Chapter 67 Pediatric Sarcomas of Bone
Etiology and Epidemiology
The incidence of Ewing’s sarcoma is approximately 2.8 cases per million in children younger than 15 years of age.1 Generally, the disease occurs in the teenage years during the adolescent growth spurt.2 The age of onset is variable, however; approximately 30% of cases occur in the first decade of life and 10% occur in the third decade. More males than females are affected. There is a predilection for whites, in whom the disease is more frequent than Asians. It rarely occurs in black children.
The cause of Ewing’s sarcoma is unknown, and it does not appear to be induced by any known agents.
Osteosarcoma also is primarily a disease of adolescents and young adults; a different type of osteosarcoma linked to Paget’s disease occurs in older adults. This section focuses on osteosarcoma in the younger age-group. There is limited understanding of the etiology of osteosarcoma. The peak incidence coincides with a period of rapid bone growth, suggesting that a correlation exists between this phenomenon and the evolution of osteosarcoma. Furthermore, osteosarcoma is known to be induced by radiation therapy, particularly in children with retinoblastoma or other genetic abnormalities.3
Biologic Characteristics and Molecular Biology
The histogenesis of Ewing’s sarcoma has been controversial. The tumor was first described as an endothelioma of bone. It is now thought to be of neural origin, specifically from postganglionic, parasympathetic, primordial cells.4,5 Previously, extraosseous Ewing’s sarcoma and malignant peripheral neuroectodermal tumor (PNET) were considered separate entities from Ewing’s sarcoma of bone and were treated differently; they are now believed to be from the same family of tumors, however.5 This is confirmed by their similar characteristics. Both exhibit identical chromosomal translocations, t11:22 (q24;q12), and more than 85% of patients share a common surface antigen, MIC2.6–8 Ewing’s sarcoma, atypical Ewing’s sarcoma, and PNET of bone exist in the spectrum within this family, from the most undifferentiated tumors to those with neural differentiation. PNET can be differentiated from Ewing’s sarcoma by the presence of a globular growth pattern, neuron-specific enolase (NSE) positivity, and Homer Wright rosettes.
Osteosarcoma is associated with inactivation of the retinoblastoma tumor suppressor gene (13q14), which occurs in approximately one-third of cases.9 Other genetic abnormalities include translocations, gene amplification, and abnormal TP53 function.10,11
Pathology and Pathway of Spread
Ewing’s sarcoma is an undifferentiated blue round cell tumor usually of the bone. Its pathologic appearance is a monomorphic pattern of densely packed, small, round, malignant cells with hyperchromatic nuclei and varying amounts of cytoplasm.12 Immunohistochemical studies reveal cell-surface glycoprotein p30/32 MIC2 (CD 99) and vimentin, HBA-71, and β2-microglobulin positivity. Occasionally, cytokeratin and neuron-specific enolase (NSE) are positive. These studies can help differentiate Ewing’s sarcoma from other small round cell malignant tumors of childhood. Approximately 87% of cases within the family of Ewing’s tumor are Ewing’s sarcoma of bone.13 The remainder are peripheral primitive neuroectodermal tumors (PNETs) or extraosseous Ewing’s sarcoma.
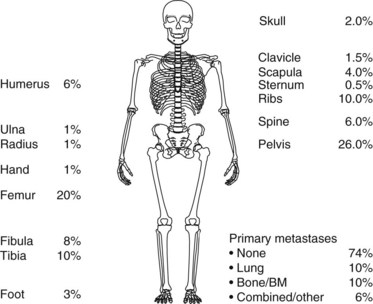
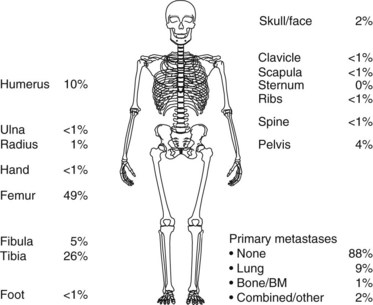
About 75 % percent of patients with Ewing’s sarcoma present with localized disease at diagnosis (Fig. 67-1). Approximately 80% of children experience distant metastases if treated with only local therapy, however. This suggests that in the majority of cases, there are unidentifiable micrometastases present at diagnosis. The most common site of metastasis is the lung, followed by bone. Other distant sites include bone marrow, soft tissues, and, rarely, the liver or central nervous system (see Fig. 67-1).
Osteosarcoma is derived from bone-forming mesenchyme, and is described as a malignant sarcomatous stroma associated with the production of osteoid bone, its defining histopathologic feature.12 The most common types in the pediatric population are the conventional osteosarcomas, including osteoblastic, chondroblastic, and fibroblastic types. Each type has varying amounts of osteoid formation and a different predominant component. There is no difference in outcome or treatment recommendations among these different types. Other types of less common osteosarcomas include telangiectatic, small cell, juxtacortical, periosteal, and high-grade surface sarcomas.
Approximately 90% of children with osteosarcoma present with localized disease at diagnosis (Fig. 67-2). However, if only the primary tumor is treated, about 90% will experience metastatic disease.12
Clinical Manifestations, Patient Evaluation, and Staging
Ewing’s sarcoma patients present in general with localized pain, swelling, and a palpable mass. The most common primary tumor sites are illustrated in Figure 67-1. In Ewing’s sarcoma, plain radiographs show a lytic, destructive lesion, most typically of the diaphysis, with or without a soft tissue mass. Codman’s triangle may form from the elevated periosteal reaction. An “onion skin” effect, derived from the development of parallel, multilaminar, periosteal reactions, is typically seen.
Osteosarcoma patients present with similar signs and symptoms, in general, localized pain, swelling, and a palpable mass. The frequency with which osteosarcoma occurs within the different regions of the body is illustrated in Figure 67-2. Plain radiographs of patients with osteosarcoma typically show sclerotic or lytic lesions of the metaphysis. The elevated periosteal reaction may cause Codman’s triangle to form. In osteosarcoma, periosteal new bone formation may be present, with the blastic component showing a bony sunburst pattern.
The evaluation for Ewing’s sarcoma and osteosarcoma patients is similar.14 A complete history and physical examination are performed, with particular attention to the duration of symptoms, the presence of pain, difficulty of function, neurologic symptoms, and the location and size of the mass. Studies commonly obtained to evaluate the extent of disease include routine blood work, urine analysis, bone scanning, plain radiographs, and computed tomography (CT) scanning or magnetic resonance imaging (MRI) of the primary region. A chest CT scan should be obtained to rule out lung metastases. If the chest radiograph is negative and the CT scan is positive for subtle anomalies, an excision may be needed for accurate staging and treatment recommendations. If available, a fluorodeoxyglucose (FDG) PET scan should be performed. An electrocardiogram and echocardiogram are included in the evaluation before chemotherapy is initiated. In the case of Ewing’s sarcoma, a bone marrow biopsy is obtained. A summary of staging and follow up investigations is given in Table 67-1.
TABLE 67-1 Staging Investigations at Diagnosis in Osteosarcoma and Ewing’s Sarcoma
| Investigation | Diagnosis | Follow-up |
|---|---|---|
| Radiograph in two planes, whole bone with adjacent joints | + | + |
| MRI and/or CT, affected bone(s) and adjacent joints | + | + |
| Biopsy: material for histologic and molecular biologic testing | + | |
| Thoracic CT (lung window) | + | + |
| Bone marrow biopsy and aspirates (in Ewing’s sarcoma): microscopy (molecular biology still investigational) | + | |
| Whole-body technetium-99 m bone scan | + | + |
| FDG-PET scan | + + | + + |
CT, Computed tomography; FDG-PET, fluorine-18 fluorodeoxyglucose positron emission tomography; MRI, magnetic resonance imaging; +, mandatory; ++, indicated, if available.
In osteosarcoma, available staging systems are those of the Musculoskeletal Tumor Society and, lately, of the International Union Against Cancer and American Joint Committee on Cancer (UICC/AJCC).15,16
The prognosis for patients with Ewing’s sarcoma is dependent on a number of factors, the most important of which is the presence or absence of metastatic disease. Other prognostic factors include the site of the lesion and its size.17 Furthermore, the histologic response to chemotherapy is an extremely good predictor of outcome.
The prognostic features of osteosarcoma are similar, namely, the presence or absence of metastatic disease, the tumor location and size, whether complete resection of the primary tumor was possible, and the response to adjuvant chemotherapy.18–21
Treatment of Ewing’s Sarcoma
Primary Therapy
Historically, patients with Ewing’s sarcoma received treatment to the primary lesion only; cure rates were less than 20%. In the early 1960s, single-institution studies began to show an improved outcome with the addition of adjuvant chemotherapy.22–24 Today, the standard treatment consists of local therapy and neoadjuvant and adjuvant polychemotherapy. Local therapy consists of surgery or radiotherapy, or a combination of both. Concerning systemic therapy, a number of randomized trials have been performed to assess the value of different drug combinations.
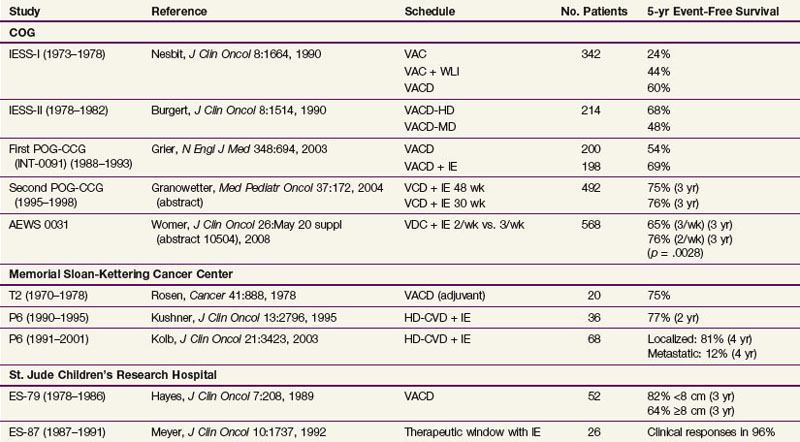
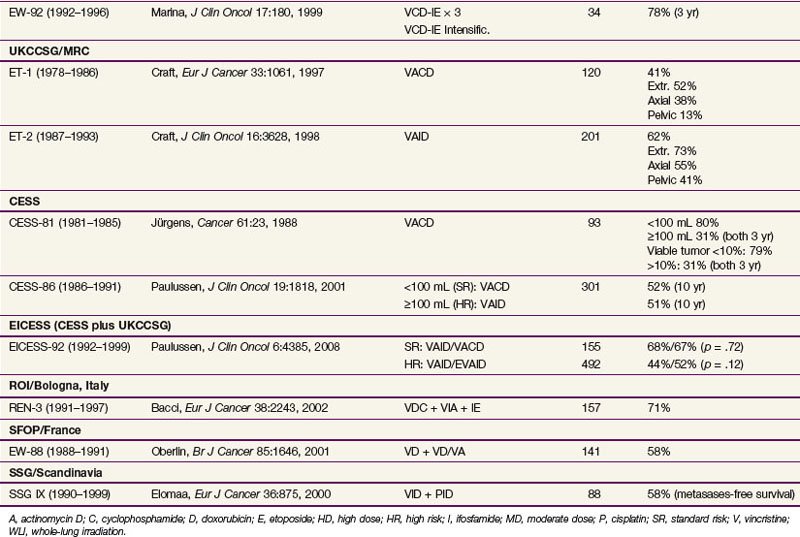
The first Intergroup Ewing Sarcoma Study (IESS) investigated the use of polychemotherapy from 1973 until 1978. Patients received radiation therapy to the primary lesion and were randomized among three adjuvant chemotherapy treatment arms: vincristine, actinomycin D, and cyclophosphamide (VAC); VAC plus doxorubicin (trade name Adriamycin, so the regimen is known as VACA); or VAC plus bilateral pulmonary radiation therapy.25 This study showed a significant improvement in all parameters with the addition of doxorubicin. Furthermore, VAC plus bilateral lung irradiation, although less effective than VACA, showed superior results to VAC alone. As a consequence of these results, doxorubicin was considered to be an essential drug in further trials. The importance of doxorubicin and also of high-intensity initial treatment was further highlighted by a systematic meta-analysis of clinical trials by Smith and colleagues.26 In addition to the standard VACA regime, an additional benefit of the use of ifosfamide and etoposide has been discussed.
The third large intergoup study, INT-0091, investigated the addition of etoposide and ifosfamide to VACA; at 5 years, data showed a statistically significant benefit from the addition of these two drugs.27 In the European Cooperative Intergroup Ewing Sarcoma Study EICESS-92, the value of the single agents was evaluated. In standard-risk patients (localized disease and tumor volume of <100 mL), VACA was randomized against VAIA (ifosfamide instead of cyclophosphamide). There was no significant difference in event-free survival rates between the groups. In high-risk patients (larger tumors or metastatic disease), VAIA was randomized against VAIA plus etoposide; again, no significant difference in event-free survival rates was observed, but a marginal benefit was demonstrated for patients with large localized disease with the use of etoposide.28 In 2008, the American Children’s Oncology Group (COG) study AEWS0031, comparing three-weekly doses versus two-weekly doses of vincristine, doxorubicin, cyclophosphamide, ifosfamide-etoposide (VDC-IE), showed an impressive 10% 3-year event-free survival benefit for two-weekly doses of chemotherapy, a regimen that is becoming standard in the United States.29 A further treatment intensification strategy is the use of high-dose chemotherapy with autologous hematopoietic stem cell rescue. Because of its toxicity, this treatment is mainly used for very high risk patients. In the ongoing Euro-EWING 99 trial, in patients who respond poorly as shown by histologic testing, the use of high-dose chemotherapy with busulfan is randomized against conventional therapy or, in metastatic patients with lung metastases only, against conventional chemotherapy and whole-lung irradiation with 15 Gy or 18 Gy, depending on the age of the patient.
A summary of results of different phase III trials is given in Table 67-2.
Advanced Disease and Palliation
Children with metastatic Ewing’s disease at diagnosis continue to have a poor outcome despite the use of aggressive multiagent chemotherapy. The current overall approach in these patients is to intensify adjuvant and neoadjuvant chemotherapy. High-dose chemotherapy using busulfan or etoposide or treosulfan, and melphalan, is used for patients with bone metastases. Radiation therapy and, sometimes, surgery are used to treat the primary disease and sites of bone metastases. In patients with lung metastases only, the use of whole-lung irradiation has shown favorable results in the CESS and EICESS trials.30 Radiation therapy can be used as a palliative local measure in patients in whom curative management fails.
Local Therapy
Local therapy is an essential modality in the treatment of Ewing’s sarcoma. With systemic therapy alone, cure cannot be obtained. The modality of local therapy with best local control rates and functional outcome has been a matter of debate for some time. Essentially, local therapy can be given with surgery alone or radiotherapy alone, or a combination of both, either as preoperative or postoperative radiotherapy. To date, there have been no randomized trials comparing local therapy modalities, and no such trial is to be expected in the near future. From retrospective analyses of several groups, the impression has been that local control is improved when surgery is performed.31–34 These data are usually confounded by the fact that there is a selection bias favoring patients in whom surgery is possible. Several European and North American collaborative trials have been performed. Overall, local control rates range from 53% to 93%, with the poorer results usually reported in the earlier series.25,35–38
Definitive Radiotherapy
Patients who receive radiotherapy as the only local therapy modality usually represent an unfavorably selected group of patients. They frequently present with large tumors or tumors in unfavorable locations (e.g., vertebral tumors), or both, making radiotherapy difficult but surgery impossible. In an analysis of 1058 patients with localized Ewing’s sarcoma treated in the European Intergroup Cooperative Ewing Sarcoma Studies (EICESS) trials, 266 patients received radiotherapy alone for local treatment. Local or combined local and systemic failures in this subgroup occurred in 26% of patients, a recurrence rate that was worse than the rate following surgery with or without radiotherapy (4% to 10%).33,34 It was not possible to define a subgroup of patients in whom the use of radiotherapy alone achieved the same local control rate as surgery.
Bacci and associates39 performed a single-institution analysis of 512 patients treated in four consecutive trials. Treatment results in patients who received radiotherapy alone were worse than in patients who underwent surgery (local failure rates of 19% with radiotherapy alone, 11% with surgery and radiotherapy, and 9% with surgery alone). When different tumor sites were analyzed, radiotherapy alone was unfavorable in extremity sites but not in central tumor sites.
Definitive radiotherapy is indicated when only an intralesional resection is possible. Debulking procedures do not improve local control rates and are associated with unnecessary morbidity. In the CESS and EICESS trials33,34 and in the Bologna experience by Bacci and coworkers,39 patients who had intralesional resection followed by radiotherapy had the same local control rate as patients who had radiotherapy alone.
Postoperative Radiotherapy
Postoperative radiotherapy is always indicated following intralesional resections. These debulking procedures are not sufficient to obtain local control and should be avoided. In the CESS and EICESS trials, local control with surgery alone was excellent in all patients who had a wide resection according to the Enneking classification and showed a good response on histologic testing following initial chemotherapy (defined as <10% viable tumor cells in the resected specimen).40 Only one local failure in 101 patients occurred in this subgroup. Patients with wide resection and a poor histologic testing response were at higher risk of local failure (12%). This local failure rate was improved with the use of postoperative radiotherapy (6%).33 There was also a trend toward benefit with the use of postoperative irradiation in patients who had a marginal resection. Bacci and colleagues39 observed no improvement with postoperative radiotherapy following wide or marginal resection (local failure rate of 7% without postoperative radiotherapy and 6% with postoperative radiotherapy).
Preoperative Radiotherapy
The systematic use of preoperative radiotherapy was incorporated into the EICESS-92 trial. The governing objective was to sterilize the tumor compartment before surgery and thereby potentially reduce the rate of dissemination during surgery. With growing experience with the use of preoperative radiotherapy, it was used in this trial when narrow resection margins were expected. More than 40% of the EICESS patients were treated with preoperative radiotherapy. In an analysis of the data from these 246 patients, however, a reduction in systemic failure could not be demonstrated.33 On the other hand, the local control rate following preoperative radiotherapy was excellent, with only 5% of the patients experiencing local or combined local and systemic failures.
Irradiation Techniques/Tolerance
Radiation Dose and Fractionation
In order to control Ewing’s sarcoma, a radiation dose above 40 Gy is necessary. In the St. Jude’s Children’s Research Hospital experience with the use of lower radiation doses, a high rate of local recurrence was observed.37 Although a clear dose-response correlation at doses above 40 Gy has not been established, for definitive radiotherapy, doses between 55 Gy and 60 Gy are usually given. When surgery precedes or follows radiotherapy, the doses range between 45 Gy and 55 Gy, depending on the individual risk factors (i.e., resection margins and response).
Usually, conventional fractionation with daily fractions of 1.8 Gy to 2 Gy is given. In the CESS-86 and EICESS-92 trials, hyperfractionated radiotherapy with a twice-daily dose of 1.6 Gy was also applied; after a total of 22.4 Gy had been given, a 10-day break was scheduled to permit the administration of chemotherapy. There has been no difference in local control rates between the two different fractionation groups.41
Target Volume
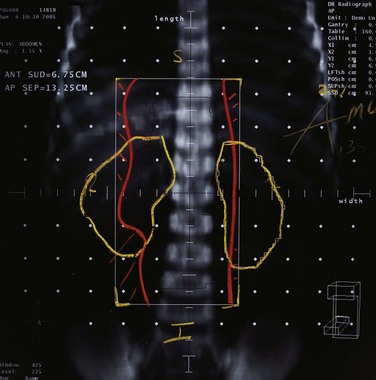
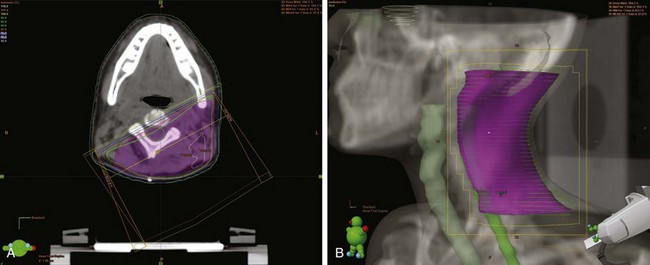
In terms of volume, previous treatment recommendations were to include the entire bone. The Pediatric Oncology Group (POG) series and other series confirmed that local failures occur generally within the high-dose irradiation volume.38 In a randomized trial, the treatment of the whole tumor-bearing compartment showed no better results than irradiation to the tumor and an additional safety margin.42 The planning target volume is defined, therefore, as the initial tumor extent on MRI with an additional margin of 2 cm. In tumors protruding into preformed cavities (i.e., the thorax or pelvis) without infiltration, the residual intracavitary tumor volume following chemotherapy is used for treatment planning. Surgically contaminated areas, scars, and drainage sites must be included in the radiation fields. Circumferential irradiation of extremities should be avoided in order to reduce the risk of lymphedema. In growing children, growth plates must be considered. They should either be fully included in the radiation field up to 30 Gy, or they should not be included at all. A dose gradient through the epiphysis results in asymmetric growth and may lead to functional deficits. Similarly, vertebral bodies should either be fully included or spared from the radiation field. Three-dimensional conformal radiotherapy should be given in patients with Ewing’s sarcoma. In selected cases (i.e., in vertebral tumors), intensity-modulated radiotherapy (IMRT) or proton therapy may be beneficial. Figures 67-3 and 67-4 show an example of treatment planning.
Irradiation of Lung Metastases
The benefit of lung irradiation in Ewing’s sarcoma tumors has been shown in the randomized IESS-I trial.25 In patients without radiologic evidence of metastases (in the era before CT became available), the best results were observed with VACA, although VAC plus lung irradiation was significantly better than VAC alone. In an analysis of the EICESS-92 trial, patients with lung metastases alone showed a trend toward a better outcome with the use of radiotherapy.43 The recommended dose is 15 to 20 Gy in 1.5-Gy daily fractions. In a CT-based treatment plan, doses above the prescribed dose in the lung should be avoided; without CT-based planning, the lung correction factor should be considered. Opposing radiation portals should include both lungs down to the diaphragmatic recess. Treatment in deep inspiration may help to reduce the volume of irradiated liver, stomach, and upper kidneys. Doxorubicin and actinomycin D must not be given during whole-lung radiotherapy because of the increased risk of pneumonitis, and actinomycin D should also be avoided after lung irradiation because of the risk of recall pneumonitis. Lung radiotherapy is therefore best given after completion of conventional chemotherapy. In the context of a busulfan-containing regimen, lung irradiation is obsolete.
Radiation of Bone Metastases
Radiotherapy is indicated in patients presenting with bone metastases. In an analysis of metastatic patients treated in the EURO-EWING 99 trial, there was a significant improvement in the 3-year event-free survival rate in patients who received local therapy to the primary tumor and to all metastatic sites compared with patients who received local therapy to the primary tumor only or no local therapy at all (39% vs. 17% vs. 14%).44 In a multivariate analysis that considered patient age, volume of the primary tumor, number of bone metastases, type of therapy (i.e., high-dose chemotherapy and local therapy to the primary tumor and/or to extrapulmonary metastases), significant factors associated with a poor prognosis were no local therapy to the primary tumor and/or extrapulmonary metastases and no high-dose chemotherapy. When bone metastases are few, radiotherapy can be given to all initially involved sites with doses of about 50 Gy, depending on surrounding critical structures. Irradiation of more than 50% of the estimated bone marrow volume can result in significant myelosuppression and accentuate the toxicity of chemotherapy. Therefore, in patients presenting with multiple bone metastases, precluding irradiation of all sites, radiotherapy can be given to sites of residual tumor seen on imaging or PET scanning. Following high-dose busulfan therapy, two cases of myelopathy have been reported with the application of 50 Gy in conventional fractionation to vertebral sites. In this context, a dose reduction is necessary.
Treatment of Osteosarcoma
Primary Therapy
The overall current recommended approach to treatment of osteosarcoma is surgical resection of the primary tumor either by amputation or a limb-sparing procedure. Local treatment is preceded by neoadjuvant chemotherapy and followed by postoperative chemotherapy. Irradiation is used in patients in whom complete surgical resection cannot be obtained and as palliation in patients with inoperable metastases. Historically, patients with osteosarcoma were also treated with local therapy alone, either surgery or radiation therapy. Single-institution studies showed an improvement in overall survival rates among patients receiving adjuvant chemotherapy compared with historic controls.20,45,46 Two prospective, randomized studies confirmed this, one at the University of California Los Angeles and one national multi-institution study.47,48 Currently, the initial chemotherapeutic agents used include methotrexate, cisplatin, doxorubicin, and ifosfamide. Patients with tumor necrosis of more than 90% have the best outcome.19,20 If the primary tumor shows only minimal response to neoadjuvant chemotherapy, many believe the chemotherapy agents should be altered for the postoperative therapy, but this has not been proved. Currently, between 60% and 70% of patients with localized osteosarcoma are cured.
Advanced Disease and Palliation
The overall approach to metastatic disease with osteosarcoma is with multiagent chemotherapy. The overall prognosis depends on the location and number of metastases, whether complete surgical resection of all tumor tissue was possible, and the response to chemotherapy.49,50 Patients with metastasis only in the lung or a limited number of metastases who can undergo complete surgical resection fare better; the 3-year survival rate is approximately 50%, compared with 20% to 30% for more advanced disease.
Radiation therapy to the lungs for metastases, when appropriate, can be given to a dose of 15 to 20 Gy to the whole lung. The use of whole-lung radiation therapy in the setting of metastatic disease is controversial. Whole-lung radiation therapy as prophylaxis against metastatic disease is also controversial and is usually not recommended. Randomized trials of the European Organization for Research and Treatment of Cancer (EORTC) showed that prophylactic whole-lung radiation therapy compared well with chemotherapy.51 However, no benefit was shown in the U.S. studies.52
Radiation therapy may be incorporated into the treatment of osseous metastatic sites not amenable to surgical resection. In an analysis of the prognosis of relapsing patients treated in the Cooperative Osteosarcoma Study Group (COSS) trials, results were best when metastases could be surgically removed. Radiotherapy of inoperable sites, though, was associated with an improved survival rate compared with that of patients who received no local therapy.53
The use of 153Sm-EDTMP (153Samarium ethylenediaminetetramethylene phosphonic acid) therapy has been investigated in unresectable osteosarcoma (for the primary tumor and metastases). This therapeutic beta-emitting isotope, which also emits a gamma photon to permit imaging and dosimetry, localizes to osteoblastic lesions with very high tumor to nontumor ratios.54 Although it is not yet possible to make any definitive statements about the long-term efficacy of this treatment modality, a multimodal concept of high-activity 153Sm-EDTMP in combination with external beam radiation therapy, polychemotherapy, and autologous hematopoietic progenitor cell support for unresectable osteosarcoma seems to be promising.55,56
Local Therapy
Surgery is the mainstay of local treatment of osteosarcoma. Surgical resection is performed either by amputation or a limb-sparing approach and has a 5% local failure rate. Although not well studied and rarely used, the addition of radiation therapy in limb-sparing approaches is a possibility and can be considered in special circumstances. Osteosarcoma is usually considered to be a radioresistant tumor. In vitro studies, however, show that it has a radiosensitivity similar to that of other human tumor cell lines.57 Also, there have been case reports of patients with inoperable or residual tumor treated with radiation doses ranging between 50 Gy and 70 Gy who remained in continuous remission.58 In an analysis of osteosarcoma patients refusing surgery but receiving systemic therapy and local irradiation, the local progression-free survival rate at 5 years was 56%.59 The survival rate was about 90% at 5 years in the subgroup of irradiated patients responding to chemotherapy. DeLaney and associates60 reported on 41 patients with osteosarcoma with no surgery or close or positive resection margins. The respective local control rates at 5 years were 40%, 78%, and 78%, respectively.60 Although surgery is the local treatment of choice, cure can also be achieved with irradiation in inoperable tumors or patients refusing surgery.
Future Possibilities and Challenges
New radiation therapy approaches to the treatment of pediatric bone sarcomas include the use of brachytherapy, intraoperative radiation therapy, proton therapy, and intensity-modulation therapy. Some of these modalities are currently under investigation and development.61,62
14 Halperin E, Constine L, Tarbell N, et al. Osteosarcoma. In: Halperin E, Constine L, Tarbell N, Kun L, editors. Pediatric Radiation Oncology. ed 3. New York: Raven Press; 1997:267-289.
15 Enneking WF. A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res. 1986;204:9.
16 Sobin LH, Wittekind C, editors. TNM Classification of Malignant Tumours, ed 6, New York: Wiley-Liss, 2002.
17 Sauer R, Jurgens H, Burgers J, et al. Prognostic factors in the treatment of Ewing’s sarcoma. Radiother Oncol. 1987;10:101.
18 Link MP, Goorin AM, Horowitz M, et al. Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the multi-institutional osteosarcoma study. Clin Orthop Relat Res. 1991;270:8.
19 Glasser D, Lane J, Huvos A, et al. Survival, prognosis and therapeutic response in osteogenic sarcoma. Cancer. 1991;69:698.
20 Myers PA, Heller G, Healey J, et al. Chemotherapy for nonmetastatic osteogenic sarcoma. The Memorial Sloan-Kettering experience. J Clin Oncol. 1992;10:5.
21 Bielack S, Kempf-Bielack B, Delling G, et al. Prognostic factors of high-grade osteosarcoma of the extremities or trunk. An analysis of 1702 patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2002;20:776.
25 Nesbit M, Gehan E, Burget E, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone. A long-term follow-up of the first intergroup study. J Clin Oncol. 1990;8:1664.
26 Smith MA, Ungerleider RS, Horowitz ME, Simon R. Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing’s sarcoma. J Natl Cancer Inst. 1991;83(20):1460.
27 Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694.
28 Paulussen M, Craft AW, Lewis I, et al. Results of the EICESS-92 study. Two randomized trials of Ewing’s sarcoma treatment. Cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol. 2008;26(27):4385.
29 Womer RB, West DC, Krailo MD, et al. Randomized comparison of every-two-week v. every-three-week chemotherapy in Ewing sarcoma family tumors (ESFT). J Clin Oncol. 26, 2008. (May 20 suppl; abstract 10504)
30 Paulussen M, Ahrens S, Burdach S, et al. Primary metastatic (stage IV) Ewing tumor. Survival analysis of 171 patients from the EICESS studies. Ann Oncol. 1998;9:275.
31 Bacci F, Ferrari S, Longhi A, et al. Role of surgery in local treatment of Ewing’s sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep. 2004;11:111.
32 Sailer S, Harmon D, Mankin H, et al. Ewing’s sarcoma. Surgical resection as a prognostic factor. Int J Radiat Oncol Biol Phys. 1988;15:43.
33 Schuck A, Ahrens S, Paulussen M, et al. Local therapy in localized Ewing tumors. Results of 1,058 patients treated in the CESS 81, CESS 86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2003;55:168.
34 Schuck A, Hofmann J, Rube C, et al. Radiotherapy in Ewing’s sarcoma and PNET of the chest wall. Results of the trials CESS 81, CESS 86 and EICESS 92. Int J Radiat Oncol Biol Phys. 1998;42:1001.
35 Burget E, Nesbit M, Garnsey L, et al. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone. Intergroup Study IESS-II. J Clin Oncol. 1990;8:1514.
36 Craft A, Cotterill S, Malcolm A, et al. Ifosfamide-containing chemotherapy in Ewing’s sarcoma. The Second United Kingdom Children’s Cancer Study Group and the Medical Research Council Ewing’s Tumor Study. J Clin Oncol. 1998;16:3628.
37 Arai Y, Kun LE, Brooks MT, et al. Ewing’s sarcoma. Local tumor control and patterns of failure following limited-volume radiation therapy. Int J Radiat Oncol. 1991;21:1501.
38 Donaldson S, Shuster J, Andreozzi C. The Pediatric Oncology Group (POG) experience in Ewing’s sarcoma of bone. Med Pediatr Oncol. 1989;17:283.
39 Bacci G, Longhi A, Briccoli A, et al. The role of surgical margins in the treatment of Ewing’s sarcoma family tumors. Experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol. 2006;65:766.
40 Salzer-Kuntschik M, Delling G, Beron G, et al. Morphological grades of regression in osteosarcoma after polychemotherapy—study COSS 80. J Cancer Res Clin Oncol. 1983;106(Suppl):21.
41 Dunst J, Jurgens H, Sauer R, et al. Radiation therapy in Ewing’s sarcoma. An update of the CESS 86 trial. Int J Radiat Oncol Biol Phys. 1995;32:919.
42 Donaldson S, Torrey M, Link A, et al. A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma. End results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys. 1998;42:125.
43 Bölling T, Schuck A, Paulussen M, et al. Whole lung irradiation in patients with exclusively pulmonary metastases of Ewing tumors. Strahlenther Onkol. 2008;24:193.
44 Häusler J, Ranft A, Bölling T, et al. The value of local treatment in patients with primary extrapulmonary metastastic Ewing sarcoma. Cancer. 2010;116:443-450.
49 Karger L, Zoubek A, Potschger U, et al. Primary metastatic osteosarcoma. Presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2003;21:2011.
50 Temeck B, Wexler L, Steinberg S, et al. Metastasectomy for sarcomatous pediatric histologies. Results and prognostic factors. [Abstract.]. Ann Thorac Surg. 1995;59:1385.
51 Burgers JM, van Glabbeke M, Bussan A, et al. Osteosarcoma of the limbs. Report of the EORTC-SIOP 03 Trial 20781 investigating the value of adjuvant treatment with chemotherapy and/or prophylactic lung irradiation. Cancer. 1988;61:124.
52 Rab GY, Ivins JC, Childs CSJr, et al. Elective whole lung irradiation in the treatment of osteogenic sarcoma. Cancer. 1976;38:939.
53 Kempf-Bielack B, Bielack S, Jürgens H, et al. Osteosarcoma relapse after combined modality therapy. An analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol. 2005;20:559.
54 Anderson PM, Wiseman GA, Dispenzieri A, et al. High dose samarium-153 ethylene diamine tetramethylene phosphonate. Low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. J Clin Oncol. 2002;20:189.
55 Franzius C, Bielack S, Flege S, et al. High-activity Samarium-153-EDTMP therapy followed by autologous peripheral blood stem cell support in unresectable osteosarcoma. Nuklearmedizin. 2001;40:215.
56 Franzius C, Schuck A, Bielack S. High-dose Samarium-153-ethylene diamine tetramethylene phosphonate. Low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. J Clin Oncol. 2002;20:1953.
57 Phillips TL, Sheline GE. Radiation therapy of malignant bone tumors. Radiology. 1969;92:1537-1545.
59 Machak GN, Tkachev SI, Solovyev YN, et al. Neoadjuvant chemotherapy and local radiotherapy for high grade osteosarcoma of the extremities. Mayo Clin Proc. 2003;78:147.
60 DeLaney T, Park L, Goldberg S, et al. Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys. 2005;61:492.
61 Yamamuro T, Kotoura Y. Intraoperative radiation therapy for osteosarcoma. In: Humphrey G, editor. Osteosarcoma in Adolescents and Young Adults. Boston: Kluwer; 1993:178.
62 Chauvel P. Osteosarcomas and adult soft tissue sarcomas. Is there a place for high LET radiation therapy? Ann Oncol. 1992;3:S107.
1 Gurney J, Severson R, Davis S, et al. Incidence of cancer in children in the United States. Cancer. 1995;75:2186.
2 Dorfman H, Czerniak B. Bone cancers. Cancer. 1995;75:203.
3 Newton WAJr, Meadows AT, Shimada H, et al. Bone sarcomas as second malignant neoplasms following childhood cancer. Cancer. 1991;67:193.
4 Thiele C. Biology of pediatric peripheral neuroectodermal tumors. Cancer Metastasis Rev. 1991;10:311.
5 Cavazzana AO, Miser JS, Jefferson J, et al. Experimental evidence for a neural origin of Ewing’s sarcoma of bone. Am J Pathol. 1987;127:507.
6 Ambros IM, Ambros PF, Strehl S, et al. MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67:1886.
7 Zucman J, Delattre O, Desmaze C, et al. Cloning and characterization of the Ewing’s sarcoma and peripheral neuroepithelioma t(11;22) translocation breakpoints. Genes Chromosom Cancer. 1992;5:271.
8 Truc-Carl C, Aurias A, Mugneret F, et al. Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet Cytogenet. 1988;32:229.
9 Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643.
10 Tebbi CK, Gaeta J. Osteosarcoma. Pediatr Ann. 1988;17:285.
11 Landanyi M, Cha C, Lewis R, et al. MDM gene amplification in metastatic osteosarcoma. Cancer Res. 1993;53:16.
12 Link M, Grier H, Donaldson S. Sarcomas of bone. In: Fernbach D, Vietti T, editors. Clinical Pediatric Oncology. St. Louis: Mosby-Year Book; 1991:545.
13 Horowitz M, Malawer M, Woo S, et al. Ewing’s sarcoma family of tumors. Ewing;s sarcoma of bone and soft tissue and the peripheral primitive neuroectodermal tumors. In: Pizzo P, editor. Pediatric Oncology. Philadelphia: Lippincott-Raven; 1997:831.
14 Halperin E, Constine L, Tarbell N, et al. Osteosarcoma. In: Halperin E, Constine L, Tarbell N, Kun L, editors. Pediatric Radiation Oncology. ed 3. New York: Raven Press; 1997:267-289.
15 Enneking WF. A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res. 1986;204:9.
16 Sobin LH, Wittekind C, editors. TNM Classification of Malignant Tumours, ed 6, New York: Wiley-Liss, 2002.
17 Sauer R, Jurgens H, Burgers J, et al. Prognostic factors in the treatment of Ewing’s sarcoma. Radiother Oncol. 1987;10:101.
18 Link MP, Goorin AM, Horowitz M, et al. Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the multi-institutional osteosarcoma study. Clin Orthop Relat Res. 1991;270:8.
19 Glasser D, Lane J, Huvos A, et al. Survival, prognosis and therapeutic response in osteogenic sarcoma. Cancer. 1991;69:698.
20 Myers PA, Heller G, Healey J, et al. Chemotherapy for nonmetastatic osteogenic sarcoma. The Memorial Sloan-Kettering experience. J Clin Oncol. 1992;10:5.
21 Bielack S, Kempf-Bielack B, Delling G, et al. Prognostic factors of high-grade osteosarcoma of the extremities or trunk. An analysis of 1702 patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2002;20:776.
22 Hustu HO, Pinkel D, Pratt CB. Treatment of clinically localized Ewing’s sarcoma with radiotherapy and combination chemotherapy. Cancer. 1972;30:1522.
23 Pomeroy TC, Johnson RE. Integrated therapy of Ewing’s sarcoma. Front Radiat Ther. 1975;10:152.
24 Pomeroy TC, Johnson RE. Prognostic factors for survival in Ewing’s sarcoma. AJR Am J Roentgenol. 1975;123:898.
25 Nesbit M, Gehan E, Burget E, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone. A long-term follow-up of the first intergroup study. J Clin Oncol. 1990;8:1664.
26 Smith MA, Ungerleider RS, Horowitz ME, Simon R. Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing’s sarcoma. J Natl Cancer Inst. 1991;83(20):1460.
27 Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694.
28 Paulussen M, Craft AW, Lewis I, et al. Results of the EICESS-92 study. Two randomized trials of Ewing’s sarcoma treatment. Cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol. 2008;26(27):4385.
29 Womer RB, West DC, Krailo MD, et al. Randomized comparison of every-two-week v. every-three-week chemotherapy in Ewing sarcoma family tumors (ESFT). J Clin Oncol. 26, 2008. (May 20 suppl; abstract 10504)
30 Paulussen M, Ahrens S, Burdach S, et al. Primary metastatic (stage IV) Ewing tumor. Survival analysis of 171 patients from the EICESS studies. Ann Oncol. 1998;9:275.
31 Bacci F, Ferrari S, Longhi A, et al. Role of surgery in local treatment of Ewing’s sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep. 2004;11:111.
32 Sailer S, Harmon D, Mankin H, et al. Ewing’s sarcoma. Surgical resection as a prognostic factor. Int J Radiat Oncol Biol Phys. 1988;15:43.
33 Schuck A, Ahrens S, Paulussen M, et al. Local therapy in localized Ewing tumors. Results of 1,058 patients treated in the CESS 81, CESS 86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2003;55:168.
34 Schuck A, Hofmann J, Rube C, et al. Radiotherapy in Ewing’s sarcoma and PNET of the chest wall. Results of the trials CESS 81, CESS 86 and EICESS 92. Int J Radiat Oncol Biol Phys. 1998;42:1001.
35 Burget E, Nesbit M, Garnsey L, et al. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone. Intergroup Study IESS-II. J Clin Oncol. 1990;8:1514.
36 Craft A, Cotterill S, Malcolm A, et al. Ifosfamide-containing chemotherapy in Ewing’s sarcoma. The Second United Kingdom Children’s Cancer Study Group and the Medical Research Council Ewing’s Tumor Study. J Clin Oncol. 1998;16:3628.
37 Arai Y, Kun LE, Brooks MT, et al. Ewing’s sarcoma. Local tumor control and patterns of failure following limited-volume radiation therapy. Int J Radiat Oncol. 1991;21:1501.
38 Donaldson S, Shuster J, Andreozzi C. The Pediatric Oncology Group (POG) experience in Ewing’s sarcoma of bone. Med Pediatr Oncol. 1989;17:283.
39 Bacci G, Longhi A, Briccoli A, et al. The role of surgical margins in the treatment of Ewing’s sarcoma family tumors. Experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol. 2006;65:766.
40 Salzer-Kuntschik M, Delling G, Beron G, et al. Morphological grades of regression in osteosarcoma after polychemotherapy—study COSS 80. J Cancer Res Clin Oncol. 1983;106(Suppl):21.
41 Dunst J, Jurgens H, Sauer R, et al. Radiation therapy in Ewing’s sarcoma. An update of the CESS 86 trial. Int J Radiat Oncol Biol Phys. 1995;32:919.
42 Donaldson S, Torrey M, Link A, et al. A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma. End results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys. 1998;42:125.
43 Bölling T, Schuck A, Paulussen M, et al. Whole lung irradiation in patients with exclusively pulmonary metastases of Ewing tumors. Strahlenther Onkol. 2008;24:193.
44 Häusler J, Ranft A, Bölling T, et al. The value of local treatment in patients with primary extrapulmonary metastastic Ewing sarcoma. Cancer. 2010;116:443-450.
45 Suttow WW, Gehan EA, Dymen TPJ, et al. Multidrug adjuvant chemotherapy for osteosarcoma. Interim report of the Southwest Oncology Group studies. Cancer Treat Rep. 1978;62:265.
46 Pratt C, Champion J, Fleming I, et al. Adjuvant chemotherapy for osteosarcoma of the extremity. Cancer. 1990;65:439.
47 Eilber F, Giuliano A, Edkardt J, et al. Adjuvant chemotherapy for osteosarcoma. A randomized prospective trial. J Clin Oncol. 1987;5:21.
48 Link M, Goorin A, Miser A, et al. The effect of adjuvant chemotherapy on relapse-free survival in patient with osteosarcoma of the extremity. N Engl J Med. 1986;314:1600.
49 Karger L, Zoubek A, Potschger U, et al. Primary metastatic osteosarcoma. Presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2003;21:2011.
50 Temeck B, Wexler L, Steinberg S, et al. Metastasectomy for sarcomatous pediatric histologies. Results and prognostic factors. [Abstract.]. Ann Thorac Surg. 1995;59:1385.
51 Burgers JM, van Glabbeke M, Bussan A, et al. Osteosarcoma of the limbs. Report of the EORTC-SIOP 03 Trial 20781 investigating the value of adjuvant treatment with chemotherapy and/or prophylactic lung irradiation. Cancer. 1988;61:124.
52 Rab GY, Ivins JC, Childs CSJr, et al. Elective whole lung irradiation in the treatment of osteogenic sarcoma. Cancer. 1976;38:939.
53 Kempf-Bielack B, Bielack S, Jürgens H, et al. Osteosarcoma relapse after combined modality therapy. An analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol. 2005;20:559.
54 Anderson PM, Wiseman GA, Dispenzieri A, et al. High dose samarium-153 ethylene diamine tetramethylene phosphonate. Low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. J Clin Oncol. 2002;20:189.
55 Franzius C, Bielack S, Flege S, et al. High-activity Samarium-153-EDTMP therapy followed by autologous peripheral blood stem cell support in unresectable osteosarcoma. Nuklearmedizin. 2001;40:215.
56 Franzius C, Schuck A, Bielack S. High-dose Samarium-153-ethylene diamine tetramethylene phosphonate. Low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. J Clin Oncol. 2002;20:1953.
57 Phillips TL, Sheline GE. Radiation therapy of malignant bone tumors. Radiology. 1969;92:1537-1545.
58 Sweetnam R. Osteosarcoma. Br Med J. 1979;2:536.
59 Machak GN, Tkachev SI, Solovyev YN, et al. Neoadjuvant chemotherapy and local radiotherapy for high grade osteosarcoma of the extremities. Mayo Clin Proc. 2003;78:147.
60 DeLaney T, Park L, Goldberg S, et al. Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys. 2005;61:492.
61 Yamamuro T, Kotoura Y. Intraoperative radiation therapy for osteosarcoma. In: Humphrey G, editor. Osteosarcoma in Adolescents and Young Adults. Boston: Kluwer; 1993:178.
62 Chauvel P. Osteosarcomas and adult soft tissue sarcomas. Is there a place for high LET radiation therapy? Ann Oncol. 1992;3:S107.