CHAPTER 84 PHARMACOLOGIC SUPPORT OF CARDIAC FAILURE
Cardiovascular disease affects more than 70 million people, according to statistics from the American Heart Association in 2002. Congestive heart failure affects approximately 4.9 million of these Americans, resulting in about 1 million admissions and costs of about $27.9 billion annually.1 Admission for acute decompensated heart failure (ADHF) often results from exacerbation of pre-existing disease or following any number of events, including acute myocardial infarction, valvular disease, and arrhythmias. Additionally, patients today are older and sicker and may be undergoing cardiac and noncardiac surgery as well as developing other causes of acute heart failure such as from sepsis or pulmonary embolus. Patients with ADHF are often triaged to the medical intensive care unit (ICU); however, these patients are also presenting for urgent exploratory and elective surgery. Whether presenting with acute or chronic heart disease, this sicker population represents an increasingly difficult challenge for the surgical intensivist. Diagnosing and treating the initiating cause is imperative; the mainstay of therapy is optimal pharmacologic hemodynamic management.
PATHOPHYSIOLOGY
The pathophysiologic mechanisms leading to ADHF, as well as the goals of pharmacologic treatment, must be identified in order to successfully treat patients with ADHF. Understanding the complex nature of each specific disease process and its physiologic response is crucial for improving function and outcome. Cardiac failure is usually a result of derangement in any number of physiologic factors, including preload, afterload, contractility, heart rate, and heart rhythm.2
Increased preload is common in ADHF and is usually secondary to volume overload but can also occur with myocardial ischemia and valvular dysfunction. The body’s natural compensatory response is to increase filling pressures to improve myocardial contractility by increasing wall stress on the ventricle (moving up on the Frank-Starling curve).3 Heart failure generally causes a decrease in renal blood flow and subsequently activates the renin-angiotensin-aldosterone axis (RAAA). The end results of these compensatory mechanisms are vasoconstriction by angiotensin II with increased renal blood flow, release of aldosterone, which increases sodium absorption in exchange for potassium, and promotion of ventricular hypertrophy, fibrosis, and remodeling that ultimately leads to increased ventricular stiffness.4
Increased cardiac afterload is common in the perioperative setting due to multiple causes such as pre-existing hypertension, catecholamine surge, postoperative hypertension, and release of cytokines and inflammatory mediators. Moreover, pulmonary artery hypertension is increased due to similar causes, but can be exacerbated as well by relative hypoxic vasoconstriction and acidosis. In the failing heart, the sympathetic nervous system (SNS) is stimulated as the body acts to increase systemic vascular resistance to maintain normal perfusion to vital organs. The failing heart is further strained as it attempts to increase cardiac output against higher outflow pressures.3 The increased sympathetic tone also stimulates the release of renin, further activating the RAAA and its inherent problems in heart failure. Subsequently, there is an increase in myocardial oxygen demand, worsening sodium and water retention and a heightened potential to exacerbate lethal cardiac arrhythmias.3 Furthermore, higher plasma levels of circulating catecholamine have been correlated with worse prognosis.
Myocardial contractility is largely affected by stimulation of the SNS. Adrenergic agents increase intracellular adenosine monophosphate (cAMP), which in turn, increases calcium influx and strengthens the contraction. However, with chronically increased sympathetic tone, the failing heart becomes less responsive to circulating catecholamines, seemingly protecting the myocytes from the excessive catecholamines and their resulting inotropic and chronotropic drive. This dampened response is due to decreased sensitivity and down regulation of the β-receptors from the chronically elevated catecholamine levels that persist in congestive heart failure.2 Contractility becomes impaired and is less responsive to physiologic needs as well as to pharmacologic agents that act at the β-receptors. In addition, the failing heart responds inadequately to volume overload. The Frank-Starling mechanism is blunted, and significant increases in preload are poorly tolerated, further exacerbating congestive symptoms.
Right ventricular (RV) failure is becoming increasingly recognized as a significant cause of morbidity and mortality in the ICU.5,6 The RV is thin-walled and compliant relative to the left ventricle (LV) and is designed to function in a low-pressure, low-resistance environment. Contraction occurs in three stages: papillary muscle contraction, RV movement toward the interventricular septum, and LV contraction with twisting of the RV. There is minimal time spent in isovolemic contraction and relaxation, resulting in almost continuous flow to the lungs.5 The RV is vulnerable to elevation in pulmonary vascular resistance, which will increase the time spent in isovolemic contraction and relaxation, decreasing overall forward blood flow. The RV is perfused primarily from the right coronary artery (RCA), with perfusion occurring in systole and diastole as long as the low-pressure system remains intact. Both ventricles are dependent on movement of the interventricular septum, which can shift toward either the RV or LV, both of which can impair adequate filling and increase end diastolic pressures.
Following cardiac surgery, ventricular function is transiently impaired even in patients with normal preoperative ventricular function. This is due to several factors, including aortic cross clamping, inadequate myocardial protection, hypothermia and cardioplegia, reperfusion injury, as well as excessive levels of inotropes in the perioperative setting. There is a biphasic pattern, with initial recovery following weaning from cardiopulmonary bypass, a nadir at about 3–6 hours, and then full recovery at 8–24 hours. This pattern can obviously be delayed by poor preoperative ventricular function.7 Pharmacologic support is frequently necessary until adequate function returns.
TREATMENT
Diuretics
Diuretics are the mainstay and building block in treating patients with ADHF, as volume overload is a common occurrence. However, there are no randomized clinical trials demonstrating the efficacy of diuretics on mortality in ADHF. Nonetheless, diuretics remain an effective therapy for the volume-overloaded patient; they act by decreasing preload and intravascular volume and relieving the symptoms of dyspnea and pulmonary congestion.3 Also, hypervolemia is common in the surgical patient who has pre-existing congestive heart failure due to volume resuscitation from trauma, sepsis, major surgery, or perioperative fluid management. Loop diuretics such as furosemide are commonly used; more potent alternatives such as bumetanide or torasemide are useful in the diuretic-resistant patient. Intravenous boluses can be used, but continuous drips have been shown to be as effective and produce a more “gentle” diuresis. Continuous infusions have been shown to be less toxic and even more efficacious in patients with renal insufficiency.8 Volume status should be addressed clinically or with invasive monitoring if needed, as overdiuresis or diuresis of the normovolemic patient can cause hypotension or hypoperfusion of end organs. Diuretics may be detrimental in the face of an acute myocardial infarction or other organ dysfunction, as well as in the early postoperative setting in the presence of capillary leak and third space fluid sequestration. Other concerns include the use of high-dose diuretics, which can activate the RAAA and the SNS, and which have their own adverse long-term effects.
Vasodilators
Nitroglycerin can be an effective agent for the rapid treatment of cardiogenic pulmonary edema and ADHF and provides rapid relief of symptoms. The primary effect of nitroglycerin (NTG) is venodilation, with vasodilation occurring at higher dosing (>30 mcg/min). Nitroglycerin has minimal effects on cardiac and skeletal muscle and causes smooth muscle relaxation mainly in the venous system, allowing for increased venous capacitance. It effectively and rapidly reduces ventricular filling pressures (preload), relieving unwanted ventricular wall stress and, more importantly, reduces myocardial oxygen demand. In addition, NTG can improve coronary blood flow by reducing coronary artery resistance and prolonging diastole. The half-life of NTG is short, allowing for rapid escalation and discontinuation of the drug, but tachyphylaxis occurs and often requires persistently increasing dosage. Other unwanted side effects include headache and abdominal pain related to the powerful vasodilation.3 Volume status must be determined, and diuretics can be helpful in negating the increasing resistance to nitrates.
Nesiritide is a recombinant form of a human brain type natriuretic peptide (hBNP) that binds to vascular and endothelial receptors to increase the intracellular levels of cyclic guanosine monophosphate (cGMP). This results in smooth muscle relaxation with predominant vasodilation and some venodilation. Although considered a “natriuretic,” diuresis has not been shown to be a major effect. It may, however, potentiate the effects of other diuretics.9 Nesiritide has been shown to be effective in controlling the symptoms of congestive heart failure and lowering pulmonary capillary wedge pressures (PCWP), with a better safety profile in studies comparing it with dobutamine-based treatments. Symptomatic hypotension can occur, so nesiritide should not be used in patients who are volume depleted, hypotensive, or underperfused. At best, nesiritide appears to be better than placebo, safer than dobutamine, and equal to noninotropic regimens using diuretics and vasodilators. Furthermore, investigators have recently brought to light nesiritide’s increased incidence of renal failure and overall increased short-term risk of death in a pooled analysis of randomized controlled trials (RCT) and suggest that its use be limited to failure of traditional therapy with combination of diuresis and nitroglycerin.10
Inotropes and Vasopressors
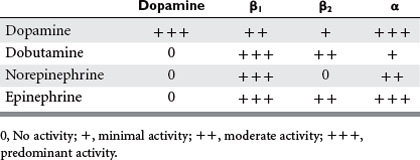
The ideal inotropic agent would increase contractility without increasing heart rate, afterload, preload, or myocardial consumption.2 The receptor-based agonists exert their effects through α1, α2, β1, β2, or dopaminergic receptor agonism, while the phosphodiesterase III inhibitors increase intracellular cAMP concentrations to enhance the contractile force in cardiac muscle (Table 1). Vasopressors, although having some intrinsic inotropic activity, are most useful in treating hypotension and maintaining an appropriate mean arterial blood pressure to ensure adequate coronary flow.
Inotropic Agents
Dobutamine is a synthetic catecholamine with predominant β1 and weak β2 agonist effects as well as weak α1 activity; its main effect is enhanced contractility and heart rate, augmenting cardiac output and stroke volume (Table 2). Systemic vasodilation occurs secondary to dobutamine’s β2 activity, affecting peripheral circulation as well as the pulmonary circulation. Although dobutamine increases myocardial oxygen consumption, this is balanced with improvement in myocardial oxygen supply by coronary vascular dilation. However, this beneficial effect only occurs if the deleterious increase in heart rate can be avoided.11 Dobutamine is contraindicated in idiopathic hypertrophic subaortic stenosis due to an increasing pressure gradient between the LV and aorta. Dobutamine should be used with caution in patients with atrial fibrillation or flutter because of its pro-arrhythmic pharmacologic effect. Its use may also be limited in the patient already taking doses of long-term beta-blockers or those with pre-existing chronic heart failure, because of the need for higher levels of medication to achieve effect. In these circumstances, combination therapy with phosphodiesterase inhibitors has been shown to be useful.
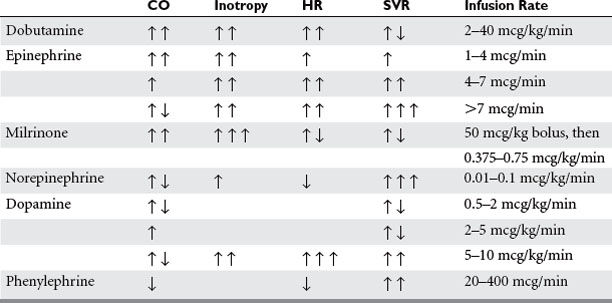
Epinephrine acts predominantly at the α1, β1, and β2 receptors, and its cardiac effects are most beneficial when used at lower doses (up to 0.02 mcg/kg/min) where the β effects predominate. Contractility is improved along with peripheral vasodilatation and increase in heart rate. The end result in the normovolemic patient is increased cardiac output and systolic pressure with decreased diastolic pressure, systemic vascular resistance (SVR), and pulmonary vascular resistance (PVR). In fact, following cardiac bypass, epinephrine produces equal increases in stroke volume as dobutamine or dopamine, with less significant tachycardia at lower dosing.2,6 At higher dosing, arrhythmias become more frequent and α activity significantly increases, which tends to encourage alternative inotropic support.
Phosphodiesterase (PDE) inhibitors are a unique class of drugs that inhibit the phosphodiesterase III isoenzyme found in myocardial and vascular smooth muscle cells, leading to increased levels of cAMP. This higher level of intracellular cAMP results in increased myocardial contractility and myocardial and vascular smooth muscle relaxation. Systemic vasodilation is also enhanced, with significant reduction in both PVR and SVR. Mean arterial blood pressure can decrease, but there is usually minimal effect on myocardial oxygen consumption and heart rate.6 This unique mechanism of action improves contractility in these situations when desensitization secondary to down regulation of β receptors has occurred. PDE inhibitors may in fact work synergistically with the β agonists, thereby decreasing the necessary concentrations of these agents and reducing their harmful side effects. The addition of even small doses of PDE inhibitor can have a large impact on cardiac index without augmenting systolic blood pressure. PDE inhibitors have been found to be beneficial for RV failure due to its inotropic ability and pulmonary vasodilatory properties (decreased RV afterload). The PDE inhibitors have a longer half-life than most inotropes but have a fairly quick onset of action. Milrinone has essentially replaced amrinone due to its improved safety profile and potency. It is 20 times as potent as amrinone and reaches peak concentration in 2 minutes, with a half-life of approximately 2–4 hours. The half-life is significantly longer than the adrenergic agents, and the risk of systemic hypotension must be taken under consideration prior to its use. It causes significantly more vasodilation compared with dobutamine at similar increases in cardiac output.12 Also, the half-life is further prolonged by its decreased elimination in patients with congestive heart failure or renal insufficiency. Other cautions of its use include ventricular and supraventricular arrhythmias, hypotension, headache, and rare occurrences of thrombocytopenia.
Vasopressors
Dopamine is a naturally occurring agent and stimulates different receptors based on serum concentration. At least part of its effect is due to release of norepinephrine from nerve terminals in myocardial cells. Dopamine is usually the drug of choice for acute heart failure with associated hypotension, also deemed cardiogenic shock. At lower doses (up to 3 mcg/kg/min), it is mainly a dopaminergic agonist. As the rate increases to 5–10 mcg/kg/min, it acts at the β1 receptor. It is in this range that cardiac contractility is stimulated, with minimal increase in heart rate, blood pressure, or SVR. Higher doses result in increasing α1 receptor stimulation.6 Because dopamine has no β2 effects, the overwhelming result is strong systemic as well as coronary vasoconstriction. At higher dosing, dopamine usually becomes limited by its profound tachycardia, arrhythmias, and coronary vasoconstriction. Hence, its use in cardiac failure is best utilized at low to moderate dosing.
Arginine vasopressin (AVP) is an endogenous hormone released from the posterior pituitary gland usually in response to changes in volume as well as blood pressure and osmolality. Its role is limited in treatment of acute heart failure but has recently been found to be useful in refractory septic shock. In early septic shock, AVP levels are high, followed by a relative depletion in circulating AVP and a relative vasoplegic state by 36 hours. Replacement of low-dose AVP to physiologic levels has been shown to restore normal vascular tone while providing paradoxical vasodilation and increased blood flow to other organs. AVP binds to motor (V1) and renal (V2) receptors, allowing some potential for renal protection, and may have pulmonary vasodilatory or rather minimal pulmonary vasoconstricting effects. Regardless, administration of low-dose AVP (0.01–0.04 unit/min) can potentially restore a catecholamine-resistant state while reducing the high dosage of additional first-line vasopressors and their added harmful effects.13
Other Agents
Thyroid hormone has been used in transplant organ donors to improve cardiac function. Sick euthyroid syndrome is being frequently diagnosed, and other nonthyroidal illnesses are relatively common in the critically ill patient. Replacement of thyroid hormone has been suggested to improve cardiac performance and enhance recovery of ventricular function after an ischemic event. In the cardiac patient, T3 hormone levels have been shown to be significantly decreased following cardiopulmonary bypass (CPB), unrelated to dilution or heparin administration. These levels then return to normal after 12–24 hours.2 Although there is some evidence that administration of T3 improves cardiac function, studies are conflicting for using T3 in the perioperative setting to wean patients from CPB.14,15 Although no adverse effects have been reported with the use of thyroid hormone in the T3-deficient patient, its use has not been clearly supported and remains relatively controversial.
Methylene blue is mentioned for its potential use in refractory shock but obviously lacks strong evidence-based studies. It is mentioned for its unique and untapped potential as a mechanism of action in sepsis or other systemic inflammatory responses (SIRS) resulting in shock. The SIRS state results in increased production of nitric oxide (NO) and subsequently smooth muscle cGMP leading to hypotension, decreased response to catecholamines, and myocardial depression. Methylene blue inhibits the production of NO and cGMP, increasing arterial blood pressure, improving myocardial function, and maintaining oxygen transport, and has been shown in a few small studies to be effective in refractory septic shock, reducing the requirement of adrenergic agents.16 There have been some reports of worsening hypoxia in those patients with acute lung injury who have been administered methylene blue, due to apparent pulmonary vasoconstriction. This appears to be lessened by lower dosing and continuous infusions.
SPECIAL CIRCUMSTANCES
Heart Failure in Septic Shock
Multiple factors contribute to the hypotensive state and relative cardiac failure associated with septic shock. Although sepsis usually results in a high output cardiac state, there is, nevertheless, a reduction in the contractile performance that is often unable to meet the metabolic demands of the tissues. RV and LV contractility is directly impaired. Preload is reduced due to a marked drop in the SVR and a loss of fluid from capillary leakage and thus poor venous return.17 Endogenous vasopressin, although initially enhanced, is quickly exhausted from prolonged intense stimulation of neurohypophyseal stores. Although dopamine, epinephrine, norepinephrine, phenylephrine, and vasopressin have been shown to increase blood pressure in septic shock, there appears to be a survival advantage specifically with norepinephrine. Both dopamine and norepinephrine are acceptable first-line agents for maintenance of normal blood pressure after adequate fluid resuscitation. Vasopressin has been shown to be useful in refractory shock and diminishing responses to first-line vasopressors. Marked depression of myocardial contractility can occur, requiring inotropic support. Dobutamine has been shown to be effective in maintaining a high normal range of cardiac output and subsequently increasing SVO2. In general, acceptable endpoints of resuscitation include establishing a mean arterial blood pressure to at least 65 mm Hg, CVP between 8 and 12 mm Hg, and SVO2 equal to or greater than 65%.18
Right Ventricular Failure
Acute RV failure had been a fairly neglected entity in the ICU until recent times. The incidence has been reported to be similar to that of LV failure and can occur in a variety of clinical settings, including heart or lung transplant, acute respiratory distress syndrome (ARDS), presence of an LV assist device (LVAD), RV infarction, positive pressure mechanical ventilation, or significant pulmonary embolism.5 Any impairment of RV preload, afterload, or LV function can result in damage to the RV. The RV is very sensitive to increased afterload. If pulmonary vascular resistance is persistently elevated, it may allow progression to RV dilation, tricuspid regurgitation, increased myocardial consumption, and worsening RV output, and initiate a degenerating cycle of RV failure. Additionally, right coronary artery perfusion of the RV, which normally continues throughout systole and diastole, degenerates into only diastolic perfusion in the presence of significant pulmonary hypertension. Increased RV end diastolic volume can also have ill effects on LV function if it is significant enough to cause interventricular septal deviation impairing LV filling. RV insufficiency during the initial few days following LVAD insertion occurs because of this leftward septal shift as the LV is unloaded.5
Maintaining higher filling pressures can restore normal hemodynamics if RV contractility, PVR, and interventricular septal function are normal.7 In fact, volume loading may be counterproductive if mean pulmonary artery pressures are already greater than 30 mm Hg. Dobutamine and PDE inhibitors such as milrinone increase RV contractility while allowing pulmonary (and systemic) vasodilation and help establish improved filling of the left ventricle by increasing right-to-left blood flow. Epinephrine and isoproterenol are also effective agents in improving RV contractility. Vasopressors are indicated to increase arterial blood pressure and improve coronary perfusion. Norepinephrine has been recommended over other α adrenergic agents to enhance right coronary artery perfusion in hypotensive patients. Inhaled NO has been shown to be effective in reducing PVR, improving oxygenation, and subsequently improving RV function due to its selective pulmonary vasodilation and improved ventilation-to-perfusion (V/Q) matching.2 Its inhaled route avoids significant effects on systemic blood pressure. However, outcome studies are lacking, and effects on mortality have not been well studied. Other provocative selective pulmonary vasodilators, such as inhaled prostacyclin with or without inhaled PDEs, have been used with anecdotal success. They boast similar pulmonary selectivity and avoidance of systemic hypotension, and are much cheaper than inhaled NO but are very labor intensive to use. Regardless of our pharmacologic treatment, the diagnosis must be determined and underlying etiology must be corrected to re-establish intrinsic RV function. Pulmonary artery monitoring and/or echocardiography can be very helpful in establishing the correct diagnosis and may aid in augmenting treatment.
Blunt Cardiac Injury
Blunt cardiac injury (BCI) infrequently requires the use of inotropic support, except in the patient with pre-existing heart disease. When it does occur, BCI results either from energy transfer from the direct blow to the chest, a deceleration-type injury, and/or a compression injury between the spine and sternum. There are no standard treatment guidelines, and care must be supportive based on injury pattern and the clinical picture. One must then familiarize oneself with all the potential causes of cardiac failure and treat accordingly. Blunt cardiac injury is a general term and encompasses all types of injury. The term “myocardial commotion” is used in the literature and occurs when no identifiable lesion exists on histology or imaging study. This is most commonly a low-impact event, such as in contact sports. Nevertheless, cardiac arrest may occur from timed impact 15–30 milliseconds before the peak of the T wave initiating ventricular fibrillation or impact during the QRS complex, causing complete heart block.19 Contusion implies a myocardial lesion and most commonly will involve the right ventricle and septum due to its proximity anteriorly. RV dysfunction can also occur, as it is sensitive to increases in RV afterload that can result from ARDS, high levels of positive-end expiratory pressure (PEEP), or severe chest trauma and pulmonary contusions. Invasive monitoring and echocardiography can help guide treatment in the hypotensive patient, the elderly, or patients with pre-existing heart disease. There are also reports of perioperative concerns including increased risk of systemic hypotension, arrhythmias, and cardiac arrest, which have been shown to persist at least up to 1 month following injury. Treatment is largely supportive, ensuring adequate preload and inotropic support. RV contusion again is common, and dysfunction must first be suspected. Treatment of increased RV afterload must be addressed, including correction of acidosis, hypoxia, and pulmonary hypertension, and avoidance of conventional mechanical ventilation and high PEEP.
1 American Heart Association. Heart and Stroke Statistical Update. Dallas, TX: American Heart Association, 2004.
2 Griffin MJ, Hines RL. Management of perioperative ventricular dysfunction. J Cardiothorac Vasc Anesth. 2001;15(1):90-106.
3 Southworth M. Treatment options for acute decompensated heart failure. Am J Health Syst Pharm. 2003;60(Suppl 4):S7-S15.
4 Ramsay JG. Cardiac management in the ICU. Chest. 1999;115(5):S138-S144.
5 Mebazza A, Karpait P, Renaud E, Algotsson L. Acute right ventricular failure—from pathophysiology to new treatments. Intern Care Med. 2004;30:185-196.
6 Doyle AR, Dhir AK, Moors AH, Latimer RD. Treatment of perioperative low cardiac output syndrome. Ann Thorac Surg. 1995;59:S3-S11.
7 Via G, Veronies R, Giuseppe M, Braschi A. The need for inotropic drugs in anesthesiology and intensive care. Ital Heart J. 2003;4(Suppl 2):S50-S60.
8 Sutter MD. New insights into decompensated heart failure. Emerg Med. 2005;37:18-25.
9 Sharma M, Teerlink JR. A rational approach to the treatment of acute heart failure: current strategies and future options. Curr Opin Cardiol. 2004;19:254-263.
10 Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293(15):1900-1905.
11 Barnard MJ, Linter SPK. Fortnightly review: acute circulatory support. BMJ. 1993;307(6895):35-41.
12 Yamani MH, Haji SA, Starling RC, Kelly L, Albert N, Knack DL, Young JB. Congestive heart failure. Am Heart J. 2001;142(6):998-1002.
13 Holmes CL, Walley KR. Vasopressin in the ICU. Curr Opin Crit Care. 2004;10:442-448.
14 Klemperer JD, Klein I, Gomez M, et al. Thyroid hormone treatment after coronary-artery bypass surgery. N Engl J Med. 1995;333(23):1522-1527.
15 Bennett-Guerrero E, Jimenez JL, White WD, et al. Cardiovascular effects of intravenous triiodothyronine in patients undergoing coronary artery bypass graft surgery: a randomized, double-blind, placebo-controlled trial. JAMA. 1996;275(9):687-692.
16 Kirov MY, Evgenov OV, Evgenov NV, et al. Infusion of methylene blue in human septic shock: a pilot, randomized, controlled study. Crit Care Med. 2001;29:1860-1867.
17 Dellinger R. Cardiovascular management of septic shock. Crit Care Med. 2003;31(3):946-955.
18 Rivers E. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368-1377.
19 Gilles O, Mustapha F, Bruno R. The heart in blunt trauma. Anesthesiology. 2001;95:544-548.