CHAPTER 157 Pharmacologic Treatment of Pain
The Phenomenon of Pain
Pain is the unpleasant physical and emotional experience associated with tissue damage—either impending or actual—and involves sensations transmitted from the periphery to the central nervous system for processing and perception. The physical phenomenon of pain signal transduction and transmission is complemented by the neuropsychological process of perceiving pain signals and their meanings. A close relationship between pain perception and culture has been identified,1 and studies demonstrate that experiences of pain intensity vary with differences in the attitudes, beliefs, and emotional and psychological state of the patient.2 Pain perception and response integrate the physiologic, cognitive, and behavioral resources necessary to promote survival when body integrity is compromised. Patients experiencing pain report higher levels of physical and emotional distress; they behave differently from persons without pain, in a way that is distinguished not just by how they feel but also by their behavior. To effectively treat pain, an appreciation of the neurophysiology and psychology of pain perception is essential.
Acute pain is nociceptive pain that is generated in reaction to focal peripheral nerve or tissue injuries such as traumatic or operative tissue damage. Acute pain usually resolves as tissues heal and inflammation subsides. Chronic pain persists beyond the usual course of injury and healing and includes persistent pain that does not respond to routine pain control measures.3 Chronic pain has many causes, but primary groups include general/mixed pain, neuropathic pain, osteoarthritis, and back and leg pain. It is estimated that between 2 and 6 million Americans cope with neuropathic pain.4 A World Health Organization study in 1997 found that 22% of primary care patients reported pain lasting more than 6 months.5 It is estimated that half of elderly people live with chronic pain.6
Pain patients consume more health care resources, including increased visits for lower priority medical problems. The exact explanation for this correlation is unclear, but it is probably due to a combination of comorbid chronic disease, psychogenic comorbidity, and chronic pain comorbidity.7 Patients with chronic back pain accrue annual health care expenses of $91 billion for treatment of all conditions.
Pain Perception and Relay
Although segmenting pain neurobiology into peripheral and central mechanisms can facilitate an understanding of the pathophysiology and treatment of pain, the pathways are interrelated, and multiple neuropeptides and neurotransmitters are involved along the signaling pathways after a painful stimulus. Conversion of a mechanical, chemical, or thermal stimulus to an electrical signal, termed transduction, is performed by nociceptive neurons. Small myelinated Aδ fibers rapidly transmit signals to the central nervous system, whereas unmyelinated C-fibers transmit signals more slowly. Transduction is facilitated by voltage-gated sodium channels, of which there are numerous subtypes. Recent research has shown that the Nav1.7 channel is involved in pain transduction abnormalities in several disorders. A gain-of-function mutation in the Nav1.7 channel is responsible for intractable pain in primary erythromelalgia and paroxysmal extreme pain disorder. A loss-of-function mutation appears key to the disorder congenital indifference to pain.8
Tissue damage releases factors, including histamine, serotonin, bradykinin, substance P, and prostaglandins, that catalyze the local inflammatory response by sensitizing already-activated nociceptors and activating dormant ones. Prostaglandin E2 (PGE2) has been demonstrated to activate ion channels in peripheral neurons. PGE2 also sensitizes dorsal horn neurons to thermal and mechanical stimuli.9 Substance P stimulates release of factors from mast cells that result in vasodilation and increased vascular permeability.
Loss of synaptic inhibition induced by inflammation or nerve injury, or both, can lead to hyperactivity of nociceptors, which is termed peripheral sensitization. Hyperexcitability of peripheral sensory neurons develops as a result of inflammation, reparatory mechanisms, and adjacent tissue reactions. It results in the metabolic activation and hyperexcitation of spinal nociceptive neurons, expansion of sensory receptive fields, and alterations in the processing of generally innocuous stimuli. Release of leukotrienes, cytokines, and neurotropic factors at the site of injury results in upregulation of sodium channels in nociceptor cells, conformational changes in transducer proteins, and increased calcium influx into second-messenger systems. The end result of this excitation is increased ectopic activity in peripheral neurons because increased voltage-gated ion channel activity leads to more subthreshold membrane potentials and a higher likelihood of ectopic action potential firing.10 The increased peripheral excitation can lead to central sensitization as the higher order neurons receive a barrage of signals from hyperactive peripheral neurons. The increased signaling is the result of temporal summation of C-fiber signals. A higher frequency of firing creates sustained excitatory postsynaptic potentials in the dorsal horn, each lasting up to 20 seconds and leading to the release of excitatory amino acids such as glutamate. Temporal summation in the central pain-projecting neurons leads to a hyperactivated state in which subsequent C-fiber input further increases the action potential strength.11
Transmission of the nociceptive signal proceeds from the second-order neurons within the substantia gelatinosa through the spinothalamic tract into the thalamus. The periaqueductal gray matter within the midbrain is a major nociceptive integration site where there are high concentrations of opioid receptors and neuroactive peptides. Other sites of involvement in pain transduction include the somatic sensory cortex, periaqueductal gray matter, periventricular gray matter, ventral medulla, nucleus raphe magnus, and limbic system. Functional magnetic resonance imaging (fMRI) studies of nociceptive stimulation show activation in the thalamus, secondary somatosensory cortex, ipsilateral insular cortex, and anterior cingulate cortex.12 fMRI studies looking at the response to opioids identify the interplay of the dorsolateral prefrontal cortex, temporal lobe, inferior parietal lobe, anterior cingulate cortex, nucleus accumbens, orbital gyrus, substantia nigra and putamen, and hippocampus (Fig. 157-1).13
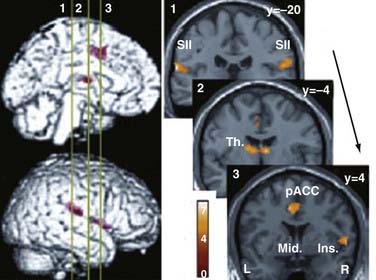
Signals transmitted to the cerebral cortex variably emerge into conscious perception. Coordinated actions among subcortical and cortical areas create the subjective, affective, and cognitive responses that determine an individual’s reaction to the pain. The insula is the most frequently activated structure in fMRI pain studies and serves as a center for the integration of neural impulses.14 Damage to the insula can lead to pain asymbolia.15 The significance of pain is conveyed through connections between the insula and the striatum to parietal association cortices and the lateral prefrontal cortices.16 Supratentorial influences can dampen the pain, as in the case of an injured person who continues activities because the circumstances require self-preservation. In these cases, central neurons downregulate the perception of pain at the level of the spinal cord and dorsal horn by releasing endogenous opioid neuropeptides (endorphins, enkephalins, dynorphins), norepinephrine, GABA, and serotonin.
Treatment of Pain
Pharmacologic Agents
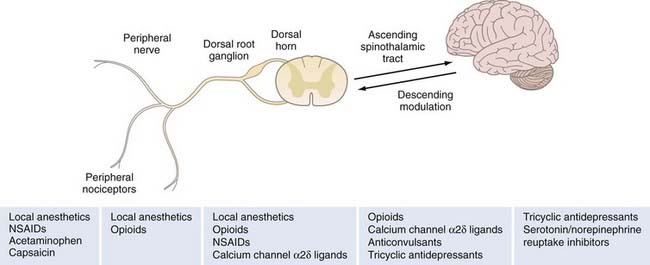
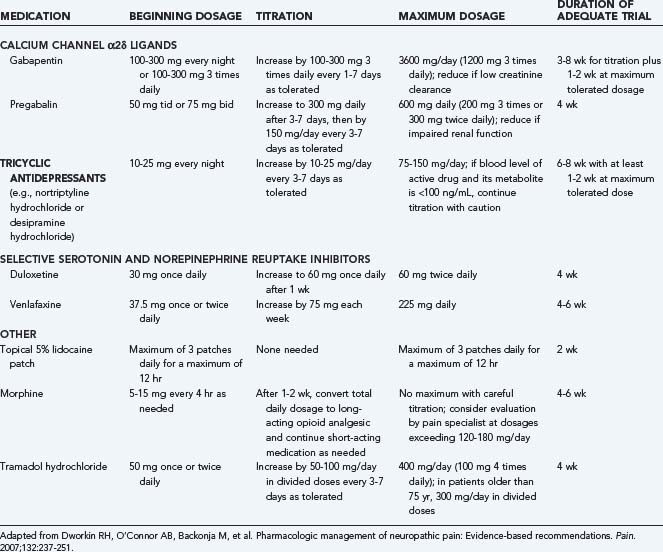
Nineteenth century physicians relied on an armamentarium of medications for pain control that included opium, morphine, codeine, and cocaine. The development of aspirin in 1897, the first compounded analgesic, revolutionized the treatment of pain. In the following century, numerous other agents were developed, with varying efficacy for a spectrum of pain conditions. Agents that are considered first line in the treatment of acute pain include opioids and nonsteroidal anti-inflammatory drugs (NSAIDs), but these agents are less effective for neuropathic pain. Recommended first-line treatment of chronic neuropathic pain includes tricyclic antidepressants (TCAs), serotonin reuptake inhibitors, calcium channel α2δ ligands, and topical lidocaine. Second- and third-line therapies such as opioid analgesics, anticonvulsants, and antidepressants are more variable in their effectiveness and have broader side effect profiles, which limits their use (Table 157-1 and Fig. 157-2).
| PAIN SOURCE | POTENTIAL AGENTS |
|---|---|
| Trauma/surgery/procedures | NSAIDs, opioids, acetaminophen, epidural analgesia |
| Acute sickle cell or cancer pain exacerbations | Opioids, NSAIDs, acetaminophen, steroids |
| Chronic low back pain | NSAIDs, opioids, muscle relaxants |
| Diabetic peripheral neuropathy | Antidepressants, anticonvulsants, capsaicin |
| Trigeminal neuralgia | Carbamazepine, baclofen, phenytoin |
| Postherpetic neuralgia | Antidepressants |
| Phantom limb pain | Carbamazepine, mexiletine, epidural analgesia |
| Cancer/HIV disease | Multiple combinations |
HIV, human immunodeficiency virus; NSAIDs, nonsteroidal anti-inflammatory drugs.
Acetaminophen
Although acetaminophen is often grouped with the NSAIDs because of its mild side effect profile and its effectiveness in analgesia and antipyresis, it does not produce an anti-inflammatory effect. The medication’s mechanism of action is still poorly understood but is believed to be related to a decrease in prostaglandin production through reduction of the oxidized form of COX and selective inhibition of prostaglandin H2 synthase.17–19 Additional research suggests that an acetaminophen metabolite may block the uptake of anandamide and consequently inhibit vanilloid receptor activation in nociceptors. There also appears to be a central effect on COX-3 inhibition leading to decreased PGE2 concentrations within the brain.20 Adverse effects include hepatotoxicity, most often associated with intentional overdose and patients with a history of alcohol abuse. Acetaminophen is only a weak inhibitor of platelet COX-1, and therefore the platelet inhibition effect is more limited than that with NSAIDs.
Opioids
Opioids are one of the most effective classes of medications available to treat both acute and chronic pain and are therefore a mainstay for both types of pain. In one study examining the treatment of chronic central pain, patients’ pain intensity scores were significantly lower after the administration of opioids than after placebo.21 Opioids act to suppress pain through mu-receptor activation on primary afferent nerve fibers, dorsal horn neurons, and supraspinal pain center neurons. Presynaptic activation by bound opioids reduces neurotransmitter release, whereas postsynaptic activation causes hyperpolarization through increased conductance of potassium.22 Opioids were originally thought to be ineffective for neuropathic pain,23 but more recent trials have demonstrated that pain intensity scores were significantly lower after opioid administration than after placebo for neuropathic pain.24
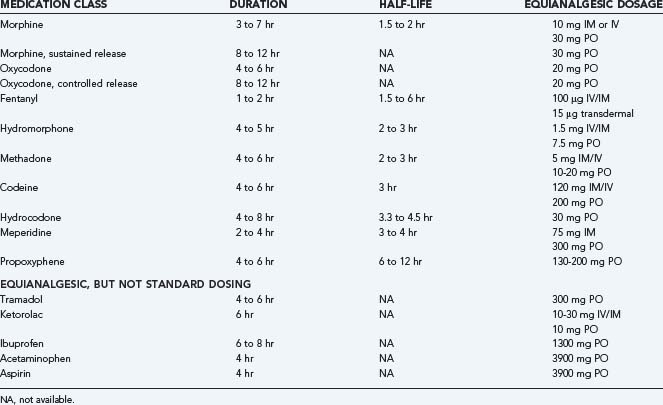
The most common reasons that patients discontinue opioid therapy are inadequate analgesia and intolerable side effects.25 In one prospective study, 17% of patients withdrew because of adverse events and 8% because of lack of efficacy (Table 157-2).26 In general, side effects can be addressed by decreasing the dose or changing the route of administration. Opioid rotation, in which the opioid is combined with other medications in a complementary strategy, or using a second medication targeting the side effect can also alleviate side effects.27 The most commonly prescribed opioid medications are morphine, codeine, oxycodone, hydrocodone, fentanyl, alfentanil, meperidine, propoxyphene, and methadone. Each varies in side effect profile, drug-drug interactions, and therapeutic effects. All members of the opioid class cross the placenta, although no teratogenic effects have been observed. If used during labor, the medications can cause neonatal depression. Interestingly, in Japan, opioids are rarely prescribed because other medications appear to provide satisfactory levels of relief.28
| SIDE EFFECT | MECHANISM |
|---|---|
| Neurological | Delirium, hallucination, euphoria, sedation/drowsiness, dizziness, diminished psychomotor performance, seizures, headaches, muscle rigidity, hyperalgesia, paradoxical pain |
| Cardiopulmonary | Cardiac dysrhythmias (bradycardia/long QTc, torsades de pointes), hypotension, decreased systemic vascular resistance, noncardiogenic pulmonary edema, respiratory depression |
| Gastrointestinal | Nausea and vomiting, constipation, gastroesophageal reflux disease, xerostomia, biliary duct obstruction |
| Genitourinary | Altered kidney function, urinary retention, peripheral edema |
| Endocrinologic | Hypogonadism, sexual dysfunction, osteoporosis |
Tramadol is an atypical opioid related to codeine. It exerts some of the pain-relieving properties of opioids while having a more tolerable side effect profile by acting as a norepinephrine and serotonin reuptake inhibitor. Its primary metabolite, O-desmethyltramadol, has mu-opioid agonist activity, which provides tramadol a potency about 10% of morphine. Tramadol has been shown to cut pain levels by more than 50% in the treatment of neuropathic pain.29 Adverse effects include nausea, sweating, vomiting, dry mouth, constipation, dizziness, sedation, and incoordination. There are also smaller risks of a reduced seizure threshold, respiratory depression, withdrawal symptoms, and serotonin syndrome. Caution should be exercised in prescribing tramadol to elderly patients because it can cause cognitive impairment. It has been demonstrated to have a much lower risk for abuse than other opioid drugs,30 and it does not have the gastrointestinal side effects associated with NSAIDs (Table 157-3).
Ion Channel Blockers
Medications that block voltage-gated sodium channels, including lidocaine, amitriptyline, lamotrigine, and carbamazepine, raise the threshold for generating an action potential in peripheral nociceptors. Although these medications appear to have effects on some categories of chronic pain, this relief is not across the board for all types of pain. Lamotrigine has been shown to be most effective for trigeminal neuralgia31 and less effective for human immunodeficiency virus (HIV) neuropathy, intractable neuropathic pain,32 and spinal cord injury–related pain. A meta-analysis demonstrated that there was no statistically significant benefit of treatment with lamotrigine for central poststroke pain or diabetic neuropathy, but anecdotal improvements are reported.33 Similarly, carbamazepine was shown to be ineffective for acute herpes zoster but was effective in treating trigeminal neuralgia,34 diabetic neuropathy, postherpetic neuralgia, and other neuropathic pain.35 The adverse effects of ion channel blockers include drowsiness, dizziness, constipation, nausea, ataxia, rash, and headaches. Several medications, including carbamazepine, require surveillance monitoring of serum drug levels. Because of the less convincing results in trials with these medications, they are best used in patients who have failed first- and second-line medications.
Tricyclic Antidepressants
TCAs were the first medications approved by the U.S. Food and Drug Administration (FDA) for the treatment of neuropathic pain. Notably, their analgesic effect appears to be separate from their antidepressant action in that it is evoked at a lower dosage. Numerous studies have demonstrated the effectiveness of TCAs for postherpetic neuralgia and painful diabetic peripheral neuropathy. TCAs were no more effective than placebo in trials for HIV neuropathy, spinal cord injury, cisplatin neuropathy, neuropathic cancer pain, phantom limb pain, and chronic lumbar root pain. Patient response to TCAs is highly variable, and no more than 40% to 60% of patients achieve partial relief of pain with these agents.36 Adverse effects include sedation, dry mouth, blurred vision, weight gain, and urinary retention. Major adverse effects of TCAs include cardiac toxicity, overdose risk, and potentiation of cytochrome P-450. Nortriptyline was associated with sinus tachycardia and increased ventricular ectopy in one trial of patients with depression and ischemic heart disease. Before administration, a patient’s cardiac history should be reviewed, and in some cases, cardiac function studies should be performed. Monitoring of drug levels is not usually indicated if the dosage is less than 150 mg/day.
Calcium Channel α2δ Ligands
GABAergic agents were introduced in the mid-1970s, with gabapentin first synthesized in 1977 and approved initially by the FDA for the treatment of complex partial seizures. Pregabalin was developed in 1989 and approved by the FDA in 2004. Their mechanism of action involves calcium channel α2δ subunit binding within the central nervous system. This results in a stabilizing effect on cell membranes, glutamate suppression, suppression of substance P transmission, influence on NMDA receptor sites, modulation of GABA receptors, and increased GABA inhibition.37–40 Studies show little benefit of gabapentin in the treatment of acute pain, but GABAergic agents are highly effective in the treatment of chronic neuropathic pain. In one study, 42% of participants improved versus 19% with placebo.41 Pregabalin was the first medication approved by the FDA for the treatment of fibromyalgia syndrome. Adverse effects associated with GABAergic agents include dizziness, somnolence, headache, diarrhea, confusion, and nausea. The main dose-limiting effects are sedation and dizziness.
Acute Pain
Treatment of acute pain is geared toward disruption of the inflammatory mediators that contribute to peripheral nociceptive excitability and to the interruption of signal conduction and transmission from the periphery to the central nervous system. To this end, a combination of NSAIDs, opioids, and local anesthetics can be used. NSAIDs downregulate prostaglandin synthesis peripherally and centrally to decrease inflammatory mediators. Local anesthetics decrease or prevent nerve excitability by blocking voltage-gated ion channel activation. Epidural, intrathecal, and systemic opioids act at the site of interneurons to inhibit synaptic transmission of peripheral signals. Patient-controlled analgesia has been demonstrated to result in improved outcomes when compared with nurse-administered parenteral opioids.42 A strategy of multimodal therapy is recommended to address the pain cascade at several sites; this approach has been shown to convey improved postoperative pain control and reduced analgesia-related adverse effects. In both children and adults, postoperative acute pain prevention should begin in the preoperative period with an emphasis on educating patients about the procedure and shaping their expectations (Table 157-4).
| MEDICATION | DOSING | MAXIMUM DOSE |
|---|---|---|
| Acetaminophen | 325-1000 mg PO q4-6h | 1 g/dose, 4 g/24h |
| Parenteral NSAID | ||
| Ketorolac | 30-60 mg IM or 15-30 mg IV q6h | 120 mg/day |
| Oral NSAIDs | ||
| Ketorolac | 10 mg PO q4-6h | 40 mg/day; <5 days total |
| Celecoxib | 200 mg PO bid | 400 mg/day |
| Ibuprofen | 400-600 mg PO q4-6h | 2400 mg/day |
| Naproxen | 250-500 mg PO bid | 1250 mg/day |
| Parenteral Opioids | ||
| Fentanyl | 50-100 µg IV q1-2h prn or 0.5-1.5 µg/kg/hr by IV infusion | |
| Morphine | 2.5-10 mg SC/IM/IV q2-6h prn or 0.8-10 mg/hr IV | |
| Hydromorphone | 1-4 mg SC/IM/IV q4-6h | |
| Meperidine | 50-150 mg SC/IM q3-4h | |
| Methadone | 2.5-5 mg SC/IM/IV q8-12h prn | |
| Oral Opioids | ||
| Morphine | 10-30 mg PO/SL q3-4h prn or 15-30 mg ER PO q8-12h | |
| Hydromorphone | 2-8 mg PO q3-4h prn | |
| Methadone | 2.5-10 mg PO q8-12h prn | |
| Codeine | 15-60 mg PO q4-6h | 60 mg/dose |
| Hydrocodone | 5-20 mg PO q4-6h | 8 tablets in 24 hr |
| Oxycodone | 5-30 mg PO q4h prn or 10-160 mg ER PO q12h | |
| Tramadol | 50-100 mg PO q4-6h prn | Maximum: 400 mg/day; 300 mg/day if >75 years old |
| Benzodiazepines | ||
| Diazepam | 2-10 mg PO divided bid-qid | 30 mg/8 hr |
| Lorazepam | 2-3 mg/day PO/IM/IV divided bid-tid | 10 mg/day |
ER, extended release; NSAID, nonsteroidal anti-inflammatory drug; prn, as needed; SL, sublingual.
Acute Pain in Children
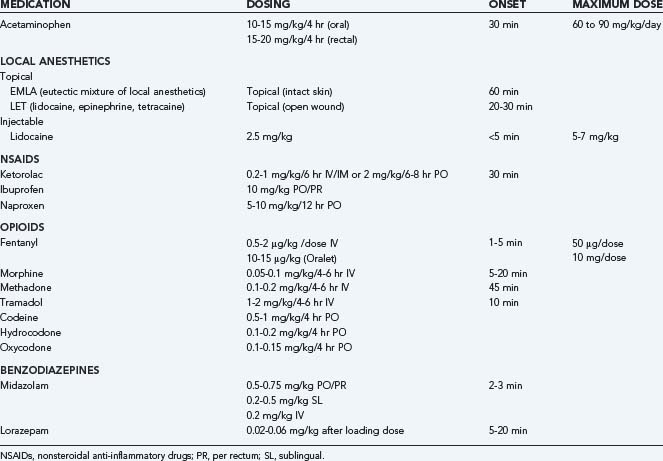
Pain in children has historically been underestimated, and it was not until the 1980s that studies quantified the prevalence of postoperative pain in pediatric patients and demonstrated that infants have a nearly equivalent distribution of nociceptors as adults.43–45 Because infants and children cannot report their pain clearly, clinicians and parents must be observant of signs—including irritability, apathy, muscle rigidity, guarding, crying—that a child is experiencing pain. Scheduling doses of medications so that pain levels do not escalate and the use of nonmedical pain control measures (see later) facilitate a child’s comfort. Use of long-acting local anesthetics such as bupivacaine and providing analgesics on a dosing schedule help prevent pain beyond the child’s control. Topical local agents can make peripheral procedures more tolerable. Eutectic mixture of local anesthetics (EMLA) cream contains lidocaine and prilocaine, and onset of anesthesia is achieved within 60 minutes. Oral and intravenous NSAIDs and acetaminophen are the mainstays of treatment of mild to moderate pain in children and have both anti-inflammatory and analgesic effects. Mild to moderate pain can usually be treated with acetaminophen and NSAIDs, whereas severe pain may require opioid agents. Opioid use remains controversial in children, in part because of risk for respiratory depression and side effects, including sedation, nausea, and vomiting. However, opioids remain the drug of choice for severe pain and mechanically ventilated patients. Studies demonstrate successful control of acute pain with patient- or nurse-controlled analgesia in children as young as 6 years.46,47 Anxiolytic medications such as midazolam can complement analgesics. In neonates, the clinician must take differences in blood-brain permeability and metabolism into account when selecting pain control regimens. Furthermore, children with pulmonary or neurological abnormalities may have altered thresholds for sedation and respiratory depression (Table 157-5).48
Nonmedical interventions offer significant pain relief in children as well. Successful techniques include distraction through playing games, touch, playing music, muscular relaxation, and guided imagination.49 Minimizing the duration of the procedures and any delays can also reduce a child’s pain and anxiety, as can controlling the level of noise and light in patient care areas.50 The majority of American pediatric anesthesia centers offer modalities of complementary and alternative pain treatments, including biofeedback, guided imagery, hypnosis, massage, relaxation therapy, acupuncture, art therapy, meditation, therapeutic touch, music therapy, self-help groups, herbal medicine, yoga, Tai Chi, and chiropractic care.51 Some studies show a link to shorter postoperative hospital stays52 and reduced pain and anxiety53 related to these nonmedical interventions.
Chronic Pain
In chronic pain, afferent function is altered in a persistent way as a result of peripheral and central nerve sensitization, and different management strategies are required. Although several of the medication classes used for acute pain are effective for chronic pain, other classes are far more optimal. Common agents used include NSAIDs, TCAs, ion channel blockers, and opioids. A meta-analysis evaluating the efficacy of several classes of medications identified the number needed to treat for beneficial response (Table 157-6).54 It should be noted that none of these agents were completely effective, so a strategy of sequential trials of medication should be applied to identify each patient’s most effective pharmacologic regimen (Table 157-7).55,56
| MEDICATION CLASS | NUMBER NEEDED TO TREAT* |
|---|---|
| Tricyclic antidepressants | 2.4 |
| Carbamazepine | 2.5 |
| Tramadol | 3.5 |
| Gabapentin | 3.7 |
| Selective serotonin reuptake inhibitor | 6.7 |
| Dextromethorphan | 1.9 |
| Levodopa | 3.4 |
| Capsaicin | 5.9 |
| Mexiletine | 38 |
* Number needed to treat is an epidemiologic calculation of treatment effectiveness and is defined as the number of patients who need to be treated to see benefit from the treatment in one patient.
Management of chronic pain is best accomplished through a multimodal approach that addresses a patient’s behavior and lifestyle in addition to providing medical management. A multidisciplinary pain team model views pharmacotherapy not as a cure but as an integral component of a more comprehensive approach that involves physical therapy, psychiatry, invasive procedures, and complementary and alternative medicine interventions. A Japanese example of multidisciplinary treatment involves targeting identified pain behavior through patient education about their diagnosis, treatment strategies, influence of disuse, role of medications, incorporation of therapeutic exercise, long- and short-term goal setting, and cognitive and behavioral techniques, including relaxation techniques, reframing, breathing techniques, and positive imaging with the aim of reducing fears related to pain.57 Furthermore, the addition of antidepressants, anxiolytics, and physical therapy can improve quality of life, especially physical and emotional function. Pharmacotherapy alone has an effect on reducing pain intensity without a demonstrable improvement in quality of life and degree of patient activity.55
Tolerance, Addiction, and Withdrawal
Over time, many patients will need an increased dosage of a drug to produce the same level of analgesia that existed previously. Tolerance is suspected when a reduced physiologic effect is observed with constant dosing. Analgesic tolerance is not always evident during opioid treatment and is not to be confused with addiction, which is a dysfunctional craving for a drug action driven by physiologic and psychological factors.58 Tolerance occurs in a minority of patients, and dose escalation can be required to continue to address patients’ pain needs.59 This can be signaled by the decreased duration of analgesia that each dose provides a patient. Physical dependence on a substance is a state of adaptation in which a withdrawal syndrome can be provoked if the substance is decreased or discontinued or an antagonist is administered. Physical dependence is a normal adaptation to the drug, reinforced by continued use. Chronic pain patients may exhibit end-of-dose failure in which the pain worsens between doses of the opioid medications. Those taking medications on an as-needed basis may also experience intermittent withdrawal symptoms. The most important symptoms of opioid withdrawal are increased preexisting pain, deep bone pain, and diffuse muscles aches.
Addiction is the compulsive use of a substance despite physical harm. Many addicts become tolerant of their chosen drug with habitual use, but addiction is characterized by dysfunctional use behavior that includes impaired control over drug use, compulsive use, and continued use despite harm and craving. Pseudoaddiction results when pain is treated with an underpotent agent. This state leads patients to manifest signs of drug seeking that can be misinterpreted as signs of addiction.60,61
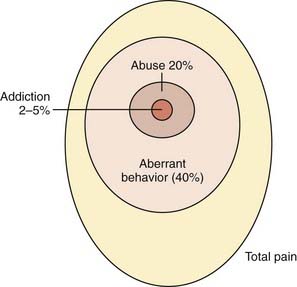
Addiction and diversion of opioid medications are a concern for clinicians prescribing chronic opioid therapy. However, of 2042 patients in a 17-study meta-analysis, in only 1 patient did signs of opioid addiction (“drug-seeking behavior”) develop, which translates to an addiction rate of 0.05% (Fig. 157-3).62 Misuse and abuse of medications used for the treatment of chronic pain have increased in the past decade. Thirty percent of illicit drug users report nonmedical prescription drug use; nearly half occur in young people aged 12 to 25.63 The National Household Survey on Drug Abuse indicated a statistically significant increase in nonmedical use of opioids between 2000 and 2001,64 but only a small proportion of this increase appears to involve patients with no previous abuse history who are receiving new opioid prescriptions. Several studies have revealed that patients without a history of substance abuse problems do not abuse the opioids prescribed to them for relief of chronic pain, and in some cases, patients’ fear of addiction developing leads to self-discontinuation of opioids.65 In one study, more than 97% of patients who abuse opioids had a previous history of abusing opiates, alcohol, or other drugs.66 The most commonly abused opioids are oxycodone, hydrocodone, hydromorphone, morphine, and methadone.
Despite these studies, clinicians are hesitant to prescribe opioids for numerous reasons: they are unfamiliar with dosing or with monitoring urine drug tests, have concerns about side effects, have concerns about illegal diversion and about causing addiction, or are apprehensive about regulatory scrutiny or about relying on patients to gauge their own pain intensity.67 Enacting a set policy for opioid treatment can alleviate many of these concerns:
A history of opioid abuse alone should not rule out prescribing these medications to a patient. Rather, the clinician may choose to proceed with very close monitoring.68 A survey of clinicians identified a belief that high daily opioid doses predispose to addiction,71 but this has not been demonstrated in studies. In general, younger patients and males have greater potential for problems.72 Other risks for potential abuse include a low functional level, excessive opioid needs, exaggeration of pain, deception or lying, doctor shopping, prescription forgery, frequent telephone calls or patient visits, and multiple medication allergies/intolerances.58
American Society of Anesthesiologists Task Force on Acute Pain Management. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on acute pain management. Anesthesiology. 2004;100:1573-1581.
Astuto M, Rosano G, Rizzo G, et al. Methodologies for the treatment of acute and chronic nononcologic pain in children. Minerva Anestesiol. 2007;73:459-465.
Becerra L, Harter K, Gonzalez RG, et al. Functional magnetic resonance imaging measures of the effects of morphine on central nervous system circuitry in opioid-naive healthy volunteers. Anesth Analg. 2006;103:208-216.
Brislin RP, Rose JB. Pediatric acute pain management. Anesthesiol Clin. 2005;23:789-814.
Carr DB, Goudas LC. Acute pain. Lancet. 1999;353:2051-2058.
Descartes R. De Homine. Leyden: Moyardus and Leffen; 1662.
Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237-251.
Gilron I. Gabapentin and pregabalin for chronic neuropathic and early postsurgical pain: current evidence and future directions. Curr Opin Anaesthesiol. 2007;20:456-472.
Gottschalk A, Smith D. New concepts in acute pain therapy: preemptive analgesia. Am Fam Physician. 2001;63:1979-1984.
Krafte DS, Bannon AW. Sodium channels and nociception: recent concepts and therapeutic opportunities. Curr Opin Pharmacol. 2008;8:50-56.
Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971-979.
Qiu Y, Noguchi Y, Honda M, et al. Brain processing of the signals ascending through unmyelinated C fibers in humans: an event-related functional magnetic resonance imaging study. Cereb Cortex. 2006;16:1289-1295.
Rathmell JP, Wu CL, Sinatra RS, et al. Acute post-surgical pain management: a critical appraisal of current practice. Reg Anesth Pain Med. 2006;31(4):1-42.
Scholtz J, Woolf C. Can we conquer pain? Nat Neurosci. 2002:1062-1067.
Sullivan MJL. Toward a biopsychomotor conceptualization of pain: implications for research and intervention. Clin J Pain. 2008;24:281-290.
Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician. 2006;9:1-39.
Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6:432-442.
Woolf CJ, Mannion R. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959-1964.
Zacny J, Bigelow G, Compton P, et al. College on Problems of Drug Dependence taskforce on prescription opioid non-medical use and abuse: position statement. Drug Alcohol Depend. 2003;69:215-232.
1 Bates MS. Ethnicity and pain: a biocultural model. Soc Sci Med. 1987;24:47-50.
2 Kitahara M, Kojima KK, Ohmura A. Efficacy of interdisciplinary treatment for chronic nonmalignant pain patients in Japan. Clin J Pain. 2006;22:647-655.
3 Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician. 2006;9:1-39.
4 Foley KM. Opioids and chronic neuropathic pain. N Engl J Med. 2003;348:1279-1281.
5 Gureje O, Von Korff M, Simon GE, et al. Persistent pain and well-being: a World Health Organization study in primary care. JAMA. 1998;280:147-151.
6 Helme RD, Gibson SJ. The epidemiology of pain in elderly people. Clin Geriatr Med. 2001;17:417-431.
7 Von Korff M, Lin EHB, Fenton JJ, et al. Frequency and priority of pain patients’ health care use. Clin J Pain. 2007;23:400-408.
8 Krafte DS, Bannon AW. Sodium channels and nociception: recent concepts and therapeutic opportunities. Curr Opin Pharmacol. 2008;8:50-56.
9 Zeilhofer HU. Loss of glycinergic and GABAergic inhibition in chronic pain—contributions of inflammation and microglia. Int Immunopharmacol. 2008;8:182-187.
10 Waxman SG. The molecular pathophysiology of pain: abnormal expression of sodium channel genes and its contribution to hyperexcitability of primary sensory neurons. Pain. 1999;6:S133-S140.
11 Schwartzman RJ, Gorthusen J, Kiefer TR, et al. Neuropathic central pain: epidemiology, etiology, and treatment options. Arch Neurol. 2001;58:1547-1550.
12 Qiu Y, Noguchi Y, Honda M, et al. Brain processing of the signals ascending through unmyelinated C fibers in humans: an event-related functional magnetic resonance imaging study. Cereb Cortex. 2006;16:1289-1295.
13 Becerra L, Harter K, Gonzalez RG, et al. Functional magnetic resonance imaging measures of the effects of morphine on central nervous system circuitry in opioid-naive healthy volunteers. Anesth Analg. 2006;103:208-216.
14 Brooks JCW, Tracey I. The insula: a multidimensional integration site for pain. Pain. 2007;128:1-2.
15 Greenspan JD, Lee RR, Lenz FA. Pain sensitivity alterations as a function of lesion location in the parasylvian cortex. Pain. 1999;81:273-282.
16 Mesulam MM, Mufson EJ. Insula of the old world monkey: III. Efferent cortical output and comments on function. J Comp Neurol. 1982;212:38-52.
17 Remy C, Marret E, Bonnet F. State of the art of paracetamol in acute pain therapy. Curr Opin Anaesthesiol. 2006;19:562-565.
18 Kis B, Snipes JA, Busija DW. Acetaminophen and the cyclooxygenase-3 puzzle: sorting out facts, fictions, and uncertainties. J Pharmacol Exp Ther. 2005;315:1-7.
19 Aronoff DM, Oates JA, Boutaud O. New insights into the mechanism of action of acetaminophen: its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases. Clin Pharmacol Ther. 2006;79:9-19.
20 Botting R, Ayoub SS. COX-3 and the mechanism of action of paracetamol/acetaminophen. Prostaglandins Leukot Essent Fatty Acids. 2005;72(2):85-87.
21 Eisenberg E, McNicol E, Carr DB. Opioids for neuropathic pain. Cochrane Database Syst Rev. 2006;3:CD006146.
22 Hollingshead J, Dühmke RM, Cornblath DR. Tramadol for neuropathic pain. Cochrane Database Syst Rev. 2006;3:CD003726.
23 Arner S, Meyerson BA. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11-23.
24 Eisenberg E, McNicol E, Carr DB. Opioids for neuropathic pain. Cochrane Database Syst Rev. 2006;3:CD006146.
25 Cherny N, Ripamonti C, Pereira J, et al. Strategies to manage the adverse effects of oral morphine: an evidence-based report. J Clin Oncol. 2001;19:2542-2554.
26 Rowbotham MC, Lindsey CD. How effective is long-term opioid therapy for chronic noncancer pain? Clin J Pain. 2007;23:300-302.
27 Harris JD. Management of expected and unexpected opioid-related side effects. Clin J Pain. 2008;24:S8-S13.
28 Kitahara M, Kojima KK, Ohmura A. Efficacy of interdisciplinary treatment for chronic nonmalignant pain patients in Japan. Clin J Pain. 2006;22:647-655.
29 Hollingshead J, Dühmke RM, Cornblath DR. Tramadol for neuropathic pain. Cochrane Database Syst Rev. 2006;3:CD003726.
30 Desmeules JA. The tramadol option. Eur J Pain. 2000;4(suppl A):15-21.
31 Wiffen PJ, Rees J. Lamotrigine for acute and chronic pain. Cochrane Database Syst Rev. 2007;2:CD006044.
32 McCleane G. 200 mg daily of lamotrigine has no analgesic effect in neuropathic pain: a randomised, double-blind, placebo controlled trial. Pain. 1999;83(1):105-107.
33 Eisenberg E, Lurie Y, Braker C, et al. Lamotrigine reduces painful diabetic neuropathy: a randomized, controlled study. Neurology. 2001;57:505-509.
34 Wiffen PJ, McQuay HJ, Moore RA. Carbamazepine for acute and chronic pain. Cochrane Database Syst Rev. 2005;3:CD005451.
35 Wiffen P, Collins S, McQuay H, et al. Anticonvulsant drugs for acute and chronic pain. Cochrane Database Syst Rev. 2005;3:CD001133.
36 Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237-251.
37 Woolf CJ, Mannion R. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959-1964.
38 Bennett M, Simpson K. Gabapentin in the treatment of neuropathic pain. Palliat Med. 2004;18:5-11.
39 Gilron I. Is gabapentin a “broad-spectrum” analgesic? Anesthesiology. 2002;97:537-539.
40 Sutton KG, Martin DJ, Pinnock RD, et al. Gabapentin inhibits high-threshold calcium channel currents in cultured rat dorsal root ganglion neurones. Br J Pharmacol. 2002;135:257-265.
41 Wiffen PJ, McQuay HJ, Edwards JE, et al. Gabapentin for acute and chronic pain. Cochrane Database Syst Rev. 2005;3:CD005452.
42 Rathmell JP, Wu CL, Sinatra RS, et al. Acute post-surgical pain management: a critical appraisal of current practice. Reg Anesth Pain Med. 2006;31(4):1-42.
43 Mather L, Mackie J. The incidence of postoperative pain in children. Pain. 1983;15:271-282.
44 Johnston CC, Abbott FV, Gray-Donald K, et al. A survey of pain in hospitalized patients aged 4-14 years. Clin J Pain. 1992;8:154-163.
45 Anand K. The biology of pain perception in newborn infants. In: Tyler D, Krane E, editors. Advances in Pain Research and Therapy: Pediatric Pain. New York: Raven Press; 1990:113-122.
46 Berde CB, Lehn BM, Yee JD, et al. Patient-controlled analgesia in children and adolescents: a randomized, prospective comparison with intramuscular administration of morphine for postoperative analgesia. J Pediatr. 1991;118:460-466.
47 Brislin RP, Rose JB. Pediatric acute pain management. Anesthesiol Clin. 2005;23:789-814.
48 Purcell-Jones G, Dormon F, Sumner E. The use of opioids in neonates. A retrospective study of 933 cases. Anaesthesia. 1987;42:1316-1320.
49 Astuto M, Rosano G, Rizzo G, et al. Methodologies for the treatment of acute and chronic nononcologic pain in children. Minerva Anestesiol. 2007;73:459-465.
50 Mencía S, López-Herce J, Freddi N. Analgesia and sedation in children: practical approach for the most frequent situations. J Pediatr (Rio J). 2007;83(suppl 2):S71-S82.
51 Lin YC, Lee AC, Kemper KJ, et al. Use of complementary and alternative medicine in pediatric pain management service: a survey. Pain Med. 2005;6:452-458.
52 Lambert S. The effects of hypnosis/guided imagery on the postoperative course of children. J Dev Behav Pediatr. 1996;17:307-310.
53 Zeltzer LK, Tsao JC, Stelling C, et al. A phase I study on the feasibility and acceptability of an acupuncture/hypnosis intervention for chronic pediatric pain. J Pain Symptom Manage. 2002;24:437-446.
54 Sindrup SH, Jensen TS. Pharmacologic treatment of pain in polyneuropathy. Neurology. 2000;55:915-920.
55 Dworkin RH, Backonja M, Rowbotham MC, et al. Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Arch Neurol. 2003;60:1524-1534.
56 Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237-251.
57 Kitahara M, Kojima KK, Ohmura A. Efficacy of interdisciplinary treatment for chronic nonmalignant pain patients in Japan. Clin J Pain. 2006;22:647-655.
58 Trescot AM, Boswell MV, Atluri SL, et al. Opioid guidelines in the management of chronic non-cancer pain. Pain Physician. 2006;9:1-39.
59 Højsted S, Sjøgren P. An update on the role of opioids in the management of chronic pain of nonmalignant origin. Curr Opin Anaesthesiol. 2007;20:451-455.
60 Weissman DE, Haddox JD. Opioid pseudoaddiction—an iatrogenic syndrome. Pain. 1989;36:363-366.
61 Mitra S, Sinatra RS. Perioperative management of acute pain in the opioid-dependent patient. Anesthesiology. 2004;101:212-227.
62 Noble M, Tregear SJ, Treadwell JR, et al. Long-term opioid therapy for chronic noncancer pain: a systematic review and meta-analysis of efficacy and safety. J Pain Symptom Manage. 2008;35:214-228.
63 Butler SF, Venuti SW, Benoit C, et al. Internet surveillance: content analysis and monitoring of product-specific internet prescription opioid abuse-related postings. Clin J Pain. 2007;23:619-628.
64 Zacny J, Bigelow G, Compton P, et al. College on Problems of Drug Dependence taskforce on prescription opioid non-medical use and abuse: position statement. Drug Alcohol Depend. 2003;69:215-232.
65 Rowbotham MC, Lindsey CD. How effective is long-term opioid therapy for chronic noncancer pain? Clin J Pain. 2007;23:300-302.
66 Cicero TJ, Adams EH, Geller A, et al. A postmarketing surveillance program to monitor Ultram (tramadol hydrochloride) abuse in the United States. Drug Alcohol Depend. 1999;57:7-22.
67 Lin JJ, Alfandre D, Moore C. Physician attitudes toward opioid prescribing for patients with persistent noncancer pain. Clin J Pain. 2007;23:799-803.
68 Kornbluth ID, Freedman MK, Holding MY, et al. Interventions in chronic pain management. 4. Monitoring progress and compliance in chronic pain management. Arch Phys Med Rehabil. 2008;89(suppl 1):S51-S55.
69 Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6:432-442.
70 Butler SF, Fernandez K, Benoit C, et al. Validation of the revised Screener and Opioid Assessment for Patients with Pain (SOAPP-R). J Pain. 2008;9(4):360-372.
71 Turk DC. Clinicians’ attitudes about prolonged use of opioids and the issue of patient heterogeneity. J Pain Symptom Manage. 1996;11:218-230.
72 Katz NP, Sherburne S, Beach M, et al. Behavioral monitoring and urine toxicology testing in patients receiving long-term opioid therapy. Anesth Analg. 2003;97:1097-1102.






