Chapter 71 Pediatric Leukemias and Lymphomas
Epidemiology
ALL accounts for 75% of childhood leukemia and is diagnosed in 2500 to 3000 children in the United States annually1,2 with a peak age incidence between 2 and 5 years. The incidence of ALL is higher among boys than girls. This difference is more pronounced in pubertal children3 and particularly evident in cases of T-cell ALL. In the United States, ALL occurs more commonly among white children than among African-American children.4
Etiology
A single etiology of ALL has not been identified; however, several predisposing risk factors include genetics, environmental factors, viral infection, and immunodeficiency. Genetics appears to play a role in the cause of ALL in some children as evidenced by the association between specific chromosomal abnormalities and childhood ALL. The most common constitutional chromosomal abnormality associated with childhood leukemia is trisomy 21 or Down syndrome5; less common chromosomal abnormalities include Kleinfelter’s syndrome and trisomy G syndrome.6 The majority of cases of leukemia in children cannot be attributed to any known factor.
ALL is believed to develop from the malignant transformation of a single abnormal progenitor cell that then expands indefinitely. In pediatric ALL, evidence suggests that it occurs in committed lymphoid precursors. In contrast, in Philadelphia chromosome–positive ALL and in pediatric acute myelogenous leukemia (AML) this event may occur earlier, because mutations are observed in multiple cell lineages.7,8 The sequence of events resulting in malignant transformation is likely to be multifactorial. Leukemia can develop along any point in the normal stages of lymphoid development, which reflects the fact that ALL is a biologically heterogeneous disorder.
Molecular Biology and Classification
ALL has been classified according to morphologic, immunologic, cytogenetic, biochemical, and molecular genetic characteristics. The most widely accepted morphologic classification of ALL is that proposed by the French-American-British (FAB) Cooperative Working Group.9,10 The FAB system defines three categories of lymphoblasts:
Immunophenotyping
The immunobiologic studies of ALL cells indicate that leukemic transformation and clonal expansion occurs at different stages of lymphocyte maturation. The use of monoclonal antibodies has classified leukemias; however, these monoclonal antibodies are not purely lineage specific and so the term lineage associated is used. In 80% to 85% of cases lymphoblasts are found to have surface markers consistent with B-cell lineage, although very few have surface immunoglobulin (found on mature B cells). Most cases of B-cell lineage ALL in children and adolescents are found to have the common ALL antigen (CALLA) CD10 on their cell surfaces.11,12 Patients with B-cell precursor ALL whose lymphoblasts express the common ALL antigen (CALLA) or CD10 on their cell surfaces have a more favorable prognosis than those who do not, primarily owing to the strong association of CD10 negativity with rearrangements of the MLL gene on chromosome 11.13,14 Children with mature B-cell ALL (characterized by the presence of mature B-cell antigens, including surface immunoglobulin) have a poorer prognosis than those of earlier B-cell lineage if treated with standard ALL therapies; such patients are more appropriately treated on regimens for patients with advanced-stage Burkitt’s lymphoma. Ten to 15 percent of children will have ALL that is of T-cell origin. In contrast to B-cell precursor ALL, T-cell ALL is more frequently associated with older age at diagnosis, a higher presenting leukocyte count, and bulky extramedullary disease. Extramedullary involvement can include an anterior mediastinal mass, lymphadenopathy, hepatosplenomegaly, and overt CNS involvement.15
Cytogenetics
Advances in cytogenetics have led to an increasing understanding of the biology of ALL.16 Cytogenetic abnormalities identified in ALL include abnormalities in chromosomal number (ploidy) as well as those in chromosomal structure. Ploidy is determined either by counting the modal number of chromosomes in a preparation of a metaphase karyotype or by measuring DNA content by flow cytometry. Hyperdiploidy exists when there are more than 46 chromosomes. Most cases of ALL are diploid or hyperdiploid. Ploidy in childhood B-lineage leukemia has prognostic significance.17,18,19 Children with higher ploidy, greater than 50 chromosomes, have a better prognosis than other children with ALL, including those with 47 to 50 chromosomes; those with 51 to 65 chromosomes (higher hyperdiploid) with specific trisomies (including chromosomes 4, 10, and 17) appear to have an especially favorable prognosis.20,21,22 Those patients who have hyperdiploid leukemia with favorable trisomies also tend to have other favorable prognostic features (see later). Hypodiploidy (modal chromosomal number less than 44) is associated with inferior outcomes, especially among those with near-haploid ALL, with 24 to 28 chromosomes.23,24,25
Structural chromosomal abnormalities occur in ALL leukemia cells. The most common of these abnormalities are translocations. Translocations lead to alterations in the regulation of oncogenes. The activation of oncogenes and the loss of tumor suppressor genes are examples of the altered regulation believed to be involved in the evolution of leukemia. The most frequently identified translocation in ALL is t(12;21) and leads to the TEL-AML1 fusion protein. This occurs in approximately 20% of cases (nearly exclusively in those with B-lineage phenotype) and is associated with a favorable prognosis.26,27 In contrast, t(9;22) and t(4;11) translocations are associated with early treatment failure.19,28
The t(9;22)(q34;q11) translocation results in the formation of the Philadelphia (Ph) chromosome and is found in 5% of childhood ALL. Children with Ph chromosome–positive ALL have a poor response to standard therapy but may fare better with intensive regimens that include the use of a tyrosine kinase inhibitor such as imatinib.29,30,31,32,33
Presentation and Evaluation
At the time of initial diagnosis the evaluation of patients includes cerebrospinal fluid examination, evaluation of uric acid levels, liver function tests, renal function tests, coagulation screening, chest radiography, evaluation for infection, and echocardiography (Table 71-1). Manifestations of CNS involvement such as headache, lethargy, papilledema, and cranial neuropathies (most often the third, fourth, sixth, and seventh) are infrequently present at diagnosis. CNS involvement is divided into three categories, based on the number of WBCs and the presence of lymphoblasts on the Cytospin. CNS-1 is defined as no evidence of lymphoblasts in the cerebrospinal fluid. CNS-2 is defined as fewer than 5 WBCs per microliter with blasts present in the Cytospin, and CNS-3 is defined as 5 or more WBCs per microliter with blasts in the Cytospin or the presence of cranial nerve palsy. CNS-3 involvement of the CNS at diagnosis is found in less than 5% of children with ALL.34
* Bone marrow studies: morphology, cytogenetics, immunophenotype, cytochemistry.
† Therapeutic lumbar puncture performed for diagnosis concurrent with administration of intrathecal chemotherapy.
Prognostic Factors
Several clinical and biologic factors determined at diagnosis have been identified and shown to have prognostic significance (Table 71-2). These prognostic factors are used to assign patients to risk strata that tailor the treatment. These factors have been integral in the development of all modern therapeutic trials. Although many of these factors have shown prognostic importance, not all are currently used to determine risk stratification.
TABLE 71-2 Prognostic Factors in Acute Lymphoblastic Leukemia
The presenting leukocyte counts and age at diagnosis are uniformly accepted as prognostic features. Children with the highest initial WBC count have a poorer prognosis.35,36 Infants younger than 1 year of age have the worst prognosis, and adolescents also have a poorer event-free survival than younger children, except those younger than age 1 year.37,38 Children older than 10 often have other poor risk features, including T-cell phenotype.38 Infants with ALL have shown the worst outcome, with an event-free survival of 10% to 20%.37 Infants also have other poor-risk features, including very high presenting WBC count, presence of CNS leukemia, massive organomegaly, and a poor day 14 response to induction chemotherapy.37 Infant ALL has unique biologic features, including chromosomal abnormalities that are associated with a worse prognosis. Structural abnormalities of chromosome 11, such as rearrangement of band q23, within the MLL/ALL1 gene are often present.39,40 The t(4;11) translocation is commonly seen, and many have leukemia cells that coexpress myeloid markers (CD15).37,39,41 This finding suggests that infant ALL arises in a multipotent precursor cell. Chromosomal abnormalities in the leukemia cells also have prognostic significance. The presence of the Ph chromosome and of MLL rearrangements (even in noninfant patients) is associated with a higher risk of relapse. T-cell immunophenotype and the presence of CNS disease at diagnosis are also considered high-risk features. Early responses to initial therapy are also prognostic. These include the time to achieve remission and the decrease in peripheral blast count after administration of corticosteroids, when longer time to remission or poor response to initial corticosteroid therapy is associated with worse outcomes. Rapid morphologic clearance of blasts in the marrow 7 to 14 days after the initiation of multiple-agent chemotherapy has been associated with more favorable outcomes.
Nearly all current leukemia protocols include measurements of minimal residual disease at the end of induction chemotherapy. Multiple-parameter flow cytometry and polymerase chain reaction assays of leukemia-specific gene arrangements have been used to assess submicroscopic levels of minimal residual disease soon after the initiation of therapy.42,43,44,45 High levels of minimal residual disease at the end of the first month of therapy and other early time points has been associated with a very high risk of subsequent relapse.46,47,48,49,50
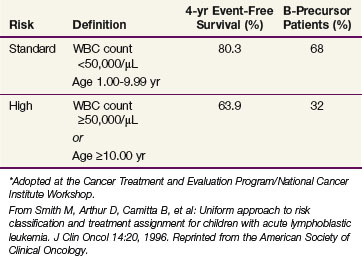
As therapy for ALL has become more intensive and the outcomes have improved, many of these factors have lost statistical significance as prognostic factors. In addition, the definitions of high leukocyte counts and age varied among cooperative groups. In order to allow comparisons of results, the National Cancer Institute held a workshop to develop a uniform set of prognostic factors.51 The age and WBC count criteria for childhood ALL agreed on at the Cancer Treatment and Evaluation Program/National Cancer Institute Workshop are shown in Table 71-3.
Treatment
The foregoing prognostic indicators are used to stratify children with ALL into risk groups. The stratification is dependent on prompt, accurate evaluation and sophisticated techniques. To accomplish this, as well as to treat patients with the increased intensity of current therapy, evaluation and treatment is most appropriately carried out in recognized pediatric oncology centers. The treatments for the different risk groups vary in intensity and by institution. However, the framework for all risk groups includes four key elements: remission induction, CNS preventative therapy, consolidation, and maintenance (Fig. 71-1).
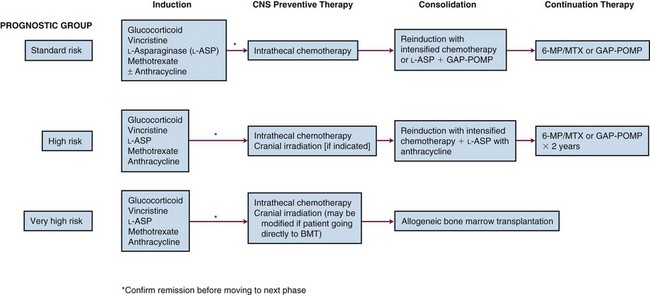
Induction treatment is designed to achieve complete remission, defined as no evidence of leukemia. Peripheral blood cell counts must be within the normal range, and the bone marrow must be of normal cellularity and contain less than 5% lymphoblasts. There should be no evidence of CNS disease on examination of the cerebrospinal fluid, and there must be no extramedullary disease. Remission can be achieved in 85% of children with ALL using vincristine and a glucocorticoid; however, the addition of L-asparaginase and/or the addition of an anthracycline will improve the remission induction rate to 95%.52 In general, most current regimens utilize a three- or four-drug induction phase, including vincristine, a corticosteroid, and L-asparaginase plus or minus an anthracycline.
The prevention of CNS disease is one of the major elements of successful ALL treatment. The approach to CNS prophylaxis has evolved over the decades since it was first incorporated into ALL therapy in the 1970s. Craniospinal irradiation and cranial irradiation plus intrathecal methotrexate reduced the relapse rate in the CNS from 50% to less than 10%.53 To avoid the myelosuppression and the effects on spinal growth, intrathecal methotrexate was substituted for spinal irradiation as part of the standard CNS preventative therapy. Intrathecal therapy has been shown to successfully prevent CNS relapse in standard-risk patients and avoids the neurocognitive sequelae and secondary tumors that can result from cranial irradiation. Intrathecal chemotherapy in conjunction with intensive systemic therapy has also provided adequate CNS prevention for some children with high-risk features.
Once complete remission has been achieved, consolidation and maintenance therapy are used to provide continued cytoreduction of the leukemic cell burden without permitting the emergence of drug-resistant clones. Components of the consolidation phase vary according to risk group, with more intensive treatments used for patients classified as high or very-high risk. The continuation phase consists of less-intensive, low-dose chemotherapy. Nearly all regimens include weekly methotrexate and daily 6-mercaptopurine during this phase. Some protocols also use intermittent pulses of vincristine and corticosteroid in addition to methotrexate and 6-mercaptopurine.54 Total duration of therapy is typically 2 to 3 years. On some regimens, boys receive a longer maintenance phase than girls.
Indications for Radiation Therapy
In most regimens, CNS prophylaxis for patients at lower risk is achieved with systemic and intrathecal chemotherapy without cranial irradiation. Children with high-risk features are at an increased risk of CNS relapse and, historically, have received prophylactic cranial irradiation. These features include a presenting WBC count of 50,000/µL or greater; those with WBC counts over 100,000/µL are at particularly high risk of CNS relapse. Additional high-risk features that are indications on some treatment protocols for cranial irradiation are T-cell phenotype, Ph chromosome–positive ALL, and the presence of t(4;11). Infants younger than age 12 months with 11q23 abnormalities are at very high risk of CNS relapse but because of their young age are usually treated without cranial irradiation, using intensified systemic and intrathecal chemotherapy to treat the CNS. Historically, the standard dose for high-risk ALL cranial prophylaxis had been 1800 cGy; however, published trials from the Berlin-Frankfurt-Munster group used 1200 cGy in patients with CNS-1 disease with very good results.12 The standard dose for prophylactic cranial irradiation in those high-risk patients still treated with irradiation is now 1200 cGy.
Currently, fewer than 20% of children with ALL are treated with prophylactic cranial irradiation. Investigators are continuing to evaluate whether cranial irradiation can be eliminated in a greater proportion of patients, especially in very young children in whom the toxicities from irradiation are the most significant. Some protocols have omitted cranial irradiation entirely, using strategies that intensify the use of intrathecal chemotherapy and CNS-penetrant systemic chemotherapy, such as high-dose methotrexate.55,56 In one such study from St. Jude Children’s Research Hospital, the 5-year cumulative risk of isolated CNS relapse was 2.7% and that of any CNS relapse (isolated or combined with bone marrow) was 3.9%, although the risk of relapse was higher in some high-risk patient subsets.56
Children who present with CNS-3 disease at diagnosis, regardless of the other features of their disease, are deemed at high risk and are considered to have meningeal leukemia. The treatment of these patients varies, and some protocols continue to employ craniospinal irradiation or cranial irradiation in addition to intrathecal chemotherapy. Irradiation is not administered until the patient is in remission, including clearance of the cerebrospinal fluid. After achievement of complete remission, 1800-cGy cranial irradiation has been administered early in the therapy.12
Isolated CNS relapse is rare with the current approach to ALL treatment. The treatment depends on the time from first remission as well as the extent of prior CNS therapy. Patients with an isolated CNS relapse occurring 18 months or more after initial diagnosis have an event-free survival of approximately 80% when treated with intensive systemic chemotherapy and cranial or craniospinal irradiation. A dose of 1800 cGy to the cranial field has been shown to be effective in such patients.57 Patients with earlier CNS relapses, or those with T-cell phenotype or a history of cranial irradiation prior to CNS relapse, may be considered for stem cell transplant once a second remission is achieved. In that situation the total dose of craniospinal irradiation may need to be modified depending on timing of the transplant.
Cranial nerve palsies can be seen in patients with meningeal leukemia. Urgent RT to the base of the skull initially to reverse the palsy is often given, with full cranial irradiation given at the scheduled time for CNS treatment. Doses between 1000 and 1200 cGy are used. Although the use of higher doses have been reported, no dose response was observed for higher doses.58
Testicular Leukemia
Testicular involvement that is clinically apparent at diagnosis is infrequent; however, occult disease is found in 25% of boys.59 Overt involvement is manifest as painless testicular enlargement, which may be unilateral or bilateral. Testicular biopsies during maintenance or at the conclusion of all therapy have been abandoned because the biopsy results did not accurately predict eventual relapse.17,60,61 Some protocols have used prophylactic bilateral testicular irradiation, although this approach has not been widely used because the current systemic therapy protocols have lessened the rate of testicular relapse and testicular irradiation causes permanent sterility.62,63
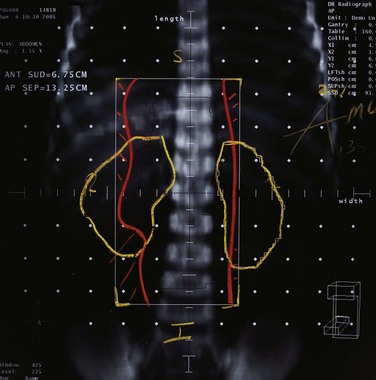
Testicular relapse occurs in less than 5% of cases in the current era of intensive therapy.64,65 Both systemic therapy as well as local RT, directed at the testes, are used to treat testicular relapse. Re-induction therapy is needed as well as CNS-directed treatment. Bilateral testicular irradiation is used in doses of 2400 to 2600 cGy in 200-cGy fractions.66 The treatment is delivered with the child supine in the frog-leg position with the soles of the feet touching. The penis is taped or secured out of the RT field. A bolus dose may be needed to ensure adequate dose to the testes, or electron boost therapy can be used. Radiation is delivered to the entire scrotal contents.
Late Sequelae
The CNS sequelae are of particular concern. Neurocognitive functional impairment has been observed in ALL survivors. Lower doses of cranial irradiation, for example, 1200 cGy rather than 1800 cGy in high-risk patients, and the elimination of cranial irradiation from standard-risk patients should help lessen the late sequelae. However, in a prospective randomized trial comparing 1800 cGy with no cranial irradiation, all patients received intrathecal and intravenous methotrexate and the irradiated patients had no worse neurocognitive effects.67 Children irradiated when younger than 8 years of age and particularly when younger than 5 years of age appear to experience the most severe neurocognitive sequelae. Leukoencephalopathy is a devastating and fortunately uncommon event in the current era. It is characterized by multifocal demyelination. Although this occurs most commonly in patients who have received higher cumulative doses of RT as well as higher cumulative doses of intrathecal and systemic methotrexate (e.g., those with recurrent meningeal leukemia), this severe form of neurotoxicity has been observed in children who received no cranial irradiation.68,69
Neuroendocrine sequelae involving the hypothalamic-pituitary axis have been observed in ALL survivors treated with cranial irradiation. Most commonly seen is growth hormone deficiency, which appears to be dose related in that children treated with 1800 cGy have reduced growth hormone levels compared with healthy controls but not as low as levels as those treated with 2400 cGy.70 However, growth delay and short stature have also been seen in unirradiated ALL survivors, suggesting that the cause is likely to be multifactorial.
The risk of second tumors in ALL survivors is estimated to be between 3% and 12% at 5 to 24 years after the initial diagnosis.71,72 Second solid tumors have most commonly occurred in patients treated with cranial irradiation and are within or near the RT field. However, there are reports of ALL patients not irradiated who developed secondary CNS tumors.73 The most common hematopoietic tumors are AML and myelodysplastic syndrome.71,72
Testicular irradiation results in sterility. Some studies have shown elevated follicle-stimulating hormone and luteinizing hormone levels, decreased testosterone levels, and delays in sexual maturation, suggesting that the endocrine function of the testes may be adversely affected at doses of 2400 cGy and higher.74,75 Supplemental or replacement androgens may be required.
Acute Myelogenous Leukemia
AML comprises a group of hematologic malignancies that arise from the precursors of the myeloid, monocytoid, erythroid, and megakaryocytic cell lineages. AML represents 15% to 25% of childhood leukemia but accounts for over 30% of deaths from leukemia. The classification used in AML is the FAB schema, which defines seven subtypes of AML (M1 to M7), characterized by morphologic, histochemical, immunophenotypic, and cytogenetic features (Table 71-4). An M0 subtype has been used for an acute undifferentiated leukemia.76
TABLE 71-4 Morphology of Acute Myelogenous Leukemia: French-American-British (FAB) Classification System
| FAB Type | Name |
|---|---|
| M1 | Acute myeloblastic: no maturation |
| M2 | Acute myeloblastic with maturation |
| M3 | Acute promyelocytic: hypergranular type |
| M3v | Acute promyelocytic: microgranular variant |
| M4 | Acute myelomonocytic |
| M4Eo | Acute myelomonocytic with eosinophilia |
| M5a | Acute monocytic |
| M5b | Acute monocytic with differentiation |
| M6 | Acute erythroleukemia |
| M7 | Acute megakaryoblastic |
Treatment of AML involves first stabilization of the patient at diagnosis, aggressive supportive care to treat potential complications such disseminated intravascular coagulation, infectious complications, tumor lysis syndrome, and hyperleukostasis. Induction of remission involves multiagent chemotherapy. Induction is followed by postremission consolidation, CNS prophylaxis, and, in some studies, maintenance chemotherapy. Induction therapy for AML most commonly includes cytarabine coupled with an anthracycline plus or minus additional agents such as etoposide, thioguanine, or dexamethasone.77,78 Treatment of M3 AML, a disease characterized by promyelocytic morphology and the presence of t(15;17), includes all-trans-retinoic acid in addition to cytotoxic chemotherapy.
Postremission therapy, particularly with increased dose intensity of cytarabine, resulted in lower relapse rates and longer remission duration.79,80 CNS chemoprophylaxis is used for all patients. Children with the highest risk of a CNS relapse are those with M4 or M5 AML or with very high presenting peripheral blast counts. CNS relapse is generally followed by bone marrow relapse. With the use of prophylaxis in all patients with AML, the incidence of CNS relapse is 5%.79,81
Extramedullary disease outside the CNS can also occur, such as the development of chloromas, most commonly in M4 and M5 subtypes. There was no benefit to the use of local RT to treat chloromas, in addition to systemic therapy in children.82 If a chloroma is causing significant morbidity, such as vision loss or spinal cord compression, RT is indicated.
Because the use of intensified chemotherapy has improved the event-free survival in children with AML, this raised the question as to whether even greater dose intensification, such as with myeloablative therapy and stem cell rescue, could improve the event-free survival even more. Randomized trials have shown that children with AML who undergo allogeneic stem cell transplants from human leukocyte antigen–matched siblings have an improved relapse-free survival.83,84,85–89 Postremission consolidation therapy with allogeneic bone marrow transplantation has become the standard of care for children with high-risk AML based on the presence of certain molecular features and/or a poor response to initial therapy. Total-body irradiation is often used as a part of the myeloablative regimen for bone marrow transplantation.
Non-Hodgkin’s Lymphoma
Lymphomas in children comprise the third most common malignancy of childhood, superceded only by acute lymphoblastic leukemia and brain tumors. In children younger than age 15 years, 60% of cases are NHL. If one includes children through age 18, Hodgkin’s disease is slightly more frequent.90 NHL is diagnosed annually in the United States in approximately 500 children. The median age at diagnosis is 10 years; NHL is less frequent in children younger than age 3 years. There is no bimodal age distribution; the frequency increases with increasing age. For unknown reasons, NHL is more common in the white population than in blacks and two to three times more frequent in males than in females.1 Specific populations at increased risk of developing NHL include children with congenital immunodeficiency disorders such as Wiskott-Aldrich syndrome, ataxia-telangiectasia, and X-linked lymphoproliferative syndrome.91
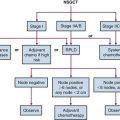
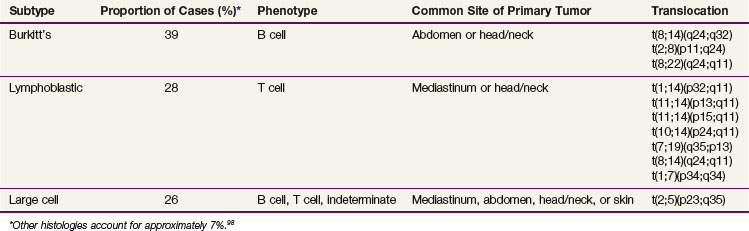
In contrast to NHL in adults, NHL in children is primarily aggressive, with diffuse tumors and frequently extranodal presentation. There are three main subtypes: Burkitt’s, lymphoblastic, and large cell (diffuse large B-cell lymphoma and anaplastic large cell lymphoma)92 (Table 71-5). The staging system used in childhood NHL is the Murphy/St. Jude’s Children’s Research Hospital staging system, which incorporates extent of disease and clinical patterns93 (Table 71-6). Treatment of childhood NHL is based on the stage, immunophenotype, and histology.
| Stage | Description |
|---|---|
| I |
Burkitt’s lymphoma, historically referred to as small non–cleaved cell lymphoma in the National Cancer Institute’s Working Formulation, makes up 39% of childhood NHL and is of B-cell immunophenotype. These lymphomas present in the abdomen or in the head and neck. The epidemiology of Burkitt’s lymphoma is of great interest because of its major differences in incidence and clinical features based on geography. For example, it is the most common childhood malignancy in equatorial Africa yet is very rare in Japan.94 In Africa, the endemic subtype occurs and is characterized by younger age at diagnosis and frequent involvement of the jaw, abdomen, orbit, and paraspinal area.94 The sporadic subtype, which occurs in the United States and Western Europe, is characterized by older median age at diagnosis and frequent involvement of the abdomen, nasopharynx, and bone marrow. These tumors are characterized by translocations that result in the juxtaposition of the CMYC gene and an immunoglobulin gene; there are breakpoint differences within these translocations that have also been reported to vary with geography.94 The association of Epstein-Barr virus with Burkitt’s lymphoma varies with geography; Epstein-Barr virus is highly associated with the endemic (African) subtype.94
The treatment of Burkitt’s lymphoma uses cyclophosphamide-based chemotherapy, with the addition of agents including vincristine (Oncovin), prednisone, doxorubicin (Adriamycin), and methotrexate. Children with advanced-stage Burkitt’s lymphoma have an improved outcome with the addition of cytarabine or high-dose methotrexate. Children with bone marrow or CNS involvement have an improved outcome, with more intensive regimens resulting in an 80% to 90% event-free survival even for patients with advanced-stage disease.95–98,99
Lymphoblastic lymphomas make up approximately 29% of childhood NHL. Children with lymphoblastic lymphoma typically present with advanced-stage disease (i.e., mediastinal mass which in some cases is associated with bone marrow involvement); however, some children do present with limited-stage disease. These lymphomas share morphologic, immunophenotypic, and cytogenetic features with acute lymphoblastic leukemia. The majority (~90%) are characterized by a T-cell immunophenotype and are designated as precursor T-lymphoblastic leukemia/lymphoma in the World Health Organization classification system; the remainder have a precursor B-cell immunophenotype.92 The distinction between ALL and lymphoblastic lymphoma is made arbitrarily on the basis of degree of bone marrow involvement. Those with greater than 25% replacement of the marrow by lymphoblasts are considered to have ALL, whereas those with a lesser degree of involvement are considered to have advanced stage (IV) lymphoblastic lymphoma. Whether there is a biologic distinction between lymphoblastic leukemia and lymphoblastic lymphoma is somewhat controversial; however, there appear to be subtle differences in the molecular biology, cytogenetics, and immunophenotypic markers between these two entities.93,100,101 The treatment of lymphoblastic lymphoma in children uses multiple-agent chemotherapy comprising drugs similar to those used for T-cell ALL, which include vincristine, prednisone, L-asparaginase, doxorubicin, cytarabine, cyclophosphamide, and high-dose methotrexate. The total duration of therapy generally ranges from 2 to 3 years, although shorter durations have been attempted. With contemporary treatment protocols, between 80% to 85% of patients are long-term event-free survivors.102–104
Large cell lymphomas represent approximately 27% of childhood lymphomas. Forty to 50 percent are T-cell anaplastic large cell lymphomas (ALCL, CD30+ and often associated with t(2;5),105 30% to 40% are diffuse large B-cell lymphomas (DLBCL), which include the mediastinal large B-cell lymphomas (MLBCL), and the remainder are nonanaplastic mature T-cell lymphomas.106 Historically, the treatment of childhood NHL in the United States has been based on histology. However, there has been a shift toward using an immunophenotype-directed approach, which has been the standard practice in Europe for many years. With this approach, children with DLBCL are treated with regimens for children with Burkitt’s lymphoma and achieve a comparable outcome.96 The MLBCLs have a slightly poorer prognosis than the DLBCLs. Those with ALCL are treated with either cyclophosphamide, hydroxydaunomycin (doxorubicin), vincristine (Oncovin), and prednisone (CHOP)-based regimens or regimens derived from Burkitt’s lymphoma regimens.107,108,109,110 A randomized comparison of ACOP versus APO (doxorubicin [Adriamycin], vincristine [Oncovin], prednisone with or without cyclophosphamide) showed no significant difference in outcome.110,111 The optimal approach for treatment of anaplastic NHL in children has not been established. Approximately 70% of children are long-term event-free survivors regardless of approach.108,109
The role of local RT in the management of NHL in children has been more limited because of the improved outcomes with more effective chemotherapy. In a prospective randomized trial evaluating the need for local RT in early-stage NHL,112 no benefit was found for the addition of local RT. A second trial,113 built on the results of the first, confirmed this. Although children with primary lymphoma of bone were not randomized in the first trial and all received local RT, in the second trial RT was not given and there were no local relapses. Based on these results, the current management of localized NHL of bone includes chemotherapy but does not use consolidative local RT. Children with advanced-stage lymphoma do not benefit from involved-field RT.114
Cranial irradiation has been eliminated as a component of CNS prophylaxis in the management of children with NHL.97,104 RT is used for some cases of relapse or refractory disease and as a palliative modality. Children with persistent disease after chemotherapy or who experience relapse are often considered for high-dose chemotherapy protocols with stem cell rescue if they have chemosensitive disease.115 Consolidative RT can be considered either before or after transplant in some cases. There are no prospective randomized trials in pediatrics to confirm the benefit of this approach.
Urgent RT is rare, but there are occasions when it is considered. Mediastinal masses in children can cause significant airway compromise or superior vena cava compression. If a tissue diagnosis cannot be established to allow administration of systemic therapy, urgent RT may be considered. It should be noted that if one treats empirically with systemic therapy for the most likely diagnosis, the correct treatment is given in the vast majority of cases.116 Radiation oncologists are often called to be aware of children with newly diagnosed mediastinal masses. If RT is given, only a few fractions are needed to achieve stabilization and allow appropriate systemic therapy to be initiated once a histologic diagnosis is made.
9 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451-458.
10 Bennett JM, Catovsky D, Daniel MT, et al. The morphological classification of acute lymphoblastic leukaemia. Concordance among observers and clinical correlations. Br J Haematol. 1981;47:553-561.
11 Abshire TC, Buchanan GR, Jackson JF, et al. Morphologic, immunologic and cytogenetic studies in children with acute lymphoblastic leukemia at diagnosis and relapse. A Pediatric Oncology Group study. Leukemia. 1992;6:357-362.
18 Trueworthy R, Shuster J, Look T, et al. Ploidy of lymphoblasts is the strongest predictor of treatment outcome in B-progenitor cell acute lymphoblastic leukemia of childhood. A Pediatric Oncology Group study. J Clin Oncol. 1992;10:606-613.
19 Chromosomal abnormalities and their clinical significance in acute lymphoblastic leukemia. Third International Workshop on Chromosomes in Leukemia. Cancer Res. 1983;43:868-873.
20 Pui CH, Carroll AJ, Head D, et al. Near-triploid and near-tetraploid acute lymphoblastic leukemia of childhood. Blood. 1990;76:590-596.
21 Rubin CM, Le Beau MM. Cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1991;13:202-216.
25 Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112-1115.
26 Loh ML, Goldwasser MA, Silverman LB, et al. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood. 2006;107:4508-4513.
28 Bloomfield CD, Goldman AI, Alimena G, et al. Chromosomal abnormalities identify high-risk and low-risk patients with acute lymphoblastic leukemia. Blood. 1986;67:415-420.
29 Behm FG, Raimondi SC, Frestedt JL, et al. Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood. 1996;87:2870-2877.
31 Fletcher JA, Lynch EA, Kimball VM, et al. Translocation (9;22) is associated with extremely poor prognosis in intensively treated children with acute lymphoblastic leukemia. Blood. 1991;77:435-439.
32 Uckun FM, Nachman JB, Sather HN, et al. Clinical significance of Philadelphia chromosome positive pediatric acute lymphoblastic leukemia in the context of contemporary intensive therapies. A report from the Children’s Cancer Group. Cancer. 1998;83:2030-2039.
34 Bleyer WA. Central nervous system leukemia. Pediatr Clin North Am. 1988;35:789-814.
41 Katz F, Simpson E, Lam G, Gibbons B. Rearrangement of T-cell receptor and immunoglobulin heavy chain genes in childhood acute mixed lineage leukaemia. Leuk Res. 1988;12:955-960.
42 van Dongen JJ, Seriu T, Panzer-Grumayer ER, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352:1731-1738.
43 Coustan-Smith E, Behm FG, Sanchez J, et al. Immunological detection of minimal residual disease in children with acute lymphoblastic leukaemia. Lancet. 1998;351:550-554.
46 Cave H, van der Werff ten Bosch J, Suciu S, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer—Childhood Leukemia Cooperative Group. N Engl J Med. 1998;339:591-598.
47 Coustan-Smith E, Sancho J, Hancock ML, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood. 2000;96:2691-2696.
49 Panzer-Grumayer ER, Schneider M, Panzer S, et al. Rapid molecular response during early induction chemotherapy predicts a good outcome in childhood acute lymphoblastic leukemia. Blood. 2000;95:790-794.
51 Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14:18-24.
54 Bleyer WA, Sather HN, Nickerson HJ, et al. Monthly pulses of vincristine and prednisone prevent bone marrow and testicular relapse in low-risk childhood acute lymphoblastic leukemia. A report of the CCG-161 study by the Children’s Cancer Study Group. J Clin Oncol. 1991;9:1012-1021.
56 Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730-2741.
57 Miller DR, Coccia PF, Bleyer WA, et al. Early response to induction therapy as a predictor of disease-free survival and late recurrence of childhood acute lymphoblastic leukemia. A report from the Children’s Cancer Study Group. J Clin Oncol. 1989;7:1807-1815.
63 Nesbit ME, Sather H, Robison LL, et al. Sanctuary therapy. A randomized trial of 724 children with previously untreated acute lymphoblastic leukemia: A report from the Children’s Cancer Study Group. Cancer Res. 1982;42:674-680.
67 Waber DP, Shapiro BL, Carpentieri SC, et al. Excellent therapeutic efficacy and minimal late neurotoxicity in children treated with 18 grays of cranial radiation therapy for high-risk acute lymphoblastic leukemia. A 7-year follow-up study of the Dana-Farber Cancer Institute Consortium Protocol 87-01. Cancer. 2001;92:15-22.
69 Rubinstein LJ, Herman MM, Long TF, Wilbur JR. Disseminated necrotizing leukoencephalopathy. A complication of treated central nervous system leukemia and lymphoma. Cancer. 1975;35:291-305.
71 Neglia JP, Meadows AT, Robison LL, et al. Second neoplasms after acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;325:1330-1336.
72 Kimball Dalton VM, Gelber RD, Li F, et al. Second malignancies in patients treated for childhood acute lymphoblastic leukemia. J Clin Oncol. 1998;16:2848-2853.
73 Nygaard R, Garwicz S, Haldorsen T, et al. Second malignant neoplasms in patients treated for childhood leukemia. A population-based cohort study from the Nordic countries. The Nordic Society of Pediatric Oncology and Hematology (NOPHO). Acta Paediatr Scand. 1991;80:1220-1228.
75 Brauner R, Czernichow P, Cramer P, et al. Leydig-cell function in children after direct testicular irradiation for acute lymphoblastic leukemia. N Engl J Med. 1983;309:25-28.
79 Creutzig U, Ritter J, Schellong G. Identification of two risk groups in childhood acute myelogenous leukemia after therapy intensification in study AML-BFM-83 as compared with study AML-BFM-78. AML-BFM Study Group. Blood. 1990;75:1932-1940.
81 Woods WG, Kobrinsky N, Buckley JD, et al. Timed-sequential induction therapy improves postremission outcome in acute myeloid leukemia. A report from the Children’s Cancer Group. Blood. 1996;87:4979-4989.
83 Dahl GV, Kalwinsky DK, Mirro JJr, et al. Allogeneic bone marrow transplantation in a program of intensive sequential chemotherapy for children and young adults with acute nonlymphocytic leukemia in first remission. J Clin Oncol. 1990;8:295-303.
84 Cassileth PA, Harrington DP, Appelbaum FR, et al. Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in first remission. N Engl J Med. 1998;339:1649-1656.
93 Murphy SB. Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas. Dissimilarities from lymphomas in adults. Semin Oncol. 1980;7:332-339.
94 Magrath IT, Bhati K. Pathogenesis of small noncleaved cell lymphomas (Burkitt’s lymphoma. In: Magrath IT, editor. The Non-Hodgkin’s Lymphomas. London: Arnold; 1997:385-409.
95 Patte C, Auperin A, Michon J, et al. The Société Française d’Oncologie Pédiatrique LMB89 protocol. Highly effective multiagent chemotherapy tailored to the tumor burden and initial response in 561 unselected children with B-cell lymphomas and L3 leukemia. Blood. 2001;97:3370-3379.
96 Patte C, Auperin A, Gerrard M, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents. It is possible to reduce treatment for the early responding patients. Blood. 2007;109:2773-2780.
97 Cairo MS, Gerrard M, Sposto R, et al. Results of a randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007;109:2736-2743.
98 Reiter A, Schrappe M, Tiemann M, et al. Improved treatment results in childhood B-cell neoplasms with tailored intensification of therapy. A report of the Berlin-Frankfurt-Munster Group Trial NHL-BFM 90. Blood. 1999;94:3294-3306.
100 Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms. A proposal from the International Lymphoma Study Group. Blood. 1994;84:1361-1392.
102 Reiter A, Schrappe M, Ludwig WD, et al. Intensive ALL-type therapy without local radiotherapy provides a 90% event-free survival for children with T-cell lymphoblastic lymphoma. A BFM group report. Blood. 2000;95:416-421.
103 Sandlund JT, Pui CH, Zhou Y, et al. Effective treatment of advanced-stage childhood lymphoblastic lymphoma without prophylactic cranial irradiation. Results of St Jude NHL13 study. Leukemia. 2009;23(6):1127-1130.
104 Burkhardt B, Woessmann W, Zimmermann M, et al. Impact of cranial radiotherapy on central nervous system prophylaxis in children and adolescents with central nervous system-negative stage III or IV lymphoblastic lymphoma. J Clin Oncol. 2006;24:491-499.
107 Reiter A, Schrappe M, Tiemann M, et al. Successful treatment strategy for Ki-1 anaplastic large-cell lymphoma of childhood. A prospective analysis of 62 patients enrolled in three consecutive Berlin-Frankfurt-Munster group studies. J Clin Oncol. 1994;12:899-908.
109 Seidemann K, Tiemann M, Schrappe M, et al. Short-pulse B-non-Hodgkin lymphoma-type chemotherapy is efficacious treatment for pediatric anaplastic large cell lymphoma. A report of the Berlin-Frankfurt-Munster Group Trial NHL-BFM 90. Blood. 2001;97:3699-3706.
110 Laver JH, Mahmoud H, Pick TE, et al. Results of a randomized phase III trial in children and adolescents with advanced stage diffuse large cell non-Hodgkin’s lymphoma. A Pediatric Oncology Group study. Leuk Lymphoma. 2002;43:105-109.
111 Hutchison RE, Berard CW, Shuster JJ, et al. B-cell lineage confers a favorable outcome among children and adolescents with large-cell lymphoma. A Pediatric Oncology Group study. J Clin Oncol. 1995;13(8):2023-2032.
112 Link MP, Shuster JJ, Donaldson SS, et al. Treatment of children and young adults with early-stage non-Hodgkin’s lymphoma. N Engl J Med. 1997;337:1259-1266.
1 Gurney JG, Severson RK, Davis S, Robison LL. Incidence of cancer in children in the United States. Sex-, race-, and 1-year age-specific rates by histologic type. Cancer. 1995;75:2186-2195.
2 Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer J Clin. 2000;50:7-33.
3 Fraumeni JFJr, Wagoner JK. Changing sex differentials in leukemia. Public Health Rep. 1964;79:1093-1100.
4 Pollock BH, DeBaun MR, Camitta BM, et al. Racial differences in the survival of childhood B-precursor acute lymphoblastic leukemia. A Pediatric Oncology Group Study. J Clin Oncol. 2000;18:813-823.
5 Dordelmann M, Schrappe M, Reiter A, et al. Down’s syndrome in childhood acute lymphoblastic leukemia. Clinical characteristics and treatment outcome in four consecutive BFM trials. Berlin-Frankfurt-Munster Group. Leukemia. 1998;12:645-651.
6 Muts-Homsma SJ, Muller HP, Geraedst JP. Klinefelter’s syndrome and acute non-lymphocytic leukemia. Blut. 1982;44:15-20.
7 Schenk TM, Keyhani A, Bottcher S, et al. Multilineage involvement of Philadelphia chromosome positive acute lymphoblastic leukemia. Leukemia. 1998;12:666-674.
8 Kasprzyk A, Harrison CJ, Secker-Walker LM. Investigation of clonal involvement of myeloid cells in Philadelphia-positive and high hyperdiploid acute lymphoblastic leukemia. Leukemia. 1999;13:2000-2006.
9 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451-458.
10 Bennett JM, Catovsky D, Daniel MT, et al. The morphological classification of acute lymphoblastic leukaemia. Concordance among observers and clinical correlations. Br J Haematol. 1981;47:553-561.
11 Abshire TC, Buchanan GR, Jackson JF, et al. Morphologic, immunologic and cytogenetic studies in children with acute lymphoblastic leukemia at diagnosis and relapse. A Pediatric Oncology Group study. Leukemia. 1992;6:357-362.
12 Reiter A, Schrappe M, Ludwig WD, et al. Chemotherapy in 998 unselected childhood acute lymphoblastic leukemia patients. Results and conclusions of the multicenter trial ALL-BFM 86. Blood. 1994;84:3122-3133.
13 Greaves MF. Differentiation-linked leukemogenesis in lymphocytes. Science. 1986;234:697-704.
14 Crist W, Boyett J, Roper M, et al. Pre-B cell leukemia responds poorly to treatment. A Pediatric Oncology Group study. Blood. 1984;63:407-414.
15 Hann IM, Richards SM, Eden OB, Hill FG. Analysis of the immunophenotype of children treated on the Medical Research Council United Kingdom Acute Lymphoblastic Leukaemia Trial XI (MRC UKALLXI). Medical Research Council Childhood Leukaemia Working Party. Leukemia. 1998;12:1249-1255.
16 Chessels JM, Swansbury GJ, Reeves B, et al. Cytogenetics and prognosis in childhood lymphoblastic leukaemia. Results of MRC UKALL X. Medical Research Council Working Party in Childhood Leukaemia. Br J Haematol. 1997;99:93-100.
17 Dahl GV, Rivera G, Pui CH, et al. A novel treatment of childhood lymphoblastic non-Hodgkin’s lymphoma. Early and intermittent use of teniposide plus cytarabine. Blood. 1985;66:1110-1114.
18 Trueworthy R, Shuster J, Look T, et al. Ploidy of lymphoblasts is the strongest predictor of treatment outcome in B-progenitor cell acute lymphoblastic leukemia of childhood. A Pediatric Oncology Group study. J Clin Oncol. 1992;10:606-613.
19 Chromosomal abnormalities and their clinical significance in acute lymphoblastic leukemia. Third International Workshop on Chromosomes in Leukemia. Cancer Res. 1983;43:868-873.
20 Pui CH, Carroll AJ, Head D, et al. Near-triploid and near-tetraploid acute lymphoblastic leukemia of childhood. Blood. 1990;76:590-596.
21 Rubin CM, Le Beau MM. Cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1991;13:202-216.
22 Heerema NA, Sather HN, Sensel MG, et al. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (>50 chromosomes). J Clin Oncol. 2000;18:1876-1887.
23 Brodeur GM, Williams DL, Look AT, et al. Near-haploid acute lymphoblastic leukemia. A unique subgroup with a poor prognosis? Blood. 1981;58:14-19.
24 Heerema NA, Nachman JB, Sather HN, et al. Hypodiploidy with less than 45 chromosomes confers adverse risk in childhood acute lymphoblastic leukemia. A report from the Children’s Cancer Group. Blood. 1999;94:4036-4045.
25 Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112-1115.
26 Loh ML, Goldwasser MA, Silverman LB, et al. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood. 2006;107:4508-4513.
27 Rubnitz JE, Wichlan D, Devidas M, et al. Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia. A Children’s Oncology Group study. J Clin Oncol. 2008;26:2186-2191.
28 Bloomfield CD, Goldman AI, Alimena G, et al. Chromosomal abnormalities identify high-risk and low-risk patients with acute lymphoblastic leukemia. Blood. 1986;67:415-420.
29 Behm FG, Raimondi SC, Frestedt JL, et al. Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood. 1996;87:2870-2877.
30 Fleming ID, Turk PS, Murphy SB, et al. Surgical implications of primary gastrointestinal lymphoma of childhood. Arch Surg. 1990;125:252-256.
31 Fletcher JA, Lynch EA, Kimball VM, et al. Translocation (9;22) is associated with extremely poor prognosis in intensively treated children with acute lymphoblastic leukemia. Blood. 1991;77:435-439.
32 Uckun FM, Nachman JB, Sather HN, et al. Clinical significance of Philadelphia chromosome positive pediatric acute lymphoblastic leukemia in the context of contemporary intensive therapies. A report from the Children’s Cancer Group. Cancer. 1998;83:2030-2039.
33 Colleoni GW, Bridge JA, Garicochea B, et al. ATIC-ALK: A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am J Pathol. 2000;156:781-789.
34 Bleyer WA. Central nervous system leukemia. Pediatr Clin North Am. 1988;35:789-814.
35 Simone JV, Verzosa MS, Rudy JA. Initial features and prognosis in 363 children with acute lymphocytic leukemia. Cancer. 1975;36:2099-2108.
36 Sather HN. Age at diagnosis in childhood acute lymphoblastic leukemia. Med Pediatr Oncol. 1986;14:166-172.
37 Reaman G, Zeltzer P, Bleyer WA, et al. Acute lymphoblastic leukemia in infants less than one year of age. A cumulative experience of the Children’s Cancer Study Group. J Clin Oncol. 1985;3:1513-1521.
38 Crist W, Pullen J, Boyett J, et al. Acute lymphoid leukemia in adolescents. Clinical and biologic features predict a poor prognosis—a Pediatric Oncology Group Study. J Clin Oncol. 1988;6:34-43.
39 Ludwig WD, Bartram CR, Harbott J, et al. Phenotypic and genotypic heterogeneity in infant acute leukemia. I. Acute lymphoblastic leukemia. Leukemia. 1989;3:431-439.
40 Katz F, Malcolm S, Gibbons B, et al. Cellular and molecular studies on infant null acute lymphoblastic leukemia. Blood. 1988;71:1438-1447.
41 Katz F, Simpson E, Lam G, Gibbons B. Rearrangement of T-cell receptor and immunoglobulin heavy chain genes in childhood acute mixed lineage leukaemia. Leuk Res. 1988;12:955-960.
42 van Dongen JJ, Seriu T, Panzer-Grumayer ER, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352:1731-1738.
43 Coustan-Smith E, Behm FG, Sanchez J, et al. Immunological detection of minimal residual disease in children with acute lymphoblastic leukaemia. Lancet. 1998;351:550-554.
44 Weir EG, Cowan K, LeBeau P, Borowitz MJ. A limited antibody panel can distinguish B-precursor acute lymphoblastic leukemia from normal B precursors with four color flow cytometry. Implications for residual disease detection. Leukemia. 1999;13:558-567.
45 Li A, Zhou J, Zuckerman D, et al. Sequence analysis of clonal immunoglobulin and T-cell receptor gene rearrangements in children with acute lymphoblastic leukemia at diagnosis and at relapse. Implications for pathogenesis and for the clinical utility of PCR-based methods of minimal residual disease detection. Blood. 2003;102:4520-4526.
46 Cave H, van der Werff ten Bosch J, Suciu S, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer—Childhood Leukemia Cooperative Group. N Engl J Med. 1998;339:591-598.
47 Coustan-Smith E, Sancho J, Hancock ML, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood. 2000;96:2691-2696.
48 Nyvold C, Madsen HO, Ryder LP, et al. Precise quantification of minimal residual disease at day 29 allows identification of children with acute lymphoblastic leukemia and an excellent outcome. Blood. 2002;99:1253-1258.
49 Panzer-Grumayer ER, Schneider M, Panzer S, et al. Rapid molecular response during early induction chemotherapy predicts a good outcome in childhood acute lymphoblastic leukemia. Blood. 2000;95:790-794.
50 Zhou J, Goldwasser MA, Li A, et al. Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood. 2007;110:1607-1611.
51 Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14:18-24.
52 Ortega JA, Nesbit MEJr, Donaldson MH, et al. L-Asparaginase, vincristine, and prednisone for induction of first remission in acute lymphocytic leukemia. Cancer Res. 1977;37:535-540.
53 Aur RJ, Simone JV, Hustu HO, Verzosa MS. A comparative study of central nervous system irradiation and intensive chemotherapy early in remission of childhood acute lymphocytic leukemia. Cancer. 1972;29:381-391.
54 Bleyer WA, Sather HN, Nickerson HJ, et al. Monthly pulses of vincristine and prednisone prevent bone marrow and testicular relapse in low-risk childhood acute lymphoblastic leukemia. A report of the CCG-161 study by the Children’s Cancer Study Group. J Clin Oncol. 1991;9:1012-1021.
55 Uyttebroeck A, Suciu S, Laureys G, et al. Treatment of childhood T-cell lymphoblastic lymphoma according to the strategy for acute lymphoblastic leukaemia, without radiotherapy. Long term results of the EORTC CLG 58881 trial. Eur J Cancer. 2008;44:840-846.
56 Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730-2741.
57 Miller DR, Coccia PF, Bleyer WA, et al. Early response to induction therapy as a predictor of disease-free survival and late recurrence of childhood acute lymphoblastic leukemia. A report from the Children’s Cancer Study Group. J Clin Oncol. 1989;7:1807-1815.
58 Ha CS, Chung WK, Koller CA, Cox JD. Role of radiation therapy to the brain in leukemic patients with cranial nerve palsies in the absence of radiological findings. Leuk Lymphoma. 1999;32:497-503.
59 Kim TH, Hargreaves HK, Brynes RK, et al. Pretreatment testicular biopsy in childhood acute lymphocytic leukaemia. Lancet. 1981;2:657-658.
60 Nachman J, Palmer NF, Sather HN, et al. Open-wedge testicular biopsy in childhood acute lymphoblastic leukemia after two years of maintenance therapy. Diagnostic accuracy and influence on outcome—a report from Children’s Cancer Study Group. Blood. 1990;75:1051-1055.
61 Kim TH, Hargreaves HK, Chan WC, et al. Sequential testicular biopsies in childhood acute lymphocytic leukemia. Cancer. 1986;57:1038-1041.
62 Kay H, Rankin A. Testicular irradiation in leukaemia. Lancet. 1981;2:1115.
63 Nesbit ME, Sather H, Robison LL, et al. Sanctuary therapy. A randomized trial of 724 children with previously untreated acute lymphoblastic leukemia: A Report from Children’s Cancer Study Group. Cancer Res. 1982;42:674-680.
64 Cáp J, Foltinová A, Misíková Z. Prognostic significance of testicular relapse in boys with acute lymphoblastic leukemia. Neoplasma. 1992;39:115-118.
65 Dordelmann M, Reiter A, Zimmermann M, et al. Intermediate dose methotrexate is as effective as high dose methotrexate in preventing isolated testicular relapse in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 1998;20:444-450.
66 Mirro JJr, Wharam MD, Kaizer H, et al. Testicular leukemic relapse. Rate of regression and persistent disease after radiation therapy. J Pediatr. 1981;99:439-440.
67 Waber DP, Shapiro BL, Carpentieri SC, et al. Excellent therapeutic efficacy and minimal late neurotoxicity in children treated with 18 grays of cranial radiation therapy for high-risk acute lymphoblastic leukemia. A 7-year follow-up study of the Dana-Farber Cancer Institute Consortium Protocol 87-01. Cancer. 2001;92:15-22.
68 Laxmi SN, Takahashi S, Matsumoto K, et al. Treatment-related disseminated necrotizing leukoencephalopathy with characteristic contrast enhancement of the white matter. Radiat Med. 1996;14:303-307.
69 Rubinstein LJ, Herman MM, Long TF, Wilbur JR. Disseminated necrotizing leukoencephalopathy. A complication of treated central nervous system leukemia and lymphoma. Cancer. 1975;35:291-305.
70 Stubberfield TG, Byrne GC, Jones TW. Growth and growth hormone secretion after treatment for acute lymphoblastic leukemia in childhood. 18-Gy versus 24-Gy cranial irradiation. J Pediatr Hematol Oncol. 1995;17:167-171.
71 Neglia JP, Meadows AT, Robison LL, et al. Second neoplasms after acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;325:1330-1336.
72 Kimball Dalton VM, Gelber RD, Li F, et al. Second malignancies in patients treated for childhood acute lymphoblastic leukemia. J Clin Oncol. 1998;16:2848-2853.
73 Nygaard R, Garwicz S, Haldorsen T, et al. Second malignant neoplasms in patients treated for childhood leukemia. A population-based cohort study from the Nordic countries. The Nordic Society of Pediatric Oncology and Hematology (NOPHO). Acta Paediatr Scand. 1991;80:1220-1228.
74 Blatt J, Sherins RJ, Niebrugge D, et al. Leydig cell function in boys following treatment for testicular relapse of acute lymphoblastic leukemia. J Clin Oncol. 1985;3:1227-1231.
75 Brauner R, Czernichow P, Cramer P, et al. Leydig-cell function in children after direct testicular irradiation for acute lymphoblastic leukemia. N Engl J Med. 1983;309:25-28.
76 Bennett JM, Catovsky D, Daniel MT, et al. Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-MO). Br J Haematol. 1991;78:325-329.
77 Woods WG, Kobrinsky N, Buckley J, et al. Intensively timed induction therapy followed by autologous or allogeneic bone marrow transplantation for children with acute myeloid leukemia or myelodysplastic syndrome. A Children’s Cancer Group pilot study. J Clin Oncol. 1993;11:1448-1457.
78 Pui CH, Mahmoud HH, Wiley JM, et al. Recombinant urate oxidase for the prophylaxis or treatment of hyperuricemia in patients with leukemia or lymphoma. J Clin Oncol. 2001;19:697-704.
79 Creutzig U, Ritter J, Schellong G. Identification of two risk groups in childhood acute myelogenous leukemia after therapy intensification in study AML-BFM-83 as compared with study AML-BFM-78. AML-BFM Study Group. Blood. 1990;75:1932-1940.
80 Ravindranath Y, Steuber CP, Krischer J, et al. High-dose cytarabine for intensification of early therapy of childhood acute myeloid leukemia. A Pediatric Oncology Group study. J Clin Oncol. 1991;9:572-580.
81 Woods WG, Kobrinsky N, Buckley JD, et al. Timed-sequential induction therapy improves postremission outcome in acute myeloid leukemia. A report from the Children’s Cancer Group. Blood. 1998;87:4979-4989.
82 Dusenbery K, Arthur D, Howells W. Granulocytic sarcomas (chloromas) in pediatric patients with newly diagnosed acute myeloid leukemia. Proc Am Soc Clin Oncol. 1996;15:369A.
83 Dahl GV, Kalwinsky DK, Mirro JJr, et al. Allogeneic bone marrow transplantation in a program of intensive sequential chemotherapy for children and young adults with acute nonlymphocytic leukemia in first remission. J Clin Oncol. 1990;8:295-303.
84 Cassileth PA, Harrington DP, Appelbaum FR, et al. Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in first remission. N Engl J Med. 1998;339:1649-1656.
85 Appelbaum FR. Allogeneic hematopoietic stem cell transplantation for acute leukemia. Semin Oncol. 1997;24:114-123.
86 Dinndorf P, Bunin N. Bone marrow transplantation for children with acute myelogenous leukemia. J Pediatr Hematol Oncol. 1995;17:211-224.
87 Frassoni F, Labopin M, Gluckman E, et al. Results of allogeneic bone marrow transplantation for acute leukemia have improved in Europe with time—a report of the Acute Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. 1996;17:13-18.
88 Graus F, Saiz A, Sierra J, et al. Neurologic complications of autologous and allogeneic bone marrow transplantation in patients with leukemia. A comparative study. Neurology. 1996;46:1004-1009.
89 Zittoun R, Suciu S, Watson M, et al. Quality of life in patients with acute myelogenous leukemia in prolonged first complete remission after bone marrow transplantation (allogeneic or autologous) or chemotherapy. A cross-sectional study of the EORTC-GIMEMA AML 8A trial. Bone Marrow Transplant. 1997;20:307-315.
90 Ries LAG, Miller BA, Hankey BF. SEER Cancer Statistics Review, 1973-1991. Bethesda, MD: National Institutes of Health; 1994.
91 Sandlund JT, Downing JR, Crist WM. Non-Hodgkin’s lymphoma in childhood. N Engl J Med. 1996;334:1238-1248.
92 Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001.
93 Murphy SB. Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas. Dissimilarities from lymphomas in adults. Semin Oncol. 1980;7:332-339.
94 Magrath IT, Bhati K. Pathogenesis of small noncleaved cell lymphomas (Burkitt’s lymphoma). In: Magrath IT, editor. The Non-Hodgkin’s Lymphomas. London: Arnold; 1997:385-409.
95 Patte C, Auperin A, Michon J, et al. The Société Française d’Oncologie Pédiatrique LMB89 protocol. Highly effective multiagent chemotherapy tailored to the tumor burden and initial response in 561 unselected children with B-cell lymphomas and L3 leukemia. Blood. 2001;97:3370-3379.
96 Patte C, Auperin A, Gerrard M, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents. It is possible to reduce treatment for the early responding patients. Blood. 2007;109:2773-2780.
97 Cairo MS, Gerrard M, Sposto R, et al. Results of a randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007;109:2736-2743.
98 Reiter A, Schrappe M, Tiemann M, et al. Improved treatment results in childhood B-cell neoplasms with tailored intensification of therapy. A report of the Berlin-Frankfurt-Munster Group Trial NHL-BFM 90. Blood. 1999;94:3294-3306.
99 Woessmann W, Seidemann K, Mann G, et al. The impact of the methotrexate administration schedule and dose in the treatment of children and adolescents with B-cell neoplasms. A report of the BFM Group Study NHL-BFM95. Blood. 2005;105:948-958.
100 Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms. A proposal from the International Lymphoma Study Group. Blood. 1994;84:1361-1392.
101 Bernard A, Boumsell L, Reinherz EL, et al. Cell surface characterization of malignant T cells from lymphoblastic lymphoma using monoclonal antibodies. Evidence for phenotypic differences between malignant T cells from patients with acute lymphoblastic leukemia and lymphoblastic lymphoma. Blood. 1981;57:1105-1110.
102 Reiter A, Schrappe M, Ludwig WD, et al. Intensive ALL-type therapy without local radiotherapy provides a 90% event-free survival for children with T-cell lymphoblastic lymphoma. A BFM group report. Blood. 2000;95:416-421.
103 Sandlund JT, Pui CH, Zhou Y, et al. Effective treatment of advanced-stage childhood lymphoblastic lymphoma without prophylactic cranial irradiation. Results of St. Jude NHL13 study. Leukemia. 2009;23:1127-1130.
104 Burkhardt B, Woessmann W, Zimmermann M, et al. Impact of cranial radiotherapy on central nervous system prophylaxis in children and adolescents with central nervous system-negative stage III or IV lymphoblastic lymphoma. J Clin Oncol. 2006;24:491-499.
105 Kaneko Y, Frizzera G, Edamura S, et al. A novel translocation, t(2;5)(p23;q35), in childhood phagocytic large T-cell lymphoma mimicking malignant histiocytosis. Blood. 1989;73:806-813.
106 Sandlund JT. Should adolescents with NHL be treated as old children or young adults? Hematology Am Soc Hematol Educ Program. 2007;2007:297-303.
107 Reiter A, Schrappe M, Tiemann M, et al. Successful treatment strategy for Ki-1 anaplastic large-cell lymphoma of childhood. A prospective analysis of 62 patients enrolled in three consecutive Berlin-Frankfurt-Munster group studies. J Clin Oncol. 1994;12:899-908.
108 Laver JH, Kraveka JM, Hutchison RE, et al. Advanced-stage large-cell lymphoma in children and adolescents. Results of a randomized trial incorporating intermediate-dose methotrexate and high-dose cytarabine in the maintenance phase of the APO regimen. A Pediatric Oncology Group phase III trial. J Clin Oncol. 2005;23:541-547.
109 Seidemann K, Tiemann M, Schrappe M, et al. Short-pulse B-non-Hodgkin lymphoma-type chemotherapy is efficacious treatment for pediatric anaplastic large cell lymphoma. A report of the Berlin-Frankfurt-Munster Group Trial NHL-BFM 90. Blood. 2001;97:3699-3706.
110 Laver JH, Mahmoud H, Pick TE, et al. Results of a randomized phase III trial in children and adolescents with advanced stage diffuse large cell non-Hodgkin’s lymphoma. A Pediatric Oncology Group study. Leuk Lymphoma. 2002;43:105-109.
111 Hutchison RE, Berard CW, Shuster JJ, et al. B-cell lineage confers a favorable outcome among children and adolescents with large-cell lymphoma. A Pediatric Oncology Group study. J Clin Oncol. 1995;13:2023-2032.
112 Link MP, Shuster JJ, Donaldson SS, et al. Treatment of children and young adults with early-stage non-Hodgkin’s lymphoma. N Engl J Med. 1997;337:1259-1266.
113 Suryanarayan K, Shuster JJ, Donaldson SS, et al. Treatment of localized primary non-Hodgkin’s lymphoma of bone in children. A Pediatric Oncology Group study. J Clin Oncol. 1999;17:456-459.
114 Murphy SB, Hustu HO. A randomized trial of combined modality therapy of childhood non-Hodgkin’s lymphoma. Cancer. 1980;45:630-637.
115 Griffin TC, Weitzman S, Weinstein H, et al. A study of rituximab and ifosfamide, carboplatin, and etoposide chemotherapy in children with recurrent/refractory B-cell (CD20+) non-Hodgkin lymphoma and mature B-cell acute lymphoblastic leukemia. A report from the Children’s Oncology Group. Pediatr Blood Cancer. 2009;52:177-181.
116 Loeffler JS, Leopold KA, Recht A, et al. Emergency prebiopsy radiation for mediastinal masses. Impact on subsequent pathologic diagnosis and outcome. J Clin Oncol. 1986;4:716-721.