5 Pediatrics / Strabismus
Anatomy
At birth, diameter of eye is 66% that of adult eye
Eye enlarges until 2 years of age, then further growth in puberty (Table 5-1)
| Ocular dimensions | Infant (mm) | Adult (mm) |
|---|---|---|
| Axial length | 17 | 24 |
| Corneal diameter | 9.5–10.5 | 12 |
| Corneal radius of curvature | 6.6–7.4 | 7.4–8.4 |
| Scleral thickness | Half of adult |
Infants have variable levels of astigmatism
Majority of children are hyperopic; increases up to 7 years of age, then diminishes
Eye color darkens during first few months of life
Dilator pupillae poorly developed at birth
Physiology
Visual acuity levels (see Strabismus section)
Decreased vision in infants and children
History
family history, complications in pregnancy, perinatal problems
Orbital Disorders
Congenital Anomalies
Anophthalmos
Bilateral absence of eye due to failure of primary optic vesicle to form; extremely rare
Cryptophthalmos
Failure of differentiation of lid and anterior eye structures
Partial or complete absence of the eyebrow, palpebral fissure, eyelashes, and conjunctiva
Attenuation of the levator, orbicularis, tarsus, and conjunctiva
Infections
Benign Lesions
Dermoid Cyst (Choristoma)
Arises from dermal elements (neural crest origin)
Lined by keratinizing epithelium with dermal appendages
Most common orbital mass in childhood
Usually located in superotemporal quadrant near brow, often adjacent to bony suture
Epidermoid Cyst (Choristoma)
Arises from epidermal elements
Lipodermoid
Solid tumor usually located beneath the conjunctiva over lateral surface of globe
May appear similar to prolapsed orbital fat, prolapsed lacrimal gland, or lymphoma
Teratoma
Rare, cystic tumor arising from 2 or more germinal layers
Usually composed of ectoderm along with either endoderm or mesoderm (or both)
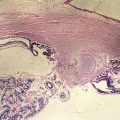
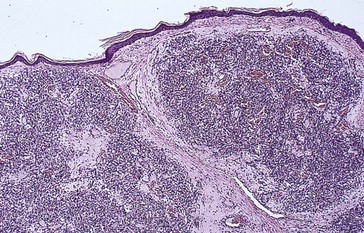
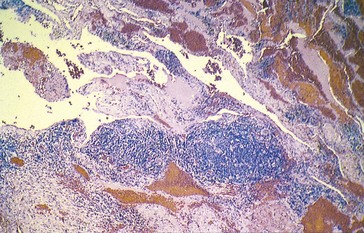
Capillary Hemangioma
Most common benign tumor of the orbit in children
Spontaneous involution over the next few years
Predilection for the superior nasal quadrant of the orbit and medial upper eyelid
Diffuse irregular mass of plump endothelial cells and small vascular channels
Findings
Pathology
numerous blood-filled channels lined by endothelium; little contribution from larger vessels or stroma; unencapsulated (Figure 5-1)
Complications
High-output congestive heart failure can occur with multiple visceral capillary hemangiomas
Lymphangioma
Rare, lymphatic-filled choristoma
Appears in first decade of life
Involves the eyelids, conjunctiva, and deeper orbital tissues
Fibrous Dysplasia
Tumor of fibrous connective tissue, cartilage, and bone
Progressive disease of childhood and young adulthood
Monostotic (in young adults) or polyostotic
Polyostotic
multiple bones involved; can cause narrowing of optic canal and lacrimal drainage system
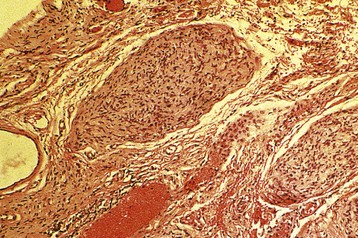
Neurofibroma
18% have neurofibromatosis (NF) type 1
Nearly all adults with NF 1 have neurofibromas
Plexiform neurofibroma
most commonly involves the upper lid; tortuous, fibrous cords infiltrate orbital tissues
Optic Nerve Glioma (Grade I Astrocytoma)
Considered pilocytic astrocytoma of the juvenile type
Slow-growing hamartoma derived from interstitial cells, astroglia, and oligodendroglia
Usually appears during first decade of life
Malignant Neoplasms
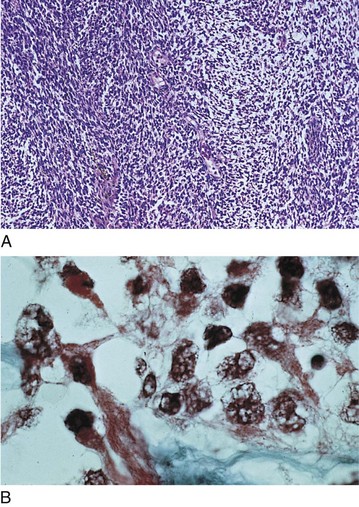
Rhabdomyosarcoma
Most common primary orbital malignancy of children
Most common soft tissue malignancy of childhood
Most common mesenchymal tumor of orbit
Malignant spindle cell tumor with loose myxomatous matrix
Average age at diagnosis is 8 years old (90% before age 16)
Unilateral; tends to involve superonasal portion of orbit
Types
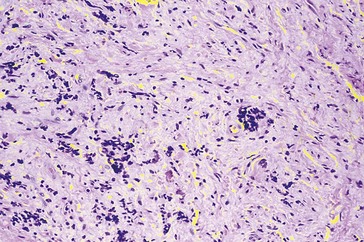
Neuroblastoma
Most common metastatic orbital tumor of childhood
Usually originates in the adrenal gland or sympathetic ganglion chain, also mediastinum or neck
40% develop orbital metastases
Average age of presentation with metastatic neuroblastoma to orbit is 2 years old
Ewing’s Sarcoma
Second most frequent metastatic tumor to the orbit
Primary intermedullary malignancy of bone; originates in long bones of extremities or axial skeleton
Histiocytosis X
Children under age 2 with multifocal disease have a poor prognosis (50% survival rate)
Findings
most frequent orbital presentation is lytic defect of orbital roof causing progressive proptosis
Craniofacial Disorders
Structural development of head and face occur during 4th–8th week of gestation
Ocular motility disturbances occur in 75% of patients with craniofacial disorders
Syndromes
Crouzon’s Syndrome (Craniofacial Dysostosis) (autosomal dominant [AD] or Sporadic)
Absence of forward development of the cranium and midface
Lid Disorders
Ankyloblepharon
Partial or complete fusion of lid margins; usually temporal, often bilateral
Blepharophimosis
Horizontally and vertically shortened palpebral fissures with poor levator function
Coloboma
Embryologic cleft involving lid margin; unilateral or bilateral; partial or full thickness
Congenital Blepharoptosis
Droopy eyelid; 75% unilateral; nonhereditary
Congenital Ectropion
Eversion of eyelid margin due to vertical shortening of anterior lamella
Congenital Entropion
Distichiasis
Partial or complete accessory row of eyelashes growing from or posterior to meibomian orifices
Epiblepharon
Usually occurs in lower lid and resolves spontaneously
Rarely requires surgery (excision of skin and muscle for significant trichiasis)
Lacrimal Disorders
Dacryocystocele
Presents at birth as bluish swelling inferior and nasal to medial canthus
Infection (dacryocystitis) develops if condition does not resolve spontaneously
Nasolacrimal Duct Obstruction (NLDO)
Up to 5% of infants have obstruction of the NLD, usually due to membrane covering valve of Hasner
Most open spontaneously within 4–6 weeks of birth; 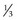 are bilateral
are bilateral
Conjunctival Disorders
Conjunctivitis
Ophthalmia Neonatorum
Conjunctivitis within first month of life
Papillary conjunctivitis (no follicular reaction in neonate due to immaturity of immune system)
Etiology
Other Infections
Age dependent; more common in younger children (<3 years old)
Vernal Keratoconjunctivitis
Form of seasonal (warm months), allergic conjunctivitis
Male > female (2 : 1); onset by age 10 years, lasts 2–10 years, usually resolves by puberty
Associated with atopic dermatitis (75%) or family history of atopy (66%)
Ligneous
Rare, bilateral, pseudomembranous conjunctivitis in children; commonly young girls
Etiology
appears to be exaggerated response to tissue injury following infection, surgery, or trauma
Kawasaki’s Disease (Mucocutaneous Lymph Node Syndrome)
Systemic childhood inflammatory disease / vasculitis with prominent mucocutaneous manifestations
Occurs in children <5 years old
More common among individuals of Japanese descent
Epidemics suggest exposure to causal agent; siblings have 10∞ increased risk
More than 50% of familial cases occur within 10 days after onset of 1st case
Corneal Disorders
Megalocornea
Horizontal diameter of cornea greater than 12 mm in newborn (>13 mm in adult)
Microcornea
Corneal diameter less than 9 mm in newborn (<10 mm in adult)
Posterior Keratoconus
Discrete posterior corneal indentation with stromal haze and thinning
Nonprogressive, usually central and unilateral
Anterior Segment Dysgenesis (Mesodermal Dysgenesis Syndromes)
Bilateral, congenital, hereditary disorders affecting anterior segment structures
Metabolic Disorders (Mucopolysaccharidoses, Mucolipidoses)
Congenital Hereditary Endothelial Dystrophy (CHED)
Rare, bilateral; mapped to chromosome 20p
Onset at or shortly after birth
Corneal clouding due to edema from defect of corneal endothelium and Descemet’s membrane
Congenital Hereditary Stromal Dystrophy (CHSD) (AD)
Rare, nonprogressive, diffuse opacification of superficial cornea
Dermoid
No hereditary pattern; 25% bilateral
Types
Other Causes of Corneal Opacity
Riley-day syndrome (familial dysautonomia; AR)
autonomic nervous system dysfunction due to block of NE production; Eastern European Jews
Infections
Syphilis
Other findings
Hutchinson’s triad
interstitial keratitis, Hutchinson’s teeth, deafness
Iris Disorders
Aniridia
Bilateral absence of iris, commonly a rudimentary iris stump exists
Hereditary or sporadic; mapped to chromosome 11p13 (PAX6)
Types
Coloboma
Iris sector defect due to incomplete closure of embryonic fissure; usually located inferonasal
May have other colobomas (lid, ciliary body, choroid, retina, and optic nerve)
Congenital Iris Ectropion
Persistent Pupillary Membrane
Remnants of anterior tunica vasculosa lentis that appear as fine iris strands
Primary Iris Cysts
Due to spontaneous separation of pigmented and nonpigmented epithelium
Occur anywhere between pupil and ciliary body
Brushfield’s Spots
Focal areas of iris stromal hyperplasia surrounded by relative hypoplasia
Appear as ring of peripheral, elevated, white-gray spots (10–20/eye)
Occur in 85% of Down syndrome patients
May be found in normal individuals (Kunkmann-Wolffian bodies)
Juvenile Xanthogranuloma (JXG; Nevoxanthoendothelioma)
Histiocytic proliferation usually of skin
Yellow-orange nodules appear before 1 year of age
Orange because of vascularity (red) combined with high lipid content (yellow)
May involve iris (may cause spontaneous hyphema)
May involve muscles, salivary glands, stomach, and other internal organs
Rarely associated with an orbital granuloma (which causes proptosis)
Lens Disorders
Congenital Anomalies
Chicken tracks (epicapsular star)
brown or golden flecks on anterior lens capsule; remnant of anterior tunica vasculosa lentis
Lenticonus
Congenital Cataracts
Characteristics
Types
classified by location or etiology
 hereditary;
hereditary;  associated with systemic syndromes;
associated with systemic syndromes;  of unknown origin
of unknown originEtiology of bilateral cataracts
Specific entities
Diagnosis of bilateral cataracts
if AD pattern determined, no workup is necessary
Etiology of unilateral cataracts
DDx of congenital cataracts and glaucoma
Lowe’s syndrome, rubella, Hallermann-Streiff syndrome
Prognosis
Glaucoma
Childhood Glaucoma
Several types of glaucoma typically categorized by age of onset
Infantile (between 3 months and 3 years of age)
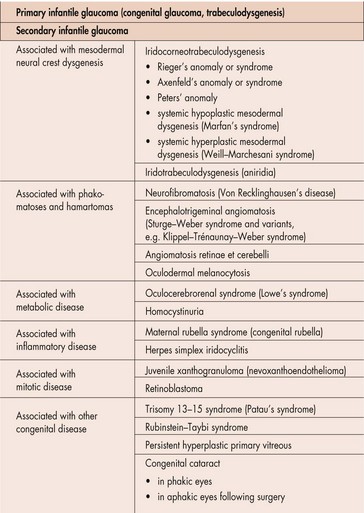
Primary Congenital Glaucoma
Diagnosis
Treatment
definitive treatment is surgical; medication is a temporizing measure
 years of age; TM incised under direct gonioscopic visualization; requires clear cornea; 77% success rate
years of age; TM incised under direct gonioscopic visualization; requires clear cornea; 77% success rate years old, or if goniotomy fails twice; Schlemm’s canal is entered via an external incision, and the trabeculotome rotates into the AC and tears the TM; 77% success rate
years old, or if goniotomy fails twice; Schlemm’s canal is entered via an external incision, and the trabeculotome rotates into the AC and tears the TM; 77% success rateUveitis
Anterior Uveitis
Juvenile Rheumatoid Arthritis (JRA)
Types (Table 5-2)
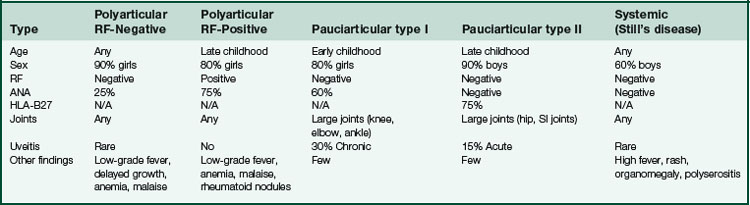
Other findings (30%)
arthritis, fever, lymphadenopathy, maculopapular rash, myocarditis, hepatosplenomegaly
Posterior Uveitis
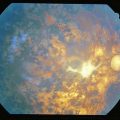
Toxoplasmosis
Due to infection with Toxoplasma gondii, usually congenital (maternal infection during gestation)
Most common cause of posterior uveitis (25%); 98% congenital
Most common cause of pediatric uveitis (50% of posterior uveitis in children)
Tachyzoite form is responsible for inflammation
Findings
inactive chorioretinal scar in posterior pole, often in macula (Figure 5-7); active focal white fluffy lesion (‘headlight in fog’ appearance) occurs adjacent to old scar with granulomatous uveitis and vitritis; may have white spots along arterioles (Kyrieleis’ plaques); may have microphthalmia, nystagmus, strabismus
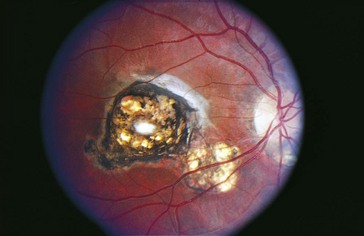
Other findings
Treatment
Toxocariasis (Table 5-3)
Due to infection with 2nd-stage larval form of dog roundworm Toxocara canis (ocular larva migrans)
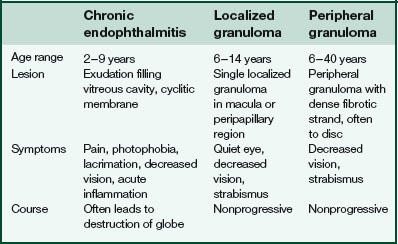
Acquired by ingestion of contaminated soil
Visceral larva migrans
fever, lymphadenopathy, hepatomegaly, pneumonitis, eosinophilia, no eye involvement
Metabolic Disorders
Box 5-2 Ocular manifestations of childhood cerebral degenerations
Retinal Disorders
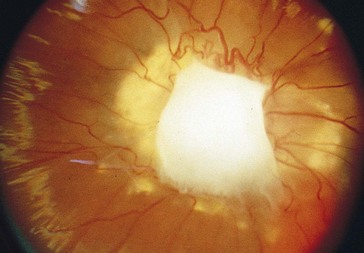
Persistent Hyperplastic Primary Vitreous (PHPV)
Due to incomplete regression of tunica vasculosa lentis and primary vitreous
Retinopathy of Prematurity (Retrolental Fibroplasia)
2 phases
Classification
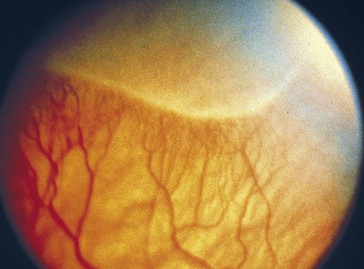
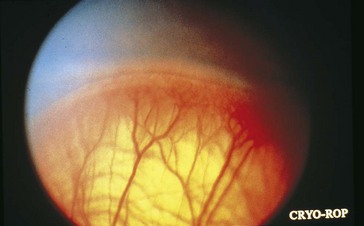
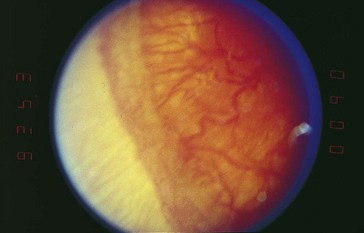
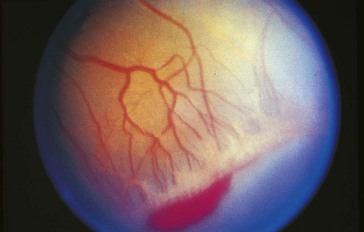
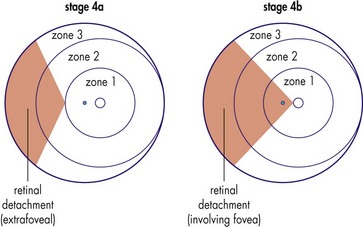
Threshold disease
(level at which 50% go blind without treatment) = stage 3 in zone 1 or 2 with Plus disease and at least 5 contiguous or 8 cumulative clock hours of involvement; usually develops at 27 weeks’ postgestation (Figure 5-15)
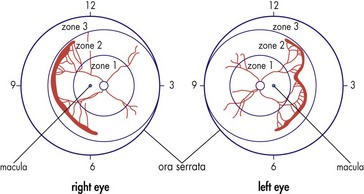
Aggressive posterior (AP) ROP or Rush disease
plus disease in zone 1 or posterior zone 2; rapidly progressive
DDx of peripheral vascular changes and retinal dragging
FEVR, incontinentia pigmenti (Bloch-Sulzberger syndrome), X-linked retinoschisis
Diagnosis
screen infants <1250 g, on supplemental oxygen during first 7 days of life, or <27 weeks’ gestation
Treatment
observation, laser, cryo, surgery
Prognosis
depends on extent of disease; most cases resolve spontaneously without visible residua
Complications
Major clinical study
Cryotherapy for ROP Study (Cryo-ROP)
The more posterior the zone and the greater the extent of stage 3+ ROP, the poorer is the outcome
After 10 years, treated eyes were much less likely than control eyes to be blind
Cryotherapy preserves visual acuity in eyes with threshold disease
Prethreshold disease: examine every week until regression or threshold disease develops
Threshold disease: perform cryotherapy within 72 hours of diagnosis
Laser photocoagulation or cryotherapy to avascular retina, 90% regression rate
Stage 4 disease: 60% reattachment rate with scleral buckle; 5% obtain useful vision
Familial Exudative Vitreoretinopathy (FEVR) (AD or X-Linked Recessive)
Mapped to chromosome 11q13-q23 (EVR1), 11p13-p12 (EVR3)
Most are unaware that they have this disorder
Findings
similar to ROP (Figures 5-16 and 5-17)
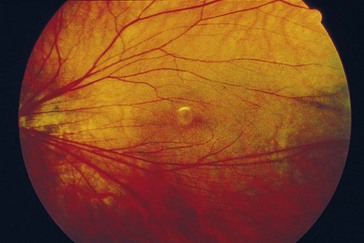
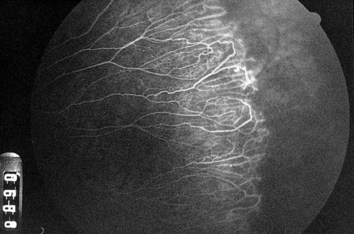
Coats’ Disease (Leber’s Miliary Aneurysms)
Nonhereditary, proliferative exudative vascular disease
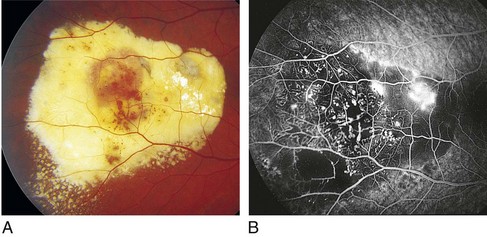
Inherited Retinal Diseases
Fundus Flavimaculatus (AR)
Stargardt’s Disease
AR or less often AD; mapped to chromosomes 1p21-p22 (STGD1), 4p (STGD4), 6q14 (STGD3)
Juvenile macular degeneration with flecks
Most common hereditary macular dystrophy
Onset in first 2 decades of life with decreased vision
Findings
bilateral pisciform, yellow-white flecks at level of RPE; change with time (new ones appear, others disappear); beaten-metal appearance of fundus; foveal atrophy; bull’s-eye maculopathy; may have salt and pepper pigmentary changes of peripheral retina (Figure 5-19)
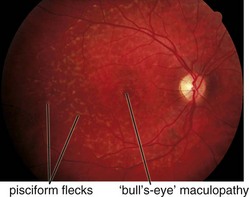
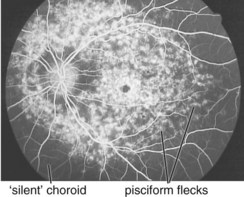
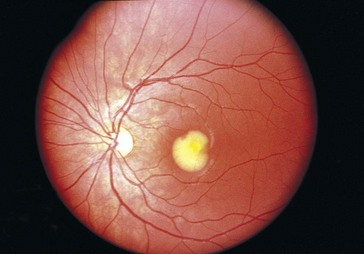
Best’s Disease (Vitelliform Dystrophy) (AD)
Variable penetrance; mapped to chromosome 11q13 (VMD2 [bestrophin])
Second most common hereditary macular dystrophy
Progressive with onset in 1st decade of life
Associated with strabismus and hyperopia
Findings
Familial Drusen (Doyne’s Honeycomb Dystrophy) (AD)
Mapped to chromosome 2p16-p21 (EFEMP1)
Small yellow-white, round to oval deposits on Bruch’s membrane; decreased vision after age 40
Maternal Inherited Diabetes and Deafness (MIDD)
Mitochondrial disease (maternal inheritance); point mutation at position 3243 of maternal mtDNA
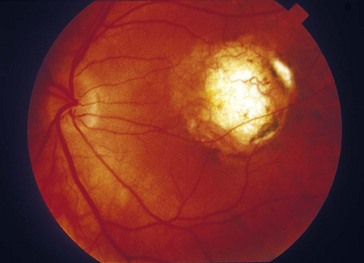
North Carolina Macular Dystrophy (AD)
Mapped to chromosome 6q14-q16 (MCDR1)
Onset in 1st decade with drusen progressing to chorioretinal atrophy with staphyloma of macula (Figure 5-22)
Pseudoinflammatory Macular Dystrophy (Sorsby’s) (AD)
Mapped to chromosome 22q12-q13 (SFD [TIMP-3])
Atrophy, edema, hemorrhage, and exudate
Decreased acuity and color vision occurs between ages 40 and 50
Pattern Dystrophies (Usually AD)
Group of diseases with central pigmentary disturbance, good central vision, normal ERG, abnormal EOG
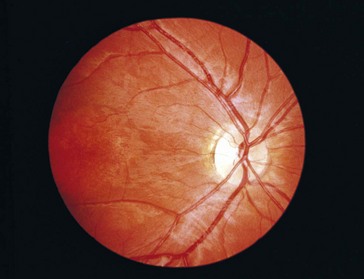
Progressive Cone Dystrophy
Progressive dysfunction of cones with normal rod function
Onset during first 3 decades of life with decreased vision, dyschromatopsia, photophobia
Findings
decreased vision (to 20/200), decreased color vision, central scotoma, nystagmus (25%), NFL loss, optic atrophy, macular degeneration (granular appearance early, then beaten-metal appearance); can have fine, golden subretinal deposits, bull’s-eye maculopathy (Figure 5-23); can develop a pattern that mimics Stargardt’s or fundus flavimaculatus
Stationary Cone Disorders
Tapetoretinal Degeneration
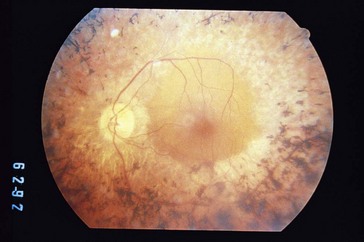
Retinitis Pigmentosa (RP)
Group of progressive dystrophies caused by abnormal photoreceptor protein production
Most common hereditary degeneration, incidence 1 : 5000
RP type I (rod–cone)
RP Variants
Leber’s congenital amaurosis (AR)
mapped to chromosomes 1p31 (LCA2), 14q11 (RPGRIP1), 17p13 (LCA1,LCA4), 19q13 (CRX)
RP Syndromes
Usher’s syndrome (AR)
most common syndrome associated with RP; mapped to chromosome 1q41 (USH2A)
Refsum’s disease (AR)
deficiency of phytanic acid oxidase interferes with fatty acid metabolism
Phytanic acid accumulates in RPE cells, sensory retina deteriorates; onset in childhood
Mapped to chromosomes 7q21-q22 (PEX1), 10p15-p12 (PNYH)
Bassen-Kornzweig syndrome (AR)
hereditary abetalipoproteinemia; mapped to chromosome 4q24 (MTP)
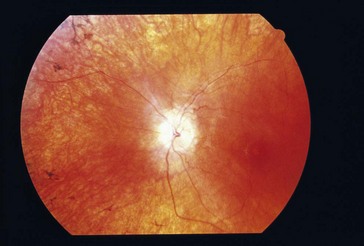
Chronic progressive external ophthalmoplegia (CPEO)
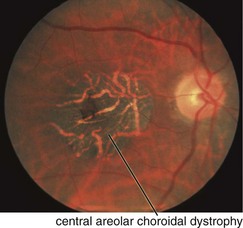
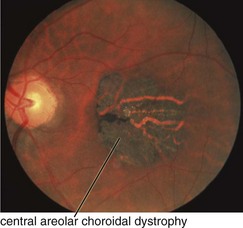
Central Areolar Choroidal Dystrophy (AD)
Mapped to chromosome 6p (RDS [peripherin]), 17p13 (CACD)
Decreased vision in 4th decade
Findings
RPE mottling in macula progressing to geographic atrophy (choroidal vessels visible) (Figures 5-26, 5-27)
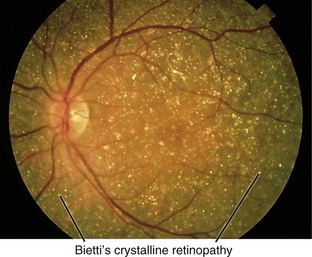
Bietti’s Crystalline Retinopathy (AR)
Congenital Stationary Night Blindness (CSNB)
Poor night vision (nyctalopia)
Group of nonprogressive rod disorders classified by fundus appearance
Abnormal fundus
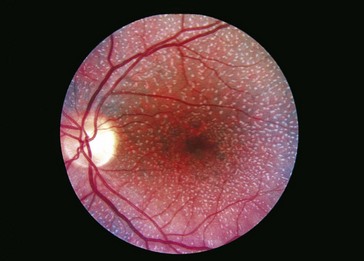
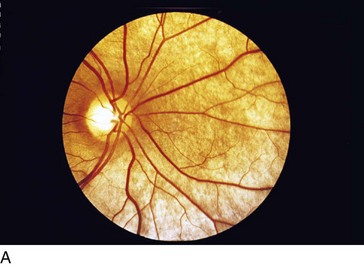
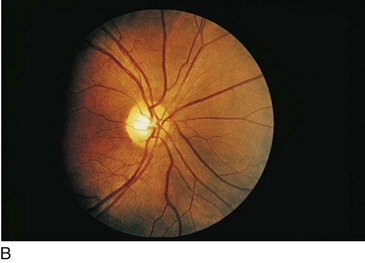
Choroideremia (X-Linked Recessive)
Progressive disorder of choriocapillaris; considered a form of rod–cone degeneration
Findings
early – degeneration of RPE and choriocapillaris in periphery (scalloped RPE atrophy); late – absence of RPE and choriocapillaris except in macula (Figure 5-31)
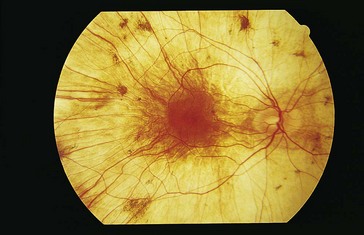
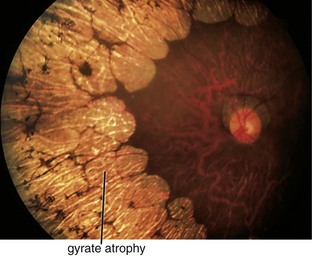
Gyrate Atrophy (AR)
Progressive retinal degeneration; starts peripherally and spreads toward posterior pole
Onset by 2nd decade of life with decreased vision, nyctalopia, and constricted visual fields
Findings
scalloped areas of absent choriocapillaris and RPE in periphery with abrupt transition between normal and atrophic areas (Figure 5-32); eventually lose choriocapillaris and medium-sized choroidal vessels; myopia (90%), cataracts, vitreous degeneration, CME