[level-membership-for-basic-science-category]
Pharmaceutical nanotechnology and nanomedicines
Yvonne Perrie
Chapter contents
Applications of pharmaceutical nanotechnology
Rationale for polymer conjugation
Polymer-drug conjugates case studies
Antibodies and antibody-drug conjugates
Dendrimer systems – case studies
Polymeric micelles – case studies
Nanosized drug particles and drug nanocrystals
Liposomes and bilayer vesicles
Clinical application of liposomes
Formulation design considerations for liposomes
Key points
Introduction
In general terms, pharmaceutical nanotechnology is a term applied to the design, characterization and production of pharmaceutical materials, structures and products that have one or more dimension between approximately 1 and 100 nm. However this classification remains open to debate, and a degree of ambiguity remains over what is considered nanotechnology, particularly regarding the size range considered. Currently an internationally accepted definition of nanotechnology is lacking and frequently particles in larger size ranges are considered as nanotechnology, e.g. the United States Food and Drug Administration (FDA) often considers 1000 nm as an appropraite upper limit regarding the screening of materials for consideration as nanotechnology. However, with the use of nanotechnology growing, both the European Medicines Agency and the FDA continue to refine their regulatory guidelines concerning nanotechnology in recognition of the key properties that nanomedicines can offer, i.e. their small size and high surface area to volume ratio. Yet it is generally agreed that using size alone as the defining factor for nanomedicines may be misleading. It is also useful to consider if a product exhibits different physical, chemical or biological properties that are attributed to its dimensions, even if these dimensions fall outside the nanoscale range. It is important to be aware that the functional effects, of the product, such as bioavailability, toxicity and/or potency may be influenced by the product’s dimensions.
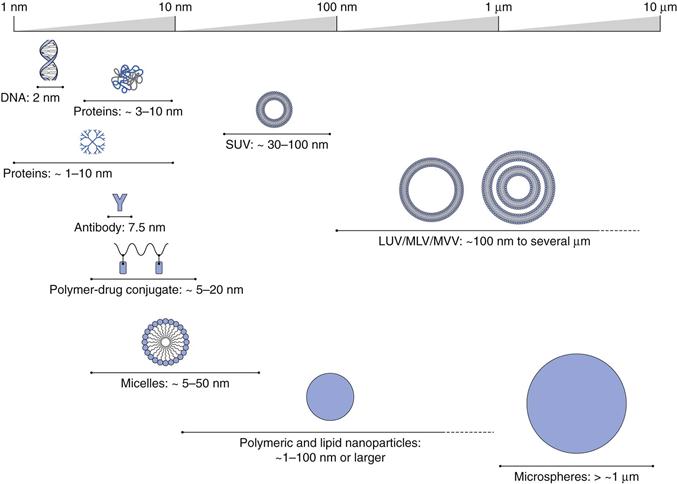
By applying this general definition, pharmaceutical nanotechnology can encompass many systems from macromolecules, such as antibodies (e.g. Herceptin®) and polymer-protein conjugates (e.g. PegIntron®) to nanoscale particles (e.g. Emend®), through to colloidal and particulate constructs in the nano size range, such as liposomal formulations (e.g. Ambisome®) and nanoparticle systems (e.g. Abraxane®). Examples of the range of such pharmaceutical nanotechnologies, sometimes referred to nanomedicines, are shown in Figure 45.1.
Applications of pharmaceutical nanotechnology
The application of nanotechnology encompasses the formulation and development of nanomedicines to improve drug potency and efficacy and the use of nanomaterials in tissue engineering and implants to fabricate structures to support tissue regeneration within the body. Nanotechnology can also include the development of devices in the nano-range such as implantable sensory systems (nanodiagnostics) for improved diagnostic measurements. This chapter will focus on the use of nanotechnology in drug formulation, which can offer particular advantages including:
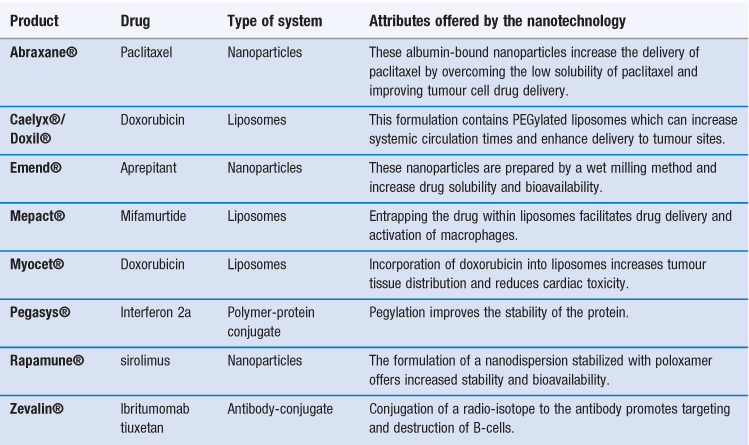
There are already several products authorized for clinical use that can be classified as nanomedicines (Table 45.1). Indeed some of these products have been approved for several years, having been originally approved for registration prior to the recognized classification of nanotechnology products. This includes some of the liposome-based products which have been licensed for clinical use since the mid-1990s. For example, the liposome formulation of doxorubicin (licensed in the US as Doxil® and marketed within Europe as Caelyx®) was the first liposomal product approved by the FDA. This product is a suspension of polyethylene glycol-coated-liposomes entrapping doxorubicin. Due to the ability of the liposome formulation to enhance targeting of doxorubicin to tumour sites, it was first developed for the treatment of AIDS-related Kaposi’s sarcoma, and is now licensed for other anti-tumour indications including metastatic breast cancer, advanced ovarian cancer and relapsed/refractory multiple myeloma. Table 45.1 shows that nanotechnology systems can offer a variety of attributes and each of these types of system is discussed in further detail in this chapter.
Polymer-drug conjugates
To improve the drug solubility and/or the delivery of drugs, drug molecules may be conjugated to polymers producing polymer-drug conjugates. These polymer-drug conjugates are considered as new chemical entities in their own right and, as their overall size is generally below 100 nm, these systems can be classified within the general area of nanotechnology. To build polymeric-drug conjugates there is a large range of synthetic polymers that can be produced having appropriate quality and stability attributes, and they can be custom-made to have distinct characteristics, including specified molecular weight, size, charge, etc. As these polymers are synthetic they are generally less immunogenic than naturally derived macromolecules. For the production of polymer-drug conjugates for parenteral administration, water soluble polymers are used.
A polymer-drug conjugate can be described as being built of three basic components:
A water soluble polymer backbone.
This can include synthetic polymers such as poly(ethyleneglycol) (PEG), poly(ethyleneimine) (PEI), poly(vinylpyrrolidone) (PVP) and polyvinylalcohol (PVA), poly(glutamic acid) (PGA), and hydroxypropylmethacrylate (HPMA) copolymers. Alternatively natural polymers such as dextran, chitosans, hyaluronic acid and proteins can be used. Of the polymers, PEG is the most widely used; it is approved by the FDA for human use and offers properties including low immunogenicity, antigenicity and toxicity. PEG chains also offer high hydration and flexibility which is useful in improving solubility and drug delivery. Another important property of PEG is their low polydispersity (in terms of molecular weight). The ease with which PEG can be modified and conjugated to drugs and proteins also offers an advantage. However, conjugation of PEG to proteins may in some instances reduce their biological activity so the conjugation site of the PEG on the protein is an important consideration. Within clinically approved products PEG molecular weights of 5000 to 40 000 Da are used.
A linker group.
Whilst a drug can be directly covalently bonded to a polymer, it is more common to attach the drug via a linker or spacer group, to help avoid the therapeutic action of the drug being blocked by the polymer. The linker can also be designed to be cleaved under certain conditions, such as changes in pH, enzymatic degradation or hydrolysis. This property can be used to promote the triggered release of the drug from the polymer conjugate under suitable conditions, thereby enhancing drug targeting. Examples of linker groups that can be used include amine, carbamate and ester groups, with an amide linker being the most common option.
Drug.
Commonly drugs delivered using these conjugates are those used in anti-cancer chemotherapy, such as doxorubicin and paclitaxel. This is because polymer conjugates can improve delivery and reduce unwanted side effects for these drugs which have narrow therapeutic windows. A second group of drugs that benefits from formulating as polymer conjugates is proteins e.g. L-asparaginase or interferons. Generally, proteins suffer from short half-lives and low stability after administration into the body. By conjugating proteins to polymers it is possible to increase their half-life by protecting the proteins from enzyme degradation and reducing clearance rates.
In addition to the three components of the system, targeting groups can be added to the polymer-conjugate with the aim of enhancing specificity and cellular uptake. However, there are no licensed products currently on the market which adopt this active targeting method. Examples of polymer-drug conjugates on the market are given in Table 45.2. As can be seen, PEG is used in several of these formulations as the polymer backbone and the majority of the systems are used to deliver protein therapeutics.
Table 45.2
| Name | Polymer-Drug | Indication |
| Adagen® | PEG-adenosine deaminase | SCID syndrome |
| Cimzia® | PEG-anti TNF Fab’ fragment | Crohn’s disease and rheumatoid arthritis |
| Oncaspar® | PEG-asparaginase | Acute lymphoblastic leukaemia |
| PEG-Intron® | PEG-Interferon 2b | Hepatitis C |
| Pegasys® | PEG-Interferon 2a | Hepatitis B and Hepatitis C |
| Neulastra® | PEG-Granulocyte colony-stimulating factor | Prevention of neutropenia associated with cancer chemotherapy |
| Macugen® | PEG-anti-VEGF aptamer | Age-related macular degeneration |
| Somavert® | PEG-growth hormone receptor antagonist | Acromegaly |
| Zinostatin Stimalamer® | Syren-maleic anhydride copolymer-Neocarzinostatin | Hepatocellular carcinoma |
Rationale for polymer conjugation
As noted above, the conjugation of drugs to polymers can improve the therapeutic action of the drug by improving solubility, protecting the drug from enzyme degradation, enhancing plasma circulation times and/or enhancing drug targeting. This is achieved through various actions.
Improving solubility
The conjugation of low-solubility drugs (e.g. paclitaxel, camptothecin or palatinate derivatives) to water soluble polymers can enhance the solubility of the overall system. For example OPAXIO®, a polymer-conjugate currently under development, comprising paclitaxel conjugated to poly-(L)-glutamic acid. The paclitaxel-conjugate has enhanced solubility compared to paclitaxel, therefore the conjugate can be administered without further solubilising agents.
Enhancing bioavailability and plasma half-life
The increased hydrodynamic volume of the polymer-drug conjugate compared to the free drug can reduce excretion rates via the kidneys. The renal clearance of compounds from the circulation is dictated by their molecular weight, with clearance rates decreasing with increasing molecular weight up to a threshold of around 45 kDa. Above 45 kDa, renal excretion cannot occur and larger polymers are more susceptible to clearance by the mononuclear phagocytic system (MPS). So, for example, the conjugation of molecules such as paclitaxel (~850 Da), and proteins such as interferon (~20 kDa) to water-soluble polymers increases their overall molecular weight enhancing drug circulation times and reducing kidney clearance rates.
Protecting against degradation after administration
The polymeric chains in the polymer-drug conjugate can also prevent the approach of antibodies and proteolytic enzymes to the drug. Water-soluble polymers become strongly hydrated and these hydrated polymer strands can promote steric hindrance, and block enzymes and antibodies reaching the drug. This protects the drug from degradation and enhances their plasma half-life and bioavailability. This is of particular advantage to protein-based therapeutic agents that are rapidly degraded by enzymes. However, it has been reported that antibodies against PEG can be generated in vivo and these can remove and neutralize PEG-conjugate products.
Reducing aggregation, immunogenicity and antigenicity
The hydrophilic coating offered by the polymers to the conjugate compound is the key to this property. The hydrated polymer chains can mask the hydrophobic regions in the protein, improve solubility and provide a steric shield that can help prevent protein-protein association, and reduce aggregation. For example, the native proteins in Neulastra® and PEG-Intron® have a high tendency to aggregate, however, PEG conjugation (referred to as PEGylation or pegylation) of these proteins can reduce aggregation and subsequently reduce associated immunogenic and antigenic problems. As already noted, the presence of the hydrated polymer in the conjugate can reduce antibody interactions, also reducing immunogenicity. PEGylation of proteins can also help stabilize proteins during lyophilization so helping to produce products with acceptable storage conditions.
Promoting targeting to specific organs, tissue or cells
By conjugation of drugs or proteins to water-soluble polymers, not only can their half-lives be improved, but the specific accumulation of the drug or protein can also be promoted in certain tissues. This can be achieved through the use of targeting groups or the phenomenon known as the enhanced permeability and retention (EPR) effect. This EPR effect can be described as passive targeting, whereby the distribution of the conjugate is dictated by local physiological conditions at the target site. Normally after a drug enters the systemic circulation, the drug is required to cross the endothelial lining of the vasculature before it can reach the target site. In most parts of the body, the endothelial lining is continuous with the endothelial cells situated on a basal membrane, and tight junctions between adjacent cells. This makes transport across this barrier difficult. However, the structure of the blood capillary wall varies in different organs and tissues, with three general types of endothelial cells being recognized:
Continuous.
Continuous endothelial cells are the most common. These cells have tight junctions and a continuous basement membrane. Continuous endothelial lining is found in areas, such as capillaries in the brain, lung and muscles.
Fenestrated.
This type of endothelial cells have gaps of between 20 and 80 nm between them, and this can allow the passage of small molecules out of the systemic circulation. Fenestrated endothelial linings are found in the capillaries in the kidneys and gastrointestinal tract.
Sinusoidal.
Here there are gaps between the endothelial cells of up to 150 nm. The basement membrane is either discontinuous, as in the capillaries in the spleen, or absent altogether, as in the case of the capillaries in the liver.
Additionally, the integrity of the endothelial barrier can be disturbed by inflammatory processes or by tumour growth. This can result in defective hypervasculature, leading to endothelial fenestrations as large as 200 to 300 nm being present in the endothelial lining. In addition, due to rapid tumour growth, deficient lymphatic drainage is also an issue. This modified permeability of the endothelium at such sites allow nanomedicines, including polymer-conjugates, to escape from the central circulation into the tumour site, where they are retained due to the poor tumour lymphatic drainage. This phenomenon is the EPR effect described above. Thus, conjugation of a drug or protein to an appropriate soluble polymer will result in a construct with a large hydrodynamic volume which will have reduced kidney excretion and therefore an enhanced systemic circulation time. Due to the EPR effect the polymer conjugates can escape from the systemic circulation into tumour sites where they will accumulate, enhancing drug action at the tumour site and reducing unwanted side effects elsewhere in the body.
The use of targeting groups to dictate the distribution of drugs and drug carriers can also be considered to promote targeting to specific site. This is commonly referred to as active targeting. Here, the designer of the drug-delivery system is relying on the interactions between a targeting group, which can be covalently attached to the polymer, and a corresponding receptor to facilitate the targeting of the system to a specific site. Examples of targeting groups include the use of antibodies due to their ability to specifically recognize and bind specific antigens (see below) and folate to target folate receptors which are overexpressed in tumour cells. Similarly, lectins are over-expressed on the surface of many tumour cells and can be targeted via the use of glycoproteins.
Polymer-drug conjugates case studies
OPAXIO® – a small-molecule conjugate.
In this polymer-drug conjugate, paclitaxel is conjugated to poly-L-glutamic acid (PGA) via an ester linker. Conjugating paclitaxel to the water-soluble PGA overcomes the poor aqueous solubility of paclitaxel, and the congugate can be infused into the body without the addition of solvents. This conjugate has a high drug content (~37% w/w) and is stable in the circulation and, while it remains bound to the polymer, paclitaxel is inactive. Due to its construct, the conjugate can passively target tumour sites via the EPR effect. The drug is then released intracellularly via degradation of PGA by lysosomal proteases, and the ester linker is degraded by esterases or acid hydrolysis. OPAXIO® is currently undergoing clinical trials as a potential treatment for non-small cell lung cancer and ovarian cancer.
Oncaspar® – a protein conjugate.
In this conjugate L-asparaginase is bound to non-biodegradable monomethoxyl poly(ethylene glycol) (5000 g/mol) via an amide linker. This conjugate is used in the treatment of acute lymphoblastic leukaemia and its mechanism of action is based on selective killing of leukemic cells due to the depletion of plasma asparagine. Asparaginase is an enzyme which breaks downs the amino acid, L-asparagine. This interferes with the growth of malignant cells which, unlike most healthy cells, are unable to synthesize L-asparagine for their metabolism. PEGylation of the enzyme enhances the circulation time of the enzyme, allowing less frequent dosing. It can also be given to patients with a history of hypersensitivity to native L-asparaginase.
Antibodies and antibody-drug conjugates
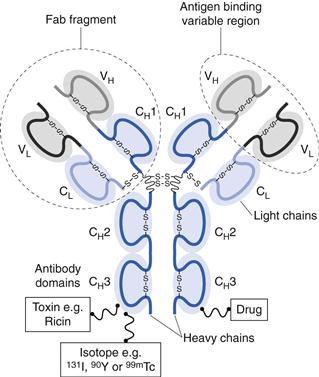
Antibodies are large ‘Y’-shaped protein macromolecules produced by B cells. Antibodies can specifically bind to a range of pathogens, including bacteria and viruses, through their ability to specifically bind to antigens. There are several classes (or isotypes) of antibodies with the five main types being IgG, IgA, IgM, IgE and IgD (Ig for immunoglobulin). Of these, IgG is the most abundant in the body. The basic structure of IgG comprises two identical heavy (50 kDa) polypeptide chains and two identical light (23 kDa) polypeptide chains. These chains are held together by disulphide bonds. The ability of antibodies to specifically target antigens is due to their amino acid sequence at the tips of the protein, which are the antigen binding sites (Fig. 45.2).
Antibody therapies
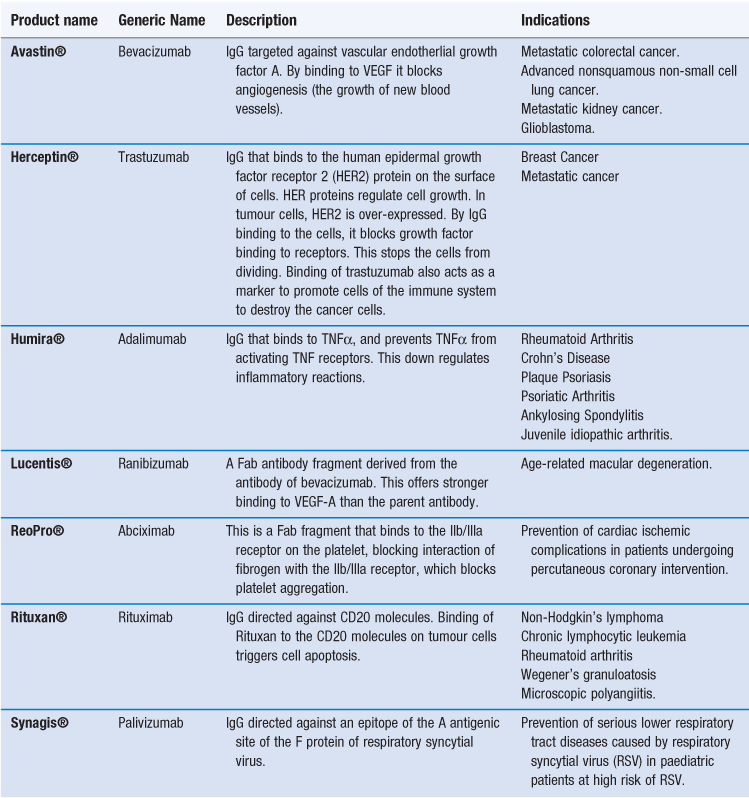
Given their ability to target a range of specific antigens and cell types, antibodies can be used for drug targeting, either as drugs in their own right or as targeting groups for drugs or delivery systems. To achieve this, IgG monoclonal antibodies (mAbs) have been developed. There are already several monoclonal antibodies available as therapeutic agents (Table 45.3). As is the case with other nanotechnology systems, these therapies are generally focused on oncology. In the case of antibody therapies, the antibody is designed to actively target tumour cells. By the antibody binding to tumour cells, cell death can then occur via two types of mechanisms:
Alternatively, the antibody can bind to a protein and thereby block its ability to bind to a receptor, as in the case of Humira® which binds to TNF-α, thereby blocking its ability to bind to activate TNF receptors. In addition to whole antibodies, antibody fragments can be used. Fab fragments of monoclonal antibodies retain the targeting specificity of whole monoclonal antibodies, and may even offer stronger binding, however, the fragments can be produced more economically. Examples of antibody fragments licensed for use include Reopro® and Lucentis® (Table 45.3).
Antibody conjugates
Similar to polymer-drug conjugates, antibodies can also function as carriers for drugs and other agents, including radioisotopes and toxins. These antibody conjugates are sometimes referred to as immunoconjugates. By conjugating a molecule to an appropriate monoclonal antibody, the molecules actively target the drug to the required site of action. Unfortunately, most antibody conjugates have a relatively low capacity for drugs, however, their high target specificity allows them to be used effectively in the clinical environment. Examples of antibody conjugates are given in Table 45.4. These either target radioisotopes, which can be used to promote cell death (e.g. Bexxar® and Zevalin®), or they can be used for diagnostic imaging (e.g. ProstaScint®). Alternatively, the antibodies can be used to target toxins which can kill cells (e.g. Mylotarg®).
Dendrimers
Dendrimers are highly branched polymeric, star-shaped macromolecules which can be prepared in the nanosize range. Dendrimers are built by a controlled chemical synthesis and there are three main elements to dendrimers (Fig. 45.3):
By varying the construction of the dendrimer around the core unit, dendrimers of different shapes and sizes can be built, which can offer the ability to carry drugs within the construct, or drugs can be conjugated to the surface of the dendrimer. The surface of the dendrimer can also be modified with targeting groups, or a hydrophilic coating to enhance solubility. Depending on their design dendrimers can be built to be biodegradable or non-biodegradable, similar to the polymeric-drug conjugates. These branched polymeric structures are synthesized by step-wise addition of layers of polymer branching, referred to as generations (termed G1, G2, etc.). In general, as the number of layers or generations increases, the structure of the dendrimer moves from the open structures of the low generation dendrimer to an increasingly more globular and dense structure. In all cases, the resulting constructs are built to have a specific size, a high degree of molecular uniformity and a narrow molecular weight distribution. Whilst dendrimers can be considered as an evolution of branched polymers, they offer the advantages of being capable of preparation with a very narrow size distribution. Further, the large number of peripheral groups on the exterior surface of the dendrimer, which increase exponentially with each generation added, allows for higher drug-loading capacities compared to the linear or branched polymers used in polymer-drug conjugates.
Applications of dendrimers
In general, the attributes offered by polymer-conjugates discussed above apply to dendrimers; dendrimers can be designed to increase drug solubility and bioavailability, and can enhance drug delivery and targeting. The additional advantages of dendrimers over polymer conjugates include their near monodisperse size-range and high drug-loading capacity. Dendrimers has been investigated for their potential to encapsulate drug molecules within the construct or by conjugation of the drug to the dendrimer. Encapsulation of the drug within the dendrimer can be exploited to protect labile drugs which are quickly degraded. Similarly, dendrimers can act as a solubilizing agent for low solubility drugs (similar to micelles) by encapsulating the drug within the dendrimer construct that offers a hydrophobic core and a hydrophilic exterior. The drug can be entrapped within the dendrimer either by simple physical entrapment, or by non-bonding interactions, such as electrostatic interactions. When the drug is encapsulated within the dendrimer, controlled drug release can be achieved by designing the dendrimer to have triggered degradation. However, the ability to incorporate drugs within the system is heavily dictated by the architecture of the dendrimer.
Alternatively, drug molecules can also be loaded onto the surface of the dendrimers via electrostatic interactions or via conjugation of the drug to the surface groups on the dendrimer. Conjugation of the drug to the exterior of the dendrimer generally offers high drug loading due to the large number of peripheral end groups available on each dendrimer molecule. Drugs can be covalently attached through hydrolysable or biodegradable linkers, thereby offering greater control over drug release compared to electrostatic interactions. However, the addition of molecules to the surface of the dendrimer can also influence the dendrimer properties. For example, the addition of PEG to the surface of dendrimers allows for their pharmacokinetic profile to be modified, with clearance via the liver being reduced and plasma circulation time increased. This can promote passive dendrimer-drug targeting via the EPR effect, similar to PEGylated-protein constructs. Alternatively, active targeting can be considered by the conjugation of targeting groups to the surface of the dendrimer (Fig. 45.3).
Dendrimer systems – case studies
VivaGel®.
This formulation, being developed by Starpharma, is a microbicide gel which uses dendrimers. In this formulation, the dendrimer is the active ingredient in its own right rather than being used as a delivery system. The dendrimer has antiviral properties due to its ability to bind to viruses and thereby blocking their ability to infect cells.
Starburst® dendrimers.
Whilst not approved for clinical use, there are a range of commercially available dendrimers such as polyamidoamine (PAMAM; Starburst®) and poly(propylenemine) (Astramol®) dendrimers which have been widely studied for drug delivery.
Micelle systems
Micelles are widely used to formulate low solubility drugs in colloidal solutions. Micelles form due to the ability of surfactant molecules to self-assemble into micelles in an aqueous environment. Whilst not a new type of formulation (see Chapter 5), their size (often < 100 nm) means that micelles can be considered within the nanotechnology classification. As a consequence of the micellar structure, which offers a hydrophobic core and a hydrophilic surface, micelles are commonly used as solubilizing agents. For example, Fugizone® is a mixed micellar formulation which is employed to solubilize Amphotericin B, an anti-fungal agent used to treat invasive fungal infections, such as systemic candidiasis and histoplasmosis.
Polymeric micelles
Copolymers with surfactant characteristics can also be used to formulate micelles. Micelles formed from copolymers tend to have a relatively narrow size distribution compared to standard surfactant micelles. Generally, they also have a lower critical micelle concentration (CMC) and are more stable. Due to their low CMCs, polymeric micelles are relatively insensitive to dilution, thus preventing their rapid dissociation and enhancing their circulation time compared to surfactant micelles. Polymeric micelles are built from copolymers with hydrophobic components comprising poly(propylene oxide), poly(D,L-lactic acid), poly(ε-caprolactone), poly(L-aspartate) and poloxamers. For the hydrophilic component, which forms the outer hydrophilic shell of the micelle, PEG is commonly used. The use of PEG as the hydrophilic component supports the formation of the micelles. The hydrated PEG surface created on the micelles enhances their plasma half-life by promoting steric hindrance and blocking enzymes and antibodies reaching the drug, thereby offering protection to the drug and blocking interactions with the mononuclear phagocytic system (MPS). As the micelles are sufficiently large (>50 kDa) to avoid renal excretion yet small enough (<200 nm) to avoid clearance by the liver and spleen, they are able to promote the specific accumulation of the micelles at tumour sites and sites of inflammation due to passive targeting.
As for dendrimers, the outer surface of the polymeric micelles can be further functionalized with targeting groups (such as folate, sugar residues or proteins) to promote their application in drug delivery and targeting. The attachment of monoclonal antibodies to reactive groups incorporated in the hydrophilic coating of polymeric micelles has also been investigated and shown to promote specific interaction of the micelles with antigens corresponding to the antibodies. These micelles are often referred to as immunomicelles.
Polymeric micelles – case studies
Estrasorb®.
This is an estradiol topical formulation designed to deliver estradiol to the blood circulation following topical application. Estradiol hemihydrate is encapsulated in a micellar nanoparticle drug delivery system (which comprises soybean oil, water, polysorbate 80 and ethanol). The formulation is used to treat menopausal symptoms, including hot flushes and night sweats.
Genexol-PM®.
This is a polymeric micelle formulation of paclitaxel prepared using methoxy-PEG-poly(D,L-lactide) which is approved in South Korea for metastatic breast cancer. In vivo anti-tumour efficacy of the micellar formulation has been shown to be significantly higher than that of Taxol®.
Solid nanoparticles
Solid nanoparticles are solid constructs in the nanometre range, and can be prepared by a number of different manufacturing methods which generally involve either size reduction of particles (e.g. by milling) to within the nanoparticle range, or molecular agglomeration (e.g. by precipitation methods) to form nanoparticles. The former is used to prepare drug particles in the nanosize range where there is no carrier material added, whilst the latter is more commonly used to prepare nanoparticle carriers in which drug is loaded.
Nanosized drug particles and drug nanocrystals
Reducing drug particles to within the nanosize range substantially increases the total surface area of the system, hence increasing the solubility of the drug. This attribute can be exploited to improve the dissolution and bioavailability of drugs delivered by the oral route. The use of nanosized drug particles in oral drug delivery can also avoid variations in drug bioavailability caused by the fed/fasted state of a patient. However, nanosized drug particles, due to their high surface area and subsequent high interfacial energy tend to be unstable and prone to particle aggregation. To reduce this problem, surface active agents can be used, as in the case of NanoCrystal Technology, developed by Elan Corporation. NanoCrystals are prepared from 100% drug with no carrier, and stabilizers (non-ionic and anionic surfactants) are added during the size-reduction process to improve stability. Within nanoparticle systems, the drug can be either crystalline or amorphous. The small particle size increases dissolution and saturation solubility. For oral delivery, nanocrystals are normally formulated into tablets or capsules. With these systems, a key consideration is drug loading, as a high nanocrystal loading in tablets could result in contact between nanocrystals promoting potential fusion of the crystals during tablet compression. There are several drug nanoparticle products on the market which exploit this NanoCrystal Technology (Table 45.5).
Table 45.5
| Product | Drug | Attributes |
| Emend® | Aprepitant | An oral capsule form of the poorly soluble drug, Aprepitant, which is only absorbed in the upper gastrointestinal tract. Therefore the rapid dissolution offered by the nanocrystals, supports fast absorption and increased bioavailability. |
| Megace ES® | Megesterol acetate | The ES in the product title stands for Enhanced Stability. This product is a liquid dosage form and the nanosized version of the drug allows for the drug to be formulated in less volume. The reduced dose volume, improved dissolution times, and enhanced bioavailability are beneficial in drug compliance. |
| Rapamume® | Sirolimus | This is an oral tablet of the poorly soluble drug which is an immunosuppressant. The oral tablet offers higher bioavailability and can be more user friendly compared to a liquid product. |
| TriCor® | Fenofibrate | Generally, fenofibrate uptake is from the gut lumen region therefore bioavailability is influenced by the patient’s fed/non-fed state. By formulating as nanocrystals, the lipophilic drug has improved solubility therefore uptake of the drug is not influenced by solubilization of the drug in food components. |
Solid polymeric nanoparticles
In addition to their production by size reduction of drug particles, solid nanoparticles can be formed from polymers with the drug incorporated within the polymer matrix or associated onto the particle surface. As such, the delivery system can be loaded with a wide range of drugs (e.g. water-soluble and low-solubility drugs, small and large molecular weight drugs, small molecules and proteins) and can offer protection to the drug. Incorporation of the drug into solid polymeric nanoparticles also allows for modified drug biodistribution as the drug pharmacokinetic profile will be dictated by the properties of the nanoparticle attributes rather than those of the drug.
Polymeric nanoparticles are generally formulated from natural or synthetic polymers with the most commonly studied polymers being those which are biodegradable, such as poly(lactide-co-glycolide) (PLGA), polylactic acid (PLA), polycaprolactone (PCL) and polysaccharides (particularly chitosan). The advantage of these polymers is that they are well characterized and used in a range of clinical products, particularly PLGA. In terms of drug delivery, the main areas in which polymeric nanoparticles are being considered is for their ability to promote passive targeting of drugs to tumour sites via the EPR effect. Similar to the other nanotechnology systems discussed, the surface coating of PEG to these nanoparticles produces so called ‘stealth’ nanoparticles with the hydrated PEG surface coating prohibiting protein and antibody binding, thereby reducing recognition and clearance from the circulation. By increasing plasma circulation time of the polymeric nanoparticles, this supports their accumulation at sites of leaky vasculature including tumour sites. To formulate these ‘stealth’ systems, PEG-PLGA copolymers are often employed. Alternatively, active targeting of these systems can be achieved by the attachment of targeting groups to the nanoparticles.
Solid-lipid nanoparticles
These are nanoparticles made from solid (high melting point) lipids dispersed in an aqueous phase. Examples of lipids used include solid triglycerides, saturated phospholipids and fatty acids which are well tolerated by the body. Due to their composition, they are sometimes described as solidified o/w emulsions in which the oil globule is replaced by solidified lipid. Much like the solid polymeric nanoparticles, solid-lipid nanoparticles can be used as drug delivery systems with the drug being incorporated within the lipid matrix of the particle or by attaching the drug to the lipid nanoparticle surface. Lipid particles normally have a size greater than 50 nm and can be prepared on a large-scale by homogenization to disperse the lipid into an aqueous environment.
Solid lipid nanoparticle dispersions have been developed for parenteral, oral, ocular, dermal and cosmetic applications. As with the polymeric nanoparticles, PEG coating of these systems has been shown to passively target tumour sites via the EPR effect. To actively target cancer cells, covalent coupling of targeting groups such as ferritin or galactose to the lipids used in the formulation has been investigated. Currently, there are a range of cosmetic products which use lipid nanoparticles loaded with cosmetic components such as: ascorbyl palmitate, beta-carotene and co-enzyme Q10. As these are all lipophilic in nature, they are efficiently incorporated within lipid nanoparticles.
Protein nanoparticles
In addition to polymers and lipids, nanoparticles can also be prepared from proteins. The first commercial product based on protein nanotechnology was Abraxane® (nabTM-paclitaxel). Abraxane® is approved for the treatment of breast cancer in patients who do not respond to combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy. Abraxane® consists of 130 nm particles of albumin-bound paclitaxel. The drug, paclitaxel, has a low water solubility and requires addition of solubility-enhancing agents to allow its clinical use. Prior to the development of Abraxane®, paclitaxel was only available as Taxol®. This is a liquid product with paclitaxel solubilized in polyethoxylated castor oil (Cremophor® EL) and ethanol. However, this formulation requires special infusion sets, prolonged infusion times and has toxicity issues associated with its use. By incorporating paclitaxel into albumin nanoparticles, the albumin functions to coat the paclitaxel and provide colloidal stabilization to the drug. This circumvents both the low solubility and the Cremophor®-associated side effects.
Targeting mechanisms of Abraxane®
The albumin within the nanoparticle-albumin technology used in Abraxane serves as more than a solubility enhancing agent: albumin also promotes active targeting of the paclitaxel to tumour cells. As highlighted above, drug targeting of nanoparticles to tumours may be enhanced as a result of the EPR effect; the dense and highly permeable endothelial microvascular structure of tumours (which is a result of angiogenesis) allows large macromolecules and nanoparticles to leak into the underlying tumour tissue. The impaired lymphatic drainage at the tumour site slows drainage of these nanoparticles and macromolecules from the tumour site, resulting in the particles becoming trapped.
In the case of Abraxane®, a second mechanism has also been associated with the targeting and uptake of the albumin nanoparticles to tumour sites. This is the Albumin-activated (glycoprotein) gp60 pathway. Within the body, albumin is able to transport hydrophobic molecules, such as vitamins, hormones and other plasma constituents, across the endothelial lining and out of the blood circulation. This is achieved by albumin binding to gp60 albumin receptors found on the surface of vasculature endothelial cells. These gp60 receptors are then responsible for the transport of albumin across blood vessel walls. The binding of albumin to the gp60 receptors, activates the membrane protein caveolin-1. The activation of caveolin-1 subsequently results in the internalization of the cell membrane and the formation of transcytotic vesicles (known as caveolae). These caveolae then transport their contents across the endothelial cell cytoplasm, and release their contents into the cell’s interstitium. In the case of tumours, this transport system is thought to be upregulated. Therefore Abraxane® is able to exploit this albumin-activated gp60 transport mechanism to target the tumour site. After entering the systemic circulation, the albumin-bound paclitaxel can bind to the gp60 albumin receptors and be carried across the endothelial cells via transcytosis in the same way as albumin.
After crossing the endothelial lining, the drug must cross the tumour cell membrane and enter the tumour cells. Once the albumin-bound paclitaxel reaches the interstitium, the albumin may bind to an extracellular matrix glycoprotein known as SPARC (secreted protein acid and rich in cysteine) which is also over-expressed in tumour cells. This can trigger the release of the paclitaxel from the albumin, allowing the free drug to diffuse to the nucleus of tumour cells initiating cell death. Given that this mechanism of tumour targeting is an attribute of the albumin carrier system and not the drug, it is conceivable it may be applied to the delivery of other low solubility anti-cancer agents.
Inorganic nanoparticles
Nanoparticles can be fabricated from inorganic materials including metal oxides, metal sulphides, carbon nanotubes, calcium phosphate and ceramics. These nanoparticles are generally not biodegradable and so have a more limited application. Abdoscan® is an example (albeit no longer available in Europe) of a metal oxide nanoparticle product. Abdoscan® can be used for magnetic resonance imaging (MRI) diagnostics of the bowel, as it is a superparamagnetic iron oxide nanoparticle formulation which is administered orally. The particles have a mean diameter no less than 300 nm. It is a negative contrast agent utilized for peroral bowel MRI diagnosis due to its high magnetic signal strength and decreased cytotoxicity. The particles are suspended in viscosity increasing agents, such as starch, to prevent aggregation of the particles in-vivo.
Liposomes and bilayer vesicles
Liposomes are closed spherical vesicles consisting of an aqueous core surrounded by one or more bilayer membranes (lamellae) alternating with aqueous compartments (Fig. 45.4). These bilayer membranes can be composed of natural or synthetic amphiphilic lipids, and commonly phospholipids are employed for the formulation of liposomes, however a range of amphiphilic lipids can be used. Liposomes form when the lipids (which are surface active with a hydrophilic head group and a hydrophobic chain(s) at opposing ends) are exposed to an aqueous environment. Under appropriate lipid-to-water ratio and temperature, the lipids will arrange into bilayer vesicles. Unlike micelle formulation, which form spontaneously, energy must be added to the system to drive the formation of liposomes.
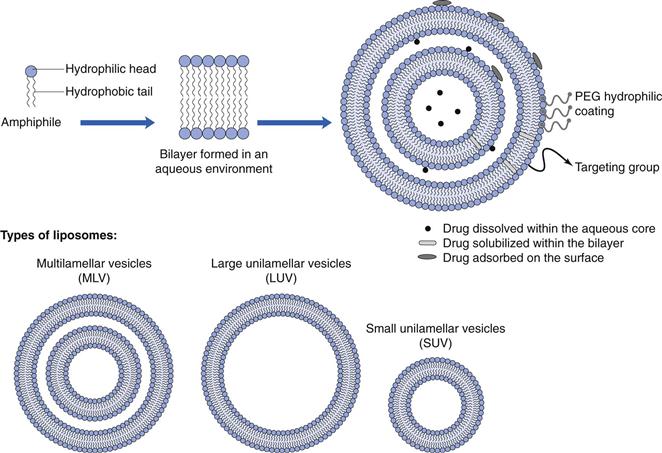
Due to their nature, liposomes are able to carry both water-soluble and lipophilic moieties, with the water-soluble drugs being incorporated within the aqueous compartments and lipid-soluble drugs incorporated within the bilayer (similar to the solubilization of drugs within the core of micelles). In addition, some drugs and molecules can be adsorbed onto the surface of the liposomes through electrostatic interactions, e.g. nucleic acids and many proteins are anionic in nature and can be electrostatically bound to the surface of cationic liposomes.
Liposomes can be formulated in a range of diameters from around 30 nm up to several micrometres, therefore they can be considered as nanotechnology. Generally, liposomes are classified based on their size and number of lamellae (Fig. 45.4).
Small unilamellar vesicles (SUV).
These are single-bilayer vesicles, around 30 to 100 nm in size. These are generally easier to prepare in a homogeneous size range compared to other types of vesicles and are the most commonly used in clinically approved products. Due to their small size there is a low ratio of internal aqueous volume per mole of lipid.
Large unilamellar vesicles (LUV).
These are large single-bilayer vesicles of 100 nm and greater. These vesicles offer a larger aqueous compartment compared with SUVs.
Multilamellar vesicles (MLV).
These vesicles have multiple concentric bilayers and are 100 nm to several micrometers in size, depending on their composition and their method of preparation. Their low aqueous volume (due to the multiple bilayers) reduces their capacity for carrying water-soluble drugs.
Multivesicular vesicles (MVV).
MVV are of similar size to MLVs, however rather than having multiple concentric bilayers, they have vesicles within vesicles.
Clinical application of liposomes
Examples of liposome formulations on the market are given in Table 45.6. These include formulations which are prescribed for the treatment of certain cancers (e.g. Caelyx® and DaunoXome®), systemic fungal infections (e.g. AmBisome®), vaccines (e.g. Inflexal V®) and macular degeneration (e.g. Visudyne®); the majority of the products are designed for intravenous injection.

These formulations exploit the ability of liposomes to control the pharmacokinetic profile of their incorporated drug. Early in the development of liposomes as drug delivery vehicles it was established that intravenously injected liposomes interact with blood opsonins, which cause their removal from the blood circulation at rates that are dictated by vesicle size, lipid composition and surface charge. Due to this interaction with opsonins, liposomes end up (via opsonin recognition by appropriate cell receptors) in the tissues of the mononuclear phagocytic system (MPS), mostly fixed to macrophages in the liver and spleen. This property allows liposomes to be used for passive targeting of such sites. Alternatively, when administered via the intramuscular or subcutaneous routes, liposomes reach the lymphatic system including the local lymph nodes. This provides the opportunity for liposomes to be used for the delivery of vaccines. However to use liposomes for drug delivery to sites other than the MPS, the stability of liposomes in blood or interstitial fluids, vesicle clearance rates, and tissue distribution are key factors.
Formulation considerations that can improve this aspect include coating with hydrophilic polymers, such as polysialic acids and PEG; these polymers provide a hydrophilic surface which helps to repel opsonins and thus contribute to longer vesicle circulation times and, therefore, provide opportunities for the liposomes to interact with target cells other than those of the MPS. By avoiding MPS uptake, liposomes can accumulate in pathological sites with leaky vasculature including tumour sites and sites of inflammation through the EPR effect.
The application of liposomes in cancer chemotherapy
There are several liposome products designed for the delivery of cancer chemotherapy, and they can promote improved drug delivery via a range of different mechanisms. For example Myocet® is a liposomal formulation of doxorubicin. The liposomes within Myocet® are ~180 nm in size and composed of egg phosphatidylcholine and cholesterol. Due to their larger size and lack of PEG coating, the liposomes in Myocet are rapidly taken by the MPS. This is thought to create a ‘MPS depot’ which produces a slow release of the drug into the blood circulation, mimicking a slow transfusion.
Alternatively, liposomes can be formulated to exploit the EPR effect thereby allowing them to target anti-cancer agents to tumour sites via passive targeting. To achieve this, the liposomes can be prepared with a PEGylated surface coating, as in Caelyx® (also marketed as Doxil®). Within this formulation the liposomes are less than 100 nm in size and incorporate PEG2000 – distearoylphosphatidyl ethanolamine (DSPE) within their bilayer, which provides a hydrophilic PEG coating on the surface of the liposomes. This hydrophilic coating inhibits opsonization of the liposomes, avoids their clearance by the MPS, and therefore increases their circulation half-life. Subsequently, the liposomes are able to accumulate at tumour sites via the EPR effect. Indeed targeting via the EPR effect is driven by the plasma concentration of liposomes. The liposomes retained in the tumour subsequently breakdown and release doxorubicin which acts locally on cells.
DaunoXome®, a liposome formulation of daunorubicin, is also able to selectively deliver the entrapped drug to tumour sites. Unlike Caelyx®, the liposomes in DaunoXome® do not have a PEGylated hydrophilic coating. DaunoXome® liposomes are formulated from high transition temperature lipids and cholesterol, which makes the liposome bilayer resistant to opsonization. This, combined with their small size (45 nm), supports prolonged blood residence times and tumour targeting.
The application of liposomes in the treatment of systemic fungal infections
AmBisome is an antifungal preparation, administered by intravenous injection, consisting of amphotericin B incorporated in liposomes which are ~80 nm in diameter. Amphotericin B is generally the first line drug used for the treatment of life-threatening systemic fungal and protozoal infections. However, its application is limited by its associated adverse side effects. There are three lipid-based formulations, with AmBisome® being the only liposome formulation and it has an aqueous core within a bilayer system. Within AmBisome®, the liposomes are composed of hydrogenated soy phosphatidylcholine, cholesterol, distearoylphosphatidylglycerol (DSPG) and α-tocopherol. In these liposomes the amphotericin B is intercalated within the liposomal membrane due to its low solubility.
Abelcet® and Amphotec® are two alternative lipid-based formulations of amphotericin B. Abelcet® is formulated from dimyristolphosphatidylcholine (DMPC) and dimyristol phosphatidylglycerol (DMPG), and the resultant constructs are ribbon-like complexes which are around 1.6 to 11 µm in diameter. Amphotec® consists of a complex of amphotericin B with cholesteryl sulfate which are disc-like in structure, with a diameter around 100–140 nm. Given the difference in their physical structures these three formulations have different pharmacokinetic profiles, including difference in clearance rates and volume of distribution, and therefore may not be considered as directly interchangeable.
Liposomal delivery of vaccines
In addition to their ability to passively target tumour sites, the natural tendency for liposomes to be taken up by phagocytic cells can be exploited to target cells of the immune system, such as dendritic cells, and thereby enhance the delivery of antigens. Indeed a range of bilayer vesicle systems have been shown to induce humoral and cellular immunity to a wide range of antigens. This is particular useful for the delivery of soluble antigens which have a good safety profile but are weak at inducing immune responses. There are currently two liposome-type constructs, which are built from viral components and phospholipids, that are licensed as vaccine delivery systems (Table 45.6). These virosomes use viral components, such as influenza haemaglutinin, which enhances antigen delivery and targeting to the immune cells.
Sustained drug release from liposomes
Sustained drug release, multi-vesicle liposomes have been developed for clinical use. Both DepoCyte® and DepoDur® comprise large multivesiclular vesicles which achieve sustained drug release through their slow clearance from the administration site and their slow breakdown. In the case of DepoDur®, this is administered as a single epidural injection and can give relief from post-operative pain for up to 48 hours.
Formulation design considerations for liposomes
When designing liposomes for drug delivery, in addition to consideration of the vesicle size and lamellarity, the lipid bilayer composition can play an important role in dictating the properties of the vesicles, and there is a wide range of options available for formulating liposomes.
Choice of lipid
Most clinically approved products use phosphatidylcholines (e.g. egg or soy phosphatidylcholine; dioleoyl phosphatidylcholine; dimyristoyl phosphatidylcholine) as the lipid within the formulation. It is used to give a structural framework in the liposome bilayers (Table 45.6). By varying the structure of the lipid fatty acid tails the characteristics of the liposomes can be manipulated. By increasing the length of the carbon tails and the degree of saturation of the lipids the liposomes can be designed to have a more rigid and less permeable bilayer. This is useful for improving drug retention and avoiding opsonization. For example DaunoXome is formulated using distearoyl phosphatidylcholine (DSPC). DSPC has two fatty acid tails which are 18 carbons in length and both are fully saturated. This allows strong hydrophobic interactions between the lipid tails and dense packaging of them within the bilayer, which enhances the stability of the liposomes.
In addition to phospholipids, a range of other lipid/amphiphilic molecules can be used to form liposomes. These include, for example, non-ionic surfactants and amphiphilic synthetic block copolymers which form vesicles referred to as niosomes and polymersomes, respectively. There are also bilosomes which are synthetic vesicles that include bile salts within the vesicle bilayer. Whilst each of these variations on liposomes may offer some differences in physicochemical characteristics, they all offer the bilayer structure which can incorporate drugs within the aqueous phase or within the bilayer depending on the drug attributes.
The key factor that dictates the aggregation of amphiphile molecules into bilayer vesicles rather than, for example micelles or liquid crystals, is the molecular shape of the amphiphile as this will influence their geometrical packaging within a given solution environment. The shape of an amphiphile may be expressed as its critical packing parameter (CPP), which is related to the ratio of the volume of the molecule (v) to the area of the headgroup (S0) and the hydrophobic chain length (lc), i.e.:
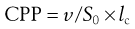 (45.1)
(45.1)
Amphiphile molecules with a CPP of less than 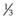 (i.e. a cone shape) tend to form micelles, as in the case of, for example, sodium dodecyl (lauryl) sulphate. Amphiphiles with a CPP of between
(i.e. a cone shape) tend to form micelles, as in the case of, for example, sodium dodecyl (lauryl) sulphate. Amphiphiles with a CPP of between 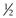 and 1 (a truncated cone, e.g. phosphatidylcholine) will form vesicles. If the CPP of the molecule increases to above 1 (e.g. dioleoylphosphatidyethanolamine; DOPE) inverted micelles tend to form. Whilst the use of CPP can provide a useful approach to predicting which structure amphiphiles may form, it is important to consider that changes in pH, temperature and lipid concentration can also have an influence. With lipid mixtures, the overall CPP of the system must be considered.
and 1 (a truncated cone, e.g. phosphatidylcholine) will form vesicles. If the CPP of the molecule increases to above 1 (e.g. dioleoylphosphatidyethanolamine; DOPE) inverted micelles tend to form. Whilst the use of CPP can provide a useful approach to predicting which structure amphiphiles may form, it is important to consider that changes in pH, temperature and lipid concentration can also have an influence. With lipid mixtures, the overall CPP of the system must be considered.
Cholesterol content
Cholesterol is also a common component of the liposome formulations. Although cholesterol does not form bilayers on its own, it can be incorporated into liposome bilayers at concentrations up to 50% of total lipid. The presence of cholesterol in the bilayer can reduce the bilayer permeability of the liposomes and thus improve drug retention within liposomes. This has been attributed to the ability of cholesterol to reduce the mobility of the phospholipids and improve lipid packing within the bilayer.
Surface characteristics
A third consideration of the liposomes is their surface properties. In addition to considering a PEGylated hydrophilic coating, liposomes can be formulated to have an anionic or cationic surface charge. Cationic liposomes are currently being considered for the delivery of nucleic acid based therapies such as DNA and siRNA, which are anionic and can electrostatically bind to cationic liposomes. Cationic liposomes are also being considered for the delivery of sub-unit vaccines which are often anionic in nature. CAF01 is a cationic vesicle formulation, developed by Statens Serum Institut, Denmark. It is currently in clinical trials as a vaccine for the prevention of tuberculosis.
The conjugation of targeting groups to the surface of liposomes to promote their active targeting has also been investigated. Similar to the polymer-based systems, targeting groups such as folate or glycoproteins for targeting of tumour cells, or the attachment of antibodies to liposomes have been investigated. Presently none of these are used clinically.
Drug characteristics
In addition to the design of the liposomal bilayer, the characteristics of the drug to be loaded into the vesicles require consideration. Drugs with high aqueous solubility (log P < ~1.7) can be incorporated and retained within the aqueous compartments of the liposomes whereas lipophilic drugs (log P > 5) are incorporated and retained within the liposome bilayers. Drugs with intermediate log P values are more difficult to incorporate within liposomes as they can partition between the aqueous phase and the bilayers resulting in loss of the drug from the liposomes.
To address this issue and improve drug loading, remote drug loading has been developed. This is used in some marketed products, such as Caelyx® and DaunoXome®. In these formulations, the liposomes are first prepared within an aqueous core containing ammonium sulphate. This results in an ammonium sulphate gradient across the liposome bilayer. When doxorubicin is added to the external aqueous phase, surrounding the liposomes during manufacture, doxorubicin diffuses across the liposomal membrane and enters the internal aqueous compartment of the liposomes. When inside the liposomes, due to the concentration of ammonium sulphate, the doxorubicin forms a drug-sulphate complex and becomes trapped within the vesicles. Using this method, high drug loading efficiencies of over 90% can be achieved.
Microcapsules and microspheres
Even though, as the name suggests, these particles are generally in the micrometre size range and therefore can fall outside the nanoparticle definition, these systems are worth consideration given their clinical application in drug delivery. They are essentially spherical particles that can be manufactured to be solid or porous (often termed microparticles or microspheres) or alternatively they can be hollow in nature (microcapsules). Their composition is basically the same as nanoparticles in that they can be formulated from polymers, lipids, proteins, etc. Polymers such as PLA and PLGA are commonly used. Drugs can be incorporated within the matrix systems, and therefore drug release is dictated by the degradation rate of the matrix. There are a number of polymeric microparticles approved for clinical use. Examples include the following:
Ongoing developments
Within the field of nanotechnology there are a large number of formulation options to enhance the delivery of a drug. There are several systems that are already licensed for clinical use, and many more in various stages of clinical trials. The majority of these systems are being considered for their application in oncology. However, nanotechnology is not limited to this area and the application of these delivery systems is also being considered for peptide-based therapies and the delivery of DNA and siRNA. Given the range of functional attributes of nanotechnology, it is likely that the application of this evolving technology will continue to expand providing new and reformulated therapeutics which offer better clinical outcomes and improved patient-focused formulations.
Bibliography
1. Duncan R. Polymer therapeutics as nanomedicines: new perspectives. Current Opinion in Biotechnology. 2011;22(4):392–501.
2. Gregoriadis G, Perrie Y. Liposomes. In: Encyclopedia of Life Sciences (ELS). Chichester: John Wiley & Sons, Ltd; 2010.
3. Lee CL, MacKay JA, Frechet JMJ, Szoka FC. Designing dendrimers for biological applications. Nature Biotechnology. 2005;23:1517–1526.
4. Perrie Y, Rades T. FASTtrack Pharmaceutics – Drug Delivery and Targeting. London: Pharmaceutical Press; 2010.
5. Uchegbu IF, ed. Synthetic Surfactant Vesicles, Niosomes and Other Non-Phospholipid Vesicular Systems. Amsterdam: Harwood Academic Publishers; 2000.
6. Veronese FM, ed. PEGylated Protein Drugs: Basic Science and Clinical Applications. Germany: Birkhäuser Verlag; 2009.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Pharmaceutical nanotechnology and nanomedicines
Yvonne Perrie
Chapter contents
Applications of pharmaceutical nanotechnology
Rationale for polymer conjugation
Polymer-drug conjugates case studies
Antibodies and antibody-drug conjugates
Dendrimer systems – case studies
Polymeric micelles – case studies
Nanosized drug particles and drug nanocrystals
Liposomes and bilayer vesicles
Clinical application of liposomes
Formulation design considerations for liposomes
Key points
Introduction
In general terms, pharmaceutical nanotechnology is a term applied to the design, characterization and production of pharmaceutical materials, structures and products that have one or more dimension between approximately 1 and 100 nm. However this classification remains open to debate, and a degree of ambiguity remains over what is considered nanotechnology, particularly regarding the size range considered. Currently an internationally accepted definition of nanotechnology is lacking and frequently particles in larger size ranges are considered as nanotechnology, e.g. the United States Food and Drug Administration (FDA) often considers 1000 nm as an appropraite upper limit regarding the screening of materials for consideration as nanotechnology. However, with the use of nanotechnology growing, both the European Medicines Agency and the FDA continue to refine their regulatory guidelines concerning nanotechnology in recognition of the key properties that nanomedicines can offer, i.e. their small size and high surface area to volume ratio. Yet it is generally agreed that using size alone as the defining factor for nanomedicines may be misleading. It is also useful to consider if a product exhibits different physical, chemical or biological properties that are attributed to its dimensions, even if these dimensions fall outside the nanoscale range. It is important to be aware that the functional effects, of the product, such as bioavailability, toxicity and/or potency may be influenced by the product’s dimensions.
By applying this general definition, pharmaceutical nanotechnology can encompass many systems from macromolecules, such as antibodies (e.g. Herceptin®) and polymer-protein conjugates (e.g. PegIntron®) to nanoscale particles (e.g. Emend®), through to colloidal and particulate constructs in the nano size range, such as liposomal formulations (e.g. Ambisome®) and nanoparticle systems (e.g. Abraxane®). Examples of the range of such pharmaceutical nanotechnologies, sometimes referred to nanomedicines, are shown in Figure 45.1.
Applications of pharmaceutical nanotechnology
The application of nanotechnology encompasses the formulation and development of nanomedicines to improve drug potency and efficacy and the use of nanomaterials in tissue engineering and implants to fabricate structures to support tissue regeneration within the body. Nanotechnology can also include the development of devices in the nano-range such as implantable sensory systems (nanodiagnostics) for improved diagnostic measurements. This chapter will focus on the use of nanotechnology in drug formulation, which can offer particular advantages including:
There are already several products authorized for clinical use that can be classified as nanomedicines (Table 45.1). Indeed some of these products have been approved for several years, having been originally approved for registration prior to the recognized classification of nanotechnology products. This includes some of the liposome-based products which have been licensed for clinical use since the mid-1990s. For example, the liposome formulation of doxorubicin (licensed in the US as Doxil® and marketed within Europe as Caelyx®) was the first liposomal product approved by the FDA. This product is a suspension of polyethylene glycol-coated-liposomes entrapping doxorubicin. Due to the ability of the liposome formulation to enhance targeting of doxorubicin to tumour sites, it was first developed for the treatment of AIDS-related Kaposi’s sarcoma, and is now licensed for other anti-tumour indications including metastatic breast cancer, advanced ovarian cancer and relapsed/refractory multiple myeloma. Table 45.1 shows that nanotechnology systems can offer a variety of attributes and each of these types of system is discussed in further detail in this chapter.
Polymer-drug conjugates
To improve the drug solubility and/or the delivery of drugs, drug molecules may be conjugated to polymers producing polymer-drug conjugates. These polymer-drug conjugates are considered as new chemical entities in their own right and, as their overall size is generally below 100 nm, these systems can be classified within the general area of nanotechnology. To build polymeric-drug conjugates there is a large range of synthetic polymers that can be produced having appropriate quality and stability attributes, and they can be custom-made to have distinct characteristics, including specified molecular weight, size, charge, etc. As these polymers are synthetic they are generally less immunogenic than naturally derived macromolecules. For the production of polymer-drug conjugates for parenteral administration, water soluble polymers are used.
A polymer-drug conjugate can be described as being built of three basic components:
A water soluble polymer backbone.
This can include synthetic polymers such as poly(ethyleneglycol) (PEG), poly(ethyleneimine) (PEI), poly(vinylpyrrolidone) (PVP) and polyvinylalcohol (PVA), poly(glutamic acid) (PGA), and hydroxypropylmethacrylate (HPMA) copolymers. Alternatively natural polymers such as dextran, chitosans, hyaluronic acid and proteins can be used. Of the polymers, PEG is the most widely used; it is approved by the FDA for human use and offers properties including low immunogenicity, antigenicity and toxicity. PEG chains also offer high hydration and flexibility which is useful in improving solubility and drug delivery. Another important property of PEG is their low polydispersity (in terms of molecular weight). The ease with which PEG can be modified and conjugated to drugs and proteins also offers an advantage. However, conjugation of PEG to proteins may in some instances reduce their biological activity so the conjugation site of the PEG on the protein is an important consideration. Within clinically approved products PEG molecular weights of 5000 to 40 000 Da are used.
A linker group.
Whilst a drug can be directly covalently bonded to a polymer, it is more common to attach the drug via a linker or spacer group, to help avoid the therapeutic action of the drug being blocked by the polymer. The linker can also be designed to be cleaved under certain conditions, such as changes in pH, enzymatic degradation or hydrolysis. This property can be used to promote the triggered release of the drug from the polymer conjugate under suitable conditions, thereby enhancing drug targeting. Examples of linker groups that can be used include amine, carbamate and ester groups, with an amide linker being the most common option.
Drug.
Commonly drugs delivered using these conjugates are those used in anti-cancer chemotherapy, such as doxorubicin and paclitaxel. This is because polymer conjugates can improve delivery and reduce unwanted side effects for these drugs which have narrow therapeutic windows. A second group of drugs that benefits from formulating as polymer conjugates is proteins e.g. L-asparaginase or interferons. Generally, proteins suffer from short half-lives and low stability after administration into the body. By conjugating proteins to polymers it is possible to increase their half-life by protecting the proteins from enzyme degradation and reducing clearance rates.
In addition to the three components of the system, targeting groups can be added to the polymer-conjugate with the aim of enhancing specificity and cellular uptake. However, there are no licensed products currently on the market which adopt this active targeting method. Examples of polymer-drug conjugates on the market are given in Table 45.2. As can be seen, PEG is used in several of these formulations as the polymer backbone and the majority of the systems are used to deliver protein therapeutics.
Table 45.2
| Name | Polymer-Drug | Indication |
| Adagen® | PEG-adenosine deaminase | SCID syndrome |
| Cimzia® | PEG-anti TNF Fab’ fragment | Crohn’s disease and rheumatoid arthritis |
| Oncaspar® | PEG-asparaginase | Acute lymphoblastic leukaemia |
| PEG-Intron® | PEG-Interferon 2b | Hepatitis C |
| Pegasys® | PEG-Interferon 2a | Hepatitis B and Hepatitis C |
| Neulastra® | PEG-Granulocyte colony-stimulating factor | Prevention of neutropenia associated with cancer chemotherapy |
| Macugen® | PEG-anti-VEGF aptamer | Age-related macular degeneration |
| Somavert® | PEG-growth hormone receptor antagonist | Acromegaly |
| Zinostatin Stimalamer® | Syren-maleic anhydride copolymer-Neocarzinostatin | Hepatocellular carcinoma |
Rationale for polymer conjugation
As noted above, the conjugation of drugs to polymers can improve the therapeutic action of the drug by improving solubility, protecting the drug from enzyme degradation, enhancing plasma circulation times and/or enhancing drug targeting. This is achieved through various actions.
Improving solubility
The conjugation of low-solubility drugs (e.g. paclitaxel, camptothecin or palatinate derivatives) to water soluble polymers can enhance the solubility of the overall system. For example OPAXIO®, a polymer-conjugate currently under development, comprising paclitaxel conjugated to poly-(L)-glutamic acid. The paclitaxel-conjugate has enhanced solubility compared to paclitaxel, therefore the conjugate can be administered without further solubilising agents.
Enhancing bioavailability and plasma half-life
The increased hydrodynamic volume of the polymer-drug conjugate compared to the free drug can reduce excretion rates via the kidneys. The renal clearance of compounds from the circulation is dictated by their molecular weight, with clearance rates decreasing with increasing molecular weight up to a threshold of around 45 kDa. Above 45 kDa, renal excretion cannot occur and larger polymers are more susceptible to clearance by the mononuclear phagocytic system (MPS). So, for example, the conjugation of molecules such as paclitaxel (~850 Da), and proteins such as interferon (~20 kDa) to water-soluble polymers increases their overall molecular weight enhancing drug circulation times and reducing kidney clearance rates.
Protecting against degradation after administration
The polymeric chains in the polymer-drug conjugate can also prevent the approach of antibodies and proteolytic enzymes to the drug. Water-soluble polymers become strongly hydrated and these hydrated polymer strands can promote steric hindrance, and block enzymes and antibodies reaching the drug. This protects the drug from degradation and enhances their plasma half-life and bioavailability. This is of particular advantage to protein-based therapeutic agents that are rapidly degraded by enzymes. However, it has been reported that antibodies against PEG can be generated in vivo and these can remove and neutralize PEG-conjugate products.
Reducing aggregation, immunogenicity and antigenicity
The hydrophilic coating offered by the polymers to the conjugate compound is the key to this property. The hydrated polymer chains can mask the hydrophobic regions in the protein, improve solubility and provide a steric shield that can help prevent protein-protein association, and reduce aggregation. For example, the native proteins in Neulastra® and PEG-Intron® have a high tendency to aggregate, however, PEG conjugation (referred to as PEGylation or pegylation) of these proteins can reduce aggregation and subsequently reduce associated immunogenic and antigenic problems. As already noted, the presence of the hydrated polymer in the conjugate can reduce antibody interactions, also reducing immunogenicity. PEGylation of proteins can also help stabilize proteins during lyophilization so helping to produce products with acceptable storage conditions.
Promoting targeting to specific organs, tissue or cells
By conjugation of drugs or proteins to water-soluble polymers, not only can their half-lives be improved, but the specific accumulation of the drug or protein can also be promoted in certain tissues. This can be achieved through the use of targeting groups or the phenomenon known as the enhanced permeability and retention (EPR) effect. This EPR effect can be described as passive targeting, whereby the distribution of the conjugate is dictated by local physiological conditions at the target site. Normally after a drug enters the systemic circulation, the drug is required to cross the endothelial lining of the vasculature before it can reach the target site. In most parts of the body, the endothelial lining is continuous with the endothelial cells situated on a basal membrane, and tight junctions between adjacent cells. This makes transport across this barrier difficult. However, the structure of the blood capillary wall varies in different organs and tissues, with three general types of endothelial cells being recognized:
Continuous.
Continuous endothelial cells are the most common. These cells have tight junctions and a continuous basement membrane. Continuous endothelial lining is found in areas, such as capillaries in the brain, lung and muscles.
Fenestrated.
This type of endothelial cells have gaps of between 20 and 80 nm between them, and this can allow the passage of small molecules out of the systemic circulation. Fenestrated endothelial linings are found in the capillaries in the kidneys and gastrointestinal tract.
Sinusoidal.
Here there are gaps between the endothelial cells of up to 150 nm. The basement membrane is either discontinuous, as in the capillaries in the spleen, or absent altogether, as in the case of the capillaries in the liver.
Additionally, the integrity of the endothelial barrier can be disturbed by inflammatory processes or by tumour growth. This can result in defective hypervasculature, leading to endothelial fenestrations as large as 200 to 300 nm being present in the endothelial lining. In addition, due to rapid tumour growth, deficient lymphatic drainage is also an issue. This modified permeability of the endothelium at such sites allow nanomedicines, including polymer-conjugates, to escape from the central circulation into the tumour site, where they are retained due to the poor tumour lymphatic drainage. This phenomenon is the EPR effect described above. Thus, conjugation of a drug or protein to an appropriate soluble polymer will result in a construct with a large hydrodynamic volume which will have reduced kidney excretion and therefore an enhanced systemic circulation time. Due to the EPR effect the polymer conjugates can escape from the systemic circulation into tumour sites where they will accumulate, enhancing drug action at the tumour site and reducing unwanted side effects elsewhere in the body.
The use of targeting groups to dictate the distribution of drugs and drug carriers can also be considered to promote targeting to specific site. This is commonly referred to as active targeting. Here, the designer of the drug-delivery system is relying on the interactions between a targeting group, which can be covalently attached to the polymer, and a corresponding receptor to facilitate the targeting of the system to a specific site. Examples of targeting groups include the use of antibodies due to their ability to specifically recognize and bind specific antigens (see below) and folate to target folate receptors which are overexpressed in tumour cells. Similarly, lectins are over-expressed on the surface of many tumour cells and can be targeted via the use of glycoproteins.
Polymer-drug conjugates case studies
OPAXIO® – a small-molecule conjugate.
In this polymer-drug conjugate, paclitaxel is conjugated to poly-L-glutamic acid (PGA) via an ester linker. Conjugating paclitaxel to the water-soluble PGA overcomes the poor aqueous solubility of paclitaxel, and the congugate can be infused into the body without the addition of solvents. This conjugate has a high drug content (~37% w/w) and is stable in the circulation and, while it remains bound to the polymer, paclitaxel is inactive. Due to its construct, the conjugate can passively target tumour sites via the EPR effect. The drug is then released intracellularly via degradation of PGA by lysosomal proteases, and the ester linker is degraded by esterases or acid hydrolysis. OPAXIO® is currently undergoing clinical trials as a potential treatment for non-small cell lung cancer and ovarian cancer.
Oncaspar® – a protein conjugate.
[/not-level-membership-for-basic-science-category]
