54 Pediatric Leukemias and Lymphomas
Leukemia is the most common form of malignancy seen in the pediatric age group.1 The vast majority of childhood leukemia cases are acute, unlike those in adults. Acute lymphoblastic leukemia (ALL) is most common, accounting for 80% of all cases of childhood leukemia. Acute nonlymphoblastic leukemia, most often acute myelogenous leukemia (AML), constitutes almost 20% of cases, and chronic myelogenous leukemia (CML) approximately 3% of all leukemia cases.
Over the past two decades, an unexplained increase in leukemia rates among children younger than 15 years of age has been observed in the United States. This trend results primarily from an increase in ALL incidence, since the rates of leukemias other than ALL did not appear to increase from 1977 to 1995.2 Linet et al.3 conclude that there was no consistent change in the incidence of childhood leukemia, but rather an isolated abrupt rise in the mid-1980s. Whether these temporal trends are real events of epidemiologic importance or are merely related to changes in diagnostic and reporting methods is being debated and explored in the next generation of epidemiologic investigations.
Acute Lymphoblastic Leukemia
Epidemiology and Etiology
ALL accounts for more than 2400 of the 3250 new cases of childhood leukemia diagnosed each year in the United States.1 In the United States, childhood ALL has a peak incidence between 3 and 5 years of age, and is more common in boys than in girls and in Whites more than in African Americans.1 The frequency of ALL is increased in certain genetic disorders including Down syndrome, congenital immunodeficiency diseases (e.g., Wiskott-Aldrich syndrome, congenital hypogammaglobulinemia), and chromosomal fragility syndromes (e.g., Fanconi’s anemia, ataxia telangiectasia).4 Molecular studies of identical twins and neonatal Guthrie cards suggest a prenatal origin for acute leukemia, both lymphoid and myeloid subtypes.5,6 A number of environmental factors have been implicated with increased risk, including in utero x-ray exposure, maternal alcohol use and smoking during pregnancy, parental solvent or radiation exposure, postnatal infection and exposure to electromagnetic fields; however, none of these has been identified as a significant single etiologic agent.7
Clinical Presentation
The common presenting signs and symptoms reflect the degree of bone marrow compromise, the extent and location of leukemic cell infiltration, and the general systemic effects of these processes. Fever, the most common feature, is caused most often by the leukemia itself rather than by infection.8,9 Bleeding symptoms with bruising or petechiae and pallor with fatigue are among the common complaints resulting from the thrombocytopenia and anemia, respectively. Bone pain is common due to leukemic periosteal infiltration and marrow expansion. Lymphadenopathy and hepatosplenomegaly result from extramedullary infiltration of leukemic cells, occurring in 60% of children with ALL.8,9 An anterior mediastinal mass is present in up to 10% of newly diagnosed patients, making a chest roentgenogram crucial in the initial evaluations. Central nervous system (CNS) involvement occurs in about 5% of patients at diagnosis. Children younger than age 2 years and those with T-cell ALL have a higher incidence of CNS leukemia. Testicular leukemia manifesting as a painless enlargement of one or both testes is rare, occurring in less than 5% of patients.
Laboratory data may reveal a mild to moderate degree of anemia and thrombocytopenia along with white blood cell (WBC) counts that may be normal, decreased, or increased. A relatively small proportion of patients (14%) have hyperleukocytosis with a WBC count greater than 100,000/mm3.9 Blasts frequently, but not always, are found in the peripheral blood smear.
Diagnosis and Extent of Disease Evaluation
A thorough physical examination is required to determine the extent of the disease and detect lymphadenopathy, abdominal organomegaly, and testicular involvement. CNS involvement is suspected in the presence of cranial nerve palsies, papilledema, a history of headaches, or vomiting, although asymptomatic pleocytosis is the most common presentation of CNS ALL. The traditional definition of CNS disease requires the presence of lymphoblasts in a cytocentrifuged sample of cerebrospinal fluid (CSF) along with at least 5.0 WBC per µL of CSF or the presence of a cranial nerve palsy.10 Studies to evaluate the clinical significance of lymphoblasts in diagnostic CSF samples with less than 5.0 WBC/mL or in the setting of a traumatic tap contaminated with peripheral blood have been inconclusive, and therefore patients are treated as CNS positive.11,12 In addition, the finding of blast cells in CSF with fewer than 5.0 WBC/mL obtained during treatment is not conclusive of CNS relapse.13 Chest roentgenograms are essential to document mediastinal mass and potential risk of airway or vascular compromise before invasive procedures, sedation, or definitive therapy.
Classification
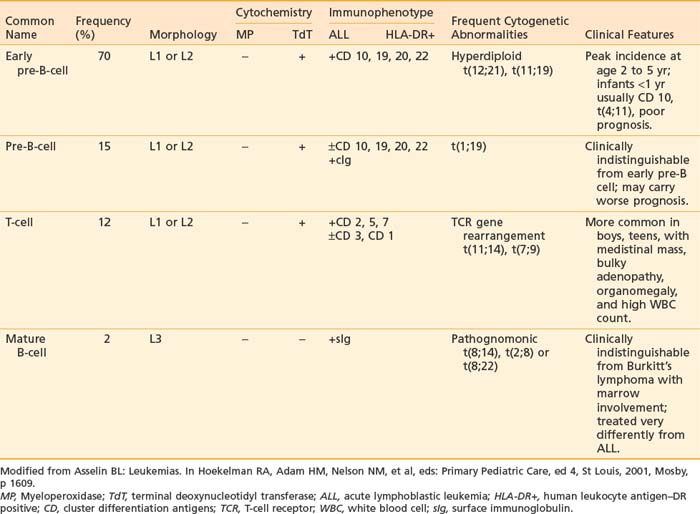
It is now well recognized that pediatric ALL is a heterogeneous disease. Originally, lymphoblasts were classified solely by morphologic criteria and cytochemical staining characteristics. These methods were sufficient to distinguish ALL from AML. More recently, classification of ALL has expanded to include immunologic and cytogenetic criteria. These criteria are useful in defining lineage and maturational stage of the leukemic clone for prognostic and therapeutic purposes as well as contributing to our understanding of important mechanisms of leukemogenesis. Classification of the different types of ALL with the characteristic phenotypes and associated immunologic and cytogenetic features is presented in Table 54-1.
Morphology
In 1976, the French-American-British Cooperative Group (FAB) devised a classification system for ALL based on the morphologic features of bone marrow lymphoblasts.14 This system divided ALL into three subtypes (L1, L2, and L3). Application of this system has been limited, because the distinction between the most common subtypes, L1 and L2 (98% of cases), is somewhat arbitrary, as results are difficult to reproduce between different observers and neither subtype correlates with immunologic, genetic, or clinical features.15,16 This stands in contrast to the L3 subtype, which predicts the mature B-cell ALL phenotype and translocations involving the MYC oncogene on chromosome 8. Thus, excluding the L3 subtype, the FAB classification system for ALL has been replaced by immunophenotyping and cytogenetics-based systems.17
Cytochemistry
Cytochemical staining properties of blast cells can be used to distinguish ALL from AML.18 Myeloperoxidase staining detects myeloperoxidase in the granules of neutrophilic, eosinophilic, and monocytic precursors. Lymphoblasts are uniformly negative, making this an important distinguishing feature of ALL versus AML. Terminal deoxynucleotidyl transferase (TdT), a DNA polymerizing enzyme, is specific to lymphoblasts of precursor B-cell or T-cell origin; mature B-cell lymphoblasts are negative, as are myeloid precursors. The lack of B- or T-cell lineage-specific cytochemical staining characteristics has limited the usefulness of these techniques. In clinical practice, they have been largely replaced by immunologic methods.
Immunophenotype
Normal hematopoietic cells undergo changes in expression of cell differentiation genes recognized as cell surface antigens as they mature from stem cells into cells of a distinct lineage. The pattern of expression of these antigens detected by flow cytometry with a panel of lineage-associated antibodies is called immunophenotype. The immunophenotype of ALL confirms the lymphoid origin and indicates lineage (B- or T-cell), as well as stage of lymphoid maturation at which the malignant transformation occurred. Current therapeutic strategies for ALL require the immunophenotype as part of the diagnostic evaluation.19
Immunophenotype studies of cell surface antigens have defined four subsets of ALL: early pre-B, pre-B, mature B-cell, and T-cell. As detailed in Table 54-1, each immunologic subtype is associated with specific clinical and cytogenetic features. The majority of ALL cases are of immature B-lineage expressing the common ALL antigen, CALLA or CD10. T-cell ALL can also be subclassified according to the stage of thymocyte maturation; however, for therapeutic purposes, only the differentiation between T-cell, mature B-cell, and B-precursor ALL is important for risk stratification and treatment assignment. More recently, measurements of these panels of immunologic markers have been adapted to allow very sensitive monitoring of minimal residual disease.20
Cytogenetics
Improvements in cytogenetic and molecular technologies now make it possible to identify abnormalities in chromosome number or structure in more than 90% of ALL cases.21 The most common abnormality, hyperdiploidy, is associated with B precursor ALL and correlates with a good prognosis. A number of these abnormalities are detectable only at the molecular level; the TEL/AMLI fusion t(12;21) is an example of such an abnormality.22 Cytogenetics and molecular genetics play an important role in the classification of leukemias and correlate significantly with the outcome of the treatment.23 It is now well recognized that the morphology, immunophenotype, and drug responsiveness of the leukemic blast cell are a direct reflection of its genetic characteristics.24,25
Prognostic Factors
Interest in prognostic factors arose in the late 1970s as therapy became successful in many patients who had ALL. Pediatric oncologists began looking for common features among groups of patients who did well compared with patients whose disease relapsed. Through retrospective analysis of disease-free survival (DFS), certain features present at the time of diagnosis of ALL were identified that were useful in predicting which patients had a good, fair, or poor prognosis.10,26,27 The initial WBC count and the age of the child at diagnosis have been the two most reliable indicators of prognosis, both for duration of remission and survival.26,28
Higher leukocyte counts, more frequent in T-cell disease and infants, are strongly correlated with a worse prognosis, although the biologic rationale for this is not clear. Children who are younger than 1 year of age or older than 10 years of age have a relatively poor prognosis compared to those in the intermediate age group. The majority of infants have 11q23 rearrangements, primarily the t(4;11) translocation, which creates an MLL-AF4 fusion gene and is associated with hyperleukocytosis and a poor prognosis.29
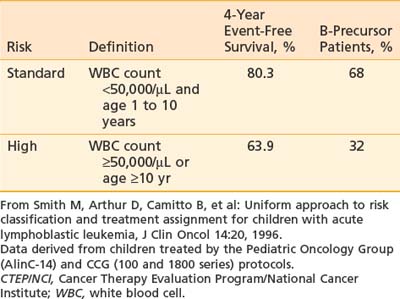
The prognostic importance of other clinical features such as gender, race, degree of organomegaly, presence of a mediastinal mass, initial platelet count, and DNA index is not reproducible among different study groups. As a result, the National Cancer Institute (NCI) sponsored a workshop in September 1993 to define uniform criteria to be used for risk-based treatment assignment for children with ALL. Workshop participants agreed on uniform age and WBC count criteria (Table 54-2) and a common set of prognostic factors to be evaluated prospectively including cytogenetics, early response to treatment, and minimal residual disease status.28 Current ALL risk classification schemas of the major cooperative groups use these NCI criteria for age and WBC count to define standard- and high-risk groups, and for purposes of treatment intensity many groups further subdivide into a low-risk group if they have favorable cytogenetics such as hyperploidy, trisomies 4, 10, and 17 or telAML1, and early response to therapy (such as a day 15 marrow) and low minimal residual disease (MRD) measured at the end of induction. Patients are considered higher risk if they have CNS or testicular disease at diagnosis. A very high-risk group is defined by the presence of MLL gene rearrangement, Philadelphia chromosome, minimal residual disease at the end of induction, or induction failure.30
Risk-Targeted Therapy
The Childhood Cancer Survivor Study recently published the results of their 25-year follow-up among 4,000 survivors of ALL diagnosed before the age of 21 years.31 Significant findings include an excess cumulative mortality of 13% due to recurrent ALL and second neoplasms. In comparison to the general population and a sibling cohort, survivors reported more frequent chronic health problems, mental health issues, and functional limitations with lower rates of college graduation, employment, and marriage. The risk of death or adverse outcome was greatest among survivors treated with radiation. These results highlight the need for ongoing multidisciplinary care for the growing numbers of leukemia survivors, but also the need for continued research and refinements of therapy to avoid such late effects.
Common Elements of Therapy
Most treatment regimens for B-precursor ALL divide therapy into four phases: initial remission induction, intensification (consolidation), CNS prophylaxis, and continuation (maintenance) therapy. Induction treatment typically includes prednisone, vincristine, and asparaginase and intrathecal therapy. With these drug regimens, complete remission is achieved in 97% of children.32 Treatment for prevention of overt CNS disease is initiated early and continues at intervals throughout the other phases of therapy. Most centers continue treatment for 2.5 to 3 years, which includes 1 to 2 years of maintenance therapy with nercapto, mtx, and variable number of pulses of vincristine and corticosteroids.
Mature B-Cell ALL
Similar to Burkitt’s lymphoma, mature B-cell ALL is more common in boys, with a median age of onset of 8 years. It frequently is associated with bulky abdominal tumor and a very high rate of cell proliferation, factors that can result in the metabolic complications associated with tumor lysis syndrome. When treated with traditional ALL therapy, patients with B-cell ALL had a grim prognosis. This changed dramatically with the development of short-term (6- to 8-month) regimens of extremely intensive therapy that incorporate fractionated high-dose cyclophosphamide, high-dose methotrexate, and cytarabine.33–35 These regimens have been adopted by virtually all centers with event-free survival (EFS) of up to 80%. Some groups are looking at the benefits of addition of ifosfamide, etoposide, or both in these regimens36,37 but at the cost of significant toxicity. Rituximab, a chimeric monoclonal antibody to CD-20, has shown remarkable response rates when added to chemotherapy of adults with non-hodgkins lymphoma (NHL) without additional toxicities. The efficacy, toxicity, and pharmacology are currently being evaluated in children with B-cell disease in a Children’s Oncology Group (COG) trial.
T-Cell ALL
Historically, T-cell ALL in children has been associated with a worse prognosis than in other subtypes of childhood ALL. Patients with T-ALL are more likely to present with high-risk features of older age, high leukocyte count, mediastinal mass, bulky organomegaly, and lymphadenopathy often termed “lymphomatous presentation,” as well as testicular involvement. T-cell ALL can be successfully treated according to the same regimens as B-lineage ALL, although, regardless of presence of other risk features, these patients require treatment on a high-risk arm.38,39 High doses of methotrexate (5 grams/m2) appear to improve the outcome in patients with T-cell ALL.38,40 Nelarabine, a prodrug of Ara G, has been shown in early phase clinical trials to have selective cytotoxicity for T-lineage lymphoblasts and has been incorporated into an ongoing study of COG to evaluate its safety and efficacy when combined with multiagent chemotherapy.41
ALL in Infants
Among children younger than 1 year, in whom the prognosis is poor, 80% have rearrangements of the MLL gene.27,42 Most cooperative groups have designed treatment regimens specifically for infants, which include high-dose cytarabine, high-dose methotrexate, etoposide, and cyclophosphamide combined with elements of high-risk ALL therapy such as vincristine, corticosteroid, asparaginase, and anthracyclines. Allogeneic bone marrow transplantation (BMT) is considered in first remission when a matched-related donor is available. Overall, the cure rates are low with DFS of 40% to 60%. Recent biologic studies of cases of infant ALL with MLL gene rearrangements have shown high levels of FLT 3 expression. This observation has created great interest in development of FLT 3 tyrosine kinase inhibitors such as lestaurtinib (CEP 701) as a treatment to improve outcome in infant ALL.43,44
The Evolving Role of Radiotherapy
Historically, the role of radiotherapy in ALL has been in the management of extramedullary disease. Trials that compared its efficacy to that of alternative therapy for both prophylaxis and treatment of extramedullary leukemia addressed the growing concern about the potential long-term toxic effects of radiation in young children.45–48 The findings of these trials have resulted in a reduction in the use of radiotherapy in the treatment of extramedullary pediatric ALL. More recently, radiation has played a significant role in BMT cytoreductive therapy designed for patients with a poor prognosis or relapse.
Central Nervous System Prophylaxis
With the advent of effective CNS prophylaxis, the survival rates associated with ALL improved dramatically. Improving the quality of life in the increasing number of survivors became a main focus of CNS prophylaxis trials of the 1980s. Ways to modify the delivery of CNS radiotherapy to potentially decrease the incidence and severity of treatment-related late toxicity were explored. Reduction of the cranial radiation therapy (CRT) dose from 24 Gy to 18 Gy was tested successfully in the Children’s Cancer Group (CCG) 143 trial (1974 to 1975).49
Of note is a recent collaborative meta-analysis of pre-1993 trials performed to clarify the relative effects on relapse and survival of different types of therapies directed at the CNS in childhood ALL.50 Radiotherapy and long-term intrathecal therapy gave similar outcomes, with no significant difference in EFS despite random assignment of treatment to 2848 patients, 1001 of whom suffered relapse or death. Intravenous methotrexate reduced non-CNS rather than CNS relapses, and hence the addition of intravenous methotrexate to a treatment regimen including radiotherapy or long-term intrathecal therapy improved EFS with a 17% reduction in the event rate. The EFS at 10 years in these trials was 61.9% without intravenous methotrexate and 68.1% with intravenous methotrexate. There was no significant difference in survival. No evidence was found regarding differences, between trials or between subgroups of different types of patients, in the relative effects of treatment. The conclusion was that radiotherapy could be replaced by long-term intrathecal therapy, and intravenous methotrexate gives some additional benefit by reducing non-CNS relapses. Both of these strategies have become common practice so that CRT is reserved for CNS prophylaxis in limited numbers of cases considered at high risk of CNS relapse because of young age, T-cell disease, high WBC count, or lymphomatous presentation. Additional strategies to further reduce the potential for late morbidity associated with CRT include dose reduction to 12 Gy51 and hyperfraction,52,53 but current investigative efforts have centered principally on finding effective strategies for CNS prophylaxis that do not require the use of radiotherapy.54–61
In current practice, CRT has been eliminated from CNS prophylactic therapy regimens in patients with B-precursor ALL who are between the ages of 1 and 9 years, with an initial leukocyte count of less than 50,000/mL and no evidence of lymphomatous or CNS involvement. The elimination of CRT in the treatment of “higher-risk” patients (aged >9 years and/or WBC count >50,000) remains a controversial issue. In this setting, the need for CRT as part of prophylaxis is highly dependent on the intensity of intrathecal medications and systemic medications that have CNS activity62–65 and which are administered as part of the chemotherapy regimen.
Meningeal Leukemia
Craniospinal axis radiotherapy has been shown to be successful in controlling meningeal leukemia. However, its associated toxicities both to the CNS and to the hematopoietic tissue (producing immunosuppression and thus a compromise in systemic chemotherapy delivery) have prompted investigators to develop other treatment strategies aimed at decreasing the role of radiotherapy, either by eliminating it entirely or by modifying the dosing regimen or volume, or both. Attempts at curing meningeal leukemia with chemotherapy alone, initiated in the 1970s, have generally been unsuccessful.66 Historically, regardless of the drug combination or route of administration employed, there was no convincing evidence to support the use of chemotherapy alone in the eradication of overt CNS leukemia. Although excellent remission rates are achieved with chemotherapy (both intrathecal and systemic), they were usually of short duration.66–68 However, recent data suggest that children diagnosed with B-ALL and CNS involvement (defined as L3 CSF blast, cranial nerve palsy, clinical spinal cord compression, isolated intracerebral mass, or cranial or spinal parameningeal extension) can be cured with standard intensity French-American-British/Lymphoma Malignancy B (FAB/LMB) therapy with the addition of high-dose methotrexate (8 g/m2) plus extra intrathecal therapy.69 Yet, patients with CNS involvement at diagnosis did have an inferior event-free and overall survival (OS) compared to those without CNS disease. Confirmatory trials are in progress.
On the basis of the results obtained to date for chemotherapy alone, CNS radiotherapy should be considered in any regimen designed for the treatment of overt meningeal leukemia, regardless of previous CNS radiation exposure, with the caveat that certain chemotherapeutic approaches may obviate the necessity of RT.35 Since the number of leukemic patients who exhibit meningeal disease is small, and since the systemic and intrathecal chemotherapy employed in the trials designed to deliver CNS radiotherapy is variable, definitive conclusions cannot yet be made with regard to the optimal dosing regimen (hyperfractionated70 versus conventional radiotherapy), field extent (craniospinal versus CRT), and timing (early versus delayed) of the radiotherapy for all scenarios of CNS disease presentation.
Recent reports of success in patients who present with CNS disease and are treated without any RT,69 with low-dose CRT (6 Gy)71 or no spinal radiation at all,72 however, do clearly challenge the necessity of spinal irradiation in meningeal leukemia, especially for chemotherapy-naive patients in whom drug-resistant clones have not yet developed. For this group of patients, CRT of 18 to 24 Gy has become a widely accepted form of CNS radiotherapy, although controversy still exists about the appropriate timing of the delivery of CRT.11 For those patients who develop a relapse either during or off therapy, CRT (craniospinal axis radiation) remains the most widely used form of CNS radiotherapy.73,74 The commonly employed total dose for the CRT is 18 to 24 Gy. Although some investigators suggest that a spinal dose of 18 Gy may be necessary for optimal CNS control, there is sufficient evidence to support the use of a lower dose of 12 Gy.75,76
A recently reported Pediatric Oncology Group (POG) trial successfully treated patients in first remission with isolated but delayed (CR1 ≥18 months) CNS relapses of B-ALL with intensive systemic chemotherapy and delayed RT (18 Gy cranial), whereas patients who relapsed within 18 months received CSI (24 Gy cranial and 15 Gy spinal).77 Currently, there is an ongoing COG study in patients with late (>18 months from diagnosis) isolated CNS relapse to evaluate the effects of intensified systemic chemotherapy with delayed reduced dose cranial radiation of 12 Gy given at 1 year postrelapse. Almost all of these patients will have been treated with systemic and intrathecal (IT) chemotherapy as primary CNS preventive therapy since RT is reserved for ∼10% to 15% of patients with highest-risk disease.78
For the small group of patients who present with symptomatic cranial nerve deficits, an immediate course of CNS radiation is a consideration. Since the goal of such therapy is to salvage nerve function, the use of as low a dose as possible to produce the desired effect is advocated. Although irradiation of the entire cranial contents is often used for this purpose, treatment to a more limited field, to include only the base of the skull, to total doses of 10 to 15 Gy, is recommended. If CRT is used, it is important that the patient be treated in a prone position, if subsequent craniospinal irradiation treatment is to be administered in that position. However, newer RT techniques, pioneered in the setting of brain tumors such as medulloblastoma, include IMRT and tomotherapy, which involve patients being treated in the supine position.79,80,80a Completion of the curative CNS radiotherapy, that is, craniospinal irradiation, may be initiated once hematologic remission has been achieved and the chance of reseeding of the CNS from medullary leukemia has been reduced. After low-dose emergent radiotherapy, an additional course of 12 to 18 Gy to the cranial contents is administered.
Techniques of Central Nervous System Radiotherapy
Cranial Radiation Therapy
When CRT is indicated, the intent is to deliver a full dose to the subarachnoid spaces, including the superficial meninges and the majority of the retina and orbital apex. By providing a 1- to 2-cm field margin around the skull and restricting the beam energy to 6 MeV or less (necessary for treatment of the lateral meninges, which are close to the skull surface), one can deliver a full dose as planned with opposed lateral photon beams.81 To assure adequate margin inferiorly on the base of the skull, the bottom edge of the second cervical vertebra is used. Special attention is needed when designing the blocking to ensure an adequate margin on the cribriform plate and temporal fossa. Because the subarachnoid space extends around the optic nerves and the retina is anatomically an extension of the nervous system, both can be involved by leukemia independently or in association with other CNS sites. Consequently it is standard practice to include the optic nerves, posterior retina, and orbital apex in the treatment field. Several techniques allow one to encompass the posterior orbit and globe while sparing the sensitive anterior aspect of the globe and lens. One approach uses inferior rotation of the gantry (i.e., angling the beam posteriorly) to achieve a parallel anterior margin at the bony orbital rim. The block or multileaf collimator margin for this approach is identified by fiducial field markers placed at the bony orbital rim during simulation, or simply by identifying this structure during a CT simulation.80b
Craniospinal Axis Irradiation
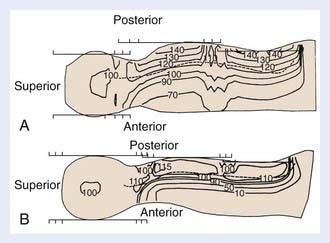
Photon therapy is most widely used for the spine fields; however, electrons may be used when the depth of the anterior surface of the spinal cord is situated within the maximum range of available electrons, after the bone attenuation is accounted for. Although the dosimetry of electrons in regions of inhomogeneities is quite complex, the sparing of anterior structures can be substantial, as shown in a comparison of the dose distributions generated with photon and with electron spinal fields (Fig. 54-1). The spine, indicated on the illustration by a dashed line, is adequately covered using either modality. The electrons, however, deliver less than 10% of the dose anteriorly, whereas the photon beam delivers 70% to 90% of the prescription dose. Depending on the location of the cranium-spine field match planes, the reduction in dose to the thyroid may be substantial. New techniques with the patient supine, and using IMRT, are appropriately employed by experienced teams.79–80 Proton beam therapy for craniospinal irradiation provides even greater sparing of the structures anterior to the spine, at the same time reducing the integral dose of radiation, the importance of which is magnified in children because of late carcinogenic effects of radiation.80 Proton beam therapy is used in the treatment of patients with medulloblastoma and other brain tumors, and is technically feasible for the treatment of the spinal axis in the setting of leukemia.80a
Craniospinal Field Matching
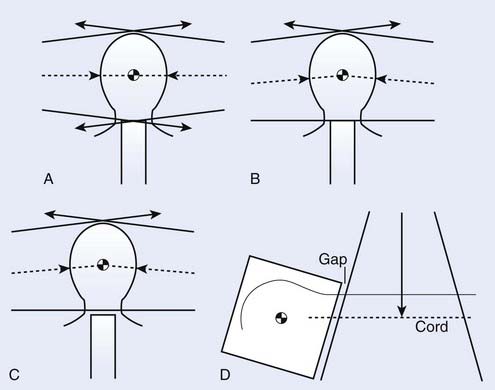
The matching of the posterior spine field with the cranial fields adds technical complexities to the alignment of the cranial fields as discussed for CRT. One approach to handling this junction problem is to accept a slight overlap because of beam divergence (Fig. 54-2A); another is to attempt to create a match plane, accomplished by pedestal rotation, which abuts to both fields (Fig. 54-2B); yet another approach is to create a gap between the cranial and spinal fields (Fig. 54-2C). In all approaches, however, the collimator is rotated for the cranial fields to create a field edge, which follows the divergence of the upper spine field (Fig. 54-2D), and the juncture where all three fields come together is shifted or moved daily to allow for an averaging out of the high-dose and low-dose regions in the junction (feathering). For any cranial field, the inferior border should never be higher than the bottom of C2 to assure complete coverage of the base of the skull; however, caudally it is limited by the presence of the shoulders. In general, the cranial field length should change by 2 to 4 cm each day of a 4-day cycle, which is equivalent to shifting the field edge by 1 to 2 cm daily.
Although gapping is a common practice, it has been shown that improved dose distributions over the junction region are achieved when fields are matched on the skin such that all three fields intersect along the mid-sagittal plane.82 Regardless of whether gapping is used, significant problems with dose inhomogeneity arise from variability in the daily treatment setup.83 When gapping is used, feathering the fields reduces the extent of inhomogeneity resultant not only from errors in the daily setup but also from the inherent underdosage in the gapped region. As noted above, IMRT (including tomotherapy) may obviate the need for such techniques by treating the entire CSI in one field, thereby avoiding the cumbersome problem of matching the fields.
Recommended Technique for Photon Craniospinal Irradiation
The problem of determining a collimator, gantry, and pedestal angle when planning photon craniospinal irradiation is quite complex.84 A recommended approach is to situate the central axes of the lateral cranial fields along the line bisecting the globes (in order to avoid divergence of the field through the contralateral lens); to rotate the collimator of the cranial fields to follow the divergence of the upper spine field (abutting the upper spine field with the cranial fields on the posterior surface of the neck using the light field); and to feather the junction daily.
Electron Spinal Fields
As previously discussed, electrons may be used to treat the spine where the maximum depth of the spine is within the range of the 80% to 90% depth dose of the most energetic electron beam available, including corrections for the bone heterogeneity. The dosimetry of the electron beams must be carefully examined, as algorithms for handling the presence of the bone heterogeneity within the field may use one-, two-, or three-dimensional corrections. With the use of computed tomography scanning to generate isodose curves, it has been reported that the 90% isodose curves shift toward the patient’s posterior surface on the order of 4 mm because of bone absorption and scatter.85 An additional 3-mm geometric margin is added to give a total shift of the 90% isodose line of 7 mm posteriorly.
Testicular Leukemia
Clinically evident testicular disease at diagnosis is a rare event.86,87 Before the advent of modern-day chemotherapy regimens, testicular relapse was a significant obstacle to cure.87,88 Because of the high rate of relapse and because of the high frequency of leukemic infiltration of testes (64% to 92%) demonstrated in autopsy series,89–91 testicular biopsies at completion of therapy became a routine practice for many investigators, as did the delivery of testicular irradiation to those boys with positive findings. Although the approach of administering presymptomatic testicular irradiation to high-risk males reduced the incidence of testicular relapse, the ultimate survival was not significantly improved.48,92–95 Since the introduction of aggressive regimens, not only have DFS rates improved, but isolated testicular relapse has become a rare event.95 As a result, testicular biopsies at completion of therapy in high-risk patients are no longer performed, and prophylactic testicular radiation is no longer necessary.
Confirmation of clinically suspected testicular leukemia is done by wedge biopsy. Bilateral biopsies should be performed, since studies have shown that involvement is bilateral.92,94 At diagnosis, testicular involvement is most often treated with systemic chemotherapy and rarely requires testicular radiation.96 Testicular relapse requires a course of testicular irradiation in combination with intensive salvage systemic chemotherapy. Because of the bilateral nature of testicular leukemia, the target volume should include the entire scrotum to encompass both the testes and epididymis. Although the exact total dose required for local control has not been established, it is clear that doses of less than 12 Gy are suboptimal,88,97 and those of 18 to 24 Gy (at 1.8 to 2 Gy/fraction) are effective.97–100 Reports of prolonged DFS after testicular relapse with aggressive salvage therapy have been encouraging, especially for those patients with an isolated testicular relapse occurring 6 months or longer after completion of therapy.101,102
Testicular radiation at doses used for ALL, independent of any contribution of chemotherapy effect, is associated with sterility and altered Leydig cell function; the latter could result in delayed sexual maturation and the need for appropriate androgen replacement.103,104
Technique of Testicular Radiotherapy
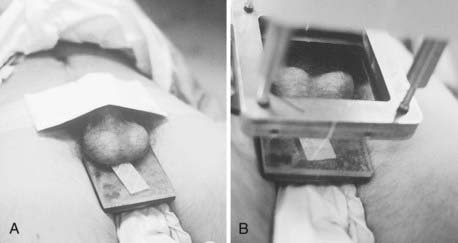
The electron beam, as administered in the energy range most often appropriate for testicular irradiation (9 to 12 MeV), offers the distinct advantage over photon modalities of a region of high-dose uniformity followed by a rapid dose falloff. There should be no more than a 10% variance in dose homogeneity within the treated volume, and the underlying perineal tissues should be spared. A polystyrene/lead combination block is used to support the testicles posteriorly and to shield the perineum (Fig. 54-3A). For electron energies of 9 to 12 MeV, 5-mm lead is adequate for shielding and the overlying 5-mm polystyrene dissipates the effect of electron backscatter. Skin apposition of the beam is achieved by angling the gantry so that the end of the cone is parallel to the treatment surface (Fig. 54-3B).
The Current Era
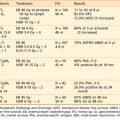
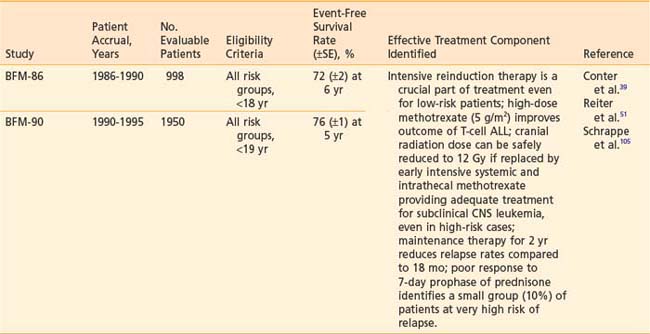
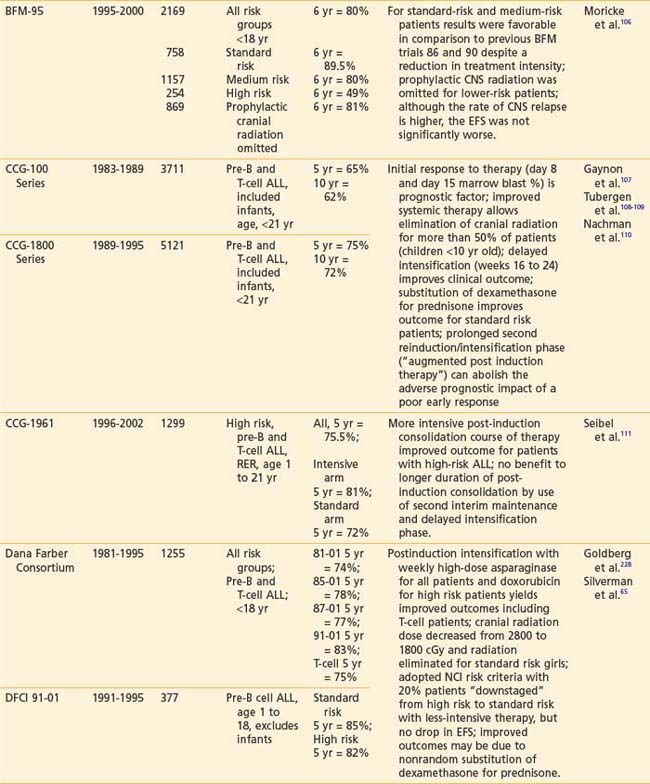
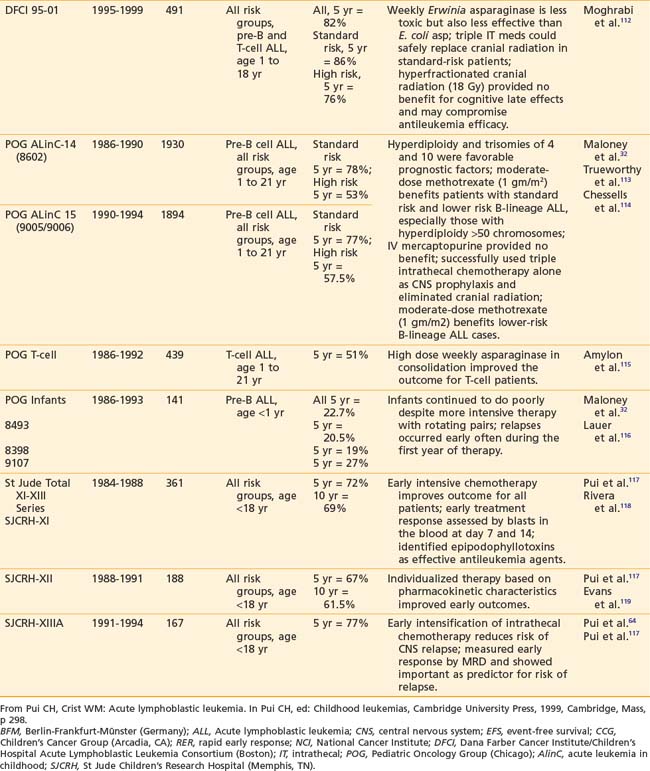
Recently reported results of major cooperative group studies demonstrate that childhood ALL is currently curable in approximately 80% of cases (Table 54-2). This level of success has resulted from the collaborative efforts of clinical oncologists and laboratory researchers over several decades. Each group has made significant contributions to the design of the sequential clinical trials that have identified increasingly effective treatments and deepened our understanding of the heterogeneous nature of leukemia. The treatment strategies responsible for these high cure rates for ALL are listed in Table 54-3. These successful treatment strategies form the basis for current ALL trials.
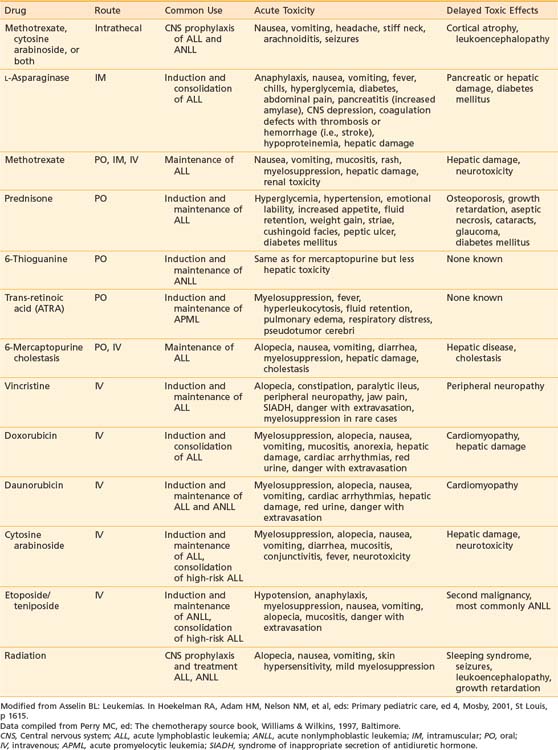
Vincristine, prednisone, methotrexate, and mercaptopurine have been the backbone to virtually all successful ALL treatments for 30 years. The dosage and schedule varies among specific protocols. Additional effective antileukemic drugs have been discovered and incorporated into the current regimens. The role and common complications of the common agents used in ALL therapy are listed in Table 54-4. The development of these intensive systemic combination chemotherapy regimens has resulted in marked improvement in outcomes especially in patients presenting with unfavorable immunophenotypic and biologic features.
The New Era: Future Directions
The evolution of curative therapy in ALL continues, with the ultimate goal of increasing the cure rate of all children with ALL, regardless of risk group, while at the same time decreasing the potential for adverse treatment-related sequelae. One direction being taken is to develop more sensitive techniques to detect MRD prospectively in patients who are apparently in complete remission according to current criteria, and thus may benefit from additional therapy to prevent relapse. Another direction is to discover new, effective antileukemic drugs and treatment approaches. Several new anticancer agents are currently being studied for ALL. They include such families as the cytokines (interferon-a, interleukin-4, and tumor necrosis factor) and the antibody-toxin conjugates.120,121 Innovative treatment regimens are being designed to better address the needs of the continuously evolving biologic subtypes and risk groups of ALL, including the use of hematopoietic growth factors to permit further intensification of therapy. Still another direction is to devise effective salvage therapy for those children who do not achieve remission or who relapse on current protocols.
Acute Nonlymphocytic Leukemia
Epidemiology and Etiology
Acute nonlymphocytic leukemia (ANLL) represents 20% of acute leukemias in children, accounting for approximately 650 of the 3250 new cases of childhood leukemia diagnosed each year in the United States.1 This is a striking contrast to the prevalence of ANLL or AML in adults, where it accounts for 80% of acute leukemias or approximately 10,000 new cases per year. The incidence of ANLL remains steady from birth to adulthood with only a slight peak in late adolescence. The median age of onset of ANLL is generally older than that of ALL, approximately 8 to 10 years.
Clinical Presentation and Diagnosis
The clinical presentation of ANLL is very similar to that of ALL. Pallor, fatigue, skin or mucosal bleeding, and fever are present in the majority of children at initial presentation.122 Infection at diagnosis is also common. Bone pain, lymphadenopathy, and marked hepatosplenomegaly are less common in ANLL than in ALL. Chloromas are solid tumor collections of immature myeloid cells that can occur in patients with ANLL. Leukemia cutis is more common in infants than older children. Almost all patients have some degree of anemia and thrombocytopenia. Twenty percent of patients present with hyperleukocytosis with a WBC count greater than 100,000/mm3. Bleeding secondary to a coagulopathy is more common in ANLL and results in an increased risk of early death from hemorrhage, especially intracranial bleeding. This is particularly true of patients with acute promyelocytic leukemia, the majority of whom have a coagulopathy at diagnosis.
The diagnosis of ANLL is confirmed by bone marrow aspirate showing greater than 25% blasts. Cytochemical staining and immunophenotyping of the leukemic blast cells are important to distinguish between the two forms of acute leukemia and help to distinguish among the different subtypes of ANLL (see Table 54-1). In young infants with Down syndrome, true ANLL must be differentiated from transient myeloproliferative syndrome.123 No single test is available to distinguish infants who have this transient myeloproliferative disorder from those who have true congenital leukemia, so conservative therapy with close monitoring for 6 to 8 weeks is the recommended approach in this situation.
Classification
The blast cell population in patients with ANLL can be characterized by morphology, cytochemistry, immunophenotype, and cytogenetics as detailed in Table 54-5. The current system of classification, defined by the FAB working group in 1976, recognizes eight subtypes of ANLL based on degree of cellular expression of granulocytic, monocytic, erythroid, or megakaryocytic features.124
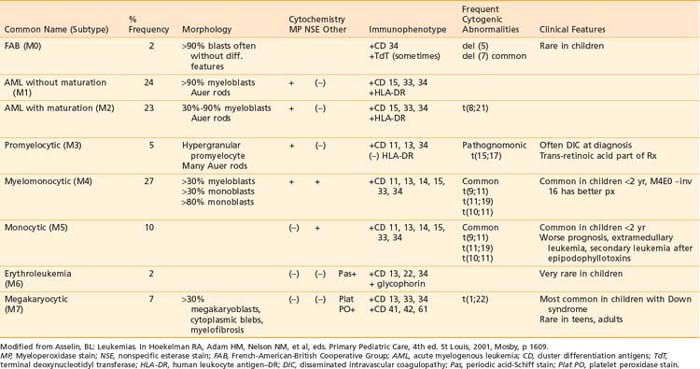
Acute myeloid leukemia (M1 and M2) is the most common form of ANLL, representing almost 50% of all cases of pediatric ANLL.125 Among patients younger than 2 years of age, the majority have either myelomonocytic or monocytic leukemia (M4 or M5 subtype) and frequently present with high WBC counts, leukemia cutis, and CNS disease.126 Acute megakaryocytic leukemia (M7) is seen most frequently in infants, especially those with DS.127
Cytogenetic abnormalities have been observed in the majority of patients with ANLL.128 Of these, the t(15;17) pattern is the only one associated with a particular FAB subtype, namely acute promyelocytic leukemia (M3). The presence of the PML-RARa fusion gene is pathognomic for M3 subtype and is thought to be crucial to pathogenesis as well.129 A unique translocation, t(1;22), has been described in infants with M7 megakaryocytic leukemia.130 The t(8;21), which produces a fusion gene of the AML1 and ETO genes from chromosomes 21 and 8, respectively, is the most common gene abnormality in AML, usually the M2 subtype, and is associated with better remission rates and prognosis.131 Translocations involving the 11q23 band with a breakpoint involving the MLL gene are common in M4 and M5 subtypes, as well as infant leukemias and secondary leukemias following epipodophyllotoxin therapy.132 Deletions of single chromosomes, either 5 or 7, are not specific to an FAB subtype but predict a poor prognosis.128,131
Prognostic Factors
The evolution of curative therapy for ANLL has not progressed as rapidly as that for ALL. Because of the relative scarcity of effective treatment options for patients with ANLL, there has been limited clinical application of the information obtained from immunophenotype or cytogenetics studies in terms of identifying prognostic indicators. Initial leukocyte count, age at diagnosis, and FAB classification appear to be the most significant factors related to treatment outcome. High WBC, age older than 14 years, infancy (age younger than 1 year), and FAB M5 subtype have been associated with worse prognosis in most studies.125,133 The era of tailored therapy to meet the specific needs of different risk groups has not been reached in AML.
Treatment
Chemotherapy
The majority of patients with ANLL are treated according to one standard therapeutic regimen. In contrast to the marked improvements in survival outcome in pediatric ALL, long-term EFS rates in ANLL have not increased dramatically over the past few decades. Although response rates may reach levels of 75% to 85%, the median duration of remission rarely exceeds 2 years.134 Relapse occurs frequently during the first 6 to 12 months on therapy, and early deaths due to treatment-related complications such as sepsis and bleeding reach levels as high as 5% to 15%. The 2-year EFS rate with current therapy remains modest, with reported values of 35% to 45%.135,136 The CCG-2961 trial results were recently published, which demonstrated a 5-year EFS of 42% and OS of 52%. This was a significant improvement in survival compared to the 44% reported between 2000 and 2002, which they attribute to increased experience and decreased toxic deaths, rather than new drugs or treatment strategies.137
Chemotherapy for all subtypes (excluding M3) consists of one to two cycles of induction therapy to achieve complete remission and postremission consolidation therapy for three to six cycles. Maintenance therapy is rarely used as it has shown clear benefit only in acute promyelocytic leukemia. Induction regimens include combinations of cytarabine and an anthracycline such as daunorubicin, mitoxantrone or idarubicin often with etoposide, thioguanine or dexamethasone. The intensity of induction therapy appears to be an important feature in the overall outcome of therapy. These regimens all produce severe myelosuppression, which is considered a key element to induce a complete remission. Current remission rates are 75% to 90%.138–140 Approximately one half of the induction failures are attributed to resistant disease and the other half to toxic deaths. Because of the intensity of therapy used to treat AML, patients should have their care coordinated by specialists in pediatric oncology and should be treated in hospitals with necessary supportive care facilities.
Postremission therapy may utilize either continued intensive chemotherapy with cytarabine-based regimens or allogeneic stem cell transplantation for the subset of patients with a matched related donor. The role of hematopoietic stem cell transplantation (HSCT) in the treatment of AML continues to evolve. Autologous transplantation achieves disease free survival rates of 55% to 60% similar to those observed with chemotherapy alone.141–144 Allogeneic HSCT has demonstrated better outcomes when compared with chemotherapy and autologous transplant, but is limited in its application because the minority of patients have a human leukocyte antigen (HLA)-matched family member to serve as donor. Several recent studies have identified favorable prognostic features that select patients with predicted DFS of 70% to 80% with chemotherapy alone.145–148 In these patients, HSCT would be considered only after a first relapse. Ongoing trials are investigating alternate donor HSCT (unrelated match, unrelated cord blood and haploidentical) for children who have poor-prognosis AML and who lack an HLA-identical sibling donor.
With growing recognition of the importance of morphology, cytogenetics, and immunology in distinguishing subtypes of ANLL with respect to the selection of chemotherapeutic agents, investigators have initiated subtype-specific therapeutic trials. The results of such trials are promising.149–159 At present, allogeneic transplantation is the recommended choice for postremission induction therapy in newly diagnosed patients with ANLL who have an available matched, related donor (unfortunately the minority of patients). The role of allogeneic transplantation using unrelated donor sources is controversial for patients in first remission.
Acute Promyelocytic Leukemia
Acute promyelocytic leukemia (APML) is the only subtype of ANLL for which there is type-specific therapy. The t(15;17) translocation, which characterizes APML blasts, produces a PML/RARa fusion gene resulting in disruption of the retinoic acid receptor and blockade of normal cellular differentiation pathways. All-trans-retinoic acid (ATRA) binds to the chimeric RARa and overcomes the differentiation blockade, resulting in cell maturation, then apoptosis.160,161 The best results have been obtained with the combination of ATRA and chemotherapy with induction rates of 90% and DFS of greater than 70%.161,162 Arsenic trioxide, also a differentiating agent, has been effective in treatment of APML, especially when resistance to ATRA develops.163 Ongoing clinical trials are designed to determine how the differentiating agents, ATRA and arsenic, can best be combined with cytotoxic drugs for the safe and effective treatment of APML.
Down Syndrome and Acute Nonlymphocytic Leukemia
Although rare in children without Down syndrome, megakaryocytic (M7) leukemia is the most common form of AML in young children with the disorder.164 Interestingly, children with Down syndrome and AML have been observed to have better responses and superior outcomes (EFS rate ∼80%) with standard AML therapy than other children with AML, even the M7 subtype. This has been shown to be a characteristic of the patient, not of the leukemia. Most treatment failures in patients with Down syndrome are a result of treatment-related mortality.165,166 Thus, increased intensity regimens and allogeneic BMT are not recommended for these patients.
Secondary Leukemia
The association between treatment with the epipodophyllotoxins, etoposide or teniposide, and development of secondary ANLL is now well established.167 The epipodophyllotoxin-induced leukemias are characterized by a short latency period (2 to 4 years) and a myelomonocytic or monocytic phenotype with a translocation involving band 11q23 with MLL gene rearrangement.168 In contrast, secondary leukemia that develops after therapy with alkylating agents is characterized by a longer latency period (∼10 years), a prodrome of myelodysplasia and monosomy 5 or 7 genotype.169 Secondary leukemias are not responsive to traditional chemotherapy and BMT holds the only hope for cure.
The Role of Radiotherapy
Chloromas, also known as myeloblastomas and granulocytic sarcomas, are extramedullary masses of malignant myeloid cells associated most commonly with ANLL (especially FAB M2), but also observed in patients with CML.170 Doses of 24 Gy, at 2 Gy per fraction, are highly effective in eradicating the local masses of malignant infiltrates refractory to chemotherapy.
Future Therapies
One of the exciting new areas of research is in the use of targeted immunotherapies.121 Gemtuzamab ozogamicin (myeloTang), a humanized anti-CD33 conjugated to calicheamicin, is an example of such an agent. Clinical trials with this immunoconjugate have shown an approximate 37% remission rate in adults with relapsed AML.171,172 In addition, a CD-45 monoclonal antibody has been radiolabeled with iodine-131 for use during BMT as a means of delivering focused bone marrow irradiation.173 Mutation of the Ras oncogene is common in AML. The Ras product must be farnesylated for it to function properly as a growth signal transduction protein. A new class of compounds called farnesyl transferase inhibitors has been developed and clinical trials are under way.174
Chronic Myelogenous Leukemia
CML is rare in childhood, representing 3% to 5% of all pediatric leukemias.1 In childhood, the disorder may appear as two distinct clinical syndromes: adult-type CML, which is virtually indistinguishable from that seen in older patients, and juvenile myelomonocytic leukemia (JMML; formerly known as juvenile CML), a disease relatively restricted to very young children that has distinct clinical, laboratory, and cytogenetic features.175
The classic adult type is characterized by the presence of the Philadelphia chromosome, a reciprocal translocation t(9;22) (q34;q11). The resultant fusion gene formed, BCR/ABL, appears to have a major role in the pathogenesis of CML. It is typically seen in adolescents, with a peak incidence between 10 and 14 years of age. Treatment and outcomes of CML in childhood parallels that of the adult patient.176 Allogeneic BMT is the only curative approach now available for patients who have CML. The disease status at the time of transplantation is the most powerful predictor of DFS. Thus, most centers recommend allo-BMT for any child who has adult-type CML in the chronic phase, preferably within 1 year from diagnosis.
The introduction of STI-571 (Gleevec) as an ABL-specific tyrosine kinase inhibitor will have a significant impact on the natural history, treatment, and survival rates of CML in the near future. As a single agent, it has shown significant activity in patients in the chronic phase who are refractory to interferon177 as well as patients in blast crisis.178 In combination with traditional chemotherapeutic agents, imatinib has shown great promise in a COG trial for treatment of Ph-positive ALL without need for HSCT.179 The optimal regimens and dosage of imatinib, dasatinib, and others are under investigation, but will likely revolutionize treatment for CML.
JMML is a clonal disorder that begins at the pluripotent stem cell level. Although the predominant abnormality is observed in the monocyte, JMML is also associated with disordered erythrocyte, platelet, and lymphocyte function.180 Common clinical features at diagnosis include age less than 2 years, persistent respiratory infection (with tachypnea, cough, wheezing), prominent lymphadenopathy, splenomegaly, skin rash (eczema, xanthomata, and café-au-lait spots), bleeding, and failure to thrive. In addition, anemia, leukocytosis, thrombocytopenia, monocytosis, and nucleated RBCs in the peripheral blood are common laboratory findings.181,182 Cytogenetic analysis may be abnormal (e.g., monosomy 7), but the Philadelphia chromosome is never found. The disease is generally more aggressive than CML with a median survival of 9 months; most patients die of infection.183 Traditional chemotherapy with single agents (e.g., busulfan, hydroxyurea) or intensive multiagent regimens have been unsuccessful. BMT is currently the standard choice of treatment of JMML. Currently, a clinical trial sponsored by the International JMML Registry is testing the efficacy and safety of farnesyl transferase inhibitors for this patient population.
Non-Hodgkin’s Lymphoma
Epidemiology and Etiology
Lymphomas, which constitute about 15% of cancer diagnoses in patients younger than 20 years of age, are the third most common form of childhood cancer. In the United States, approximately 2100 children and adolescents under age 20 years are diagnosed with lymphoma each year.1 Approximately 800 of these are NHL. The incidence peaks between 5 and 15 years of age and is higher in males than in females and in Whites than in African Americans. In younger children, NHL is more frequent than Hodgkin’s lymphoma (HL), whereas in adolescents HL is the more common type of lymphoma. Congenital immunodeficiency syndromes and acquired immunodeficiency syndrome (AIDS) are associated with an increased risk of NHL.184–186 The association of immunosuppressive drug therapy including corticosteroids and androgens with development of malignant lymphomas is well described.186 With the increasing rate of successful organ or marrow transplantation, the number of children with posttransplant lymphoproliferative disease (PTLD) represents an increasingly significant proportion of patients with NHL.187,188 Cases that are seen associated with diseases known to carry an increased risk still represent the minority of NHL diagnoses; overall, the cause is unknown in most cases.
Classification
These lymphomas are a heterogeneous group of malignancies arising from transformation of cells of lymphocytic origin, but are quite variable in terms of clinical manifestations, site of origin, histopathology, immunophenotype, and cytogenetic abnormalities. Pediatric NHL is an aggressive disease with diffuse, high grade, histologic features in which the malignant cells appear undifferentiated or poorly differentiated.189 NHL in children is characterized by disseminated disease at diagnosis, frequent extranodal involvement (including tonsils, thymus, and Peyer patches), and involvement of the bone marrow and the CNS. This is in notable contrast to the NHL seen in adults, where localized, low or intermediate grade, nodal tumors predominate.
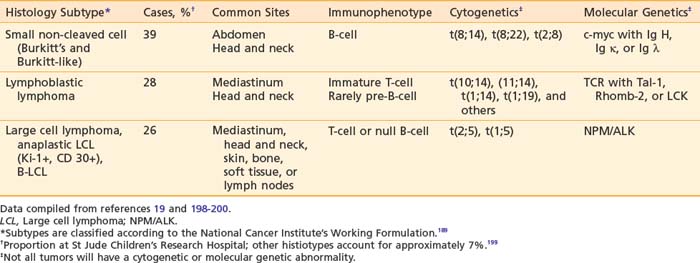
Pediatric NHL can be classified into three major categories based on predominant cell type and architectural pattern: small, noncleaved cell (Burkitt and non-Burkitt), large cell, and lymphoblastic lymphoma. All three histologic subtypes are classified as high grade according to the NCI Working Formulation for Clinical Usage.166 These three major subtypes also can be categorized further according to immunophenotype, cytogenetic abnormalities, and molecular features as shown in Table 54-6.190 Within the small noncleaved cell category are two histologic variants, the typical, uniform starry sky pattern of Burkitt lymphoma and the heterogeneous population of variable sizes designated as Burkitt-like lymphoma. Differentiation of Burkitt-like from diffuse B large cell lymphoma (DLBCL) is difficult based on histology alone and requires significant expertise and whenever possible additional cytogenetic and molecular information. DLBCL, also a mature B-cell neoplasm, presents typically in the adolescent age group with a clinical picture similar to Burkitt and Burkitt-like lymphoma, but rarely affects the bone marrow or CNS. Several chromosomal abnormalities have been shown to be pathognomic for certain subtypes of NHL. For example, the translocation t(8;14) correlates with Burkitt lymphoma191 and the nonrandom chromosomal translocation t(2;5) is expressed by the Ki-1 (CD30) positive anaplastic large cell lymphoma (T-cell immunophenotype).192 These genetic abnormalities are useful primarily in classification but not in predicting outcome or modifying therapy.193 Along with stage of disease, the classification according to histology and immunophenotype is the most important factor in determining appropriate therapy and predicting outcome.194–197 Follicular lymphoma is exceedingly rare in childhood.
Clinical Presentation
The signs and symptoms at diagnosis of NHL in pediatric patients are dependent on the anatomic site and the extent of disease.198–200 The abdomen is the most common site of disease, seen in 35% of cases, and frequently presents with nausea and vomiting; abdominal pain, swelling, or both; change in bowel habits; and ascites. The symptoms may mimic intussusception with bloody diarrhea or appendicitis since a common site of tumor infiltration is the distal ileum, appendix, or large bowel, resulting in bowel obstruction by extrinsic compression or intussusception. Primary involvement with a mediastinal mass occurs in about 25% of cases and may be accompanied by pleural effusions. Symptoms may include chest pain, dyspnea and cough, dysphagia, and swelling of the neck and face (superior vena cava syndrome). The head and neck region, including the Waldeyer lymphoid tissues and cervical lymph nodes, is involved in up to 30% of cases. Presenting features include unilateral tonsillar enlargement and orbital or jaw masses.
The presenting site of disease and thus clinical features also correlate with the histopathologic subtype as shown in Table 54-6.201 The majority of the abdominal lymphomas are small non-cleaved cell type or Burkitt’s lymphoma. In the lymphoblastic lymphomas the typical primary site is the mediastinum. Large cell lymphomas can present with an anterior mediastinal mass, abdominal primary tumor, or head and neck involvement. Anaplastic large cell lymphoma frequently involves the lymph nodes and can involve other atypical sites such as the skin, lung, thyroid, soft tissues, and bone. Relative to the other subtypes of pediatric NHL, marrow and CNS involvement are uncommon. Tumors presenting in the head and neck are seen with any of the four major histologic types.
Staging
The most widely used staging system in childhood NHL is St Jude Children’s Research Hospital Staging System, described by Murphy (Table 54-7).201 It is applicable to all histologic types of childhood lymphoma. For treatment purposes, patients often are stratified into treatment groups based on whether they have limited-stage or advanced-stage disease that corresponds to stages I/II and III/IV, respectively, of the St Jude system.
Table 54-7 The St Jude Staging System for Childhood Non-Hodgkin’s Lymphoma
| Stage | Description |
|---|---|
| I | A single tumor (extranodal) or single anatomic area (nodal), excluding the mediastinum or abdomen |
| II | A single tumor (extranodal) with regional lymph node involvement on same side of the diaphragm: |
From Murphy SB: Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas: dissimilarities from lymphomas in adults, Semin Oncol 7:332, 1980.
An alternative strategy for B-cell lymphomas (Burkitt-like SNCC, DLBCL) has been developed by the Berlin-Frankfurt-Muenster (BFM) and French Society of Pediatric Oncology which defines three clinical groups.202,203 In addition to the extent of disease and surgical resection, the relative risk of relapse is used to define subgroups of patients with low, intermediate, and high-risk disease or Group A, B, and C, respectively, that will require different treatments. This system has been adopted successfully into ongoing trials of the COG.204–206
Treatment and Results
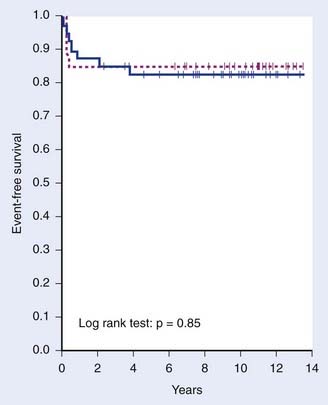
Recognition of the propensity for early widespread dissemination of pediatric NHL, regardless of stage or histology, formed the basis for development of systemic therapy regimens with combination chemotherapy and cranial nervous system prophylaxis that has dramatically improved the outcomes of children with NHL.196,198,207–211 The significant increase in EFS over two decades is demonstrated by the results of successive trials at St Jude Children’s Research Hospital (Fig. 54-4).199 Currently, cure rates of over 90% in patients with limited disease and 60% to 80% in those with extensive involvement are reported.200
Chemotherapy
In modern day regimens, chemotherapy is the primary therapeutic modality for all histologies and stages of childhood NHL. Surgery is indicated only for diagnostic purposes or in the case of an abdominal emergency such as intussusception, acute appendicitis, or intestinal obstruction.198,212 In patients who present with life-threatening situations such as airway obstruction, superior vena cava syndrome, or spinal cord compression, low-dose focal RT may be helpful in alleviating critical symptoms while maximal supportive care measures and specific chemotherapy are instituted.200
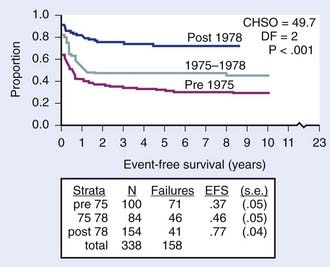
Early in the evolution of NHL therapy, it became evident that not only stage but also histologic subtype must be taken into consideration in selecting the most appropriate treatment regimen. This was most dramatically demonstrated in a randomized trial of the four-drug COMP (cyclophosphamide, vincristine [Oncovin], methotrexate, prednisone) regimen188 versus the 10-drug LSA2L2 regimen (cyclophosphamide, vincristine, prednisone, daunomycin, methotrexate, cytarabine, thioguanine, asparaginase, BCNU, hydroxyurea), a regimen designed for treatment of ALL.194,197 The results of this CCG study confirmed the efficacy of the COMP regimen for patients with advanced-stage disease of small non-cleaved cell type, whereas patients with advanced stage lymphoblastic NHL fared significantly better with the more aggressive LSA2L2 regimen. Neither therapy was statistically superior in treatment of advanced-stage large cell lymphoma. In this trial, children with localized NHL had excellent outcomes, regardless of histology or treatment group. These findings have been confirmed with long-term follow-up as shown in Fig. 54-5.213 Today, the choice of specific chemotherapy regimens and duration of treatment are determined based on stage, histology, and immunophenotype of the disease.
Successful treatment of advanced stage small non-cleaved cell (Burkitt’s or non-Burkitt’s) consists of repetitive, dose-intensive cycles including cyclophosphamide, high-dose methotrexate, and cytarabine given over 3 to 6 months (Table 54-8). This therapy is a sharp contrast to regimens used for lymphoblastic lymphomas, which, similar to high-risk ALL therapy, use multiple chemotherapy agents and last anywhere from 24 to 36 months (see Table 54-8). The optimal therapy for advanced-stage large cell lymphoma has been difficult to define because of the biologic heterogeneity of this histologic subtype of tumors.232,234 Generally speaking, regimens that include cyclophosphamide, vincristine, prednisone, and doxorubicin have been successful (see Table 54-8). In addition to intensive systemic chemotherapy, intrathecal therapy is with some exceptions an integral part of all treatment protocols for prevention of CNS disease regardless of histology.
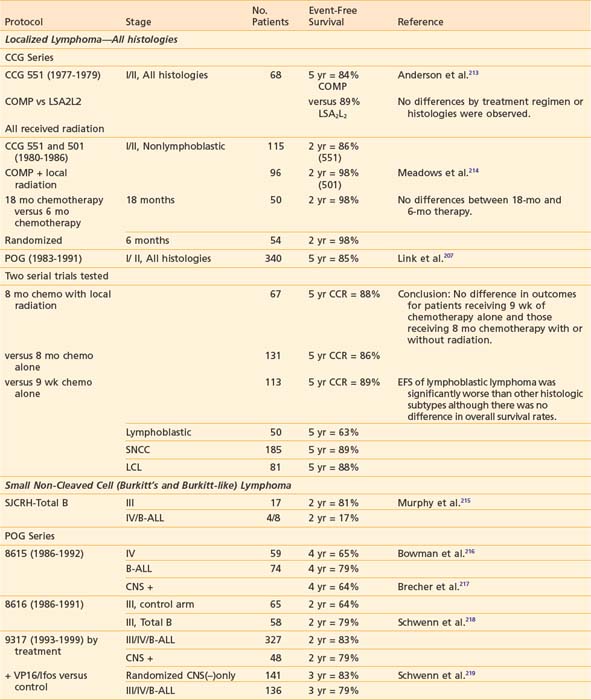
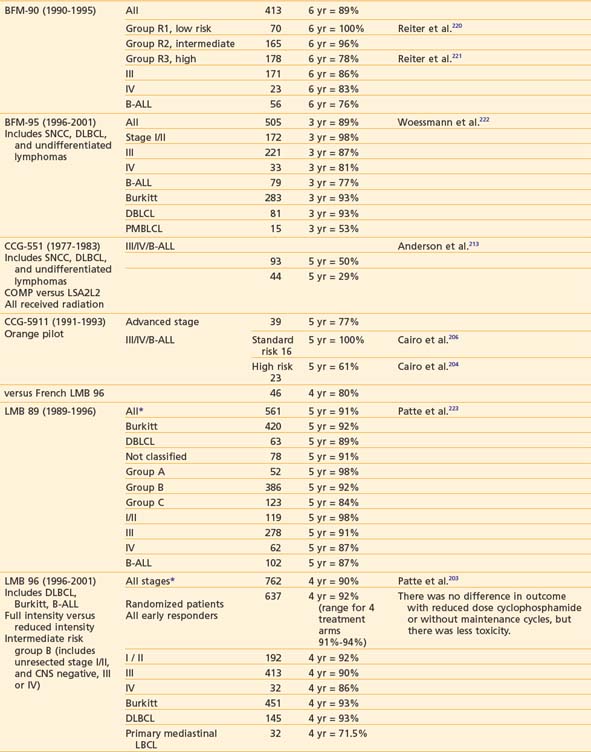
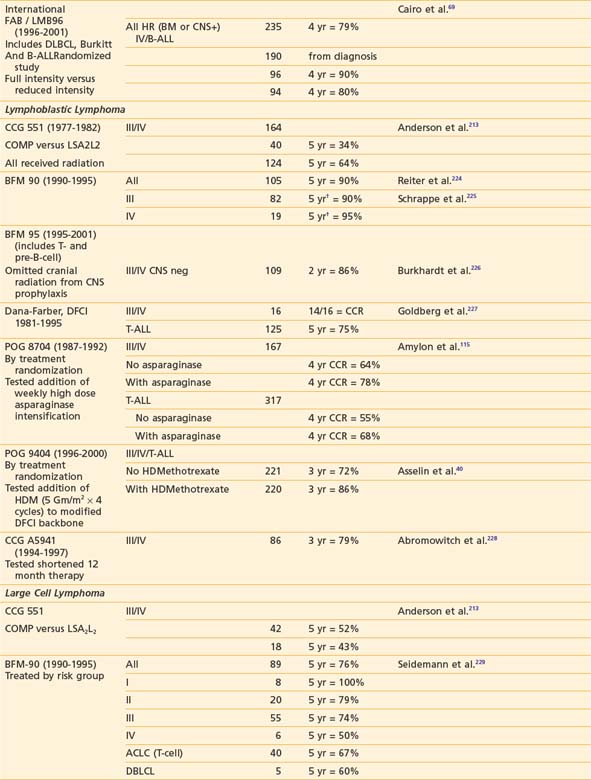
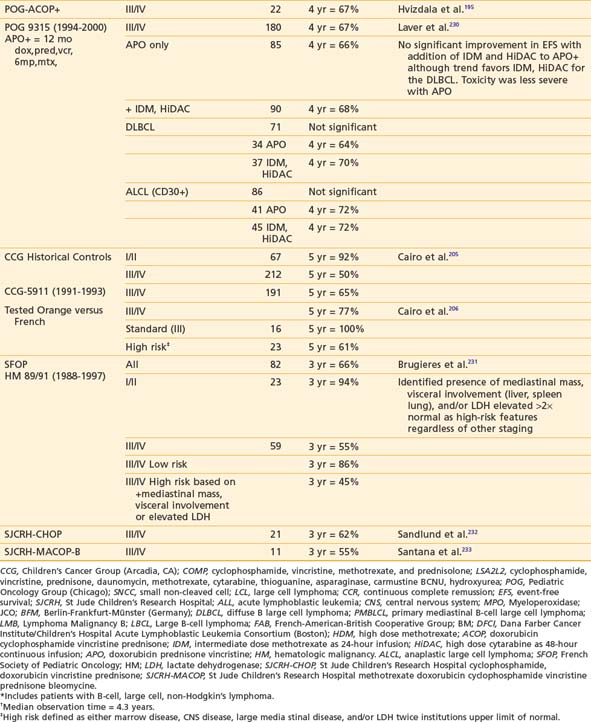
More recent data suggests that therapy based on immunophenotype may result in improved survival. The POG recently reported that among children with large cell lymphoma (LCL) treated with the same regimen, those with B-cell LCL had better outcomes than patients with non–B-cell LCL.207,235 European investigators have had excellent results in children with LCL when the treatment regimen is determined by immunophenotype (B versus T cell). Children with B-cell LCL have excellent outcomes when treated according to small non-cleaved cell regimens and those with T-cell LCL treated according to T-ALL protocols. The best therapy for the treatment of anaplastic CD30 LCL is not known; a variety of approaches such as specific anaplastic large cell lymphoma (ALCL) and B-cell specific therapy have been tried with good results.236–238 Trials to evaluate the efficacy of immunophenotype-directed therapy for pediatric large cell lymphoma are under way in Europe and the United States.
Outcomes in children diagnosed with stage I-II NHL are excellent with cure rates of 85% to 95%. The POG conducted two consecutive studies to determine if the intensity and duration of therapy could be safely reduced without decreasing the excellent prognosis. The first trial demonstrated that an 8-month chemotherapy regimen without involved field radiation of the primary sites was as effective as chemotherapy plus radiotherapy.239 In the second trial they concluded that 9 weeks of chemotherapy was adequate therapy for early-stage, nonlymphoblastic lymphoma; continuation therapy for 8 months was beneficial only in patients with lymphoblastic lymphoma.207 Although this group still had a higher relapse than patients with other histologic subtypes, salvage therapies were quite successful and OS remained good. Other groups have also observed higher relapse rates in children with early-stage lymphoblastic lymphoma compared to patients with nonlymphoblastic histology; thus, many investigators choose to treat these children with localized lymphoblastic lymphoma according to the same protocols used for ALL or advanced-stage lymphoblastic lymphoma with prolonged maintenance therapy for up to 2 years.240
The Role of Radiotherapy
The role of radiotherapy has been continuously redefined over the past two decades. With the development of effective multiagent chemotherapy regimens, local field irradiation for primary disease control is not indicated. CRT for CNS prophylaxis has also been eliminated from essentially all modern-day protocols, although it is still considered for some patients with lymphoblastic histology, or with overt CNS disease, as discussed later. Thus, radiotherapy currently is reserved primarily for the ancillary treatment of life-threatening mass effect. At very low total doses of 6 to 10 Gy, radiotherapy is often quite successful in producing rapid relief from the symptoms associated with the superior vena cava syndrome, acute respiratory distress, spinal cord compression, and orbital proptosis. Palliation of cranial nerve deficits may require higher doses of local field radiation in the range of 20 to 30 Gy.241
Primary-Site/Involved-Field Irradiation (Exclusive of Bone Primary Tumors)
Because of the excellent local control rates achieved with radiotherapy in pediatric NHL before the era of chemotherapy,211,242,243 radiotherapy to the primary site was incorporated into the design of early chemotherapy trials.194,197,244–246 Typically, doses ranging from 30 to 40 Gy were used. Because of concern for the significant acute as well as long-term toxicities observed with combined modality therapy, a reevaluation of the role of radiotherapy in pediatric NHL was undertaken. Original attempts at minimizing the potential adverse effects of RT were directed at reducing the local field dose to 20 Gy and the volume of tissue irradiated from a 5-cm to a 2- to 3-cm margin around the tumor site.214,247–249 Since results of the reduced therapy trials in patients with early-stage disease were similar to those achieved with more aggressive therapy, investigators proceeded to question the need for radiotherapy at all in the context of effective multiagent chemotherapy. The POG addressed this question of the independent contribution of radiotherapy to an effective multiagent chemotherapy regimen in children with localized NHL of all histologies exclusive of primary tumor of the bone. In a randomized trial comparing induction therapy of CHOP (cyclophosphamide, hydroxydaunomycin, vincristine [Oncovin], prednisone) alone with CHOP plus 27 Gy involved-field radiotherapy, no difference in results was observed, and a 4-year EFS rate of approximately 87% was projected for both arms.239 Although some investigators still advocate involved-field radiotherapy (IFRT) for patients with localized NHL, the vast majority have abandoned its use based on the results of this well-designed study.
To date, the use of local-field radiotherapy in advanced-stage disease has not been shown to be of benefit. In a randomized trial that studied the use of chemotherapy with or without involvement radiotherapy,209 the 2-year EFS rate in both treatment arms was only 38%, a value so low that the potential contribution of radiotherapy may have been masked by the ineffective systemic control. Nonetheless, with no concrete evidence to justify its use, because of concern for the potential of radiotherapy to compromise the effective delivery of chemotherapy in a disease that is inherently systemic, IFRT has been eliminated from the design of advanced-stage pediatric NHL trials. Recent results of the chemotherapy-alone trials tend to support this practice.215,250–252
Primary Non-Hodgkin’s Lymphoma of Bone
The high rate of systemic spread in patients with clinically apparent early-stage primary NHL of bone (PBL) has confirmed that this extranodal presentation, similar to other presentations of NHL, is a disseminated disease at diagnosis and as such requires systemic therapy.253,254 Local-field radiotherapy to the involved bone was included in the early pediatric NHL chemotherapy trials. However, because of the increased incidence of second primary bone tumors observed with the combined modality therapy,255 as well as the potential for increased morbidity in growing children, radiation is no longer a component of therapy in the setting of primary bone disease in children. This is supported by recent POG and CCG trials, which reported the experience with treatment of PBL in children for the past 15 to 20 years.256,257 Both groups observed excellent outcomes with chemotherapy regimens and therefore conclude that RT is unnecessary.
Central Nervous System Preclinical and Overt Disease
Early in the evolution of pediatric NHL therapy, CNS relapse was recognized to be an obstacle to cure in about 30% to 35% of children.196 Recent data provide insight into the frequency CNS involvement in the various histologic subtypes of NHL. In a population of 2381 children with NHL, 141 (5.9%) had CNS involvement at diagnosis. The percentage of CNS-positive patients was 8.8% for Burkitt’s, 5.4% for precursor B-lymphoblastic lymphoma, 3.3% for anaplastic large-cell lymphoma, 3.2% for T-cell-lymphoblastic lymphoma, 2.5% for diffuse large B-cell lymphoma, and 0% for primary mediastinal large B-cell NHL.258 Historically, as in leukemia, CNS prophylactic therapy was incorporated into the treatment of NHL of childhood and achieved excellent results; also, as in leukemia, CNS radiotherapy played an integral role in the early trials. The combination of CRT and intrathecal methotrexate resulted in a less than 10% incidence of CNS relapse.196 In the only randomized trial of CNS presymptomatic therapy in pediatric NHL, conducted at the St. Jude Children’s Research Hospital (SJCRH) in patients with stage II to IV NHL, 6% (1/18) of children randomly assigned to receive CRT and intrathecal methotrexate developed an isolated CNS relapse, compared with 25% (4/16) who did not receive any specific form of CNS prophylactic therapy.209 In the previously cited BFM study of the histologic subtypes which were associated with CNS involvement at diagnosis, the probability of event-free survival (EFS) at 5 years was 85% for the entire patient population with or without CNS disease, but was 64% for the CNS-positive patients.258 The presence of CNS involvement did not impact outcome for T-cell patients, but had significant negative impact on patients with Burkitt’s. Concern for the increased risk of neurotoxicity associated with the use of CRT in children prompted investigators to test the efficacy of using intrathecal chemotherapy alone for CNS prophylaxis. Results of modern chemotherapeutic trials with intrathecal chemoprophylaxis have been successful in preventing isolated CNS relapse in approximately 95% of children with NHL.213,259 Radiotherapy is thus no longer indicated for CNS prophylaxis with the possible exception of lymphoblastic (T-cell) disease, where it retains a role in some centers, and where it may also be appropriate in patients who have undergone leukemic transformation.224,260,261 Although high-dose systemic and intrathecal chemotherapy (Ara-C and methotrexate) are effectively used to treat overt CNS NHL, RT remains a consideration and is a component of treatment in some centers.202
Testicular Lymphoma
Testicular involvement at diagnosis is uncommon (estimated at 5% to 10% of children with disseminated small non-cleaved cell NHL and often associated with marrow, CNS, or other visceral involvement at diagnosis). Considered a potential sanctuary site similar to the CNS in ALLs, involvement of the testis was thought to confer a poor prognosis and thus initial treatments involved aggressive local disease control measures such as orchiectomy and scrotal irradiation. Published reports provide conflicting data on the benefit of these approaches.262,263 Intensive combination chemotherapy programs have improved the outcomes of all types of NHL even in patients with advanced stage III and IV disease (see Table 54-8), suggesting that testicular disease is curable with chemotherapy alone.264
Recent data suggest that orchiectomy and scrotal irradiation can be avoided except in small numbers of patients when testicular diagnosis proves refractory or is recurrent, and thus preserving gonadal function for the majority of patients. Some protocols, however, continue to employ scrotal irradiation for testicular involvement despite recent data that question its value.264
Future Directions
The successful treatment of pediatric NHL has been achieved by the refinement of multiagent chemotherapy protocols. Future trials will be aimed at further refinement of therapy regimens with identification of better prognostic factors265 and active agents, with the goal that patients will not be over- or undertreated. In patients with localized disease, the excellent survival rates (>90%) support the strategy of reducing therapy in our effort to minimize adverse effects without compromising efficacy.207 In patients with advanced-stage NHL, future trials must aim at better identification of patients destined to do poorly and more effective therapy regimens. The increasing understanding of molecular genetic pathogenesis of lymphomas has made it possible to develop sensitive techniques of measurement of minimal residual disease during clinical remission as a way of identifying patients at high risk of relapse as well as highly specific therapy targeted to the tumor cells.266 Recent successful trials with the use of rituximab, a monoclonal antibody directed against CD20, a B-cell antigen, demonstrate the importance of continued study of novel approaches.267,268 Future clinical trials will likely include the use of (1) immunotherapy with monoclonal antibodies269–271; (2) stem cell transplantation272; (3) antisense oligonucleotides273; and (4) cytotoxic T-lymphocytes274 or tumor vaccines.275,276
Hodgkin’s Lymphoma
Epidemiology and Biology
Overall, HL is slightly more common than NHL in children, with an annual incidence rate of 11.2/1 million children newborn to 19 years of age. For unclear reasons, the incidence rates of HL have declined slightly between 1975 and 2005.1 During this same period, the 5-year survival rate for HL has increased to 94.7%.
At least two different presentations of HL in the early age groups are apparent: a childhood form developing in patients 14 years of age or younger, and a young adult form affecting patients 15 to 34 years of age.277,278 HL is rarely diagnosed in children younger than 5 years of age in developing countries.279 In one North American clinical trial for children and adolescents with HL, 3% of patients were between newborn and 4 years of age; 12% of patients were between 5 and 9 years of age; 44% of patients were between 10 and 14 years of age, and 41% were older than 15 years of age.280 In children and younger adolescents, the disease occurs more frequently in males; in older adolescents the incidence among males and females is roughly equal. Also, most adolescent patients diagnosed with HL are White. The childhood form of HL tends to increase with increasing family size and decreasing socioeconomic status. In contrast, the young adult form of HL is associated with a higher socioeconomic status in industrialized countries. The risk for young adult HL decreases significantly with increased number of sibling size and birth order.281 The biology and natural history of HL in children is similar to that in adults, and thus will not be discussed here at length.
The large proportion of patients with HL who have high EBV antibody titers suggests that enhanced activation of EBV may precede the development of HL. Approximately 40% to 50% of HLs demonstrate EBV viral RNA or protein when assessed by in situ hybridization or immunohistochemistry with the strongest association observed in cases of mixed cellularity subtype.282 HL in the immunocompromised and in developing countries shows the strongest links to EBV, whereas EBV is most often associated with HL in young children and the elderly in industrialized countries. The incidence of EBV-associated HL also varies by ethnic background, as evidenced by its presence in 93% of Asian, 86% of Hispanic, 46% of White, and 17% of African American children with HL in one series.283 The precise role of EBV in the pathogenesis and biology of HL has not been established. EBV expression does appear to influence outcome in most series, but may be beneficial in some groups, particularly young adults.284–286
Detection and Diagnosis
Clinical presentations fall in rather predictable patterns. Most children are diagnosed on the basis of supradiaphragmatic lymph nodes, with painless cervical adenopathy in 80%. Mediastinal involvement occurs in 76% of adolescents, but in only 33% of 1- to 10-year-olds. Mediastinal disease may produce symptoms such as dyspnea, cough, or superior vena cava syndrome. Axillary adenopathy is less common.287
Clinical Staging
Imaging of the Neck and Chest
Because contemporary risk-adapted treatment protocols use response as a parameter to escalate or truncate therapy, CT evaluation of the Waldeyer lymphoid tissue and the cervical soft tissues is recommended to permit more accurate assessment of nodal response. An upright chest radiograph provides the routinely used assessment of bulky mediastinal lymphadenopathy, defined as measuring 33% or more of the maximum intrathoracic cavity at the dome of the diaphragm. Functional nuclear imaging with positron emission tomography represents an important diagnostic and monitoring modality in patients with HL that has been proven to have prognostic significance.288,289
Imaging of the Abdomen and Pelvis
Since essentially all patients are treated with chemotherapy, surgical staging with laparotomy has been abandoned. In situations where equivocal imaging abnormalities exist and require resolution for therapy planning, isolated nodal sampling may be performed. CT is also most often used to evaluate sites of infradiaphragmatic disease. Oral and intravenous contrast administration is essential to accurately diagnose visceral involvement and delineate abdominal/pelvic nodes from other infradiaphragmatic structures. Lymphomatous involvement is estimated by the size of abdominal and pelvic nodes: abdominal nodes smaller than 1 to 1.5 cm and pelvic nodes smaller than 2 to 25 cm are usually considered normal. PET imaging permits identification of disease in smaller nodes. A poorly contrasted bowel and lack of retroperitoneal fat in some patients may limit the sensitivity of CT in detecting abdominal adenopathy.290 Distinction of reactive lymph node hyperplasia from HL may also be problematic, and is more common in younger children than in adolescents.291 In these cases, MRI may provides better evaluation of fat-encased retroperitoneal lymph nodes.292
Splenic involvement occurs in 30% to 40% of patients with HL, whereas hepatic involvement is rare in the pediatric age group.287 Abnormalities on CT, MRI, or PET suggest lymphomatous involvement of the liver and spleen. Because tumor deposits in these organs may be less than one centimeter in diameter, disease status cannot be dependably assessed by organ size alone.
Classification and Staging
Histopathology and staging is described in the section on adults. Of interest in pediatric HL is that the relative distribution of the subtypes in younger children differs from that in adolescents and adults, as shown in Table 54-9, which combines data from CCG and European GPOH-95 trials.280,293,294 The distribution of stages observed in children is also somewhat different than that observed in adults, again described in Table 54-9.
Table 54-9 Clinical Characteristics of Patients Participating in Pediatric Hodgkin’s Lymphoma Trials
| No. Children, %* | No. Adults, %† | |
|---|---|---|
| No.patients | 1659 | 1912 |
| Age, yr | ||
| <10 | 269 (16.2) | |
| ≥10 | 1385 (83.5) | |
| >17 | 1912 (100) | |
| Gender | ||
| Male | 909 (54.8) | 1147 (60.0) |
| Female | 750 (45.2) | 765 (40.0) |
| Histology | ||
| Lymphocyte predominant | 161 (9.7) | 96 (5.0) |
| Mixed cellularity | 251 (15.1) | 325 (17.0) |
| Nodular sclerosing | 1210 (72.9) | 1377 (72.0) |
| Not classified and lymphocyte depleted | 37 (2.2) | 115 (6.0) |
| Stage | ||
| I | 194 (11.7) | 210 (11.0) |
| II | 922 (55.6) | 899 (47.0) |
| III | 294 (17.7) | 593 (31.0) |
| IV | 249 (15.0) | 210 (11.0) |
| B Symptoms | ||
| Present | 884 (53.3) | 612 (32.0) |
| Absent | 775 (46.7) | 1300 (68.0) |
* Data taken from Nachman et al280 and Rühl et al.294
Evolution of Therapy
When irradiation techniques and doses suitable for controlling disease in adults were used in the pediatric setting, substantial morbidities were produced.295,296 It is within this context that treatment strategies specific to HL in the pediatric age group are discussed.297 OS rates for children with HL range from 85% to 95% depending upon stage and risk features at presentation (Tables 54-10 and 54-11). The desire to cure children with minimal treatment-related toxicity provided the stimulus to limit staging procedures and reduce the intensity and types of chemotherapeutic regimens as well as the dose and volume of radiotherapy used. Historically, treatment of adults with early-stage HL often was with radiotherapy alone, reserving chemotherapy for salvage of recurrent disease.315–317 This approach was initially extended to the pediatric population.296,298,318,319 Radiation treatment regimens used primarily prescribed standard doses of 35 to 44 Gy to extended fields, regardless of the patient’s age. Impairment of soft tissue and skeletal growth including intraclavicular narrowing, shortened sitting height, decreased mandibular growth, and decreased muscle development became readily apparent as young patients matured after curative therapy.295,296 Concerns about growth impairment led to the development of clinical trials designed to specifically address the needs of children with HL. These protocols evaluated lower radiation doses (15 to 25.5 Gy) to reduced (involved) treatment fields combined with multiagent chemotherapy.* Numerous studies subsequently established the efficacy of this treatment approach, after which the primary use of standard-dose radiation was reserved for skeletally mature patients with localized disease.
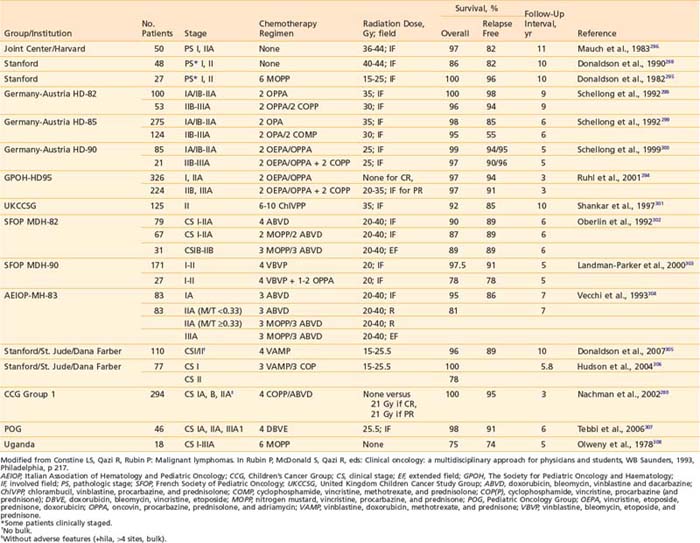
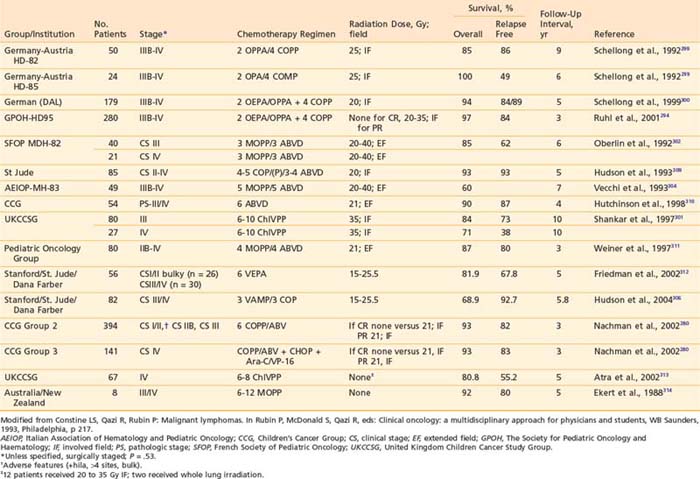
With more prolonged follow-up, reports implicating radiation as a causative factor for excess cardiovascular disease and subsequent malignancy risk in long-term pediatric survivors of HL motivated further therapeutic refinements.169,325–341 The use of standard-dose, extended-field radiation in mature adolescents with localized HL has been abandoned at most centers (as it has been for adults) because this treatment approach predisposes to a greater risk of cardiovascular disease and secondary solid tumor carcinogenesis. Contemporary risk-adapted treatment protocols have focused on further limiting radiation exposure of uninvolved tissues, especially the breast, and identifying patients for whom the addition of radiation optimizes DFS.
One means of refining risk-adapted therapy is to modify treatment according to the response to the initial few cycles of chemotherapy, so-called response-adapted therapy. This approach is based on the premise that rapid early response (RER) to chemotherapy is a predictor of eventual long-term disease control, possibly because these patients harbor biologically favorable disease. The goal of response adapted therapy is to titrate therapy by employing the prognostic value of early treatment response to minimize treatment intensity among those with RER and thereby reduce toxicity, while intensifying treatment for those with slow early response (SER), thereby improving disease control. Assessment of response in recent COG trials has been based on disease reduction as determined by CT imaging.280,307,311,342 In both early and advanced stage disease, either the number of cycles of chemotherapy, the choice or intensity of agents, or the dose of RT (including its omission entirely) can be modified based on response, and trials are in progress further testing these treatment approaches (as described below). Recent data strongly support the early response to PET as prognostic and therefore potentially useful in modifying subsequent treatment.343 However, there also are data demonstrating that PET scanning can produce false negative results, and thus additional testing of the utility of PET in modifying treatment intensity is necessary.344
In summary, devising the optimal therapeutic approach for children with HL is complicated by the increased risk for adverse treatment-related side effects. When used in children, radiotherapy doses and fields that are safely used in adults can often lead to significant musculoskeletal growth retardation and excess risk for the development of life-threatening cardiovascular dysfunction and subsequent malignancies.295,296,325–341,345,346 Because of differences in the developmental status of children of varying ages, there is, unfortunately, no single method of ideal treatment for all children. Therapy in pediatric HL continues to vary widely among investigators with regard to the modality or modalities used, as well as to the specifics of chemotherapeutic regimens and radiotherapeutic doses and techniques. Currently, most prepubertal children with HL are managed with combined multiagent chemotherapy alone or combined with low-dose, IFRT radiotherapy.
Combination Chemotherapy
The original prototypic chemotherapy regimen for HL is MOPP (mechlorethamine, vincristine [Oncovin], procarbazine, and prednisone). The well-recognized major toxicities observed with its use include an increased risk of acute leukemia,169,347 azoospermia in up to 90% of males treated at any age,348–352 and a risk of sterility in females that increases with age.353–357 The development of the ABVD (doxorubicin [Adriamycin], bleomycin, vinblastine, and dacarbazine) regimen in the 1970s provided an effective alternative to MOPP chemotherapy that was not associated with an increased risk of secondary AML or infertility.352,358,359 Compared to MOPP, ABVD produced superior treatment outcomes and lacked leukemogenic and permanent gonadal toxicity.360 For these reasons, ABVD is a standard front-line therapy for adults with newly diagnosed HL. However, reservations about potential cardiopulmonary toxicity, sequelae related to the doxorubicin and bleomycin in the combination, have influenced its use in children with HL.361,362 The severity of these complications may be exacerbated by the addition of mediastinal or mantle irradiation, in which a portion of the lungs and heart is included in the treatment field.327,329,330,346
Contemporary pediatric trials typically use ABVD or similar hybrid combinations, in risk-adapted treatment regimens prescribing fewer chemotherapy cycles for patients with favorable localized disease presentations.280,305,307,362–364 For children with advanced or unfavorable disease, ABVD or a derivative including etoposide has often been alternated or combined with alkylating agent regimens such as COPP (cyclophosphamide, vincristine [Oncovin], procarbazine, and prednisone), in an effort to reduce exposure of agents with dose-related toxicity and improve disease control.280,312,363–366
Combined Modality versus Chemotherapy Alone
Children and adolescents with HL may be cured using combination chemotherapy alone or a combined modality approach using chemotherapy and RT. Contemporary therapy produces long-term remission in 70% to 90% of patients with advanced/unfavorable HL and 85% to 100% of patients with localized favorable disease. The chemotherapy regimens still employed largely include MOPP, ABVD, or therapies derived from one of the combinations. Chemotherapy-alone trials typically prescribe more cycles of chemotherapy, whereas combined modality protocols administer low-dose IFRT in lieu of several chemotherapy cycles.280,308,313,314,367–376 The limited number of randomized pediatric trials prospectively evaluating combined modality therapy versus chemotherapy alone has not definitely established the superiority of one treatment approach over the other, although the results of some trials suggest that the addition of RT may improve outcome in unfavorable and advanced-stage patients.280,311
Consequently, debate continues among pediatric investigators regarding optimal therapy for newly diagnosed children. Treatment with chemotherapy alone eliminates the risk of radiation-associated adverse sequelae including musculoskeletal growth impairment, cardiovascular dysfunction, and solid tumor carcinogenesis. However, these sequelae are less substantial than in earlier eras because of the use of low-dose IFRT. Moreover, chemotherapy-alone protocols rely on higher cumulative doses of agents with established dose-related toxicity, especially alkylators, which may also predispose to acute and late treatment effects. The identification of prognostic features of patients who may benefit from omission or addition of RT continues to be a primary objective of contemporary pediatric trials in HL. Ongoing trials are also testing whether treatment modification (reduction or escalation) based on early response to chemotherapy using PET imaging will result in improve outcomes.364,366
For example, in the recent German GPOG-HD 95 trial, relapse-free survival was superior for patients treated with RT after partial response (93%) than for those without RT after complete response (CR) (89%). The difference was significant for patients treated for advanced-stage but not early-stage disease (Fig. 54-6).294 Similarly, in a randomized CCG trial, low-dose IFRT after an initial complete response to risk-adapted chemotherapy improved EFS but not survival.280
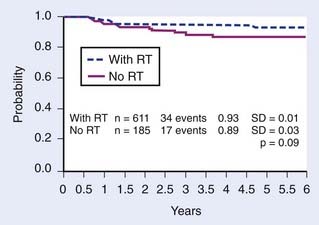
Risk-Adapted Therapy
Favorable Localized Disease
More recent pediatric trials have evaluated the efficacy of risk-adapted therapy using fewer cycles of multiagent chemotherapy and lower radiation doses and treatment volumes (15 to 25 Gy IFRT) in clinically staged patients with favorable disease presentations. A favorable presentation is defined as a limited number of sites (usually less than four) of localized “nonbulky” nodal involvement in an asymptomatic patient (without constitutional systems such as fever, sweats, or weight loss). Mediastinal masses are considered bulky if they measure 33% or more of the maximum intrathoracic cavity; the definition of peripheral lymph node bulk varies from 4 to 6 cm, depending on the study center. Most protocols prescribe two to four cycles of novel chemotherapy combinations, which omit or reduce the amount of the more toxic agents in the original MOPP and ABVD regimens.294,303,305,307,366 For example, the German-Austrian Multicenter Trial DAL-HD-90 treated favorable risk patients with two cycles of OPPA (vincristine, prednisone, procarbazine, and doxorubicin) for girls or two cycles of OEPA (procarbazine replaced by etoposide) for boys followed by 25Gy IFRT. An additional 5- to 10-Gy boost was given to sites with postchemotherapy residual mass >50 mL and/or >25% of the initial tumor volume. Five-year EFS and OS were 94% and 99.6%, respectively.300 In the subsequent German HD-95 study, RT was omitted among patients achieving a CR to chemotherapy (27% of treated patients), with the remaining patients receiving 20 to 35 Gy IFRT depending on the chemotherapy response. With a median follow-up of 38 months, the EFS was 94% among favorable risk patients.294 One potential concern regarding this protocol is the use of higher dose (≥30 Gy) IFRT in 25% of patients, primarily those with unfavorable features. This dose would be expected to produce significant late effects among some survivors, and since protocols using low-dose RT have produced good results, it is possible that this dose could be reduced without loss of efficacy.
The CCG 5942 trial protocol treated children with favorable risk disease with four to six cycles of COPP/ABV chemotherapy, with complete responders randomized to 21 Gy IFRT or no further therapy. The 3-year EFS and OS were 100% among patients receiving IFRT.280 These results and those of other studies indicate that for patients with favorable risk disease, two to four cycles chemotherapy followed by low-dose (20 to 25 Gy) IFRT is adequate to produce excellent EFS and OS. For these patients, a major focus of clinical investigation is how best to reduce treatment-related toxicity while maintaining high rates of disease control. In summary, the excellent treatment results of these trials affirm the concept that patients with localized disease could be cured with minimal therapy.
Nodular lymphocyte predominant HL (NLPHL) is recognized as a distinct clinical-pathological entity, generally with a favorable outcome, but also associated with a higher risk of late relapse and subsequent development of NHL. Sandoval et al. reported the outcome of 51 children with NLPHL, treated with a RT alone, chemotherapy alone, or combined modality therapy. Forty-eight patients (94%) were alive and disease-free at a median follow-up of 8 years (range 0.4 to 32.6).377 Three patients died, two from second cancers. Among 26 patients with NLPHL enrolled on POG clinical trials, all achieved remission after chemotherapy, RT, or combined modality therapy, with a 5-year EFS of 86.5%.378 Standard treatment for children with NLPHL is chemotherapy and low-dose IFRT, and subset analyses of NLPHL patients treated on trials using chemotherapy alone may reveal this to be adequate treatment.
Because observational studies indicate that complete surgical resection of NLPHL is curative in a proportion of children presenting with this histological subtype,379,380 ongoing trials are also prospectively evaluating this strategy as a means to reduce long-term treatment complications. For example, the COG is currently accruing patients to a single arm study in which patients with early stage single node NLPHL are observed after confirmed complete surgical resection. Patients without complete resection, and those relapsing on observation, are treated with chemotherapy alone or with low-dose IFRT, based on initial response to chemotherapy. This study should advance our understanding of the course of NLPHL postresection and overall outcome of minimal therapy.
Advanced and Unfavorable Disease
Clinical presentations associated with symptomatic disease, bulky lymphadenopathy, extranodal extension, and advanced stage (IIIB-IV) are designated unfavorable. Two treatment approaches have been used in patients with unfavorable presentations of HL. The chemotherapy prescribed in both approaches is derived from the MOPP and ABVD combinations; etoposide is also incorporated in an effort to reduce cumulative doses of alkylating agents and enhance antitumor activity. Historically, treatment approaches use combination chemotherapy administered on a twice-monthly schedule for 6 to 8 months.294,306,309,311,312,381 The second strategy uses abbreviated, dose-dense, multiagent chemotherapy delivered in a compacted schedule over 3 to 5 months.280,363,366 Myelosuppressive and nonmyelosuppressive chemotherapy is alternated in a weekly schedule with the intent to reduce the development of resistant disease and induce a rapid early response. Low-dose IFRT may be used to consolidate remission after completion of chemotherapy. Both treatment approaches produce comparable early treatment outcomes. Long-term follow-up of the compacted dose-dense approach is required to establish its superiority over conventional treatment approaches in maintaining long-term disease control.
It is notable that the use of RT in the treatment of advanced-stage pediatric HL differs substantially from the usual practice for adult HL. In both the GOPH HD-95 and CCG 5942 studies, the benefit of IFRT in reducing relapse rates was most pronounced among high-risk patients. In the HD-95 study, intermediate- and high-risk patients who received IFRT had significantly better EFS than those who did not, in spite of the latter group having achieved a better response to chemotherapy.294 In CCG 5942, IFRT was associated with an 11%, 5%, and 12% improvement in 3-year EFS among low- intermediate- and high-risk patients, respectively.280 The potential to salvage low-risk patients is used as an argument to withhold RT from this group despite the inferior EFS in its absence, whereas the ability to salvage patients with higher risk disease is less predictable. Consequently, IFRT remains a component of treatment of intermediate- and high-risk disease in most multi-institutional pediatric protocols. This is in contrast to the standard treatment for adult HL, for which trials have not found a benefit to IFRT among patients with advanced-stage disease achieving a CR after chemotherapy. The criteria for CR should be considered when interpreting the results of studies in adults, as well as the outcome for patients with PR who received IFRT. For example in the study by Aleman et al.,382 patients in PR at completion of MOPP/ABV who received IFRT had an outcome similar to those in CR who did not receive RT, suggesting the efficacy of RT for patients who do not achieve CR.
Results of Therapy
The actuarial 10-year survival rate for children with early-stage disease is reported to range from 85% to 97%, and for those with advanced-stage disease, from 70% to 90% (see Tables 54-10 and 54-11). Regardless of stage, the size of the mediastinal disease continues to influence the choice of therapy in most clinical trials. In studies in which patients are not stratified according to the mediastinal bulk, patients with residual mediastinal disease following the preselected number of chemotherapy cycles may receive higher radiation doses, obviating the ability to conclude that the bulk of disease did not influence treatment.294 The relapse-free and OS rates for children with stage III disease without B symptoms are generally excellent. Improving outcome for patients with advanced symptomatic disease, especially those with extranodal disease, remains a focus of ongoing trials.313,383
Refractory or Relapsed Disease
Historically, most treatment failures in patients with HL occur within the first 3 years. The prognosis following relapse depends on the primary therapy and timing of relapse. Salvage rates are very good if relapse develops after primary therapy with RT alone. As many as 50% to 80% of these patients can be salvaged with chemotherapy or combined modality therapy.384–389 Conversely, patients with early-stage disease who recur in areas of initial disease after chemotherapy alone may also have an excellent salvage rate without myeloablative approaches.280 Second remissions may also be achieved in up to 50% of patients who relapse after maintaining their first remission for 1 year or longer. Patients who develop refractory disease during or within 1 year of primary therapy have a very poor prognosis, particularly if they recur in irradiated sites following combined modality therapy. Intensive cytoreductive chemotherapy followed by consolidation with myeloablative therapy and hematopoietic stem cell transplantation produces the most durable remissions in these very high-risk patients.386,390,391 Overall, this approach may salvage 30% to 50% of patients with refractory disease, but acute and late treatment toxicity predisposes these patients to early mortality. IFRT to sites of recurrent disease is generally appropriate if these sites have not been previously irradiated. IFRT can be administered before the high-dose chemotherapy program in order to place patients in a minimal disease state. More often, RT is administered after the high-dose chemotherapy program in order to decrease the potential for disease progression elsewhere during the time RT is being administered, and RT-related peritransplant morbidity such as esophagitis, pneumonitis, cardiomyopathy, and veno-occlusive disease.392,393 It is noteworthy that patients treated with hematopoietic stem cell transplant may experience relapse as late as 5 years after the procedure and should be monitored for relapse as well as late treatment sequelae.
Techniques of Radiotherapy
Involved Node Radiation Therapy
Recognition that following chemotherapy, RT volumes can be restricted to lymph node regions originally involved with disease (i.e., IFRT) without compromising relapse rates has been a major advance in the management of HL. However, many contemporary clinical trials and guidelines describing IFRT recommend treating nodal structures adjacent to initially enlarged nodes, most notably inclusion of uninvolved hilar lymph nodes in cases with anterior mediastinal involvement. This raises the possibility that the effectiveness of RT could be maintained and the radiation dose to normal tissues reduced by further limiting the volume of treatment to the specific lymph nodes initially involved with disease, rather than the classically-defined lymph node regions. The premise of this involved node radiation therapy (INRT) approach is that it is not necessary to irradiate all normal-sized lymph nodes within the same region as the enlarged lymph nodes, since chemotherapy can effectively eliminate microscopic disease that may exist within these nodes. This concept is supported by the observation that among patients treated with chemotherapy alone, initially involved lymph nodes are the most common site of recurrence.394,395 In contrast to conventional IFRT, uninvolved hila are not included in the clinical target volume (CTV) for mediastinal presentations, and the length of the treated volume is not routinely extended beyond 1 cm for the planning target volume (Fig. 54-7). This approach has the potential to significantly reduce the irradiated volume of normal tissues compared to IFRT, and is to be employed in upcoming EORTC-GELA studies of adult HL.396 If successful, this could produce significant reductions in RT-related late effects.
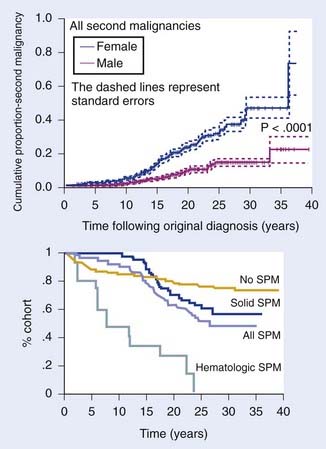
Complications of Therapy
Long-Term Effects
Long-term adverse sequelae specific to irradiation to the pediatric age group include impairment of muscle, bone, and dental development295,296,397–400 and injury to the lungs,295,401–403 heart,327,329,330,334,404 thyroid gland,405,406 and reproductive organs.349,351–357,407–409 Survivors who underwent surgical staging with splenectomy or splenic irradiation also have a life-long risk of overwhelming sepsis from encapsulated bacterial organisms.410 Cardiovascular dysfunction and secondary carcinogenesis may result in early mortality in long-term survivors.329,345,404
Acute and chronic pneumonitis, pulmonary fibrosis, and spontaneous pneumothorax are pulmonary sequelae observed in survivors of HL. Predisposing therapies include thoracic radiation and ABVD chemotherapy.295,309,401–403 The bleomycin in ABVD produces pulmonary fibrosis and may be responsible for veno-occlusive disease in rare cases. ABVD may also predispose to pneumonitis by inducing a “radiation recall” phenomenon related to the doxorubicin. Most reports describe pulmonary dysfunction in survivors treated before the era of risk-adapted therapy. Asymptomatic pulmonary dysfunction that improves over time has been commonly observed after more contemporary combined modality treatment approaches.309,402 Symptomatic pulmonary toxicity is uncommon and more often develops in patients treated with pulmonary irradiation and bleomycin.295,401,403
Chemotherapeutic agents and RT used in the treatment of pediatric HL predispose to cardiovascular disease. Cardiac dysfunction is most commonly observed after anthracycline therapy, particularly doxorubicin.404,411–413 In adults, the risk of congestive heart failure related to anthracycline-induced cardiomyopathy increases when cumulative doses exceed 550 mg/m2. Young children may be at risk for anthracycline injury at lower cumulative doses because of the drug’s adverse effect on cardiac myocyte growth. This is suggested by studies of childhood leukemia survivors who demonstrate a high frequency of abnormalities of afterload and contractility.414 Mediastinal irradiation and other chemotherapeutic agents (e.g., cyclophosphamide) may predispose pediatric Hodgkin’s survivors who received lower cumulative anthracycline doses in the range of 350 to 400 mg/m2.327,329,330,404,411–413 Acute anthracycline cardiotoxicity is rare with contemporary therapy that limits doses to 300 mg/m2 or lower. However, the risk of delayed subclinical cardiac dysfunction, which may become clinically significant with cardiac senescence, is not yet established.
Radiation also injures the pericardium and myocardium in a dose-related fashion.327,330,411–413 High-dose (35 to 44 Gy) mantle irradiation produces cardiac injury in 13% of children and adults, most commonly manifest as constrictive pericarditis or accelerated coronary atherogenesis. Stanford investigators reported that the actuarial risk of developing cardiac disease requiring pericardectomy was 4% at 17 years in a series of long-term survivors of childhood HL.330 Most patients with severe pericardial complications were irradiated before the introduction of subcarinal shielding and received little or no cardiac blocking. Premature coronary artery disease and acute myocardial infarction may also contribute to excess mortality risk in patients treated with mediastinal radiation in doses more than 30 Gy before 20 years of age.327,330,404,412,413 With the introduction of radiation techniques that reduce the radiation dose to the heart, the rates of radiation-associated cardiac injury have declined dramatically. Likewise, myocardial infarctions have not been reported in children treated with combined modality regimens using lower radiation doses and volumes and protective cardiac shielding. Again, the impact of subclinical cardiac injury after low-dose radiation may not become apparent until larger cohorts of survivors treated with these regimens begin to age.
Thyroid sequelae are common after RT for pediatric HL. Hypothyroidism, hyperthyroidism, thyroid nodules, and thyroid cancer have been observed in long-term survivors.403,405,406 Of these, hypothyroidism, particularly compensated hypothyroidism, defined as thyroid stimulating hormone (TSH) elevation in the presence of a normal thyroxine (T4) level, is the most frequently described thyroid abnormality. Risk factors for hypothyroidism include younger age at treatment and higher cumulative radiation dose. As many as 78% of patients treated with radiation doses exceeding 26 Gy demonstrate thyroid dysfunction, as indicated by elevated TSH levels.403,405,406 Because persistent TSH elevation most often signals impending gland failure and may be a carcinogenic stimulus to the gland, thyroid hormone replacement therapy should be instituted. Gland suppression with thyroid hormone replacement is also recommended for patients who develop thyroid nodules after radiation. Replacement therapy should suppress gland function, as indicated by a TSH of 0.5 to 1.5 µIU/mL. Periodic withdrawal of hormonal therapy in asymptomatic patients will permit assessment of gland recovery, which has been observed in some patients serially monitored after RT. Persistent and enlarging nodules should be monitored with ultrasound and periodic biopsies for malignant degeneration, as thyroid carcinoma is a common second malignancy following HL.406,415
The risk of gonadal injury following HL is related to the type and intensity of treatment. In males, high-dose pelvic radiation may produce a transient oligospermia or azoospermia, but spermatogenesis typically recovers.409,416 Six to eight cycles of MOPP (or similar hybrid chemotherapy containing alkylators) produces irreversible sterility in 80% to 90% of males; fertility may be preserved if treatment is limited to three or fewer cycles.417 Recovery of gonadal function 10 to 15 years following MOPP chemotherapy has been rarely reported. Anthracycline-based regimens like ABVD (or similar hybrid) are usually associated with full recovery of spermatogenesis following a temporary period of azoospermia.348,352 In contrast, testicular Leydig cell function is more resistant to the effects of antineoplastic treatment, so growth and puberty are adversely affected only in boys treated with very high cumulative doses of alkylating agent chemotherapy. Risk-adapted therapies reducing or eliminating gonadotoxic alkylating agent chemotherapy offer young men with newly diagnosed HL excellent prospects to maintain fertility.407
Young women have a higher chance of maintaining regular menses after treatment for HL compared to older women. The risk of ovarian failure after pelvic radiation is high, but can be reduced by midline ovarian transposition (oophoropexy).408 Although most young women maintain or resume menses after MOPP (or similar hybrid) chemotherapy, they are predisposed to early menopause, particularly if they also received abdominal-pelvic radiation.353–357 In a report of female Hodgkin’s disease survivors by the Late Effects Study Group, 42% of women treated with alkylating agent chemotherapy and infradiaphragmatic radiation had experienced menopause by age 31 years compared to 5% of control patients.353 Childhood Cancer Survivor Study investigators also observed that HL survivors treated with alkylating agents plus abdominopelvic radiation were at high risk of nonsurgical premature menopause, with a cumulative incidence approaching 30%.357
The overall cumulative risk of developing a subsequent malignancy following treatment for pediatric HL ranges from 7% to 8% at 15 years from diagnosis and rises to 16% to 28% by 20 years.* Tumor histologies comprise two main types: acute myeloid leukemia (including a pancytopenic myelodysplastic syndrome) and solid tumors. Secondary leukemias exhibit a relatively brief latency with a peak frequency in the first 5 to 10 years after treatment. The risk plateaus to less than 2% after 10 years from diagnosis.326,422 Clinical and treatment factors correlated with secondary leukemogenesis include older age at treatment, history of splenectomy, presentation with advanced disease, treatment with high cumulative doses of alkylating agents, and history of relapse.422 In contrast to secondary hematopoietic malignancies, the risk of developing a second solid tumor increases with the passage of time from diagnosis, with a latency usually exceeding 10 years from diagnosis. Various solid tumors have been observed, most commonly involving the breast, thyroid, bone, and soft tissues.%† In Fig. 54-7, the cumulative incidence of subsequent malignancy is provided from a multi-institutional study, and demonstrates the increased likelihood in females compared with males; this is largely, but not entirely, due to secondary breast cancer. The impact of subsequent malignancy on survival is also shown.
1 Ries LAG, Melbert D, Krapcho M, et al, editors: SEER Cancer Statistics Review, 1975–2005, National Cancer Institute. Bethesda, MD http://seer.cancer.gov/csr/1975_2005/ based on November 2007 SEER data submission, posted to the SEER web site, 2008
2 Gurney JG, Davis S, Severson RK, et al. Trends in cancer incidence among children in the US. Cancer. 1996;78:532.
3 Linet MS, Ries LAG, Smith MA, et al. Cancer surveillance series: recent trends in childhood cancer incidence and mortality in the US. J Natl Cancer Inst. 1999;91:1051.
4 Miller RW. Persons with exceptionally high risk of leukemia. Cancer Res. 1967;27:2420.
5 Greaves MF. Aetiology of acute leukemia. Lancet. 1997;349:344.
6 Wiemels JL, Ford AM, Van Wering ER, et al. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood. 1999;94:1057.
7 Spector LG, Ross JA, Robison LL, et al. Epidemiology and etiology. In Pui CH, editor: Childhood leukemias, ed 2, New York: Cambridge University Press, 2006.
8 Miller DR. Acute lymphoblastic leukemia. Pediatr Clin North Am. 1980;27:269.
9 Pui CH, Crist WM. Acute lymphoblastic leukemia. In: Pui CH, editor. Childhood leukemia. New York: Cambridge University Press, 1999.
10 Mastrangelo R, Poplack D, Bleyer WA, et al. Report and recommendations of the Rome workshop concerning poor prognosis acute lymphoblastic leukemia in children: biologic bases for staging stratification and treatment. Med Pediatr Oncol. 14, 1986.
11 Mahmoud HH, Rivera GK, Hancock ML, et al. Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med. 1993;329:314.
12 Odom LF, Wilson H, Cullen J. Significance of blasts of low-cell-count cerebrospinal fluid specimens from children with acute lymphoblastic leukemia. Cancer. 1990;66:1748.
13 Tubergen DG, Cullen JW, Boyett JM, et al. Blasts in CSF with a normal cell count do not justify alteration of therapy for acute lymphoblastic leukemia in remission: a Children’s Cancer Group study. J Clin Oncol. 1994;12:273.
14 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukemias. French-American-British Cooperative Group. Br J Haematol. 1976;33:451.
15 Bennett JM, Catovsky D, Daniel MT, et al. The morphological classification of acute lymphoblastic leukemia: concordance among observers and clinical correlations. Br J Haematol. 1981;47:553.
16 Miller DR, Leikin S, Albo V, et al. Prognostic importance of morphology (FAB classification) in childhood acute lymphoblastic leukemia (ALL). Br J Haematol. 1981;48:199.
17 Pui C. Childhood leukemias. N Engl J Med. 1995;332:1618.
18 Shaw MT. The cytochemistry of acute leukemia: a diagnostic and prognostic evaluation. Semin Oncol. 3, 1976.
19 Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood. 1993;82:343.
20 Campana D, Pui CH. Detection of minimal residual disease in acute leukemia: methodologic advances and clinical significance. Blood. 1995;85:1416.
21 Pui CH, Crist WM, Look AT. Biology and clinical significance of cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Blood. 1990;76:1449.
22 Shurtleff SA, Buijs A, Behm FG, et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995;9:1985.
23 Pui CH. Acute lymphoblastic leukemia. Pediatr Clin North Am. 1997;44:831.
24 Pinkel D. Curing children of leukemia. Cancer. 1987;59:1683.
25 Pui CH, Williams DL, Roberson PK, et al. Correlation of karyotype and immunophenotype in childhood acute lymphoblastic leukemia. J Clin Oncol. 1988;6:56.
26 Hammond D, Sather H, Nesbit M, et al. Analysis of prognostic factors in acute lymphoblastic leukemia. Med Pediatr Oncol. 1986;14:124.
27 Pui CH, Kane JR, Crist WM. Biology and treatment of infant leukemias. Leukemia. 1995;9:762.
28 Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14:18.
29 Chen CS, Sorenson PHB, Domer PH. Molecular rearrangements on chromosome 11q23 predominate in infant acute lymphoblastic leukemia and are associated with specific biologic variables and poor outcome. Blood. 1993;81:2386.
30 Schultz KR, Pullen J, et al. Risk- and response-based classification of childhood B precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group and the Children’s Oncology Group. Blood. 2007;109:926.
31 Mody R, Li S, Dover DC, et al. Twenty-five-year follow-up among survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Blood. 2008;111:5515.
32 Maloney KW, Shuster JJ, Murphy S, et al. Long-term results of treatment studies for childhood ALL: POG studies 1986-1994. Leukemia. 2000;14:2276.
33 Berg SL, Balis FM, Zimm S, et al. Phase I/II trial and pharmacokinetics of intrathecal diaziquone in refractory meningeal malignancies. J Clin Oncol. 1992;10:143.
34 Adamson PC, Balis FM, Arndt CA, et al. Intrathecal 6-mercaptopurine: preclinical pharmacology, phase I/II trial, and pharmacokinetic study. Cancer Res. 1991;51:6079.
35 Gelber RD, Sallan SE, Cohen HJ, et al. Central nervous system treatment in childhood acute lymphoblastic leukemia. Long-term follow-up of patients diagnosed between 1973 and 1985. Cancer. 1993;72:261.
36 Finkelstein JZ, Dyment PG, Hammond GD. Leukemic infiltration of the testes during bone marrow remission. Pediatrics. 1969;43:1042.
37 Nies BA, Bodey GP, Thomas LB, et al. The persistence of extramedullary leukemia infiltrates during bone marrow remission of acute leukemia. Blood. 1965;26:133.
38 Bowman WP, Shuster J, Cook B, et al. Improved survival for children with B-cell ALL and stage IV small noncleaved cell lymphoma: a POG study. J Clin Oncol. 1996;14:1252.
39 Conter V, Schrappe M, Arico M, et al. Role of cranial radiotherapy for childhood T-cell acute lymphoblastic leukemia with high WBC count and good response to prednisone. Associazione Italiana Ematologia Oncologia Pediatrica and the Berlin-Frankfurt-Munster groups. J Clin Oncol. 1997;15:2786.
40 Asselin B, Shuster J, Amylon M. Improved event-free survival with high dose methotrexate in T-cell lymphoblastic leukemia and advanced lymphoblastic lymphoma: A Pediatric Oncology Group study. Proc ASCO. 2001;20:367. Abstract
41 Buie LW, Epstein SS, Lindley CM. Nelarabine: a novel purine antimetabolite antineoplastic agent. Clin Thera. 2007;29:1887.
42 Rubnitz JE, Link MP, Shuster JJ. Frequency and prognostic significance of HRx rearrangements in infant acute lymphoblastic leukemia: a Pediatric Oncology Group study. Blood. 1994;84:570.
43 Armstrong SA, Mabon ME, Silverman LB, et al. FLT 3 mutations in childhood ALL. Blood. 2004;103:3544.
44 Brown P, Lewis M, Shurtleff S, et al. FLT 3 inhibition selectively kills childhood ALL cells with high levels of FLT 3 expression. Blood. 2005;105:812.
45 Meadows AT, Gordon J, Massari DJ. Declines in IQ scores and cognitive dysfunctions in children with acute lymphocytic leukemia treated with cranial irradiation. Lancet. 1981;2:1015.
46 Bleyer WA. Neurologic sequelae of methotrexate and ionizing radiation: a new classification. Cancer Treat Rep. 1981;65(Suppl 1):89.
47 Moss HA, Nannis ED, Poplack DG. The effects of prophylactic treatments of the central nervous system on the intellectual functioning of children with acute lymphoblastic leukemia. Am J Med. 1981;71:47.
48 Nesbit ME, Sather HN, Robison LL, et al. Sanctuary therapy: a randomized trial of 724 children with previously untreated acute lymphoblastic leukemia. Cancer Res. 1982;42:674.
49 Nesbit ME, Sather HN, Robison LL. Presymptomatic central nervous system therapy in previously untreated childhood acute lymphoblastic leukemia: Comparison of 1800 rad and 2400 rad. Lancet. 1981;1:461.
50 Clarke M, Gaynon P, Hann I, et al. CNS-directed therapy for childhood acute lymphoblastic leukemia: childhood ALL Collaborative Group overview of 43 randomized trials. J Clin Oncol. 2003;21:1798.
51 Reiter A, Schrappe M, Ludwig WD, et al. Chemotherapy in 998 unselected childhood acute lymphoblastic leukemia patients. Results and conclusions of the multicenter trial ALL-BFM 86. Blood. 1994;84:3122.
52 Waber DP, Silverman LB, Catania L, et al. Outcomes of a randomized trial of hyperfractionated cranial radiation therapy for treatment of high-risk ALL: therapeutic efficacy and neurotoxicity. J Clin Oncol. 2004;22:2701.
53 Waber DP, Turek J, Catania L, et al. Neuropsychological outcomes from a randomized trial of triple intrathecal chemotherapy compared with 18 Gy cranial radiation as CNS treatment in acute lymphoblastic leukemia: findings from Dana-Farber Cancer Institute ALL Consortium Protocol 95-01. J Clin Oncol. 2007;25:4914.
54 Littman P, Coccia P, Bleyer WA, et al. Central nervous system prophylaxis in children with low-risk acute lymphoblastic leukemia. Int J Rad Oncol Biol Phys. 1987;13:1443.
55 Sullivan MP, Chen T, Dyment PG. Equivalence of intrathecal chemotherapy and radiotherapy as central nervous system prophylaxis in children with acute lymphatic leukemia: a Pediatric Oncology Group study. Blood. 1982;60:948.
56 van Eys J, Berry D, Crist W. A comparison of two regimens for high risk acute lymphoblastic leukemia in childhood: a Pediatric Oncology Group study. Cancer. 1989;63:23.
57 Pullen J, Boyett J, Shuster J, et al. Extended triple intrathecal chemotherapy trial for prevention of CNS relapse in good-risk and poor-risk patients with B-progenitor acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol. 1993;11:839.
58 Freeman AI, Weinberg V, Brecher ML. Comparison of intermediate-dose methotrexate with cranial irradiation for the post-induction treatment of acute lymphoblastic leukemia in children. N Engl J Med. 1983;308:477.
59 Freeman AI, Boyett JM, Glicksman AS, et al. Intermediate-dose methotrexate versus cranial irradiation in childhood acute lymphoblastic leukemia: a ten-year follow-up. Med Pediatr Oncol. 1997;28:98.
60 Veerman AJ, Hahlen K, Kamps WA, et al. High cure rate with a moderately intensive treatment regimen in non-high-risk childhood acute lymphoblastic leukemia. Results of protocol ALL VI from the Dutch Childhood Leukemia Study Group. J Clin Oncol. 1996;14:911.
61 Pinkel D, Woo S. Prevention and treatment of meningeal leukemia in children. Blood. 1994;84:355.
62 Steinherz PG. Radiotherapy versus intrathecal chemotherapy for CNS prophylaxis in childhood ALL. Oncology (Huntingt). 1989;3:47.
63 Nachman J, Sather HN, Cherlow JM, et al. Response of children with high-risk acute lymphoblastic leukemia treated with and without cranial irradiation: a report from the Children’s Cancer Group. J Clin Oncol. 1998;16:920.
64 Pui CH, Mahmoud HH, Rivera GK, et al. Early intensification of intrathecal chemotherapy virtually eliminates central nervous system relapse in children with acute lymphoblastic leukemia. Blood. 1998;92:411.
65 Silverman LB, Dederck L, Gelber RD, et al. Results of Dana-Farber Cancer Institute Consortium Protocols for children with newly diagnosed acute lymphoblastic leukemia (1981-1995). Leukemia. 2000;14:2247.
66 Poplack DG, Kun LE, Magrath IT, et al. Leukemias and lymphomas of childhood. In: Devita VTJr, Hellman S, Rosenberg SA, editors. Cancer: principles and practice of oncology. Philadelphia: JB Lippincott, 1993.
67 Bleyer WA, Poplack DG. Intraventricular versus intralumbar methotrexate for central-nervous-system leukemia: Prolonged remission with the Ommaya reservoir. Med Pediatr. 1979;6:207.
68 Balis FM, Savitch JL, Bleyer WA, et al. Remission induction of meningeal leukemia with high-dose intravenous methotrexate. J Clin Oncol. 1985;3:485.
69 Cairo MS, Gerrard M, Sposto R, et al. Results of randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007;109:2736.
70 Kim TH, Ramsay NK, Steeves RA, et al. Intermittent central nervous system irradiation and intrathecal chemotherapy for central nervous system leukemia in children. Int J Radiat Biol Oncol Phys. 1987;13:1451.
71 Cherlow JM, Steinherz P, Gaynon P, et al. Cranio-spinal radiation for CNS leukemia at presentation: how high does the spinal dose need to be? Proc ASCO. 1991;10:239. Abstract
72 Patte C, Leverger G, Perel Y, et al. Results of LMB 86 and 89 protocols of the French Pediatric Oncology Society (S.F.O.P.) for children with B-cell lymphoma and leukemia with CNS involvement. Med Pediatr Oncol. 1993;21:535. Abstract
73 Land VJ, Thomas PR, Boyett JM, et al. Comparison of maintenance treatment regimens for first central nervous system relapse in children with acute lymphocytic leukemia. A Pediatric Oncology Group study. Cancer. 1985;56:81.
74 Willoughby ML. Treatment of overt meningeal leukemia in children: Results of second MRC meningeal leukemia trial. Br Med J. 1976;1:864.
75 Wells RJ, Weetman RM, Baehner RL. The impact of isolated central nervous system relapse following initial complete remission in childhood acute lymphocytic leukemia. J Pediatr. 1980;97:429.
76 Mandell LR, Steinherz P, Fuks Z. Delayed central nervous system (CNS) radiation in childhood CNS acute lymphoblastic leukemia. Cancer. 1990;66:447.
77 Barredo JC, Meenakshi D, Lauer S, et al. Isolated CNS relapse of acute lymphoblastic leukemia treated with intensive systemic chemotherapy and delayed CNS radiation: a Pediatric Oncology Group Study. J Clin Onc. 2006;24:3142.
78 Winick NJ, Smith SD, Shuster J, et al. Treatment of CNS relapse in children with acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol. 1993;11:271.
79 Parker W, Freeman C. A simple technique for cranial radiotherapy in the supine position. Radiother Oncol. 2006;78:217.
80 Panandiker A, Ning H, Likhacheva A, et al. Craniospinal irradiation with spinal IMRT to improve target homogeneity. Int J Radiat Oncol Biol Phys. 2007;68:1402.
80a Clair W, Adams J, Bues M, et al. Advantage of protons compared to conventional x-ray or IMRT in the treatment of a pediatric patient with medulloblastoma. Int J Radiat Oncol Biol Phys. 2004;58:727-734.
80b Cochran D, Yock T, Adams J, Tarbell N. Radiation dose to the lens during craniospinal irradiation—an improvement in proton radiotherapy technique. Int J Radiat Oncol Biol Phys. 2008;70:1336-1342.
81 Gillin MT, Kline RW, Kun LE. Cranial dose distribution. Int J Radiat Oncol Biol Phys. 1979;5:1903.
82 Tatcher M, Glicksman AS. Field matching considerations in craniospinal irradiation. Int J Radiat Oncol Biol Phys. 1989;17:865.
83 Holupka EJ, Humm JL, Tarbell NJ, et al. Effect of set-up error on the dose across the junction of matching cranial-spinal fields in the treatment of medulloblastoma. Int J Radiat Biol Oncol Phys. 1993;27:345.
84 Siddon RL. Solution to treatment planning problems using coordinate transformations. Med Phys. 1982;8:766.
85 Maor MH, Fields RS, Hogstrom KR, et al. Improving the therapeutic ratio of craniospinal irradiation in medulloblastoma. Int J Radiat Oncol Biol Phys. 1985;11:687.
86 Miller LP, Miller DR. Acute lymphoblastic leukemia in children: current status, controversies, and future perspective. Crit Rev Oncol Hematol. 1986;1:129.
87 Nesbit ME, Robinson LL, Ortega JA, et al. Testicular relapse in childhood acute lymphoblastic leukemia: association with pre-treatment patient characteristics and treatment. A report for Children’s Cancer Study Group. Cancer. 1980;45:2009.
88 Wong KY, Ballard ET, Strayer FH, et al. Clinical and occult testicular leukemia in long-term survivors of acute lymphoblastic leukemia. J Pediatr. 1980;96:569.
89 Finkelstein JZ, Dyment PG, Hammond GD. Leukemic infiltration of the testes during bone marrow remission. Pediatrics. 1969;43:1042.
90 Nies BA, Bodey GP, Thomas LB, et al. The persistence of extramedullary leukemia infiltrates during bone marrow remission of acute leukemia. Blood. 1965;26:133.
91 Givlier RL. Testicular involvement in leukemia and lymphoma. Cancer. 1969;23:1290.
92 Medical Research Council. Testicular disease in acute lymphoblastic leukemia in childhood. Br Med J. 1978;1:334.
93 Ortega JJ, Javier G, Toran N. Testicular infiltrates in children with acute lymphoblastic leukaemia: a prospective study. Med Pediatr Oncol. 1984;12:396.
94 Bowman WP, Aur RJA, Hustu HO, et al. Isolated testicular relapse in acute lymphocytic leukemia of childhood: categories and influence on survival. J Clin Oncol. 2, 1984.
95 Pui CH, Dahl GV, Bowman WP, et al. Elective testicular biopsy during chemotherapy for childhood leukemia is of no clinical value. Lancet. 1985;2:410.
96 Hijiya N, Liu W, Sandlund J, et al. Overt testicular disease at diagnosis of acute lymphoblastic leukemia: lack of therapeutic role of local irradiation. Leukemia. 2005;19:1399.
97 Byrd R. Testicular leukemia: incidence and management results. Med Pediatr Oncol. 1981;9:493.
98 Hustu HO, Aur RJ. Extramedullary leukemia. Clin Haematol. 1978;7:313.
99 Atkinson K, Thomas PRM, Peckham MJ. Radiosensitivity of the acute leukaemic infiltrate. Eur J Cancer. 1976;12:535.
100 Sullivan MP, Perez CA, Herson J. Radiotherapy (2500 rad) for testicular leukemia: Local control and subsequent clinical events: a Southwest Oncology Group Study. Cancer. 1980;46:508.
101 Frankel LS, Provisor A, Kletzel M, et al. Successful therapy for overt testicular relapse in childhood acute lymphocytic leukemia. Proc ASCO. 3, 1984. Abstract
102 Culbert S, Doering E, Jelden G, et al. Testicular leukemia relapse: a treatable entity. Proc ASCO. 1983;2:79. Abstract
103 Brauner R, Czernichow P, Cramer P. Leydig-cell function in children after direct testicular irradiation for acute lymphoblastic leukemia. N Engl J Med. 1983;309:25.
104 Leiper AD, Grant DB, Chessels JM. The effect of testicular irradiation on Leydig cell function in prepubertal boys with acute lymphoblastic leukemia. Arch Dis Child. 1983;58:906.
105 Schrappe M, Reiter A, Zimmerman M, et al. Long-term results of four consecutive trials in childhood ALL performed by the ALL-BFM study group from 1981 to 1995. Berlin-Frankfurt-Münster. Leukemia. 2000;14:2205.
106 Möricke A, Reiter A, Zimmermann M, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood. 2008;111:4477.
107 Gaynon PS, Trigg ME, Heerema NA, et al. Children’s Cancer Group trials in childhood acute lymphoblastic leukemia: 1983-1995. Leukemia. 2000;14:2223.
108 Tubergen DG, Gilchrist GS, O’Brien RT, et al. Improved outcome with delayed intensification for children with acute lymphoblastic leukemia and intermediate presenting features: a Children’s Cancer Group phase III trial. J Clin Oncol. 1993;11:527.
109 Tubergen DG, Gilchrist GS, O’Brien RT, et al. Prevention of CNS disease in intermediate-risk acute lymphoblastic leukemia: comparison of cranial radiation and intrathecal methotrexate and the importance of systemic therapy: a Children’s Cancer Group report. J Clin Oncol. 1993;11:520.
110 Nachman JB, Sather HN, Sensel MG, et al. Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med. 1998;338:1663.
111 Seibel NL, Steinherz PG, Sather HN, et al. Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2008;111:2548.
112 Moghrabi A, Levy DE, Asselin B, et al. Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood. 2007;109:896-904.
113 Trueworthy R, Shuster J, Look T, et al. Ploidy of lymphoblasts is the strongest predictor of treatment outcome in B-progenitor cell acute lymphoblastic leukemia of childhood: A Pediatric Oncology Group study. J Clin Oncol. 1992;10:606.
114 Chessells JM, Bailey C, Richards SM. Intensification of treatment and survival in all children with lymphoblastic leukemia: results of UK Medical Research Council trial UKALL X. Medical Research Council Working Party on Childhood Leukemia. Lancet. 1995;345:143.
115 Amylon MD, Shuster J, Pullen J, et al. Intensive high-dose asparaginase consolidation improves survival for pediatric patients with T cell acute lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: a Pediatric Oncology Group study. Leukemia. 1999;13:335.
116 Lauer SJ, Camitta BM, Leventhal BG, et al. Intensive alternating drug pairs after remission induction for treatment of infants with acute lymphoblastic leukemia: a Pediatric Oncology Group Pilot Study. J Pediatr Hematol Oncol. 1998;20:229.
117 Pui CH, Boyett JM, Rivera GK, et al. Long-term results of Total Therapy studies 11, 12 and 13A for childhood acute lymphoblastic leukemia at St Jude Children’s Research Hospital. Leukemia. 2000;14:2286.
118 Rivera GK, Raimondi SC, Hancock ML, et al. Improved outcome in childhood acute lymphoblastic leukemia with reinforced early treatment and rotational combination chemotherapy. Lancet. 1991;337:61.
119 Evans WE, Relling MV, Rodman JH, et al. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med. 1998;338:499.
120 Pui CH, Crist W. Biology and treatment of acute lymphoblastic leukemia. J Pediatr. 1994;124:491.
121 Ruffner KI, Matthews DC. Current uses of monoclonal antibodies in the treatment of acute leukemia. Semin Oncol. 2000;27:531.
122 Choi S, Simone JV. Acute nonlymphocytic leukemia in 171 children. Med Pediatr Oncol. 1976;2:119.
123 Homans AC, Verissimo AM, Vlacha V. Transient abnormal myelopoiesis of infancy associated with trisomy 21. Am J Pediatr Hematol Oncol. 1993;15:392.
124 Bennet JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103:620.
125 Lampkin BC, Woods W, Strauss R. Current status of the biology and treatment of acute non-lymphocytic leukemia in children (Report from the ANLL strategy group of Children’s Cancer Study Group). Blood. 1983;61:215.
126 Pui CH, Kalwinsky D, Schell M, et al. Acute nonlymphoblastic leukemia in infants: clinical presentation and outcome. J Clin Oncol. 1988;6:1008.
127 Windebank KP, Tefferi A, Smithson W, et al. Acute megakaryocytic leukemia (M7) in children. Mayo Clin Proc. 1989;64:1339.
128 Bloomfield CTD, de la Chapelle A. Chromosome abnormalities in acute nonlymphocytic leukemia: clinical and biologic significance. Semin Oncol. 1987;14:372.
129 Kakizuka A, Miller W, Umesuno K. Chromosome translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha gene with a novel putative transcription factor PML. Cell. 1991;66:663.
130 Carroll A, Civin C, Schneider N. The t(1;22) is nonrandom and restricted to infants with acute megakaryoblastic leukemia: a Pediatric Oncology Group study. Blood. 1991;78:748.
131 Woods WG, Nesbit ME, Buckley J, et al. Correlation of chromosome abnormalities with patient characteristics, histologic subtype and induction success in children with acute NLL. J Clin Oncol. 1985;3:3.
132 Sorenson P, Chen CS, Smith FO, et al. Molecular rearrangements of the MLL gene are present in most cases of infant acute myeloid leukemia and are strongly correlated with monocytic or myelomonocytic phenotypes. J Clin Invest. 1994;93:429.
133 Grier HE, Gelber RD, Camitta BM, et al. Prognostic factors in childhood acute myelogenous leukemia. J Clin Oncol. 1987;5:1026.
134 Champlin R, Gale RP. Acute myelogenous leukemia: Recent advances in therapy. Prolonged survival in acute myelogenous leukemia without maintenance chemotherapy. Blood. 1987;69:1551.
135 Abromowitch M, Ochs J, Pui CH, et al. Efficacy of high-dose methotrexate in childhood acute lymphocytic leukemia: analysis by contemporary risk classifications. Blood. 1988;71:866.
136 Weinstein HJ, Mayer RJ, Rosenthal DS. Chemotherapy for acute myelogenous leukemia in children and adults. VAPA update. Blood. 1983;62:315.
137 Lange BJ, Smith FO, Feusner J, et al. Outcomes in CCG-2961, a children’s oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children’s oncology group. Blood. 2008;111:1044.
138 Loeb DM, Arceci RJ. What is the optimal therapy for childhood AML? Oncol (Williston Park). 2002;16:1057.
139 Arceci RJ. Progress and controversies in the treatment of pediatric acute myelogenous leukemia. Curr Opin Hematol. 2002;9:353.
140 Hann IM, Webb DK, Gibson BE, et al. MRC trials in childhood acute myeloid leukaemia. Ann Hematol. 2004;83(Suppl 1):S108.
141 Oliansky DM, Rizzo JD, Aplan PD, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute myeloid leukemia in children: an evidence-based review. Biol Blood Marrow Transplant. 2007;13:1.
142 Woods WG, Neudorf S, Gold S, et al. A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission. Blood. 2001;97:56.
143 Stevens RF, Hann IM, Wheatley K, et al. Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council’s 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol. 1998;101:130.
144 Ravindranath Y, Yeager AM, Chang MN, et al. Autologous bone marrow transplantation versus intensive consolidation chemotherapy for acute myeloid leukemia in childhood. Pediatric Oncology Group. N Engl J Med. 1996;334:1428.
145 Bleakley M, Lau L, Shaw PJ, et al. Bone marrow transplantation for paediatric AML in first remission: a systematic review and meta-analysis. Bone Marrow Transplant. 2002;29:843.
146 Creutzig U, Reinhardt D. Current controversies: which patients with acute myeloid leukaemia should receive a bone marrow transplantation?—a European view. Br J Haematol. 2002;118:365.
147 Creutzig U, Ritter J, Zimmermann M, et al. Idarubicin improves blast cell clearance during induction therapy in children with AML: results of study AML-BFM 93. AML-BFM Study Group. Leukemia. 2001;15:348.
148 Alonzo TA, Wells RJ, Woods WG, et al. Postremission therapy for children with acute myeloid leukemia: the Children’s Cancer Group experience in the transplant era. Leukemia. 2005;19:965.
149 Hurwitz CA, Schell MJ, Pui CH, et al. Adverse prognostic features in 251 children treated for acute myeloid leukemia. Med Pediatr Oncol. 1993;21:1.
150 Odom LF, Lampkin BC, Tannous R, et al. Acute monoblastic leukemia: a unique subtype—A review from the Children’s Cancer Study Group. Leuk Res. 1990;14:1.
151 Kantarjian HM, Keating MJ, Walters RS, et al. Role of maintenance chemotherapy in acute promyelocytic leukemia. Cancer. 1987;59:1258.
152 Santana VM, Hurwitz CA, Blakley RL, et al. Complete hematologic remissions induced by 2-chlorodeoxyadenosine in children with newly diagnosed acute myeloid leukemia. Blood. 1994;84:1237.
153 Santana VM, Mirro JJr, Kearns C, et al. 2-Chlorodeoxyadenosine produces a high rate of complete hematologic remission in relapsed acute myeloid leukemia. J Clin Oncol. 1992;10:364.
154 Huang ME, Ye YC, Chen SR, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Hamatologie und Bluttransfusion. 1989;32:88.
155 Castaigne S, Chomienne C, Daniel MT, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76:1704.
156 Amadori S, Testi AM, Arico M, et al. Prospective comparative study of bone marrow transplantation and postremission chemotherapy for childhood acute myelogenous leukemia. The Associazione Italiana Ematologia ed Oncologia Pediatrica Cooperative Group. J Clin Oncol. 1993;11:1046.
157 Reiffers J, Gaspard MH, Maraninchi D, et al. Comparison of allogeneic or autologous bone marrow transplantation and chemotherapy in patients with acute myeloid leukemia in first remission: a prospective controlled trial. Br J Haematol. 1989;72:57.
158 Malik S, DeLaat C, Harris R. Allogeneic bone marrow transplantation versus autologous BMT vs chemotherapy in childhood acute myeloid leukemia. Proc ASCO. 1993;12:323. Abstract
159 Nesbit M, Burkley L, Lampkin B, et al. Comparison of allogeneic bone marrow transplantation with maintenance chemotherapy in previously untreated childhood acute non-lymphocytic leukemia. Proc ASCO. 1987;6:163. Abstract
160 Fenaux P, Chomienne C, Degos L. Acute pro-leukemia: Biology and treatment. Semin Oncol. 1997;24:92.
161 Tallman MS, Andersen JW, Schiffer CA. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337:1021.
162 Fenaux P, Chastang G, Chevret S. A randomized comparison of all-trans-retinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1999;94:1192.
163 Calleja EM, Wamell RP. Differentiating agents in pediatric malignancies: All-trans-retinoic acid and arsenic in APML. Curr Oncol Rep. 2000;2:519.
164 Zipursky A, Thorner P, DeHarven E, et al. Myelodysplasia and acute megakaryocytic leukemia in Down’s syndrome. Leuk Res. 1994;18:163.
165 Ravindranath Y, Abella E, Krischer JP, et al. Acute myeloid leukemia (AML) in Down’s syndrome is highly responsive to chemotherapy: Experience on POG AML Study 8498. Blood. 1992;80:2210.
166 Lange BJ, Kobrinsky N, Barnard DR, et al. Distinctive demography, biology and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: CCSG studies 2861 and 2891. Blood. 1998;91:608.
167 Smith MA, Rubinstein L, Andersen JR. Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins. J Clin Oncol. 1999;17:569.
168 Felix CA. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim Biophys Acta. 1998;1400:233.
169 Tucker MA, Meadows AT, Boice JD. Leukemia after therapy with alkylating agents for childhood cancer. J Natl Cancer Inst. 1987;78:459.
170 Neiman RS, Barcos M, Berard C. Granulocytic sarcoma: A clinicopathologic study of 61 biopsied cases. Cancer. 1981;48:1426.
171 Appelbaum FR. Antibody-targeted therapy for myeloid leukemia. Semin Hematol. 1999;36:2.
172 Sievers EL, Appelbaum FR, Spieberger RT, et al. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: A phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood. 1999;93:3678.
173 Matthews DC, Appelbaum FR, Eary JF, et al. Phase I study of (131) I-anti-CD45-antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood. 1999;94:1237.
174 Zujewski J, Horak ID, Bol CJ, et al. Phase I and pk study of farnesyl protein transferase inhibitor R115777 in advanced cancer. J Clin Oncol. 2000;18:927.
175 Smith KL, Johnson W. Classification of chronic myelocytic leukemia in children. Cancer. 1974;34:670.
176 Altman AJ. Chronic leukemias of childhood. Pediatr Clin North Am. 1988;35:765.
177 Druker BJ, Talpaz M, Resta D. Clinical efficacy and safety of an Abl specific tyrosine kinase inhibitor as targeted therapy for CML. Blood. 1999;94:368A.
178 Druker BJ, Kantarjian H, Sawyers CL. Activity of an Abl-specific tyrosine kinase inhibitor in patients with Bcr-Abl positive acute leukemias, including CML in blast crisis. Blood. 1999;94:697A.
179 Schultz KR, Bowman WP, Slayton W, et al. Improved early event-free survival (EFS) in children with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL) with intensive imatinib in combination with high-dose chemotherapy: Children’s Oncology Group (COG) study AALL0031. Proc Am Soc Hematol. 2007;110:9a. Abstract
180 Altman AJ, Palmer G, Baehner RL. Juvenile “chronic granulocytic” leukemia: a panmyelopathy with prominent monocytic involvement and circulating monocyte colony-forming cells. Blood. 1974;43:341.
181 Arico M, Biondi A, Pui CH. Juvenile myelomonocytic leukemia. Blood. 1997;90:479.
182 Niemeyer CM, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood. 1997;89:3534.
183 Mays JA, Neerhout RC, Bagby GC, et al. Juvenile chronic granulocytic leukemia: emphasis on cutaneous manifestations and underlying neurofibromatosis. Am J Dis Child. 1980;134:654.
184 Filipovich AH, Mathor A, Kamat D, et al. Lymphoproliferative disorders and other tumors complicating immunodeficiencies. Immunodeficiency. 1994;5:91.
185 Murphy SB, Jenson HB, McClain KL, et al. AIDS related tumors. Med Pediatr Oncol. 1997;29:381. Abstract
186 Mueller BU. Lymphoproliferative disorders and malignancies related to immunodeficiencies. In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. Philadelphia: Lippincott Williams & Wilkins, 2002.
187 Swinnen LJ. Overview of post-transplant B-cell lymphoproliferative disorders. Semin Oncol. 1999;26:21.
188 Gross TG. Low-dose chemotherapy for children with post-transplant lymphoproliferative disease. Recent Results Cancer Res. 2002;159:96.
189 National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphoma. Summary and description of a working formulation for clinical usage. The Non-Hodgkin’s Lymphoma Pathologic Classification Project. Cancer. 1982;49:2112.
190 Shad A, Magrath IT. Non-Hodgkin’s lymphoma. Pediatr Clin North Am. 1997;44:863.
191 Dalla-Favera R, Bnegni M, Erikson J, et al. Human c-myc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci. 1982;79:7824.
192 Kadin ME. Ki-1-positive anaplastic large-cell lymphoma: a clinicopathologic entity? J Clin Oncol. 1991;9:533.
193 Kurtzberg J, Graham M. Non-Hodgkin’s lymphoma: biologic classification and implications of therapy. Pediatr Clin North Am. 1991;38:443.
194 Wollner N, Burchenal HJ, Leiberman P, et al. Non-Hodgkin’s lymphoma in children: a comparative study of two modalities of therapy. Cancer. 1976;37:123.
195 Hvizdala EV, Berard C, Callihan T, et al. Non-lymphoblastic lymphoma in children: histology and stage-related response to therapy. A Pediatric Oncology Group study. J Clin Oncol. 1991;9:1889.
196 Murphy SB. Prognostic features and obstacles to cure of childhood non-Hodgkin’s lymphoma. Semin Oncol. 1977;4:265.
197 Anderson JR, Wilson JF, Jenkin RDT, et al. Childhood non-Hodgkin’s lymphoma: the results of a randomized trial comparing a 4-drug regimen (COMP) with a 10-drug regimen (LSA2-L2). N Engl J Med. 1983;308:559.
198 Sandlund JT, Downing JR, Crist WM. Non-Hodgkin’s lymphoma in childhood. N Engl J Med. 1996;331:1238.
199 Murphy SB, Fairclough DL, Hutchison RE, et al. Non-Hodgkin’s lymphomas of childhood: an analysis of the histology, staging, and response to treatment of 338 cases at a single institution. J Clin Oncol. 1989;7:186.
200 Magrath IT. Malignant non-Hodgkin’s lymphomas in children. In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. Philadelphia: Lippincott Williams & Wilkins, 2002.
201 Murphy SB. Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas: Dissimilarities from lymphoma in adults. Semin Oncol. 1980;7:332.
202 Patte C, Auperin A, Michon J, et al. The Societe Francais d’Oncologie Pediatrique LMP89 protocol: Highly effective multiagent chemotherapy tailored to the tumor burden and initial reponse in 561 unselected children with B cell lymphomas and L3 leukemia. Blood. 2001;97:3370.
203 Patte C, Auperin A, Gerrard M, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2007;109:2773.
204 Cairo MS, Sposto R, Perkins SL, et al. Burkitt’s and Burkitt-like lymphoma in children and adolescents: a review of the Children’s Cancer Group experience. Br J Haematol. 2003;120:660.
205 Cairo MS, Sposto R, Hoover-Regan M, et al. Childhood and adolescent large-cell lymphoma (LCL): a review of the Children’s Cancer Group experience. Am J Hematol. 2003;72:53.
206 Cairo MS, Krailo MD, Morse M, et al. Long-term follow-up of short intensive multiagent chemotherapy without high-dose methotrexate (‘Orange’) in children with advanced non-lymphoblastic non-Hodgkin’s lymphoma: a children’s cancer group report. Leukemia. 2002;16:594.
207 Link MP, Shuster JJ, Donaldson SS, et al. Treatment of children and young adults with early-stage non-Hodgkin’s lymphoma. N Engl J Med. 1997;337:1259.
208 Hutchison RE, Berard CW, Shuster JJ, et al. B-cell lineage confers a favorable outcome among children and adolescents with large-cell lymphoma: a Pediatric Oncology Group study. J Clin Oncol. 1995;13:2023.
209 Murphy SB, Hustu HO. A randomized trial of combined modality therapy of childhood non-Hodgkin’s lymphoma. Cancer. 1980;45:630.
210 Pinkel D, Johnson W, Aur RJ. Non-Hodgkin’s lymphoma in children. Br J Cancer. 1975;31(Suppl 2):298.
211 Sagerman RH, Wolff JA, Sitarz A, et al. Radiation therapy for lymphoma in children. Radiology. 1966;86:1096.
212 Whalen TV, La Quaglia MP. The lymphomas: an update for surgeons. Semin Pediatr Surg. 1997;6:50.
213 Anderson JR, Jenkin RDT, Wilson JF, et al. Long-term follow-up of patients treated with COMP or LSA2-L2 therapy for childhood non-Hodgkin’s lymphoma: A report of CCG-551 from the Children’s Cancer Group. J Clin Oncol. 1993;11:1024.
214 Meadows AT, Sposto R, Jenkin RDT, et al. Similar efficacy of 6 and 18 months of therapy with four drugs (COMP) for localized non-Hodgkin’s lymphoma of children: a report from the Children’s Cancer Study Group. J Clin Oncol. 1989;7:92.
215 Murphy SB, Bowman WP, Abromowitch M, et al. Results of treatment of advanced-stage Burkitt’s lymphoma and B cell (SIg+) acute lymphoblastic leukemia with high-dose fractionated cyclophosphamide and coordinated high-dose methotrexate and cytarabine. J Clin Oncol. 1986;4:1732.
216 Bowman WP, Shuster JJ, Cook B, et al. Improved survival for children with B-cell acute lymphoblastic leukemia and stage IV small noncleaved-cell lymphoma: a Pediatric Oncology Group study. J Clin Oncol. 1996;14:1252.
217 Brecher ML, Schwenn MR, Coppes MJ, et al. Fractionated cycophosphamide and back to back high dose methotrexate and cytosine arabinoside improves outcome in patients with stage III high grade small non-cleaved cell lymphomas (SNCCL): a randomized trial of the Pediatric Oncology Group. Med Pediatr Oncol. 1997;29:526.
218 Schwenn MR, Mahmoud HH, Bowman WP, et al. Successful treatment of small noncleaved cell (SNCC) lymphoma and B-cell acute lymphoblastic leukemia (B-ALL) with central nervous system (CNS) involvement: a Pediatric Oncology Group (POG) study. Proc Am Soc Clin Oncol. 2000;19:580a. Abstract
219 Schwenn MR, Mahmoud H, Bowman W, et al. The addition of vp-ifosfamide intensification did not improve event-free survival (EFS) for central nervous system (CNS) negative patients with advanced stage small noncleaved cell (SNCC) lymphoma or B-cell acute lymphoblastic leukemia (B-ALL): a Pediatric Oncology Group (POG) study. Proc Am Soc Clin Oncol. 2001;20:367a. Abstract
220 Reiter A, Schrappe M, Tiemann M, et al. Improved treatment results in childhood B-cell neoplasms with tailored intensification of therapy: a report of the BFM Group Trial NHL-BFM 90. Blood. 1999;94:3294.
221 Reiter A, Schrappe M, Ludwig WD, et al. Favorable outcome of B-cell acute lymphoblastic leukemia in childhood: a report of three consecutive studies of the BFM group. Blood. 1992;80:2471.
222 Woessmann W, Seidemann K, Mann G, et al. The impact of the methotrexate administration schedule and dose in the treatment of children and adolescents with B-cell neoplasms: a report of the BFM Group Study NHL-BFM95. Blood. 2005;105:948.
223 Patte C, Michon J, Behrendt H, et al. Results of the LMB 89 protocol for childhood B-cell lymphoma and leukemia (ALL). SIOP XXIX. Med Pediatr Oncol. 1997;29:358.
224 Reiter A, Schrappe M, Ludwig WD, et al. Intensive ALL-type therapy without local radiotherapy provides a 90% event-free survival for children with T-cell lymphoblastic lymphoma: A BFM group report. Blood. 2000;95:416.
225 Schrappe M, Tiemann M, Ludwig WD, et al. Risk-adapted therapy for lymphoblastic T-cell lymphomas: results from trials NHL-BFM 86 and 90. SIOP XXIX. Med Pediatr Oncol. 1997;29:356.
226 Burkhardt B, Woessmann W, Zimmermann M, et al. Impact of cranial radiotherapy on central nervous system prophylaxis in children and adolescents with central nervous system-negative stage III or IV lymphoblastic lymphoma. J Clin Oncol. 2006;24:491.
227 Goldberg JM, Silverman LB, Levy DE, et al. Childhood T-cell ALL: the DFCR Leukemia Consortium experience. J Clin Oncol. 2003;21:3616-3622.
228 Abromowitch M, Sposto R, Perkins S, et al. Outcome of Children’s Cancer Group (CCG) 5941: a pilot study for the treatment of newly diagnosed pediatric patients with disseminated lymphoblastic lymphoma. Proc Am Soc Clin Oncol. 2000;19:583a. Abstract
229 Seidemann K, Tiemann M, Schrappe M, et al. Short-pulse B-non-Hodgkin lymphoma-type chemotherapy is efficacious treatment for pediatric anaplastic large cell lymphoma: a report of the Berlin-Frankfurt-Munster Group trial NHL-BFM 90. Blood. 2001;97:3699.
230 Laver JH, Kraveka JM, Hutchison RE, et al. Advanced-stage large-cell lymphoma in children and adolescents: results of a randomized trial incorporating intermediate-dose methotrexate and high-dose cytarabine in the maintenance phase of the APO regimen: a Pediatric Oncology Group phase III trial. J Clin Oncol. 2005;23:541.
231 Brugieres L, Deley MC, Pacquement H, et al. CD30(+) anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French Society of Pediatric Oncology. Blood. 1998;92:3591.
232 Sandlund JT, Santana V, Abromowitch M, et al. Large cell non-Hodgkin lymphoma of childhood: clinical characteristics and outcome. Leukemia. 1994;8:30.
233 Santana VH, Abromowitch M, Sandlund JT, et al. MACOP-B treatment in children and adolescents with advanced diffuse large-cell non-Hodgkin’s lymphoma. Leukemia. 1993;7:187.
234 Sandlund JT, Pui CH, Santana VM, et al. Clinical features and treatment outcome for children with CD30+ large-cell non-Hodgkin’s lymphoma. J Clin Oncol. 1994;12:895.
235 Laver JH, Weinstein HJ, Hutchison RE, et al. Lineage-specific differences in outcome for advanced stage large cell lymphoma in children and adolescents: results of a randomized phase III Pediatric Oncology Group trial. Proc Am Soc Hematol. 2001;98:345a. Abstract
236 Reiter A, Schrappe M, Tiemann M, et al. Successful treatment strategy for Ki-1 anaplastic large cell lymphomas of childhood: a prospective analysis of 62 patients enrolled in three consecutive Berlin-Frankfurt-Münster group studies. J Clin Oncol. 1994;12:899.
237 Murphy SB. Pediatric lymphomas: Recent advances and commentary on Ki-1 positive anaplastic large cell lymphomas of childhood. Ann Oncol. 1994;5(Suppl 1):531.
238 Brugieres L, LeDeley MC, Pacquement H, et al. Anaplastic large cell lymphoma in children: analysis of 63 patients enrolled in two consecutive studies of the SFOP. Med Pediatr Oncol. 1997;29:237.
239 Link MP, Donaldson SS, Berard CW, et al. Results of treatment of childhood localized non-Hodgkin’s lymphoma with combination chemotherapy with or without radiotherapy. N Engl J Med. 1990;322:1169.
240 Patte C, Kalifa C, Flamant F, et al. Results of the LMB81 protocol a modified LSA2L2 protocol with high dose methotrexate in 84 children with non–B-cell (lymphoblastic) lymphoma. Med Pediatr Oncol. 1992;20:105.
241 Ingram LC, Fairclough DL, Furman WL, et al. Cranial nerve palsy in childhood acute lymphoblastic leukemia and non-Hodgkin’s lymphoma. Cancer. 1991;67:2262.
242 Glatstein E, Kim H, Donaldson S. Non-Hodgkin’s lymphomas: VI. Results of treatment in childhood. Cancer. 1974;34:204.
243 Jenkin RDT, Sonley MJ, Stephens CA, et al. Primary gastrointestinal tract lymphoma in childhood. Radiology. 1969;92:763.
244 Hvizdala EV, Berard C, Callihan T, et al. Lymphoblastic lymphoma in children: a randomized trial comparing LSA2-L2 with the ACOP+ therapeutic regimen: a Pediatric Oncology Group study. J Clin Oncol. 1988;6:26.
245 Weinstein HJ, Lack EE, Cassady JR. APO therapy for malignant lymphoma of large cell “histiocytic” type of childhood: analysis of treatment results for 29 patients. Blood. 1984;64:422.
246 Jenkin RD, Anderson JR, Chilcote RR, et al. The treatment of localized non-Hodgkin’s lymphoma in children: a report from the Children’s Cancer Study Group. J Clin Oncol. 1984;2:88.
247 Wollner N, Mandell L, Filippa D, et al. Primary nasal-paranasal oropharyngeal lymphoma in the pediatric age group. Cancer. 1990;65:1438.
248 Murphy SB, Hustu HO, Rivera G, et al. End results of treating children with localized non-Hodgkin’s lymphomas with a combined modality approach of lessened intensity. J Clin Oncol. 1983;1:326.
249 Jereb B, Wollner N, Kosloff C, et al. The role of local radiation in the treatment of non-Hodgkin’s lymphoma in children. Med Pediatr Oncol. 1981;9:157.
250 Schwenn MR, Blattner SR, Lynch E, et al. HiC-COM: A 2-month intensive chemotherapy regimen for children with stage III and IV Burkitt’s lymphoma and B-cell acute lymphoblastic leukemia. J Clin Oncol. 1991;9:133.
251 Dahl GV, Rivera G, Pui CH, et al. A novel treatment of childhood lymphoblastic non-Hodgkin’s lymphoma: early and intermittent use of teniposide plus cytarabine. Blood. 66, 1985.
252 Patte C, Philip T, Rodary C, et al. Improved survival rate in children with stage III and IV B cell non-Hodgkin’s lymphoma and leukemia using multi-agent chemotherapy: results of a study of 114 children from the French Pediatric Oncology Society. J Clin Oncol. 1986;4:1219.
253 Dosoretz DE, Murphy GF, Raymond AK, et al. Radiation therapy for primary lymphoma of bone. Cancer. 1983;51:44.
254 Boston MC, Dahlin DC, Ivins JC, et al. Malignant lymphoma (so-called reticulum cell sarcoma) of bone. Cancer. 1974;34:1131.
255 Loeffler JS, Tarbell NJ, Kozakewich H, et al. Primary lymphoma of bone in children: analysis of treatment results with Adriamycin, prednisone, Oncovin (APO) and local radiation therapy. J Clin Oncol. 1986;4:496.
256 Suryanarayan K, Shuster JJ, Donaldson SS, et al. Treatment of localized primary non-Hodgkin’s lymphoma of bone in children: a Pediatric Oncology Group study. J Clin Oncol. 1999;17:456.
257 Lones MA, Perkins SL, Sposto R, et al. Non-Hodgkin’s lymphoma arising in bone in children and adolescents is associated with an excellent outcome: a Children’s Cancer Group report. J Clin Oncol. 2002;20:2293.
258 Salzburg J, Burkhardt B, Zimmermann M, et al. Prevalence, clinical pattern, and outcome of CNS involvement in childhood and adolescent non-Hodgkin’s lymphoma differ by non-Hodgkin’s lymphoma subtype: a Berlin-Frankfurt-Munster Group Report. J Clin Oncol. 2007;25:3915.
259 Mandell LR, Wollner N, Fuks Z. Is cranial radiation necessary for CNS prophylaxis in pediatric NHL? Int J Radiat Oncol Biol Phys. 1987;13:359.
260 Murphy SB, Bleyer WA. Cranial irradiation is not necessary for central nervous system prophylaxis in pediatric non-Hodgkin’s lymphoma. Int J Radiat Oncol Biol. 1987;13:467.
261 Eden OB, Hann I, Imeson J, et al. Treatment of advanced stage T cell lymphoblastic lymphoma: results of the United Kingdom Children’s Cancer Study Group (UKCCSG) protocol 8503. Br J Haematol. 310, 1992.
262 Haddy TB, Sandlund JT, Magrath IT, et al. Testicular involvement in young patients with non-Hodgkin’s lymphoma. Am J Pediatr Hematol Oncol. 1988;10:224.
263 Kellie SJ, Pui CH, Murphy SB, et al. Childhood non-Hodgkin’s lymphoma involving the testis: clinical features and treatment outcome. J Clin Oncol. 1989;7:1066.
264 Dalle JH, Mechinaud F, Michon J, et al. Testicular disease in childhood B-cell non-Hodgkin’s lymphoma: the French Society of Pediatric Oncology experience. J Clin Oncol. 2001;19:2397.
265 Weitzman S, Suryanarayan K, Weinstein HJ. Pediatric non-Hodgkin’s lymphoma: Clinical and biologic prognostic factors and risk allocation. Curr Oncol Rpts. 2002;4:107.
266 Trainer KJ, Brisco MJ, Wan JH. Gene rearrangements in B- and T-lymphoproliferative disease detected by the polymerase chain reaction. Blood. 78, 1991.
267 Czuczman MS, Grillo-Lopez AJ, White CA, et al. Treatment of patients with low-grade B-cell lymphoma with the combination of chimeric anti-CD20 monoclonal antibody and CHOP chemotherapy. J Clin Oncol. 1999;17:268.
268 Milpied N, Vasseur B, Parquet N, et al. Humanized anti-CD20 monoclonal antibody (Rituximab) in post transplant B-lymphoproliferative disorder: a retrospective analysis on 32 patients. Ann Oncol. 2000;11(Suppl 1):113.
269 Maloney DG, Press OW. Newer treatments for non-Hodgkin’s lymphoma: monoclonal antibodies. Oncology. 1998;12:63.
270 Uckun FM, Reaman GH. Immunotoxins for treatment of leukemia and lymphoma. Leuk Lymphoma. 18, 1995.
271 Ansell SM, Witzig TE, Kurtin PJ, et al. Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood. 2002;99:67.
272 Kobrinsky NL, Sposto R, Narayan RS, et al. Outcomes of treatment of children and adolescents with recurrent non-Hodgkin’s lymphoma and Hodgkin’s disease with dexamethasone, etoposide, cisplatin, cytarabine, and L-Asparaginase, maintenance chemotherapy, and transplantation: Children’s Cancer Group Study CCG-5912. J Clin Oncol. 2001;19:2390.
273 McManaway ME, Neckers LM, Loke SL, et al. Tumour-specific inhibition of lymphoma growth by an antisense oligodeoxynucleotide. Lancet. 1990;335:808.
274 Rooney C, Smith C, Ng C, et al. Use of viral-specific cytotoxic lymphocytes to control Epstein-Barr virus-related lymphoproliferation. Lancet. 1995;345:9.
275 Roberts WK, Livingston PO, Agus DB, et al. Vaccination with CD20 peptides induces a biologically active, specific immune response in mice. Blood. 2002;99:3748.
276 Davis TA, Hsu FJ, Caspar CB, et al. Idiotype vaccination following ABMT can stimulate specific anti-idiotype immune responses in patients with B-cell lymphoma. Biol Blood Marrow Transplant. 2001;7:517.
277 Glaser SL, Jarrett RF. The epidemiology of Hodgkin’s disease. Baillieres Clin Haematol. 1996;9(3):401.
278 Grufferman S, Delzell E. Epidemiology of Hodgkin’s disease. Epidemiol Rev. 1984;6:76.
279 Belgaumi A, Al-Kofide A, Joseph N, et al. Hodgkin lymphoma in very young children: Clinical characteristics and outcome of treatment. Leuk Lymphoma. 2008;49:910.
280 Nachman JB, Sposto R, Herzog P, et al. Randomized comparison of low-dose involved-field radiotherapy and no radiotherapy for children with Hodgkin’s disease who achieve a complete response to chemotherapy. J Clin Oncol. 2002;20:3765.
281 Westergaard T, Melbye M, Pedersen JB, et al. Birth order, sibship size and risk of Hodgkin’s disease in children and young adults: a population-based study of 31 million person-years. Int J Cancer. 1997;72:977.
282 Glaser SL, Lin RJ, Stewart SL, et al. Epstein-Barr virus-associated Hodgkin’s disease: epidemiologic characteristics in international data. Int J Cancer. 1997;70:375.
283 Weinreb M, Day PJ, Niggli F, et al. The role of Epstein-Barr virus in Hodgkin’s disease from different geographical areas. Arch Dis Child. 1996;74:27.
284 Flavell KJ, Billingham LJ, Biddulph JP, et al. The effect of Epstein-Barr virus status on outcome in age- and sex-defined subgroups of patients with advanced Hodgkin’s disease. Ann Oncol. 2003;14:282.
285 Gandhi MK, Tellam JT, Khanna R. Epstein-Barr virus-associated Hodgkin’s lymphoma. Br J Haematol. 2004;125:267.
286 Krugmann J, Tzankov A, Gschwendtner A, et al. Longer failure-free survival interval of Epstein-Barr virus-associated classical Hodgkin’s lymphoma: a single-institution study. Mod Pathol. 2003;16:566.
287 Kaplan HS. Hodgkin’s Disease. Cambridge. MA: Harvard University Press; 1980. p 689
288 Shankar A, Fiumara F, Pinkerton R. Role of FDG PET in the management of childhood lymphomas–case proven or is the jury still out? Eur J Cancer. 2008;44:663.
289 Hines-Thomas M, Kaste SC, Hudson MM, et al. Comparison of gallium and PET scans at diagnosis and follow-up of pediatric patients with Hodgkin lymphoma. Pediatr Blood Cancer. 2008;51:198.
290 Baker LL, Parker BR, Donaldson SS, et al. Staging of Hodgkin disease in children: comparison of CT and lymphography with laparotomy. AJR Am J Roentgenol. 1990;154:1251.
291 Donaldson SS. Making choices in the staging of children with Hodgkin’s disease. Med Pediatr Oncol. 1991;19:211.
292 Hanna SL, Fletcher BD, Boulden TF, et al. MR imaging of infradiaphragmatic lymphadenopathy in children and adolescents with Hodgkin disease: comparison with lymphography and CT. J Magn Reson Imaging. 1993;3:461.
293 Cleary SF, Link MP, Donaldson SS. Hodgkin’s disease in the very young. Int J Radiat Oncol Biol Phys. 1994;28:77.
294 Ruhl U, Albrecht M, Dieckmann K, et al. Response-adapted radiotherapy in the treatment of pediatric Hodgkin’s disease: an interim report at 5 years of the German GPOH-HD 95 trial. Int J Radiat Oncol Biol Phys. 2001;51:1209.
295 Donaldson SS, Kaplan HS. Complications of treatment of Hodgkin’s disease in children. Cancer Treat Rep. 1982;66:977.
296 Mauch PM, Weinstein H, Botnick L, et al. An evaluation of long-term survival and treatment complications in children with Hodgkin’s disease. Cancer. 1983;51:925.
297 Donaldson SS, Link MP. Hodgkin’s disease. Treatment of the young child. Pediatr Clin North Am. 1991;38:457.
298 Donaldson SS, Whitaker SJ, Plowman PN, et al. Stage I-II pediatric Hodgkin’s disease: long-term follow-up demonstrates equivalent survival rates following different management schemes. J Clin Oncol. 1990;8:1128.
299 Schellong G, Bramswig JH, Hornig-Franz I. Treatment of children with Hodgkin’s disease–results of the German Pediatric Oncology Group. Ann Oncol. 1992;3(Suppl 4):73.
300 Schellong G, Potter R, Bramswig J, et al. High cure rates and reduced long-term toxicity in pediatric Hodgkin’s disease: the German-Austrian multicenter trial DAL-HD-90. The German-Austrian Pediatric Hodgkin’s Disease Study Group. J Clin Oncol. 1999;17:3736.
301 Shankar AG, Ashley S, Radford M, et al. Does histology influence outcome in childhood Hodgkin’s disease? Results from the United Kingdom Children’s Cancer Study Group. J Clin Oncol. 1997;15:2622.
302 Oberlin O, Leverger G, Pacquement H, et al. Low-dose radiation therapy and reduced chemotherapy in childhood Hodgkin’s disease: the experience of the French Society of Pediatric Oncology. J Clin Oncol. 1992;10:1602.
303 Landman-Parker J, Pacquement H, Leblanc T, et al. Localized childhood Hodgkin’s disease: response-adapted chemotherapy with etoposide, bleomycin, vinblastine, and prednisone before low-dose radiation therapy-results of the French Society of Pediatric Oncology Study MDH90. J Clin Oncol. 2000;18:1500.
304 Vecchi V, Pileri S, Burnelli R, et al. Treatment of pediatric Hodgkin disease tailored to stage, mediastinal mass, and age. An Italian (AIEOP) multicenter study on 215 patients. Cancer. 1993;72:2049.
305 Donaldson SS, Link MP, Weinstein HJ, et al. Final results of a prospective clinical trial with VAMP and low-dose involved-field radiation for children with low-risk Hodgkin’s disease. J Clin Oncol. 2007;25:332.
306 Hudson MM, Krasin M, Link MP, et al. Risk-adapted, combined-modality therapy with VAMP/COP and response-based, involved-field radiation for unfavorable pediatric Hodgkin’s disease. J Clin Oncol. 2004;22:4541.
307 Tebbi CK, Mendenhall N, London WB, et al. Treatment of stage I, IIA, IIIA1 pediatric Hodgkin disease with doxorubicin, bleomycin, vincristine and etoposide (DBVE) and radiation: a Pediatric Oncology Group (POG) study. Pediatr Blood Cancer. 2006;46:198.
308 Olweny CL, Katongole-Mbidde E, Kiire C, et al. Childhood Hodgkin’s disease in Uganda: a ten year experience. Cancer. 1978;42:787.
309 Hudson MM, Greenwald C, Thompson E, et al. Efficacy and toxicity of multiagent chemotherapy and low-dose involved-field radiotherapy in children and adolescents with Hodgkin’s disease. J Clin Oncol. 1993;11:100.
310 Hutchinson RJ, Fryer CJ, Davis PC, et al. MOPP or radiation in addition to ABVD in the treatment of pathologically staged advanced Hodgkin’s disease in children: results of the Children’s Cancer Group Phase III Trial. J Clin Oncol. 1998;16:897.
311 Weiner MA, Leventhal B, Brecher ML, et al. Randomized study of intensive MOPP-ABVD with or without low-dose total-nodal radiation therapy in the treatment of stages IIB, IIIA2, IIIB, and IV Hodgkin’s disease in pediatric patients: a Pediatric Oncology Group study. J Clin Oncol. 1997;15:2769.
312 Friedmann AM, Hudson MM, Weinstein HJ, et al. Treatment of unfavorable childhood Hodgkin’s disease with VEPA and low-dose, involved-field radiation. J Clin Oncol. 2002;20:3088.
313 Atra A, Higgs E, Capra M, et al. ChlVPP chemotherapy in children with stage IV Hodgkin’s disease: results of the UKCCSG HD 8201 and HD 9201 studies. Br J Haematol. 2002;119:647.
314 Ekert H, Waters KD, Smith PJ, et al. Treatment with MOPP or ChlVPP chemotherapy only for all stages of childhood Hodgkin’s disease. J Clin Oncol. 1988;6:1845.
315 Barrett A, Crennan E, Barnes J, et al. Treatment of clinical stage I Hodgkin’s disease by local radiation therapy alone. A United Kingdom Childrens Cancer Study Group study. Cancer. 1990;66:670.
316 Hoppe RT. Stage I-II Hodgkin’s disease: current therapeutic options and recommendations. Blood. 1983;62:32.
317 Mauch P, Tarbell N, Weinstein H, et al. Stage IA and IIA supradiaphragmatic Hodgkin’s disease: prognostic factors in surgically staged patients treated with mantle and paraaortic irradiation. J Clin Oncol. 1988;6:1576.
318 Gehan EA, Sullivan MP, Fuller LM, et al. The intergroup Hodgkin’s disease in children. A study of stages I and II. Cancer. 1990;65:1429.
319 Maity A, Goldwein JW, Lange B, et al. Comparison of high-dose and low-dose radiation with and without chemotherapy for children with Hodgkin’s disease: an analysis of the experience at the Children’s Hospital of Philadelphia and the Hospital of the University of Pennsylvania. J Clin Oncol. 1992;10:929.
320 Donaldson SS, Hudson MM, Lamborn KR, et al. VAMP and low-dose, involved-field radiation for children and adolescents with favorable, early-stage Hodgkin’s disease: results of a prospective clinical trial. J Clin Oncol. 2002;20:3081.
321 Donaldson SS, Link MP. Combined modality treatment with low-dose radiation and MOPP chemotherapy for children with Hodgkin’s disease. J Clin Oncol. 1987;5:742.
322 Dionet C, Oberlin O, Habrand JL, et al. Initial chemotherapy and low-dose radiation in limited fields in childhood Hodgkin’s disease: results of a joint cooperative study by the French Society of Pediatric Oncology (SFOP) and Hospital Saint-Louis, Paris. Int J Radiat Oncol Biol Phys. 1988;15:341.
323 Schellong G, Bramswig JH, Hornig-Franz I, et al. Hodgkin’s disease in children: combined modality treatment for stages IA, IB, and IIA. Results in 356 patients of the German/Austrian Pediatric Study Group. Ann Oncol. 1994;5(Suppl 2):113.
324 Weiner MA, Leventhal BG, Marcus R, et al. Intensive chemotherapy and low-dose radiotherapy for the treatment of advanced-stage Hodgkin’s disease in pediatric patients: a Pediatric Oncology Group study. J Clin Oncol. 1991;9:1591.
325 Beaty O, Hudson MM, Greenwald C, et al. Subsequent malignancies in children and adolescents after treatment for Hodgkin’s disease. J Clin Oncol. 1995;13:603.
326 Bhatia S, Robison LL, Oberlin O, et al. Breast cancer and other second neoplasms after childhood Hodgkin’s disease. N Engl J Med. 1996;334:745.
327 Green DM, Gingell RL, Pearce J, et al. The effect of mediastinal irradiation on cardiac function of patients treated during childhood and adolescence for Hodgkin’s disease. J Clin Oncol. 1987;5:239.
328 Green DM, Hyland A, Barcos MP, et al. Second malignant neoplasms after treatment for Hodgkin’s disease in childhood or adolescence. J Clin Oncol. 2000;18:1492.
329 Green DM, Hyland A, Chung CS, et al. Cancer and cardiac mortality among 15-year survivors of cancer diagnosed during childhood or adolescence. J Clin Oncol. 1999;17:3207.
330 Hancock SL, Donaldson SS, Hoppe RT. Cardiac disease following treatment of Hodgkin’s disease in children and adolescents. J Clin Oncol. 1993;11:1208.
331 Metayer C, Lynch CF, Clarke EA, et al. Second cancers among long-term survivors of Hodgkin’s disease diagnosed in childhood and adolescence. J Clin Oncol. 2000;18:2435.
332 Sankila R, Garwicz S, Olsen JH, et al. Risk of subsequent malignant neoplasms among 1,641 Hodgkin’s disease patients diagnosed in childhood and adolescence: a population-based cohort study in the five Nordic countries. Association of the Nordic Cancer Registries and the Nordic Society of Pediatric Hematology and Oncology. J Clin Oncol. 1996;14:1442.
333 Schellong G, Riepenhausen M, Creutzig U, et al. Low risk of secondary leukemias after chemotherapy without mechlorethamine in childhood Hodgkin’s disease. German-Austrian Pediatric Hodgkin’s Disease Group. J Clin Oncol. 1997;15:2247.
334 Scholz KH, Herrmann C, Tebbe U, et al. Myocardial infarction in young patients with Hodgkin’s disease–potential pathogenic role of radiotherapy, chemotherapy, and splenectomy. Clin Investig. 1993;71:57.
335 Swerdlow AJ, Barber JA, Hudson GV, et al. Risk of second malignancy after Hodgkin’s disease in a collaborative British cohort: the relation to age at treatment. J Clin Oncol. 2000;18:498.
336 Swerdlow AJ, Schoemaker MJ, Allerton R, et al. Lung cancer after Hodgkin’s disease: a nested case-control study of the relation to treatment. J Clin Oncol. 2001;19:1610.
337 Tarbell NJ, Gelber RD, Weinstein HJ, et al. Sex differences in risk of second malignant tumours after Hodgkin’s disease in childhood. Lancet. 1993;341:1428.
338 Tucker MA, Coleman CN, Cox RS, et al. Risk of second cancers after treatment for Hodgkin’s disease. N Engl J Med. 1988;318:76.
339 van Leeuwen FE, Klokman WJ, Veer MB, et al. Long-term risk of second malignancy in survivors of Hodgkin’s disease treated during adolescence or young adulthood. J Clin Oncol. 2000;18(3):487-497.
340 Wolden SL, Hancock SL, Carlson RW, et al. Management of breast cancer after Hodgkin’s disease. J Clin Oncol. 2000;18:765.
341 Wolden SL, Lamborn KR, Cleary SF, et al. Second cancers following pediatric Hodgkin’s disease. J Clin Oncol. 1998;16:536.
342 Schwartz CL, Constine LS, London W, et al. POG 9425: response-based, intensively timed therapy for intermediate/high stage (IS/HS) pediatric Hodgkin’s disease. Proc Am Soc Clin Oncol. 2002;21:1555. Abstr
343 Gallamini A, Hutchings M, Rigacci L, et al. Early interim 1-[18F]flouro-2-deoxy-D-glucose positron emission tomography is prognostically superior to International Prognostic Score in advanced-stage Hodgkin’s lymphoma: a report from the joint Italian-Danish study. J Clin Oncol. 2007;25:3746.
344 Picardi M, de Renzo A, Pane F, et al. Randomized comparison of consolidative radiation versus observation in bulky Hodgkin’s lymphoma with post-chemotherapy negative positron emission tomography scans. Leuk Lymphoma. 2007;48:1721.
345 Hudson MM, Poquette CA, Lee J, et al. Increased mortality after successful treatment for Hodgkin’s disease. J Clin Oncol. 1998;16(11):3592-3600.
346 LaMonte CS, Yeh SD, Straus DJ. Long-term follow-up of cardiac function in patients with Hodgkin’s disease treated with mediastinal irradiation and combination chemotherapy including doxorubicin. Cancer Treat Rep. 1986;70(4):439-444.
347 Coleman CN, Williams CJ, Flint A, et al. Hematologic neoplasia in patients treated for Hodgkin’s disease. N Engl J Med. 1977;297:1249.
348 Anselmo AP, Cartoni C, Bellantuono P, et al. Risk of infertility in patients with Hodgkin’s disease treated with ABVD vs MOPP vs ABVD/MOPP. Haematologica. 1990;75:155.
349 Chapman RM, Sutcliffe SB, Malpas JS. Male gonadal dysfunction in Hodgkin’s disease. A prospective study. JAMA. 1981;245:1323-1328.
350 Hobbie WL, Ginsberg JP, Ogle SK, et al. Fertility in males treated for Hodgkins disease with COPP/ABV hybrid. Pediatr Blood Cancer. 2005;44:193.
351 Mackie EJ, Radford M, Shalet SM. Gonadal function following chemotherapy for childhood Hodgkin’s disease. Med Pediatr Oncol. 1996;27:74.
352 Viviani S, Santoro A, Ragni G, et al. Gonadal toxicity after combination chemotherapy for Hodgkin’s disease. Comparative results of MOPP vs ABVD. Eur J Cancer Clin Oncol. 1985;21:601.
353 Byrne J, Fears TR, Gail MH, et al. Early menopause in long-term survivors of cancer during adolescence. Am J Obstet Gynecol. 1992;166:788.
354 Chemaitilly W, Mertens AC, Mitby P, et al. Acute ovarian failure in the childhood cancer survivor study. J Clin Endocrinol Metab. 2006;91:1723.
355 Horning SJ, Hoppe RT, Kaplan HS, et al. Female reproductive potential after treatment for Hodgkin’s disease. N Engl J Med. 1981;304:1377.
356 Ortin TT, Shostak CA, Donaldson SS. Gonadal status and reproductive function following treatment for Hodgkin’s disease in childhood: the Stanford experience. Int J Radiat Oncol Biol Phys. 1990;19:873.
357 Sklar CA, Mertens AC, Mitby P, et al. Premature menopause in survivors of childhood cancer: a report from the childhood cancer survivor study. J Natl Cancer Inst. 2006;98:890.
358 Bonadonna G, Santoro A. ABVD chemotherapy in the treatment of Hodgkin’s disease. Cancer Treat Rev. 1982;9:21.
359 Valagussa P, Santoro A, Fossati Bellani F, et al. Absence of treatment-induced second neoplasms after ABVD in Hodgkin’s disease. Blood. 1982;59:488.
360 Santoro A, Bonadonna G, Valagussa P, et al. Long-term results of combined chemotherapy-radiotherapy approach in Hodgkin’s disease: superiority of ABVD plus radiotherapy versus MOPP plus radiotherapy. J Clin Oncol. 1987;5:27.
361 Hudson MM. Pediatric Hodgkin’s therapy: time for a paradigm shift. J Clin Oncol. 2002;20:3755.
362 Hudson MM. Achieving cure for early stage pediatric Hodgkin disease with minimal morbidity: are we there yet? Pediatr Blood Cancer. 2006;46:122.
363 Hamilton VM, Norris C, Bunin N, et al. Cyclophosphamide-based, seven-drug hybrid and low-dose involved field radiation for the treatment of childhood and adolescent Hodgkin disease. J Pediatr Hematol Oncol. 2001;23:84.
364 Korholz D, Claviez A, Hasenclever D, et al. The concept of the GPOH-HD 2003 therapy study for pediatric Hodgkin’s disease: evolution in the tradition of the DAL/GPOH studies. Klin Padiatr. 2004;216:150.
365 Kelly KM, Hutchinson RJ, Sposto R, et al. Feasibility of upfront dose-intensive chemotherapy in children with advanced-stage Hodgkin’s lymphoma: preliminary results from the Children’s Cancer Group Study CCG-59704. Ann Oncol. 2002;13(Suppl 1):107.
366 Schwartz CL, Special issues in pediatric Hodgkin’s disease, Eur J Haematol; Suppl 66; 2005:55.
367 Baez F, Ocampo E, Conter V, et al. Treatment of childhood Hodgkin’s disease with COPP or COPP-ABV (hybrid) without radiotherapy in Nicaragua. Ann Oncol. 1997;8:247.
368 Bayle-Weisgerber C, Lemercier N, Teillet F, et al. Hodgkin’s disease in children. Results of therapy in a mixed group of 178 clinical and pathologically staged patients over 13 years. Cancer. 1984;54:215.
369 Behrendt H, Brinkhuis M, van Leeuwen EF. Treatment of childhood Hodgkin’s disease with ABVD without radiotherapy. Med Pediatr Oncol. 1996;26:244.
370 Behrendt H, van Bunningen BN, van Leeuwen EF. Treatment of Hodgkin’s disease in children with or without radiotherapy. Cancer. 1987;59:1870.
371 Jacobs P, King HS, Karabus C, et al. Hodgkin’s disease in children. A ten-year experience in South Africa. Cancer. 1984;53:210.
372 Jenkin D, Doyle J, Berry M, et al. Hodgkin’s disease in children: treatment with MOPP and low-dose, extended field irradiation without laparotomy. Late results and toxicity. Med Pediatr Oncol. 1990;18:265.
373 Lobo-Sanahuja F, Garcia I, Barrantes JC, et al. Pediatric Hodgkin’s disease in Costa Rica: twelve years’ experience of primary treatment by chemotherapy alone, without staging laparotomy. Med Pediatr Oncol. 1994;22:398.
374 Sackmann-Muriel F, Bonesana AC, Pavlovsky S, et al. Hodgkin’s disease in childhood: therapy results in Argentina. Am J Pediatr Hematol Oncol. 1981;3:247.
375 Sripada PV, Tenali SG, Vasudevan M, et al. Hybrid (COPP/ABV) therapy in childhood Hodgkin’s disease: a study of 53 cases during 1989-1993 at the Cancer Institute, Madras. Pediatr Hematol Oncol. 1995;12:333.
376 van den Berg H, Stuve W, Behrendt H. Treatment of Hodgkin’s disease in children with alternating mechlorethamine, vincristine, procarbazine, and prednisone (MOPP) and adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD) courses without radiotherapy. Med Pediatr Oncol. 1997;29:23.
377 Sandoval C, Venkateswaran L, Billups C, et al. Lymphocyte-predominant Hodgkin disease in children. J Pediatr Hematol Oncol. 2002;24:269.
378 Karayalcin G, Behm FG, Gieser PW, et al. Lymphocyte predominant Hodgkin disease: clinico-pathologic features and results of treatment–the Pediatric Oncology Group experience. Med Pediatr Oncol. 1997;29:519.
379 Mauz-Korholz C, Gorde-Grosjean S, Hasenclever D, et al. Resection alone in 58 children with limited stage, lymphocyte-predominant Hodgkin lymphoma-experience from the European network group on pediatric Hodgkin lymphoma. Cancer. 2007;110:179.
380 Pellegrino B, Terrier-Lacombe MJ, Oberlin O, et al. Lymphocyte-predominant Hodgkin’s lymphoma in children: therapeutic abstention after initial lymph node resection–a study of the French Society of Pediatric Oncology. J Clin Oncol. 2003;21:2948.
381 Hunger SP, Link MP, Donaldson SS. ABVD/MOPP and low-dose involved-field radiotherapy in pediatric Hodgkin’s disease: the Stanford experience. J Clin Oncol. 1994;12:2160.
382 Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med. 2003;348:2396.
383 Bader SB, Weinstein H, Mauch P, et al. Pediatric stage IV Hodgkin disease. Long-term survival. Cancer. 1993;72:249.
384 Akpek G, Ambinder RF, Piantadosi S, et al. Long-term results of blood and marrow transplantation for Hodgkin’s lymphoma. J Clin Oncol. 2001;19:4314.
385 Anderson JE, Litzow MR, Appelbaum FR, et al. Allogeneic, syngeneic, and autologous marrow transplantation for Hodgkin’s disease: the 21-year Seattle experience. J Clin Oncol. 1993;11:2342.
386 Baker KS, Gordon BG, Gross TG, et al. Autologous hematopoietic stem-cell transplantation for relapsed or refractory Hodgkin’s disease in children and adolescents. J Clin Oncol. 1999;17:825.
387 Desch CE, Lasala MR, Smith TJ, et al. The optimal timing of autologous bone marrow transplantation in Hodgkin’s disease patients after a chemotherapy relapse. J Clin Oncol. 1992;10:200.
388 Sureda A, Arranz R, Iriondo A, et al. Autologous stem-cell transplantation for Hodgkin’s disease: results and prognostic factors in 494 patients from the Grupo Espanol de Linfomas/Transplante Autologo de Medula Osea Spanish Cooperative Group. J Clin Oncol. 2001;19:1395.
389 Williams CD, Goldstone AH, Pearce R, et al. Autologous bone marrow transplantation for pediatric Hodgkin’s disease: a case-matched comparison with adult patients by the European Bone Marrow Transplant Group Lymphoma Registry. J Clin Oncol. 1993;11:2243.
390 Lieskovsky YE, Donaldson SS, Torres MA, et al. High-dose therapy and autologous hematopoietic stem-cell transplantation for recurrent or refractory pediatric Hodgkin’s disease: results and prognostic indices. J Clin Oncol. 2004;22:4532.
391 Schellong G, Dorffel W, Claviez A, et al. Salvage therapy of progressive and recurrent Hodgkin’s disease: results from a multicenter study of the pediatric DAL/GPOH-HD study group. J Clin Oncol. 2005;23(25):6181-6189.
392 Constine LS, Rapoport AP. Hodgkin’s disease, bone marrow transplantation, and involved field radiation therapy: coming full circle from 1902 to 1996. Int J Radiat Oncol Biol Phys. 1996;36:253.
393 Wadhwa P, Shina DC, Schenkein D, et al. Should involved-field radiation therapy be used as an adjunct to lymphoma autotransplantation? Bone Marrow Transplant. 2002;29:183.
394 Constine LS, Marcus R, Chauvenet A, et al. Patterns of failure after response-based, dose-dense therapy for intermediate/high risk pediatric Hodgkin’s disease (POG 9425). Int J Radiat Oncol Biol Phys. 2005;63:S21-S22.
395 Shahidi M, Kamangari N, Ashley S, et al. Site of relapse after chemotherapy alone for stage I and II Hodgkin’s disease. Radiother Oncol. 2006;78:1.
396 Girinsky T, van der Maazen R, Specht L, et al. Involved-node radiotherapy (INRT) in patients with early Hodgkin lymphoma: concepts and guidelines. Radiother Oncol. 2006;79:270.
397 Libshitz HI, Edeiken BS. Radiotherapy changes of the pediatric hip. AJR Am J Roentgenol. 1981;137:585.
398 Maguire A, Craft AW, Evans RG, et al. The long-term effects of treatment on the dental condition of children surviving malignant disease. Cancer. 1987;60:2570.
399 Silverman CL, Thomas PR, McAlister WH, et al. Slipped femoral capital epiphyses in irradiated children: dose, volume and age relationships. Int J Radiat Oncol Biol Phys. 1981;7:1357.
400 Willman KY, Cox RS, Donaldson SS. Radiation induced height impairment in pediatric Hodgkin’s disease. Int J Radiat Oncol Biol Phys. 1994;28:85.
401 Fryer CJ, Hutchinson RJ, Krailo M, et al. Efficacy and toxicity of 12 courses of ABVD chemotherapy followed by low-dose regional radiation in advanced Hodgkin’s disease in children: a report from the Children’s Cancer Study Group. J Clin Oncol. 1990;8:1971.
402 Marina NM, Greenwald CA, Fairclough DL, et al. Serial pulmonary function studies in children treated for newly diagnosed Hodgkin’s disease with mantle radiotherapy plus cycles of cyclophosphamide, vincristine, and procarbazine alternating with cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine. Cancer. 1995;75:1706.
403 Mefferd JM, Donaldson SS, Link MP. Pediatric Hodgkin’s disease: pulmonary, cardiac, and thyroid function following combined modality therapy. Int J Radiat Oncol Biol Phys. 1989;16:679.
404 Adams MJ, Hardenbergh PH, Constine LS, et al. Radiation-associated cardiovascular disease. Crit Rev Oncol Hematol. 2003;45:55.
405 Constine LS, Donaldson SS, McDougall IR, et al. Thyroid dysfunction after radiotherapy in children with Hodgkin’s disease. Cancer. 1984;53:878.
406 Sklar C, Whitton J, Mertens A, et al. Abnormalities of the thyroid in survivors of Hodgkin’s disease: data from the Childhood Cancer Survivor Study. J Clin Endocrinol Metab. 2000;85:3227.
407 Bramswig JH, Heimes U, Heiermann E, et al. The effects of different cumulative doses of chemotherapy on testicular function. Results in 75 patients treated for Hodgkin’s disease during childhood or adolescence. Cancer. 1990;65:1298.
408 Le Floch O, Donaldson SS, Kaplan HS. Pregnancy following oophoropexy and total nodal irradiation in women with Hodgkin’s disease. Cancer. 1976;38:2263.
409 Pedrick TJ, Hoppe RT. Recovery of spermatogenesis following pelvic irradiation for Hodgkin’s disease. Int J Radiat Oncol Biol Phys. 1986;12:117.
410 Jockovich M, Mendenhall NP, Sombeck MD, et al. Long-term complications of laparotomy in Hodgkin’s disease. Ann Surg. 1994;219:615.
411 Adams MJ, Lipshultz SE. Pathophysiology of anthracycline- and radiation-associated cardiomyopathies: implications for screening and prevention. Pediatr Blood Cancer. 2005;44:600.
412 Aleman BM, van den Belt-Dusebout AW, De Bruin ML, et al. Late cardiotoxicity after treatment for Hodgkin lymphoma. Blood. 2007;109:1878.
413 Swerdlow AJ, Higgins CD, Smith P, et al. Myocardial infarction mortality risk after treatment for Hodgkin disease: a collaborative British cohort study. J Natl Cancer Inst. 2007;99:206.
414 Lipshultz SE, Colan SD, Gelber RD, et al. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;324(12):808-815.
415 Metzger ML, Howard SC, Hudson MM, et al. Natural history of thyroid nodules in survivors of pediatric Hodgkin lymphoma. Pediatr Blood Cancer. 2006;46(3):314-319.
416 Kinsella TJ, Trivette G, Rowland J, et al. Long-term follow-up of testicular function following radiation therapy for early-stage Hodgkin’s disease. J Clin Oncol. 1989;7(6):718-724.
417 da Cunha MF, Meistrich ML, Fuller LM, et al. Recovery of spermatogenesis after treatment for Hodgkin’s disease: limiting dose of MOPP chemotherapy. J Clin Oncol. 1984;2(6):571-577.
418 Basu SK, Schwartz C, Fisher LD, et al. Unilateral and bilateral breast cancer in women surviving pediatric Hodgkin’s disease. Int J Radiat Oncol Biol Phys. 2008;72(1):34-40.
419 Bhatia S, Yasui Y, Robison LL, et al. High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin’s disease: report from the Late Effects Study Group. J Clin Oncol. 2003;21(23):4386-4394.
420 Constine LS, Tarbell N, Hudson MM, et al. Subsequent malignancies in children treated for Hodgkin’s disease: associations with gender and radiation dose. Int J Radiat Oncol Biol Phys. 2008;72(1):24-33.
421 Hodgson DC, Gilbert ES, Dores GM, et al. Long-term solid cancer risk among 5-year survivors of Hodgkin’s lymphoma. J Clin Oncol. 2007;25(12):1489-1497.
422 Hudson MM, Pratt CB. Risk of delayed second primary neoplasms after treatment of malignant lymphoma. Surg Onc Clin N Am. 1993;2:319.