Pharmacology
After reading this chapter, the student will be able to:
• Name and explain the various branches of pharmacology
• Describe administration, absorption, and distribution of drugs in the body
• Explain how biotransformation aids elimination of drugs from the body
• Describe the dose–response curve
• Describe and explain the time–response curve
• Discuss the variability of drug action
• Differentiate between adverse drug response and toxicity
• Describe the steps necessary for the development of new drugs
• Name and describe the schedules used for categorizing drugs
Introduction
HEALTHCARE APPLICATION
Examples of Drugs Used in the Various Organ Systems
| Drug | System/Condition | Effect |
| Methylphenidate | Central nervous | Stimulant |
| Digitalis | Cardiovascular | Treats congestive heart failure |
| Colchicine | Neuromuscular | Analgesic |
| Quinidine | Cardiovascular | Antiarrhythmic |
| Procainamide | Cardiovascular | Antiarrhythmic |
| Phenytoin | Central nervous | Anticonvulsant |
| Verapamil | Cardiovascular | Antianginal |
| Nitroglycerin | Cardiovascular | Antianginal |
| Acetazolamide | Renal | Diuretic |
| Levothyroxine | Endocrine | Treats hypothyroidism |
| Barbiturate | Central nervous | Sedative |
| Benzodiazepine | Central nervous | Antianxiety |
| Prednisolone | Respiratory | Inhibits inflammatory responses |
| Sodium bicarbonate | Gastrointestinal | Increases stomach pH |
| Sildenafil | Reproductive | Treats erectile dysfunction |
| Sibutramine | Obesity | Inhibits reuptake of serotonin and norepinephrine that regulate food intake |
| Somatotropin | Endocrine | Antipituitary to block IGF action on liver cells. |
Branches of Pharmacology
This chapter introduces some fundamentals of “physiological pharmacology” or “pharmacological physiology”—referring to the same anatomy, physiology, and biochemistry discussed in Chapter 2 (Chemistry of Life) and Chapter 3 (Cell Structure and Function). Although all drugs are potential poisons (substances that cause severe distress or death), when given in subtoxic doses they are medicinal tools. The goal of rational drug therapy is to aid and maintain the body and its biological systems in healthy working order. In therapy, drugs give living tissues added chemical support to prevent and to meet physical and pathological challenges to which biological systems are exposed. Pharmacology is organized into separate disciplines that describe actions of drugs:
• Pharmacodynamics addresses drug-induced responses of the physiological and biochemical systems of the body in health and disease.
• Pharmacokinetics addresses drug amounts at various sites in the body after their administration.
• Pharmacotherapeutics addresses issues associated with the choice and application of drugs to be used for disease prevention, treatment, or diagnosis.
• Toxicology is the study of the body’s response to poisons; their harmful effects, mechanisms of action, symptoms, treatment, and identification.
• Pharmacy, on the other hand, includes the preparation, compounding, dispensing of, and record keeping about therapeutic drugs, for which drug nomenclature is essential. In addition, pharmacy includes the following:
Drug Nomenclature
• Nonprescription: Over-the-counter (OTC) drugs approved by the Federal Drug Administration (FDA) and herbal supplements, not approved by the FDA
• Prescription: Drugs required by law to be prescribed by licensed practitioners (i.e., veterinarians, dentists, and physicians)
Individual drugs have three names: chemical, generic, and proprietary or trade (brand) name (Table 21.1):
TABLE 21.1
| Trade or Brand (Proprietary) Name | Generic (Nonproprietary) Name | Chemical Name | Therapeutic Class |
| Amoxil, Amoxicot, Trimox, DisperMox … | Amoxicillin | (2S,5R,6R)-6-[(R)-(–)-2-amino-2-(p-hydroxyphenyl)acetamido]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid trihydrate | Antibiotic |
| Advil, Motrin, Midol, Nuprin … | Ibuprofen | (±)-2-(p-Isobutylphenyl) propionic acid | Antiinflammatory |
| Tylenol, Anacin-3 … | Acetaminophen | N-Acetyl-p-aminophenol | Analgesic |
| Dilantin, Dilantin KAPSEALS | Phenytoin | Sodium 5,5-diphenyl-2,4 imidazolidinedione | Anticonvulsants |
| Allegra | Fexofenadine | (±)-4-[1 hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-butyl]-α,α-dimethyl benzeneacetic acid hydrochloride | Antihistamines |

• Chemical: A single accurate description of the chemical structure, which is often lengthy but is of value to synthetic chemists; for example, 3,5-dihydroxyphenylalanine
• Generic: Nonproprietary, a single name, usually indicating the relationship of a drug to a therapeutic class; for example,
• Proprietary or trade (brand) name: A legally licensed drug name assigned by manufacturers. If a drug is produced by many manufacturers the list of proprietary names can become lengthy; for example, the local anesthetic procaine has at least 24 different names
Principles of Drug Action
Drugs alter physiological activity and for them to be effective they must reach their intended target site at the appropriate concentration (Table 21.2). This involves several processes collectively called pharmacokinetics. These principles include the following:
TABLE 21.2
Drug Losses at Sites of Action Through Administration
| Route of Administration | Drug Loss From: |
| Enteral route only | Degradation in stomach |
| First-pass effect | |
| Small intestine | |
| Failure to be absorbed | |
| Binding to food or other contents | |
| Liver | |
| Secretion in bile | |
| Biotransformation | |
| Tissue binding | |
| Enteral and parenteral routes | |
| General blood circulation | Biotransformation |
| Binding to plasma proteins | |
| Distribution to body tissues | Drug too dispersed, not sufficient at site of action |
| Tissue binding | |
| Biotransformation | |
| Metabolism | |
| Excretion |
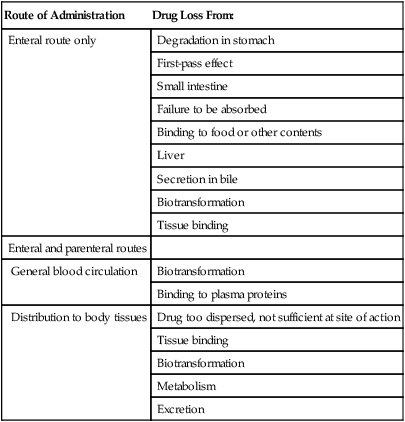
Administration
For direct local action, drugs may be given (Figure 21.1) in the following ways:
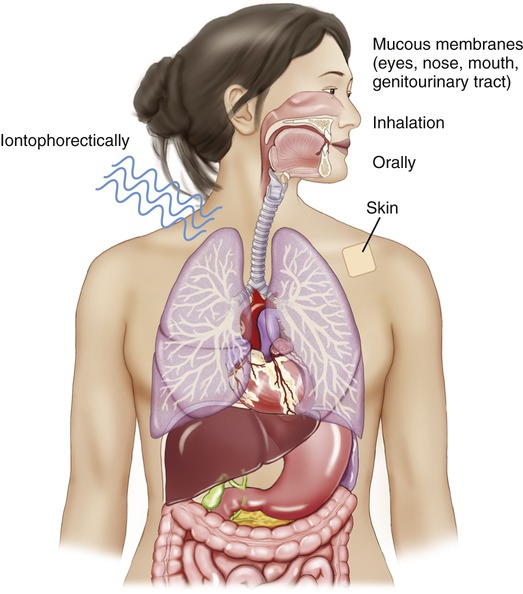
For direct administration, drugs may be administered to the skin or mucous membranes, orally, by inhalation, or iontophoretically through the skin by electrical charge to the given target.
• On mucous membranes (e.g., the nose, mouth, throat, eye, or genitourinary tract)
• Orally (limited to, e.g., osmotic cathartics, osmotic diuretics, and antacids for gastrointestinal tract infections)
• By inhalation for some respiratory and lung diseases
• Iontophoretically (in or through the skin via control of ionic [i.e., positive or negative] charges)
For systemic effects, drugs may be given (Figure 21.2) in the following ways:
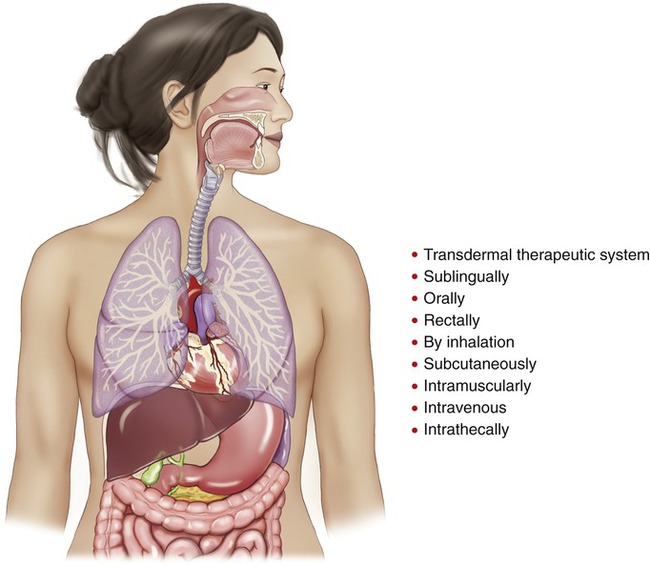
Sites through which drugs can be given to achieve a systemic effect.
• Through a transdermal therapeutic system (e.g., via a controlled slow-release drug reservoir attached to the skin)
• Sublingually: Under the tongue (e.g., nitroglycerine tablets)
• Orally: By mouth for absorption via the gastrointestinal tract
• Rectally: By suppositories in the rectum via the anus into areas of extensive vascularization and rapid blood flow. Useful in infants or in patients with loss of consciousness
• By inhalation: Into the lungs; another area of extensive vascularization and rapid blood flow. Good for direct application on huge lung tissue surface (200 m2) vascularly structured to absorb and remove materials. In addition, the lungs receive, in the course of 1 minute, the total amount of blood passing through the rest of the body during the same time interval. In brief, the lungs are the most effective absorptive area of the body
• Subcutaneously: Below the cutaneous layer. Good for slow absorption of small amounts of drug
• Intramuscularly: Rapid rate of absorption because of extensive vascularization and rapid blood flow, but less than by the intravenous route
• Intravenously: Most rapid rate of absorption. Can be modified by a port (cannulation of the great caval veins for repeated drug administrations such as in cancer chemotherapy) or by an intravenous bolus (a large volume of fluid given intravenously and rapidly at one time for an immediate effect)
• Intrathecally: Administered into the spinal subarachnoid space
Absorption
After administration, drugs must proceed to a site where they can act. Absorption describes the rate at which a drug leaves its site of administration and the extent to which it appears at the site of action. Regardless of the route of administration, the drug must be dissolved in body fluids and pass through biological barriers before it can arrive at the cellular site of action (Box 21.1). The term used to indicate the extent to which a drug reaches its site of action is bioavailability. Drugs are biotransported across these biological barriers via mechanisms discussed previously (see Chapter 2 [Chemistry of Life] and Chapter 3 [Cell Structure and Function]). For example, drugs administered orally, dermally, topically, or by inhalation must traverse the epithelium, whereas those given subcutaneously or intramuscularly must traverse the capillary wall. There are three general sites of action:
BOX 21.1 Transport of Drugs Through Biological Membranes
Transport of the various forms of drugs can be described by formulas that show:
• The percentage that will be available for successful transport through phospholipid (biological) membranes
• The percentage that will be available for excretion through the kidney
• The amounts of materials that potentially will be available for movement
• Specific information and/or general information about the substances and their mobilities
The abbreviated relationships that give this information as a percentage are as follows:
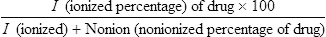
and
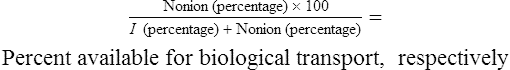
• Extracellular: Such as the decrease in blood clotting by heparin’s chemical combination with proteins to remove them from the coagulating mechanism
• Cellular: Such as the contraction of skeletal muscle by the action of acetylcholine at the nicotinic, cholinergic skeletal muscle neuromuscular receptor
• Intracellular: Such as the inhibition by sulfa drugs of bacterial growth via inhibition of intracellular components needed for bacterial growth
• Surface area: The larger the surface area is, the faster absorption will occur. For example, the large surface area of the small intestine is conducive to absorption of orally administered drugs.
• Rate of dissolution: The rate at which a drug is dissolved will determine the rate of absorption. Drugs with a high dissolution rate have a faster onset of action than those with a low dissolution rate.
• Lipid solubility: Drugs that are highly lipid soluble can more easily cross biological membranes than those with low lipid solubility.
• Blood flow: Drug absorption in areas with high blood flow is greater than in areas with decreased blood flow.
Distribution
Distribution is the transfer of drugs across biological membranes into body compartments such as the combined intracellular and extracellular fluid compartments (see Chapter 3, Cell Structure and Function). These two compartments combined make one compartment with approximately 40 liters of fluid. Drug distribution is dependent on the drug’s chemical properties and solubility, on the amount of blood flow to the target, and on the drug’s molecular size and rate of excretion, to name a few. Water-soluble drugs are readily excreted and lost unless bound. Blood flow to the site and molecular size of the drug are additional factors as well. Blood flow to the brain reveals the blood–brain barrier as another obstacle, and blood flow to the fetus must traverse the placental circulation. The placenta presents a pathway for transfer of substances from the mother to the fetus, such as nicotine and marijuana as well as alcohol.
Biotransformation
Biotransformation involves the metabolism of drugs, a mechanism by which the body inactivates drugs and engages phase I and phase II reactions. Phase I reactions convert drugs into more ionized molecules by the introduction of (or by the exposure of) already existing ionized components of the compounds (Table 21.3). Phase II reactions are synthetic reactions in which new compounds are introduced to chemically altered drugs. Some examples include conjugations of moieties with amino (–NH2), hydroxyl (OH), or sulfhydryl (SH) groups if added to the molecules (Table 21.4).
TABLE 21.3
Some Phase I Reactions of Biotransformation
| Reaction | Examples of Drugs |
| Oxidation (P450* dependent) | |
| Hydroxylation | Amphetamine, barbiturate |
| N-Dealkylation | Caffeine, morphine |
| O-Dealkylation | Codeine |
| N-Oxidation | Nicotine, acetaminophen |
| S-Oxidation | Chlorpromazine |
| Oxidation (P450 independent) | |
| Amine oxidation | Adrenaline |
| Dehydrogenation | Ethyl alcohol, chloral hydrate |
| Hydrolysis | Lidocaine, procainamide |
| Amide and ester oxidation | Aspirin, procaine |
| Reduction | Chloramphenicol, naloxone |
*P450 is a cytochrome capable of catalyzing biotransformation reactions.
TABLE 21.4
Some Phase II Reactions of Biotransformation
| Reaction | Examples of Drugs |
| Acetylation | Mescaline, sulfonamide |
| Conjugation | |
| Glutathione | Bromobenzene, ethacrynic acid |
| Glycine | Benzoic acid, salicylic acid |
| Sulfate | Methyldopa, 3-hydroxycoumarin |
| Glucuronidation | Digoxin, morphine |
| Methylation | Dopamine, histamine |
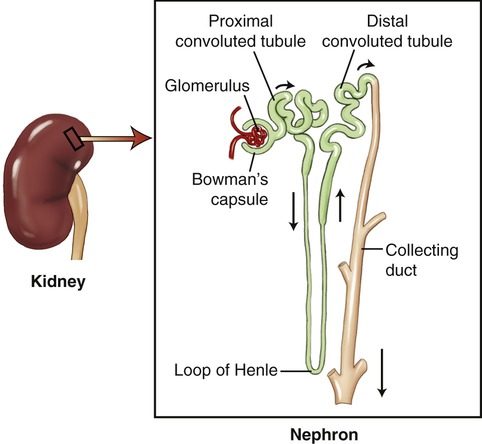
Although the liver plays the major role in drug metabolism, lungs, kidneys, and adrenal glands contribute as well. The majority of active drugs need high lipid solubility to be transported through bilipid cellular membranes. Drug metabolites also require high water solubility in order to be transported to the kidneys and eliminated in urine formed in nephrons of the kidneys (Figure 21.3). Lipid-soluble drugs can be made more water soluble by metabolic processes such as conjugation, or other metabolic processes that render the metabolite water soluble. Although metabolic processes can inactivate a drug, this is not always the case (see Medical Highlights: Prodrugs).
Responses
Dose Effects
Therapeutic Index

A high therapeutic index indicates that a drug is relatively safe, whereas a low therapeutic index indicates that the drug is relatively unsafe (Figure 21.4). Figure 21.4, B shows an overlap between the curves representing the therapeutic effect and the curve representing the lethal effect. This means that the drug dose needed to produce a therapeutic effect is very high, and may be large enough to cause death in some patients. For a drug to be safe, the highest dose necessary to produce a therapeutic effect must be substantially lower than the lowest dose required to produce death.
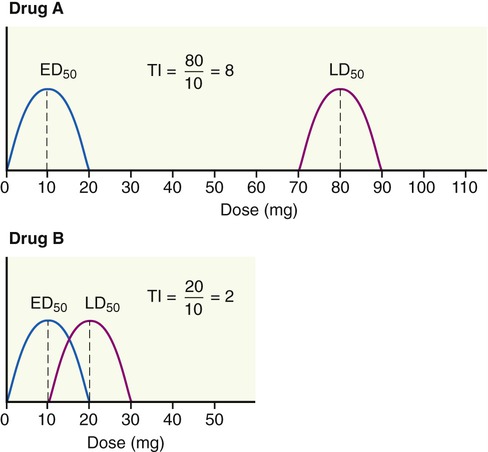
A high therapeutic index indicates that a drug is relatively safe, whereas a low therapeutic index indicates that the drug is relatively unsafe. Drug A has a high therapeutic index and is relatively safe. Drug B shows an overlap between the therapeutic effect curve and the lethal effect curve, and therefore this drug is relatively unsafe.
Individuals may vary considerably in their responsiveness to a drug. These biological variations among patients lead to pharmacological variations in the dose response of patients. Overcoming pharmacological variation requires quantitation in order to minimize errors of prediction. This begins with sampling of the collected data having a common characteristic such as age, gender, species, strain, weight, and environmental conditions. Because of biological variations patients will not necessarily respond to the same minimal effective dose (ED). However, by use of appropriate statistical methods, generalizations can be drawn with some degree of statistical probability. Normal distribution will predict that a few respond at the low dose range and a few respond at the highest doses, with most responding to a dose range in between (Figure 21.5).
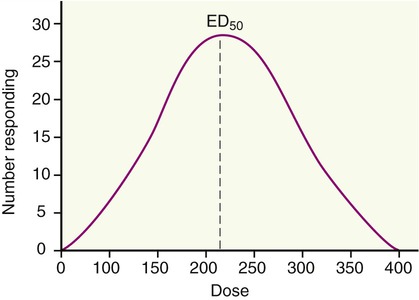
This diagram illustrates the biological variations in drug response. Normal distribution predicts that a few persons respond at the low dose range and a few respond at the highest doses, with most responding to a dose range in between. The ED50 indicates the effective dose in 50% of the tested individuals.
It is essential to know what the response to a given dose of a drug will be. Responses are either:
Time Effects
Dose response includes magnitude of response after a single dose, excluding other factors that affect concentration at the site of action (SOA), such as time. A selected time is chosen and measurements are taken to determine minimal, maximal, or any other chosen level of response for that time (Figure 21.6).
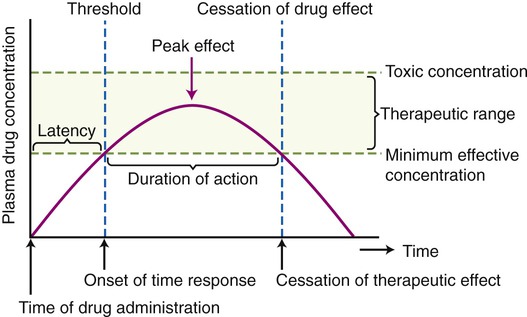
The time–response curve indicates the time of drug administration, the onset of time response (the threshold), the peak effect, the duration of action, and the cessation of drug effect. Furthermore, the diagram indicates the toxic concentration of a drug, the therapeutic range, and the minimal effective concentration of that drug.
The time response measures the following:
Antimicrobial drugs (see Chapter 22, Antimicrobial Drugs) are generally given by multiple administrations in order to achieve effective concentrations. Ongoing antimicrobial therapy should be reassessed regularly, and therapy should be appropriately adapted after initial culture results return. Dangerous negative effects can occur after toxic doses of antimicrobial drugs are accumulated or given; therefore, monitoring the patients’ vital signs as well as serum levels of drug concentration is essential. Drugs are typically withdrawn after the first sign of toxic or negative effect.
Variability
• First-pass effect: The first-pass effect is the biotransformation of a drug in the liver or gastrointestinal tract subsequent to oral administration and before entering the general circulation. Actually, the first-pass effect is the combined action of intestinal and hepatic drug-metabolizing enzymes on a drug’s bioavailability. Bioavailability is the extent to which a drug is absorbed and is available to its site of action.
• Chemical properties: Relevant chemical properties of drugs include molecular weight, ionic charge, solubility in water/lipids, and so on.
• Toxic effects: Toxic effects typically occur because of depletion of enzymes needed in detoxification reactions subsequent to the administration of a toxic dose of a drug.
• Liver and kidney diseases: Liver and kidney diseases result in an inability to metabolize or excrete drugs, respectively.
• Starvation: Starvation usually results in a decreased ability to conjugate with glycine because of depletion of that amino acid.
• Age: For example, neonates fail to metabolize drugs in the same way as adults or the elderly.
• Genetics: For example, some patients are unable to metabolize succinylcholine, resulting in succinylcholine apnea during recovery after surgery.
• Gender: For example, young males show increased sensitivity to barbiturates compared to the general population.
• Species variability: The responses of experimental animals to drugs are not always the same as those of humans.
• Circadian rhythm: The 24-hr cycle in which metabolic cycles repeat themselves. Depending on the time of day administered, a drug can have different effects due to the circadian rhythm.
Toxicity
Toxic effects are classified as:
• Acute: An effect that occurs within minutes or hours of exposure. An example is carbon monoxide, which, after a single exposure to 1.5% for 5 to 6 minutes, can be lethal
• Subacute: An effect that occurs after repeated exposure for days
• Chronic: An effect that occurs over months to years, during which time the rate of exposure exceeds the rate of elimination
Toxicology, the study of poisons, includes evaluation of the following characteristics:
Determination of drug safety (drug toxicity) is based on comparisons with other drugs or poisons to measure relative safety levels. For example, penicillin is considered safe compared with the poisonous organophosphate malathion as well as compared with the antimicrobial drug neomycin. Human drug safety evaluated by pharmacological animal testing procedures is shown in Table 21.5.
TABLE 21.5
Human Toxicity From Animal LD50 Values
| Animal LD50/kg | Degree of Toxicity | Probable LD50/70-kg Human |
| 0.1 mg | Excessive | Simple taste (less than 1mg) |
| 1.0–50 mg | High | 1 teaspoon (5.0 ml) |
| 50–500 mg | Average | 1.0 fluid ounce |
| 0.5–5.0 g | Slight | 1 fluid pint |
| 5.0–15.0 g | Nontoxic | 1 quart |
| 15.0 g | Harmless | Greater than 1 quart |
• Removing the patient from exposure to the poison source
• Identifying the poison if possible
Specific treatment for poisons (Table 21.6) is as follows:
TABLE 21.6
Specific Treatment for Selected Toxins
| Toxic agent | Specific Antidote | Mechanism of Action |
| Iron (Fe) | Sodium bicarbonate | Forms insoluble ferrous complex |
| Methanol | Ethanol | No formation of poisonous metabolite |
| Botulinum toxin | Antitoxin | Complex formed with toxin |
| Organophosphates | Pralidoxime | Removes poison from cholinesterase |
| Atropine | Pharmacological antagonism at site of toxic action | |
| Morphine and other narcotics | Naloxone | Pharmacological antagonism at site of toxic action |
| Carbon monoxide (CO) | Oxygen (O2) | Pharmacological antagonism at site of toxic action |
• In general, increase the rate of elimination of the poison and remove from the SOA via the formation of a water-soluble complex, such as by increasing the rate of bromide removal via NaCl exchange.
• For a specific poison administer the antidote; for example, in the case of cyanide poisoning administer nitrites or thiosulfate.
• For heavy metal poisoning administer a chelator, which will form a complex with the heavy metal that is then excreted; for example, in the case of mercury poisoning, administer dimercaprol (also known as British anti-Lewisite [BAL]).
• For lead poisoning administer edetate calcium disodium (CaNa2EDTA), which forms complexes with the lead and enhances its excretion 50-fold compared with uncomplexed lead.
New Drugs
Development
In addition, these investigations with plants have led to technological advances that:
• Speed the screening of natural products via improvement in separation and identification of active constituents
• Allow chemical manipulation of structures
• Develop new bioassays to identify dose–response characteristics
• Allow qualitative pharmacology procedures to identify potential prototypical pharmacological profiles
Public Safety
• Schedule I: Drugs and substances with high probability for abuse with no therapeutic use and a lack of safety controls such as opioids, heroin, lysergic acid diethylamide (LSD), marijuana, mescaline, and certain mushroom extracts (peyote).
• Schedule II: Drugs and substances with high probability for abuse and are accepted for therapeutic use or accepted for therapy under close restrictions such as raw opium, coca leaves, cocaine, morphine, methadone, amphetamines, and barbiturates.
• Schedule III: Drugs and substances with less potential for abuse than those of Schedules I and II; therapeutic drugs accepted for treatment in the United States; other agents that may have low potential for physical dependence or high psychological dependence such as nalorphine, combinations of barbiturates, and therapeutic agents with reduced concentrations of morphine, codeine, or opium.
• Schedule IV: Drugs in use in the United States with lower potential for abuse than those in Schedule III and may have reduced potential for physical and psychological dependency than those listed in Schedule III such as chloral hydrate, meprobamate, diazepam, and pentazocine.
• Schedule V: Drugs in use in the United States with lower potential for abuse (physical or psychological dependency) than those listed in Schedule IV such as codeine dihydrocodeine, opium, or atropinics.
• Any therapeutic agent to be used in humans to treat a disease
• Any combination of old and new therapeutic agents
• Any use of an approved drug for different therapeutic applications
• Any drugs used as diagnostic agents to evaluate changes in diagnostic procedures or therapeutic treatments for disease in humans
Furthermore, the IND must contain all preclinical data (see Life Application: Drug Development).
Summary
• Pharmacology is an integrated medical science of drugs and is organized into several branches: pharmacodynamics, pharmacokinetics, pharmacotherapeutics, toxicology, and pharmacy.
• Because all drugs are poisons the understanding of their nomenclature is essential in order to safely identify the correct drug for the correct use. Therapeutic chemicals are classified into nonprescription and prescription drugs, all of which have chemical, generic, and proprietary (or trade) names.
• Drugs can be administered enterally (via the digestive system) or parenterally (other than via the digestive system).
• After administration it is essential that a drug reach its site of action; therefore the drug must be dissolved in a bodily fluid and pass through biological barriers, before it can get to the cellular site of action. This involves absorption and drug distribution by the body.
• The metabolism of drugs by the body is referred to as biotransformation, which is divided into phase I and phase II reactions. The liver plays the major role in drug metabolism, but the lungs, kidneys, and adrenal glands are also involved.
• Drug clearance follows biotransformation and drugs are excreted primarily by the kidneys through urine formation via glomerular filtration, reabsorption, and tubular secretion.
• Drug administration evokes a series of responses by the human body. The factors affecting these responses include dose, time, variability, biotransformation, and toxicity.
• Dose and time response are determined by the amount of drug given, the time since administration, and the frequency of drug administration. These responses vary with age, gender, and species.
• Toxic or adverse/undesired drug effects are classified as acute, subacute, or chronic, depending on the reaction to the drug.
• Development of new drugs is an extensive process involving research and development, review of new drug application, and postmarketing monitoring, which results in the acceptance or denial of the drug by the FDA.
Review Questions
1. The branch of pharmacology that addresses drug amounts at various sites in the body after drug administration is called:
2. The generic name of a drug refers to its __________ name.
3. For a drug to have an almost immediate systemic effect it is usually applied:
4. The decrease in blood clotting by heparin occurs at which general site of action?
5. Which of the following is a phase I reaction in biotransformation?
6. All of the following are ways the kidney can use to achieve urine formation and drug clearance except:
7. Which of the following is used in the determination of a drug dose response?
8. When the drug receptors are maximally activated, this is referred to as the
9. A subacute toxic effect occurs when the adverse drug effect occurs
10. The specific antidote to botulinum toxin is:
11. The body’s metabolism of drugs is called __________.
12. The study of the body’s response to poisons and their harmful effect is referred to as __________.
13. Drug administration, absorption, distribution, and clearance are collectively called __________.
14. The ED50 is a measure of the __________ response.
15. The federal agency that approves the use of a specific drug is the __________.
16. Name and describe five different branches of pharmacology.
17. Compare and contrast dose and time response.
18. Describe five variables that influence drug response.
19. Compare and contrast nonspecific and specific treatments of suspected poisoning.
20. Discuss the public safety measures that need to be taken before a drug is approved.






