytoin), only the unbound fraction is removed by the liver. For these drugs, total drug concentrations may be lower than usual, with a decreased half-life and greater fluctuations in peak and trough levels.
Hepatic drug metabolism
The hepatic changes during pregnancy (see Chapter 12) can alter biotransformation of drugs by the liver and clearance of drugs from the maternal serum. Drugs that are primarily (greater than 70%) metabolized in their first pass through the liver (high extraction ratio) are usually cleared rapidly by the liver, because clearance of these drugs is dependent on hepatic blood flow. Hepatic elimination of drugs with high extraction ratios is generally unchanged in pregnancy. Drugs with low (less than 30%) first-pass hepatic clearance (low extraction ratio) are more dependent on liver enzyme systems. The rate of elimination of these drugs (e.g., theophylline, caffeine, and diazepam) is related to free drug levels and tends to be decreased in pregnancy with increased half-lives. Hepatic elimination of other drugs—including many antibiotics, pancuronium, phenytoin, and acetaminophen—is increased in the pregnant woman.
Some liver enzymatic processes may be slower during pregnancy, delaying drug metabolism and degradation; other processes have increased activity. The cytochrome P450 (CYP450) monooxygenase system is a group of phase I enzymes essential for hepatic metabolism of drugs. The enzymes of this system are indicated by the prefix CYP (cytochrome) followed by a series of numbers and letters, (e.g., CYP1A2, CYP3A4, CYP2D6). Activity of the following are increased during pregnancy: CYP3A4 (50% to 100%), CYP2A6 (54%), CYP2D6 (50%), and CYP2C9 (20%).5,57,72,123 Up regulation of CYP2C9 decreases plasma concentrations and increases clearance of phenytoin, nonsteroidal anti-inflammatory agents and some oral antidiabetic agents.57,68 Changes to CYP3A4 may increase the metabolism of methadone, nifedipine, and indinavir during pregnancy.102,123 However, activity of CYP1A2, which is responsible for hepatic elimination of many drugs, is decreased (33% in the first, 50% in the second, and 65% in the third trimester); activity of CYP2C19 is also decreased (50%).5,6,78,123 CYP1A2 is also involved in clearance of caffeine so decreased activity may lead to slower clearance and enhance effects during pregnancy.72,102
Activity of phase II enzymes is also altered during pregnancy, including some of the uridine 5′-diphosphatase glucuronosyltransferase (UGT) enzymes.5,102,106 For example, UGT1A4 is increased 200% in the first and second trimesters and 300% in the third trimester; UGT2B7 is increased 50% to 200% by the third trimester.6,123 Some extrahepatic enzymes, particularly cholinesterase, are also decreased, which may alter the woman’s response to neuromuscular agents.28,36,131 For example, the decrease in pseudocholinesterase during pregnancy has been enough to impair breakdown of suxamethonium with subsequent prolonged paralysis after anesthesia in some women. The relative increase in hepatic metabolism and blood flow during pregnancy (as a consequence of the increased cardiac output), may increase clearance of polar drugs.36 In general, slowly metabolized drugs that are cleared primarily by the liver tend to be cleared more slowly in pregnancy due to decreased enzymatic activity and the net decrease in liver blood flow. This may also increase the length of time potentially teratogenic intermediary metabolites remain in circulation.
Clearance and metabolism of drugs by the liver during pregnancy may also be influenced by the increases in steroid hormones that stimulate hepatic microsomal enzyme activity. For example, progesterone may induce enzyme activity for phenytoin clearance, whereas progesterone and estradiol alter hepatic elimination of caffeine and theophylline by competitive inhibition of microsomal oxidase.36,81 Estrogens have cholinergic effects and may interfere with clearance of drugs such as rifampin that are excreted into the biliary system.81 Smoking and alcohol may also induce hepatic enzymes.
Renal drug excretion
Many drugs are excreted primarily by the kidneys. Thus the increased glomerular filtration rate (GFR) in pregnancy (see Chapter 11) alters renal excretion of drugs, especially water-soluble drugs such as penicillin G, digoxin, aminoglycosides, lithium, and pancuronium. These drugs are eliminated more rapidly, leading to lower and potentially subtherapeutic blood and tissue levels.36,78,91 These effects are most prominent in late pregnancy.72 In many cases the changes are not clinically significant enough to require alteration in drug dose.36 However, this is not always the case. For example, pregnant women with paroxysmal atrial tachycardia may experience an increased frequency of attacks when taking usual doses of digoxin. These women may require up to a 50% increase in their digoxin dose during pregnancy.81 In addition, shorter dosage intervals are recommended for nifedipine and labetalol due to a threefold increase in clearance in the pregnant woman.81 The half-life of atenolol, digoxin, and peripheral myorelaxants is shorter during pregnancy.102 Because lithium is eliminated entirely by GFR, the pregnant woman may require a higher dose of this drug, especially during the third trimester.88 More rapid clearance and shorter half-lives are also seen with many penicillins, cephalosporins, aminoglycosides, and sulfonamides as a consequence of the changes in renal function during pregnancy.102 Morphine metabolites may be cleared more quickly in late pregnancy and may lead to a lowered analgesic effect.72
Alterations in drug absorption, distribution, metabolism, and excretion during the intrapartum and postpartum periods
Drug handling is further altered during the intrapartum period. GI absorption may decrease to a greater extent than seen in pregnancy. Gastric emptying is reported to decrease during labor (see Chapter 12), although this may be secondary to use of opiate agents such as meperidine and pentazocine. Administration of antacids to decrease the risk of acid aspiration with anesthesia can alter ionization by altering gastric pH, prolonging gastric emptying, or complexing with drugs, reducing their absorption. Cardiac output increases in the intrapartum period further increase the Vd and may transiently alter hepatic and renal blood flow.101 Blood flow to the lower extremities may be reduced, especially if the woman is in a supine position. This may result in a slower and more unpredictable rate of absorption of drugs given intramuscularly in the buttock or thigh.101 Plasma nonesterified fatty acids rise during labor and may compete for albumin binding sites, with an increase in free drug levels.101 Both supine positioning and oxytocin may reduce renal blood flow and delay renal excretion of drugs and drug metabolites during labor. Most analgesics and anesthetics used in labor, especially opioids, tend to be low-molecular-weight, lipid-soluble, weak bases that enhance movement across the placenta to the fetus once they enter maternal circulation. The clinical effects depend on the timing of the dose, half-life, drug physicochemical characteristics, placental blood flow, and maternal and fetal drug metabolism and excretion.79
During the immediate postpartum period, the pattern of pharmacokinetics and drug distribution is similar to that of late pregnancy and remains so until the pregnancy-induced physiologic alterations of each system return to nonpregnant status.42 During this transition, drug handling by the woman may remain altered to some extent for some drugs. Transfer of drugs to nursing infants is discussed later in this chapter in “Drugs and Lactation.”
Transfer of drugs across the placenta
Dancis suggested that in thinking of placental transfer one should “Ask not whether a maternal [substance] crosses the placenta. Ask rather, how, how much, and how fast. Ask also as to fetal need.”35 There are few compounds, endogenous or exogenous, that are unable to cross the placenta in detectable amounts given sufficient time and sensitivity of detection.20,23,35,87,91 Drugs with similar physiodynamic properties can differ in placental transfer.87,105 Plonait and Nau suggest that if no other information is available and a group of drugs is not known to have teratogenic effects, then a comparison of lipid solubility, molecular weight (MW), ionization, and degree of protein binding might assist the clinical selection of an appropriate drug for more efficient transfer (fetal drug therapy) or least efficient transfer (limited fetal exposure).105 For example, drugs that are lipid soluble have an extremely low (50 daltons) or low (51 to 599 daltons) MW, and are nonionized with low protein binding in maternal plasma cross the placenta more rapidly and in higher concentrations than drugs that are water soluble (polar), have a high MW (greater than 600 daltons), and are ionized and highly bound to protein in maternal plasma. Transfer of drugs across the placenta varies with developmental changes in the placental, and thus gestational age.121
Patterns of transfer kinetics
Three major patterns of transfer pharmacokinetics are seen: (1) complete transfer (drugs rapidly equilibrate between mother and fetus, with blood flow being the rate limiting step); (2) exceeding transfer (levels are higher in the fetus than in the mother); and (3) incomplete transfer (levels are higher in the mother than the fetus).99 These patterns are illustrated in Figure 7-2. Examples of drugs with each pattern are listed in Table 7-3.
Table 7-3
Examples of Drugs with Transplacental Kinetics*
| MODEL A OR B | MODEL C | MODEL D | MODEL E | MODEL F |
| Some barbiturates: Thiopental Pentobarbital Secobarbital Antipyrine Promethazine Ritodrine Magnesium sulfate Digoxin |
Some benzodiazepines: Diazepam Lorazepam Desmethyl-diazepam Oxazepam Valproate Salicylate Nalidixic acid Nicotine Urea Some penicillins: Ampicillin Penicillin G Methicillin |
Ascorbate Colistimethate† Furosemide Meperidine Some cephalosporins: Cephalothin Cefazolin Cephapirin Cephalexin Some aminoglycosides: Gentamycin Kanamycin |
Amide-type local anesthetic agents: Lidocaine Bupivacaine Some β-adrenoceptor blockers: Propranolol Sotalol Labetalol Dexamethasone Cimetidine Ranitidine Methadone Some sulfonamides‡ |
Heparin Quaternary ammonium compounds: Tubocurarine Succinylcholine Vecuronium Pancuronium Elementary ions: Chlorthalidone Dicloxacillin Erythromycin Nitrofurantoin |
*According to models A through F in Figure 7-2. Differentiation between models A and B, C and D, and E and F is often uncertain because of incomplete data on the initial phase of drug distribution across the placenta. Model B is rarely applied.
†Polypeptide antibiotic, with a molecular weight of 1200 daltons.
Modified from Plonait, S.L. & Nau, H. (2012). Physiochemical and structural properties regulating placental drug transfer. In R.A. Polin, W.W. Fox, & S.H. Abman (Eds.). Fetal and neonatal physiology (4th ed.). Philadelphia: Saunders.
There is a great deal of variability even among drugs with similar pharmacokinetic characteristics. However, in general, complete transfer tends to be seen with drugs that have a high pKa and MW less than 600 daltons, whereas incomplete transfer is seen with drugs that have a high pKa and MW greater than 600 daltons. Strongly ionized drugs generally have incomplete transfer, although exceptions occur. For example, ampicillin and methicillin are strongly ionized but have lipophilic side groups, so they have a complete transfer pattern.98,101 Valproate and salicylates are ionized at the normal (physiologic) pH level but rapidly cross the placenta.81
Factors influencing placental transfer
Placental transfer is influenced by the following: (1) maternal and fetal drug handling; (2) surface area of the placenta; (3) diffusing distance between maternal and fetal blood; (4) physicochemical characteristics of the drug; (5) gradients (drug concentration, electropotential [normally 20 mV], and pH) between mother and fetus; (6) degree of binding of a substance to proteins in maternal and fetal circulations; (7) concentrations of binding proteins and protein binding gradients between mother and fetus; (8) permeability of the placental barrier (primarily the syncytiotrophoblast); (9) rates of maternal and fetal blood flow through the intervillous space and villi; and (10) placental influx and efflux transporters (Table 7-4).58,68,86,98,99,102,126
Table 7-4
Factors Influencing the Distribution of Drugs and Other Substances in the Fetus
| FACTOR | POSSIBLE INFLUENCES |
| Type of drug | Factors that increase transfer include lipid solubility, low molecular weight (less than 500 to 600 daltons for lipid soluble and less than 100 daltons for polar substances), nonioinized, and unbound. |
| Amount of drug | Transfer is increased by greater maternal-to-fetal gradient, especially for drugs transferred by diffusion. |
| Membrane permeability | Diffusion of substances increases with increasing gestation and greater placental efficiency. Some drugs have greater affinity for specific fetal tissues (e.g., tetracycline in teeth, warfarin in bones, phenytoin in the fetal heart [because this is a highly lipid organ in the fetus, whereas in adults the central nervous system has a high lipid content owing to myelin sheaths], streptomycin in the optic nerve). |
| Fetal body water compartment | Drug distribution and dilution change as the total body water compartment decreases with increasing gestation. With an increased volume of distribution, peak volumes of drugs are reduced and excretion may be delayed. |
| Fetal circulation | Maternal and fetal blood flow rates will influence transfer. Upon reaching the fetal circulation, drugs may be shunted by the ductus venosus past the liver (thus missing an opportunity for detoxification), with highest concentrations of these substances in blood going to the heart and upper body. |
| Serum protein binding | Protein binding of a drug in the maternal system limits the amount of free drug available for transfer to the fetus. Binding of drugs to macromolecules in the fetal circulation may increase the maternal-to-fetal transfer by maintaining a concentration gradient from the mother to the fetus. |
| Receptor function | Functional ability of receptors on cell membranes increases with gestation, leading to increased specificity to respond to or exclude certain drugs. |
| Placental enzymes | Enzymes produced by the placenta (e.g., insulinase) may detoxify drugs and reduce transfer to the fetus. |
| Gestational age | Many of the factors identified above are altered with advancing gestation and maturity of the fetus and placenta. The size and efficiency of the placental exchange area increase with increasing gestational age. |
| Fetal pH during labor | The fetal pH is usually 0.1 to 0.15 units below that of the mother. A decrease in the fetal pH may increase the transfer of acidophilic agents from the mother to fetus. Hypoxia alters blood flow and thus drug distribution, metabolism, and excretion. For example, with hypoxia, blood flow to the liver and kidneys may be reduced with preferential flow to the brain. Albumin binding of drugs may also be reduced, resulting in more free drug in the fetal circulation. |
Diffusing distance and surface area.
Increased surface area for exchange, increased concentration gradient, and decreased diffusing distance enhance transfer across the placenta. With increasing gestation and placental growth, efficiency of the placental transfer increases as a result of the increased surface area. As the placenta matures, the distance between maternal and fetal blood decreases due to thinning of the syncytiotrophoblast and connective tissue and increases in the size and number of capillaries in the villi (see Chapter 3). Thus more drug passes to the fetus in the third trimester than in the first trimester. Transfer may be reduced with decreased surface area (small placenta, placental infarcts) or increased diffusing distance (placental edema, infection). Edema with fetal hydrops increases the diffusing distance and decreases transfer so drugs such as digoxin may be less effective in treating arrhythmias in the fetus with hydrops.71
Physiochemical characteristics and concentration gradients.
Physiochemical characteristics that influence movement across the placenta include lipid solubility, MW, degree of ionization, and protein binding. These characteristics can increase, decrease, or deter movement of potentially harmful drugs and other substances from the maternal to the fetal circulation.105 For example, placental transfer may be increased or enhanced if a substance is lipid soluble (e.g., diazepam, lipoproteins, most sedatives) or nonionized (e.g., phenobarbital), has a low MW (e.g., alcohol), or has a lower albumin binding (e.g., ampicillin). A substance will have reduced, slower, or no transport across the placenta if it has a certain charge or molecular configuration (e.g., heparin) or certain size (e.g., heparin, IgM), is altered or bound by enzymes within the placenta (e.g., amines, insulin), or is firmly and highly bound to the maternal red blood cell (e.g., carbon monoxide) or plasma proteins (e.g., dicloxacillin).
Lipid-soluble substances cross the placenta more readily than water-soluble substances, because cell membranes are made primarily of lipids that enhance diffusion. Lipid-soluble substances dissolve in the lipid membranes of the tissues separating maternal and fetal blood, primarily the syncytiotrophoblast. Polar substances cross slowly unless they have low MW and can move through pores. If the syncytiotrophoblast has a high lipid affinity for a drug (e.g., buprenorphine), the drug may be sequestered in the placenta with decreases in both maternal and fetal concentrations.120
The exact MW limit for placental transfer is unknown, although transfer of substances with MW >500 to 600 daltons is limited.105 Insulin (with an MW of more than 6000) does not cross the placenta because of its size and configuration and because of the effects of placental insulinase enzymes.36,99 Substances with higher MW tend to cross more slowly and are influenced by maternal concentrations; for example, if maternal concentrations are high, the substance is more likely to cross. These substances may also cross via active transport if a carrier is present. Immunoglobulin G (IgG) with an MW of 150,000 daltons is transported across the placenta via endocytosis and exocytosis (see Chapters 3 and 13).37 Water-soluble substances with an MW of less than 100 daltons diffuse readily; those with an MW greater than 100 diffuse more slowly. Small lipid-soluble substances (MW less than 500 to 600 daltons) cross rapidly and are blood flow–dependent; those with an MW of more than 500 to 600 daltons cross more slowly and may have incomplete transfer.20,91,120 However, MW is of limited value clinically in that most drugs have MWs less than 600 daltons. Exceptions include thyroid hormones (MW 889), insulin (MW greater than 5000), heparin (MW 20,000 to 40,000), and low-MW heparin (MW 3000 to 5000).52,68,99,102
Maternal-to-fetal concentration or electrochemical gradients also influence transfer. The maternal-fetal pH gradient influences transfer of drugs and can result in “ion trapping,” with higher concentrations of some drugs in the fetus than in the mother. Nonionized substances cross the placenta more efficiently than ionized substances.36 Normally the fetus and amniotic fluid are slightly more acidotic than the mother by 0.1 to 0.15 pH units.99 As a result of this difference, basic drugs may accumulate in the fetus and amniotic fluid with levels higher than in the mother. In the mother, a weak base tends to remain nonionized in her plasma and cross the placenta rapidly. As this weak base comes into contact with the more acidic fetal blood, the drug becomes more ionized, increasing the flow gradient from mother to fetus. The now-ionized drugs cannot return across the placenta to the mother as readily, thus becoming trapped in the fetus.20,81,99,120 For example, most opioids are weak bases that may accumulate in the fetus, which is one reason that effects of these drugs may persist in the neonatal period. The opposite effect is seen with acidic drugs: that is, these drugs are more likely to be ionized in the mother and thus not cross the placenta as readily.20 For nonionized, lipophilic drugs, the rate-limiting step for placental transfer is blood flow; for polar drugs, the rate-limiting step is diffusion.81 Hypoxia and acidosis in the fetus can influence transplacental passage of drugs. For example, local anesthetics are weak bases whose degree of ionization depends on pH. If the fetus becomes acidotic, ionization of these drugs increases with accumulation (“ion trapping”) in the fetus and neonate.47,99,102
Drug metabolites also cross the placenta to the fetus and vice versa. The majority of fetal drug elimination is by transfer back to the mother across the placenta. However, some elimination may occur by fetal metabolism, placental metabolism, or excretion into amniotic fluid.91 This transfer is also based on physicochemical characteristics, primarily ionization and lipid solubility. Most drug metabolites are polar, because polarity enhances biliary and renal elimination. Once polar substances are transferred to the fetus, elimination back across the placenta is slow and may result in accumulation in the fetus or amniotic fluid.91 In addition the fetus may metabolize a drug producing polar metabolites. These metabolites can also become trapped and accumulate in the fetus or be excreted into amniotic fluid and reswallowed by the fetus. Examples of drugs that tend to concentrate in amniotic fluid are the β-lactams, such as penicillin G, dicloxacillin, and amiodarone.36,91
Protein binding.
Drugs that are protein bound, usually to albumin or α1-acid glycoprotein, in the mother or fetus do not cross the placenta. Therefore plasma protein gradients between mother and fetus can also influence transfer. For example, ampicillin has a lower protein binding to albumin, thus increasing the amount of free drug that can cross to the fetus, and resulting in decreased maternal levels. Protein binding of dicloxacillin is 96%. Therefore dicloxacillin would be an appropriate choice to treat maternal infections, but not to treat intrauterine or fetal infection. Another example is propranolol, which is highly protein bound so it crosses much slower than atenolol, which has minimal binding.71
Maternal albumin levels fall during pregnancy and are about 15% to 20% lower than those in the fetus at term; α1-acid glycoprotein levels in the fetus are only about 37% to 40% of maternal levels.91,99,120 Thus at term, albumin (or fetal albumin macromolecule) bound drugs may be more highly bound in the fetus, whereas α1-acid glycoprotein drugs are more highly bound in the mother.91 Binding to proteins in the fetus may increase transfer of the drug by maintaining a concentration gradient across the placenta. Plasma protein binding and thus concentrations of drugs in the mother versus the fetus changes with increasing gestational age for some drugs. The decreased maternal albumin with increasing gestation increases the amount of free drug in the mother available for placental transfer. For example, diazepam binds to albumin. Albumin binding of diazepam is low in the fetus in the first half of gestation. However, in late gestation, as maternal serum albumin levels decrease, more free diazepam is present in maternal serum and, because this is a lipophilic drug, readily crosses the placenta. Thus by term, fetal diazepam levels are greater than maternal levels (Table 7-5).105 This may increase the risk of neonatal hyperbilirubinemia in that diazepam competes with bilirubin for albumin binding (see “Neonatal Pharmacokinetics”).
Table 7-5
| DRUG | PRIMARY BINDING PROTEIN | BOUND IN MOTHER (%) | BOUND IN FETUS (%) | FETUS/MOTHER TOTAL PLASMA CONCENTRATION RATIO |
| Betamethasone | AGP | 60 | 41 | 0.33 |
| Bupivacaine | AGP | 91 | 51 | 0.27 |
| Diazepam | ALB | 97 | 98.5 | 1.6 |
| Lidocaine | AGP | 64 | 24 | 0.66 |
| Mepivacaine | AGP | 55 | 36 | 0.71 |
| N-desmethyl diazepam | ALB | 95 | 97 | 1.7 |
| Phenobarbital | ALB | 41 | 36 | 1.0 |
| Phenytoin | ALB | 87 | 82 | 1.0 |
| Salicylate | ALB | 43 | 54 | 1.2 |
| Valproic acid | ALB | 73 | 88 | 1.7 |
AGP, α1-Acid glycoprotein; ALB, albumin.
From Plonait, S.L. & Nau, H. (2012). Physiochemical and structural properties regulating placental drug transfer. In R.A. Polin, W.W. Fox, & S.H. Abman (Eds.). Fetal and neonatal physiology (4th ed.). Philadelphia: Saunders.
Placental blood flow.
The rate of maternal blood flow to and through the intervillous space and fetal blood flow to and through the villi influence placental transfer, particularly of nonionized, lipophilic substances such as anticonvulsants, alcohol, and opioid derivatives. Contractions decrease transfer so drugs that cross rapidly and are given during a contraction may demonstrate decreased transfer to the fetus. Contractions may also delay clearance of drugs from the fetus.120 In addition to uterine contractions, factors that may alter uteroplacental blood flow include maternal position; anesthesia; nicotine, β-lactams, ototoxics and other drugs; emotional or physical stress; and degenerative changes within the placenta that are seen with hypertension, prolonged pregnancy, diabetes, or renal disease.71
Route of administration.
The route of administration can also affect placental transfer. For example, nalbuphine that is given intramuscularly results in fetal blood levels that are 80% of maternal venous levels; if it is given intravenously, fetal levels are three to six times greater than maternal levels.81,99 Peak drug concentrations in the fetus (and the infant, if delivered after maternal administration) do not necessarily occur at the same time as in the mother. For example, meperidine peaks at 30 to 50 minutes in the mother, but 90 minutes to 3 hours after maternal administration in the fetus as a result of transfer kinetics across the placenta. Therefore meperidine levels are highest in the infant if given to the mother 90 minutes to 3 hours before delivery; on the other hand, fentanyl is higher in the neonate than mother if given shortly before delivery. Therefore it is reasonable to give meperidine if birth is expected within the hour.
Antibiotics are some of the most commonly prescribed drugs in pregnancy. Because antibiotics cross the placenta primarily by simple diffusion, rate-limiting factors include maternal-fetal concentration gradients, MW, and other physicochemical characteristics of the drug, placental surface area, diffusing distance, and the degree to which the drug is bound to maternal plasma proteins (see Table 7-5). A general pattern of antibiotic distribution to the fetus has been described. After an intravenous dose, peak maternal serum concentrations are seen within 15 minutes then fall exponentially. Peak umbilical cord values are seen in 30 to 60 minutes followed by an exponential fall. Peak amniotic fluid levels are seen in 4 to 5 hours. This lag results from slow excretion in fetal urine due to fetal renal immaturity.114
Placental transporters.
Although many drugs cross the placenta by simple diffusion, some drugs use placental influx or efflux transporters. These transporters are located along the apical membrane of the microvillus brush border facing maternal blood in the intervillous space and on the syncytiotrophoblast basal membrane (facing the fetal connective tissue matrix and blood vessels in the villi) or fetal capillary epithelium to facilitate movement of nutrients and other physiologic substances across the placenta.44,58,91,98,102,106,120,126 Some drugs may also be transferred across the placenta by nutrient (influx) transporters, including glucose, carnitine, monoamine, and organic ion carriers.102 Nonphysiologic substances—some drugs, environmental pollutants, and toxins—if structurally similar to nutrients, may compete for nutrient transporters. The drugs that seem to be particularly able to use this method to cross the placenta include amphetamines, cocaine, cannabinoids, and nicotine and other substances from cigarettes.44,129 By successfully competing with nutrients for these transporters, nutrient transfer is reduced and fetal growth and development altered. In addition, cephalosporins, ganciclovir, and corticosteroids cross by facilitated diffusion, probably by using carriers that normally transport dipeptides, hormones, and steroids.98
The placenta also contains a variety of efflux transporters whose role is to remove steroids and other potentially toxic substances and thus protect the fetus.68,71,102,120,129,135 Originally most of the transporters were identified in studies of multidrug resistance in tumors as is reflected in some of their names. Examples of drug efflux transporters in the placenta include members of the APT-binding cassette (ABC) group such as multidrug-resistant gene protein (MRP) 1, commonly called P-glycoprotein (P-gp), MRP-2, MRP-3, and breast cancer resistance protein (BCRP).58,68,102,121,126,129,135 P-gp, MRP-2, and BCRP are located on the apical membrane of the syncytiotrophoblast cells facing maternal blood; MRP-1 and MRP-3 are on the basal or fetal side of the trophoblast (see Figure 7-3).47,68,126 Efflux transporters are also found on the gut, lung, and blood brain barrier and help protect entry of substances into the body and brain. Efflux transporters are under hormonal regulation, for example, BCRP by estradiol, and progesterone and P-gp by glucocorticoids.126 Inflammation and infection may alter regulation of these transporters.89
Each transporter appears to have specific substrates for which they provide protection. For example, studies of P-gp suggest that it exports cations, steroids, cytotoxic drugs, some antibiotics, opioids, phenobarbital, erythromycin, phenytoin, verapamil, and other substances that enter the placenta back into maternal blood, thus reducing fetal exposure.20,58,102,135 BCRP is involved in efflux of flavinoids, zidovudine, cimetidine, glyburide, nitrofurantoin, and the food-borne chemical carcinogen PhiP.58,68,102 MRP 1 and 3 prevent entry of organic anions and promote excretion and reduce entry of glutathione/glucuronide metabolites into the fetus.47 Some transporters may have bidirectional flow which can allow xenobiotics to be transported across the placenta to the fetus.68 Thus BCRP and P-gp cannot only decrease the amount of drug reaching the fetus but enhance removal of any drug reaching the fetal circulation.129 A drug that is a P-gp or BCRP substrate would be preferred for treating a maternal disorder with minimal fetal drug exposure, whereas a drug that is not a P-gp or BCRP substrate would be preferred for fetal pharmacotherapy.120
Deficiency of these transporter proteins increases the risk of teratogenesis from fetal drug and chemical exposure.58,135 Gene polymorphisms for the ABC efflux transporters may increase or decrease placental transfer of these substances and thus alter fetal susceptibility to effects of maternal drugs.58
Placental metabolism of drugs
The placenta has many enzyme systems, including various peptidases that can metabolize drugs, thus influencing transfer of specific substances from mother to fetus or fetus to mother.120 Both phase I and phase II drug metabolizing enzymes are present.106,129 Several isoforms of the CYP450 system (phase I reactions) are found in the placenta, although at levels about half of those seen in the liver. The primary CYP450 isoforms seen in the placenta are CYP1A1/1A2, although other isoforms, including CYP2E1, CYP3A4, CYP3A7, and CYP4B1, have also been reported, although with low activity.20,55,91,101,106,112,120
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
