CHAPTER 12 Pharmacogenetics
Definition
The term pharmacogenetics was introduced by Vogel in 1959 for the study of genetically determined variations that are revealed solely by the effects of drugs. Pharmacogenetics is now used to describe the influence of genes on the efficacy and side effects of drugs. Pharmacogenomics describes the interaction between drugs and the genome (i.e., multiple genes), but the two terms are often used interchangeably. Pharmacogenetics/pharmacogenomics is important because adverse drug reactions are a major cause of morbidity and mortality. It is also likely to be of increasing importance in the future, particularly as a result of the development of new drugs from information that has become available from the Human Genome Project (see Chapter 5). The human genome influences the effects of drugs in at least three ways. Pharmacokinetics describes the metabolism of drugs, including the uptake of drugs, their conversion to active metabolites, and detoxification or breakdown. Pharmacodynamics refers to the interaction between drugs and their molecular targets. An example would be the binding of a drug to its receptor. The third way relates to palliative drugs that do not act directly on the cause of a disease, but rather on its symptoms. Analgesics, for example, do not influence the cause of pain but merely the perception of pain in the brain.
Drug Metabolism
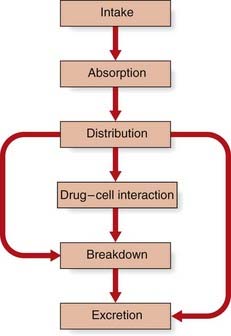
The metabolism of a drug usually follows a common sequence of events (Figure 12.1). A drug is first absorbed from the gut, passes into the bloodstream, and becomes distributed and partitioned in the various tissues and tissue fluids. Only a small proportion of the total dose of a drug will be responsible for producing a specific pharmacological effect, most of it being broken down or excreted unchanged.
Biochemical Modification
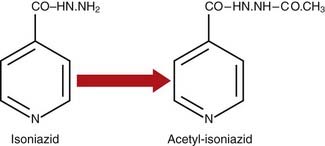
One important biochemical modification of many drugs is conjugation, which involves union with the carbohydrate glucuronic acid. Glucuronide conjugation occurs primarily in the liver. The elimination of morphine and its derivatives, such as codeine, is dependent almost entirely on this process. Isoniazid, used in the treatment of tuberculosis, and a number of other drugs, including the sulfonamides, are modified by the introduction of an acetyl group into the molecule, a process known as acetylation (Figure 12.2).
Kinetics of Drug Metabolism
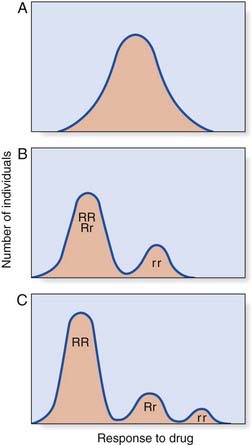
If a dose–response test is carried out on a large number of subjects, their results can be plotted. A number of different possible responses can be seen (Figure 12.3). In continuous variation, the results form a bell-shaped or unimodal distribution. With discontinuous variation the curve is bimodal or sometimes even trimodal. A discontinuous response suggests that the metabolism of the drug is under monogenic control. For example, if the normal metabolism of a drug is controlled by a dominant gene, R, and if some people are unable to metabolize the drug because they are homozygous for a recessive gene, r, there will be three classes of individual: RR, Rr, and rr. If the responses of RR and Rr are indistinguishable, a bimodal distribution will result. If RR and Rr are distinguishable, a trimodal distribution will result, each peak or mode representing a different genotype. A unimodal distribution implies that the metabolism of the drug in question is under the control of many genes—i.e., is polygenic (p. 143).
Genetic Variations Revealed by the Effects of Drugs
N-Acetyltransferase Activity
Studies in other animal species led to the cloning of the genes responsible for N-acetyltransferase activity in humans. This has revealed that there are three genes, one of which is not expressed and represents a pseudogene (NATP), one that does not exhibit differences in activity between individuals (NAT1), and a third (NAT2), mutations in which are responsible for the inherited polymorphic variation. These inherited variations in NAT2 have been reported to modify the risk of developing a number of cancers, including bladder, colorectal, breast, and lung cancer. This is thought to be through differences in acetylation of aromatic and heterocyclic amine carcinogens.
Glucose 6-Phosphate Dehydrogenase Variants
G6PD deficiency is inherited as an X-linked recessive trait (p. 114), rare in Caucasians but affecting about 10% of Afro-Caribbean males and relatively common in the Mediterranean. It is thought to be relatively common in these populations as a result of conferring increased resistance to the malarial parasite. These individuals are sensitive not only to primaquine, but also to many other compounds, including phenacetin, nitrofurantoin, and certain sulfonamides. G6PD deficiency is thought to be the first recognized pharmacogenetic disorder, having been described by Pythagoras around 500 bc.
Malignant Hyperthermia
MH susceptibility is inherited as an autosomal dominant trait affecting approximately 1 in 10,000 people. The most reliable prediction of an individual’s susceptibility status requires a muscle biopsy with in vitro muscle contracture testing in response to exposure to halothane and caffeine.
Pharmacogenetics
Maturity-Onset Diabetes of the Young
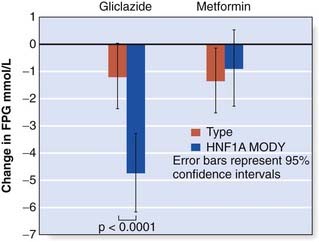
Maturity-onset diabetes of the young is a monogenic form of diabetes characterized by young age of onset (often before the age of 25 years), dominant inheritance and β-cell dysfunction (p. 236). Patients with mutations in the HNF1A or HNF4A genes are sensitive to sulfonylureas (Figure 12.4) and may experience episodes of hypoglycemia on standard doses. However, this sensitivity is advantageous at lower doses, and sulfonylureas are the recommended oral treatment in this genetic subgroup.
Neonatal Diabetes
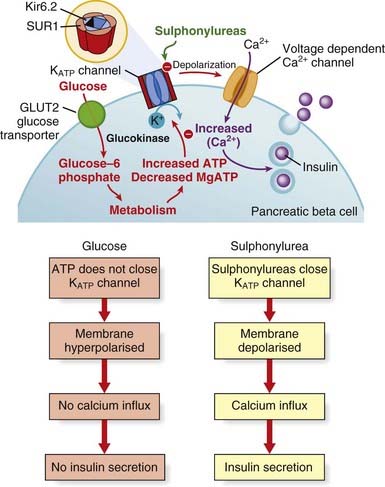
The most frequent cause of permanent neonatal diabetes is an activating mutation in the KCNJ11 or ABCC8 genes, which encode the Kir6.2 and SUR1 subunits of the ATP-sensitive potassium (K-ATP) channel in the pancreatic β cell (p. 236). The effect of such mutations is to prevent K-ATP channel closure by reducing the response to ATP. Because channel closure is the trigger for insulin secretion, these mutations result in diabetes. Defining the genetic etiology for this rare subtype of diabetes has led to improved treatment, because the majority of patients can be treated successfully with sulfonylurea tablets instead of insulin. These drugs bind to the sulfonylurea receptor subunits of the K-ATP channel to cause closure independently of ATP, thereby triggering insulin secretion (Figure 12.5). High-dose sulfonylurea therapy results in improved glycemic control with fewer hypoglycemic episodes and, for some patients, an Hb A1c level (this is a measure of glycemic control) within the normal range.
Pharmacogenomics
Pharmacogenomics is defined as the study of the interaction of an individual’s genetic makeup and response to a drug. The key distinction between pharmacogenetics and pharmacogenomics is that the former describes the study of variability in drug responses attributed to individual genes and the latter describes the study of the entire genome related to drug response. The expectation is that inherited variation at the DNA level results in functional variation in the gene products that play an essential role in determining the variability in responses, both therapeutic and adverse, to a drug. If polymorphic DNA sequence variation occurs in the coding portion or regulatory regions of genes, it is likely to result in variation in the gene product through alteration of function, activity, or level of expression. Automated analysis of genome-wide single nucleotide polymorphisms (SNPs) (p. 67) allows the possibility of identifying genes involved in drug metabolism, transport and receptors that are likely to play a role in determining the variability in efficacy, side effects and toxicity of a drug.
Efficacy
There is no doubt that the cost-effectiveness of drugs is improved if they are prescribed only to those patients likely to respond to them. Several drugs developed for the treatment of various cancers have different efficacy depending on the molecular biology of the tumour (see Table 12.1). For example, herceptin (trastuzumab) is an antibody that targets overexpression of HER2/neu protein observed in approximately one-third of patients with breast cancer. Consequently, patients are prescribed herceptin only if their tumor has been shown to overexpress HER2/neu.
Table 12.1 Examples of Drugs Effective for the Treatment of Specific Cancers
| Type of Cancer | Characteristic | Drug |
|---|---|---|
| Breast | HER2 overexpression | Herceptin (trastuzumab) |
| Chronic myeloid leukemia | t(9;22) BCR-ABL fusion | Gleevec (imatinib) |
| Non–small-cell lung cancer | EGFR activating mutation | Iressa (gefitinib) or Tarceva (erlotinib) |
| Gastrointestinal stromal tumour | KIT or PDGFRA activating mutation | Gleevec (imatinib) |
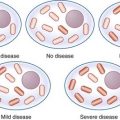
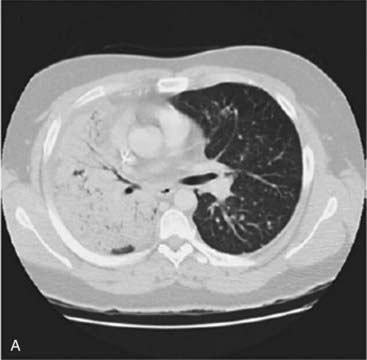
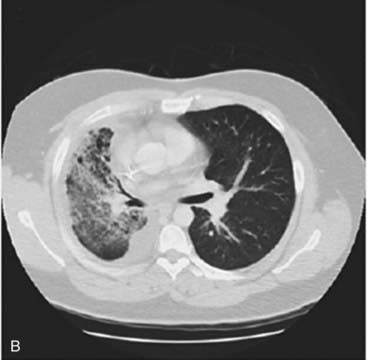
Approximately 13% of patients with non–small-cell lung cancer have an activating EGFR mutation. These mutations increase the activity of the epidermal growth factor receptor tyrosine kinase domain so that the receptor is constitutionally active in the absence of epidermal growth factor. This leads to increased proliferation, angiogenesis and metastasis. Drugs designed to block the EGFR tyrosine kinase domain and inhibit these effects have been developed. Patients with lung tumours harbouring an activating EGFR mutation can show a dramatic response to treatment with these drugs (gefitinib and erlotinib) as shown in Figure 12.6.
Beutler E. Glucose-6-phosphate dehydrogenase deficiency. N Engl J Med. 1991;324:169-174.
Review of an important ethnic pharmacogenetic polymorphism.
Goldstein DB, Tate SK, Sisodiya SM. Pharmacogenetics goes genomic. Nature Genet Rev. 2003;4:937-947.
Review of pharmacogenetics/genomics.
Neumann DA, Kimmel CA. Human variability in response in chemical exposures: measures, modelling, and risk assessment. London: CRC Press; 1998.
Newman W, Payne K. Removing barriers to a clinical pharmacogenetics service. Personalized Med. 2008;5:471-480.
A review article describing the application of pharmacogenetics to current clinical practice.
Pearson ER, Flechtner I, Njolstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. Neonatal Diabetes International Collaborative Group. N Engl J Med. 2006;355:467-477.
Pharmacogenetic treatment of monogenic diabetes.
Roses AD. Pharmacogenetics in drug discovery and development: a translational perspective. Nat Rev Drug Discovery. 2008;7:807-817.
A recent article describing the role of pharmacogenetics in drug development.
Vogel F, Buselmaier W, Reichert W, Kellerman G, Berg P, editors. Human genetic variation in response to medical and environmental agents: pharmacogenetics and ecogenetics. Berlin: Springer, 1978.
One of the early definitive outlines of the field of pharmacogenetics.
Elements