[level-membership-for-neurosurgery-category]
CHAPTER 198 Pediatric Craniopharyngioma
In 1932 Harvey Cushing stated that craniopharyngiomas were the most baffling problem confronting neurosurgeons.1 To this day, they remain challenging tumors to manage, with a wide range of possible treatment strategies. As the factors influencing treatment-related quality of life are better elucidated, the controversy over the best management of craniopharyngiomas continues.
History
The first pathologic description was from an autopsy performed in 1857, when Zenker reported a suprasellar lesion with cholesterol crystals and squamous epithelial cells. The first accurate pathologic description was by Erdheim2 in 1904. Frazier and Alpers in 1931 and Cushing in 1932 first used the term craniopharyngioma.1 In 1909 Halstead3 was the first to successfully remove a craniopharyngioma using the transsphenoidal route. A child operated on by Cushing in 1923 survived for more than 50 years.4 In 1899 Mont and Barrett suggested that these tumors arise from the hypophysial duct (Rathke’s pouch).5 The hypophysial duct is actually an invagination of the primitive stomodeum, not of the pharynx, making craniopharyngioma a misnomer.6 Rathke’s pouch forms the pars tuberalis in the development of the anterior lobe of the pituitary gland. This embryologic origin becomes important when viewing magnetic resonance imaging (MRI) scans because it is extremely uncommon not to have a portion of the tumor located within the sella turcica.
Rathke’s cleft cysts also arise from Rathke’s pouch, which is normally reduced to a neural cleft by the sixth week of embryonic life. When a Rathke’s cleft cyst persists, it has the potential to become pathologic, progressively enlarging and filling with mucoid material. Rathke’s cleft cysts and craniopharyngiomas may represent a continuum from the simplest form of Rathke’s cleft cyst to a more complex form of craniopharyngioma.7 Other entities found in this area are epithelial, epidermoid, and dermoid cysts.
Histology
There are three histologic types of craniopharyngioma: adenomatous, squamous papillary, and mixed. The adenomatous variety, which is the histologic pattern most common in the pediatric population, resembles tumors of tooth-forming tissue, having a varied histologic pattern of sheet-like epithelium, lobules, anastomosing trabeculae, cysts, and cloverleaves. The distinctive feature is a palisading layer of small cells enclosing a loose stellate reticular zone with alternating, compactly arranged squamous cells that have a tendency to whorl. The epithelium is a wet keratin, differing from the flaky keratin of epidermoid cysts, and is the hallmark of this tumor subtype. Dystrophic calcification, fibrosis, chronic inflammation, and cholesterol clefts are also prominent features.6 The squamous papillary type is rare in children but makes up approximately one third of the adult variety.8 These tumors tend to be solid rather than cystic and may occur within the third ventricle.9 They are less likely to develop calcification and do not form keratin nodules, leading to no evidence of calcification on computed tomography (CT), which is important in the differential diagnosis in adult patients. Mixed tumors contain separate foci with histologic features of both adenomatous and squamous papillary types.9
Adenomatous craniopharyngiomas tend to have finger-like projections that invade the hypothalamus. Therefore, it is common to find small foci of normal brain tissue within resected craniopharyngioma specimens.7–9 This histologic finding is not readily appreciable under the operating microscope, where the tumor capsule may seem to be distinct from the adjacent neural tissue. It is well established that the squamous papillary type, which does not have these microscopic projections, has a better clinical outcome and a lower rate of recurrence with gross total surgical removal than the adenomatous type.8,10 Other authors contend that this histologic difference is not predictive of outcome.9–11 A fibrillary gliotic reaction in the brain immediately adjacent to a craniopharyngioma may create a cleavage plane that actually aids in tumor removal.12–16 Again, there is a difference of opinion; other authors think this dense adherence makes separation from normal brain more difficult.17,18
Epidemiology
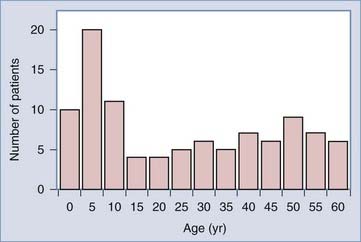
Craniopharyngiomas are the most common intracranial pediatric tumors of nonglial origin; they constitute 1.2% to 4% of all brain tumors and 6% to 9% of all pediatric brain tumors.17 There are approximately 0.5 to 2 new cases per million people per year.17 In childhood, approximately 54% of tumors in the sella-chiasmatic region are craniopharyngiomas.20 These tumors can present at any age, but there is a bimodal age distribution, with peak incidence rates in children aged 5 to 15 years and adults aged 40 to 70 years (Fig. 198-1).21 In the United States, 338 cases are expected to occur annually, with 96 patients presenting at an age younger than 14 years. Although classically thought to be more common in males, there has been no obvious gender difference in large population-based studies.21 The peak incidence in the pediatric age group is 5 to 10 years, but craniopharyngioma can occur at any age. There are no known associated genetic abnormalities or risk factors.8
Clinical Presentation
Visual impairment is common, and tumors are occasionally discovered when visual screening or parental observation results in ophthalmologic referral. One should note that very large tumors may cause minimal to no visual defects. Visual defects often go undetected until severe visual impairment and optic atrophy are present.19 Visual disturbance is noted in 37% to 68% of children at presentation.13,21–30 A formal neuro-ophthalmologic examination should be obtained preoperatively whenever possible.
Endocrine dysfunction is very common, occurring in 66% to 90% of new pediatric patients. However, it is rare for a child to be brought to medical attention specifically for an endocrine-related complaint. Short stature (defined as height less than the 3rd percentile), diabetes insipidus (7.5% to 24% of patients), and hypothyroidism (12% to 25% of patients) have been reported.6–9,11,30,31 Endocrine testing reveals deficiencies of growth hormone in up to 75% of patients, luteinizing hormone and follicle-simulating hormone in 40% of patients, adrenocorticotropic hormone (ACTH) in 25%, and thyroid-stimulating hormone in 25%. Less common presenting features are elevation of prolactin (20%), morbid obesity (11% to 18%), diplopia (8% to 11%), and disturbance of mentation.6–8,11,30,31 It should be noted that it is rare for a child with a craniopharyngioma to have clinically significant diabetes insipidus at presentation.
Imaging
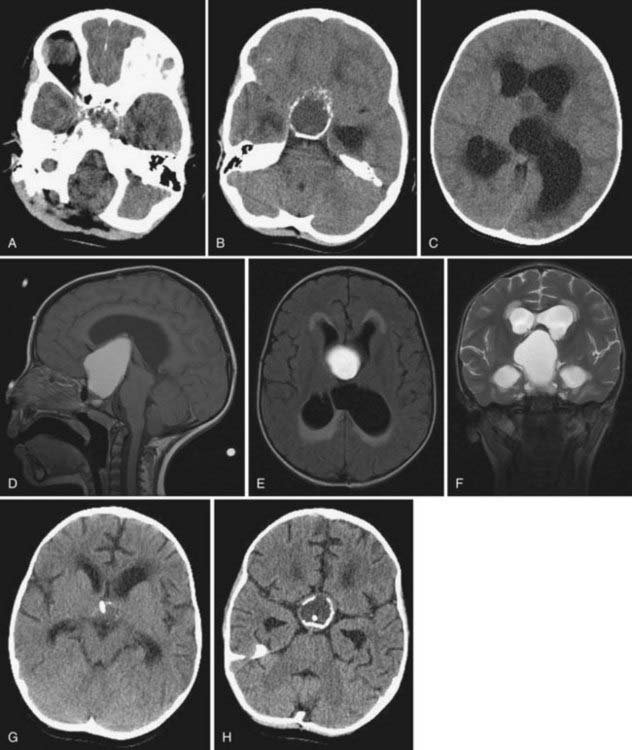
Historically, skull radiographs were very important in the diagnosis of craniopharyngioma because they demonstrated sellar enlargement and suprasellar calcification, but they are unnecessary in the modern era. MRI is required in the diagnostic evaluation and is necessary for adequate surgical planning and postoperative evaluation. Because CT scans show calcification, which occurs in 51% to 90% of pediatric craniopharyngiomas,32,33 this modality remains a useful and important part of the preoperative evaluation. CT is also superior in demonstrating the anatomy and size of the sphenoid sinus when the transsphenoidal surgical route is anticipated. The calcifications have a craggy, popcorn-like appearance, with fine eggshell lines (Fig. 198-2A to C).34 Coronal CT can facilitate the detection of intrasellar ossification.
MRI, with and without gadolinium enhancement, is critical for demonstrating the tumor’s relationship to the optic chiasm, infundibulum, hypothalamus, and major vessels (Fig. 198-3C, E, and F). The T1-weighted images should be in the axial, coronal, and sagittal planes. The postcontrast sagittal images are most useful in assessing the relationship of the tumor to the hypothalamus, providing critical information for later decisions regarding treatment options.35,36 If a patient has a cystic, enhancing suprasellar mass with calcifications, it is almost certainly an adamantinomatous type of craniopharyngioma (see Figs. 198-2 and 198-3).33
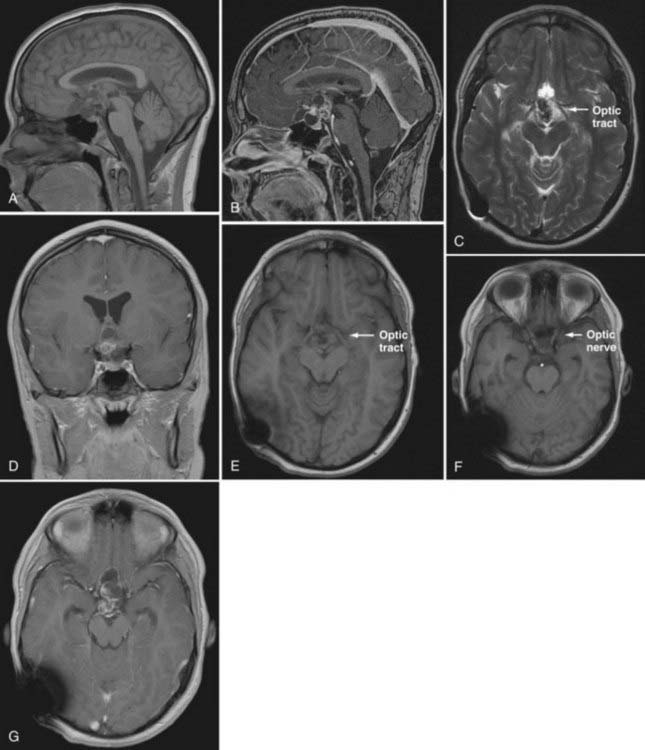
Craniopharyngiomas are typically 2- to 4-cm lesions in 58% to 76% of cases,37,38 although very large tumors have been reported.39,40 The most common morphologic finding is a predominantly cystic type of tumor in 46% to 64% of cases.30,38,41 The predominantly solid type is seen in 18% to 39% of cases, and mixed solid and cystic tumors are seen in 8% to 36% of cases.29,41 On MRI the solid portion of the tumor typically appears mottled, an indirect effect of signal dampening due to tumor calcification.42 Gadolinium produces heterogeneous enhancement of the solid tumor (see Fig. 198-3B and G). The signal intensity of cystic fluid on T1-weighted images may range from low to high. This variation and degree of intensity is the result of different protein concentrations, free methemoglobin, and cholesterol crystals.43 The walls of the cystic portions of the tumor almost always enhance with contrast (Table 198-1; see Fig. 198-3B).44
TABLE 198-1 Magnetic Resonance Imaging Characteristics of the Cystic and Solid Portions of Craniopharyngioma
| MRI SEQUENCE | TUMOR COMPONENT | DESCRIPTION |
|---|---|---|
| T1 weighted without contrast | Solid | Isointense or hypointense |
| T2 weighted without contrast | Solid | Mixed hypointensity-hyperintensity |
| T1 weighted with contrast | Solid | Homogeneous or heterogeneous enhancement |
| T1 weighted without contrast | Cystic | Hypointense |
| T2 weighted without contrast | Cystic | Hyperintense* |
| T1 weighted with contrast | Cystic | Cystic rim enhancement |
* Acute, subacute, or chronic hemorrhage; protein density; and cholesterol deposition may alter cystic imaging characteristics.
Cerebral angiography provides the best demonstration of the displacement of major vessels by tumor, but it has largely been supplanted by magnetic resonance angiography. When planning a radical surgical removal of recurrent craniopharyngioma, invasive angiography may be useful to delineate constriction of the carotid, middle, or anterior cerebral arteries and facilitate tumor dissection. Fusiform dilation of the carotid artery has been reported after radical tumor resection, and it potentially increases the risk during reoperation.35,45 Because craniopharyngiomas are relatively avascular, even modern angiography may not demonstrate the small nutrient vessels.19
There are many postoperative and intraoperative options for the follow-up of patients with craniopharyngioma. We recommend that MRI be obtained within 48 hours after surgery to document the presence or absence of residual tumor and the size of residual cysts and to minimize confusion when interpreting postsurgical changes (see Fig. 198-2G and H). If complete surgical removal is attempted, an additional CT scan is necessary because a small fleck of calcification may mean that the resection was incomplete, portending recurrent tumor growth in the follow-up period.
Differential Diagnosis
The differential diagnosis of suprasellar-sellar masses includes chiasmatic-hypothalamic glioma, germinoma, Rathke’s cleft cyst, hypothalamic hamartoma, arachnoid cyst, pituitary adenoma, and meningioma; the last two are rare in children.27 The presence of calcification on CT virtually eliminates all these diagnoses except meningioma. In children whose tumors are predominantly adamantinomatous, calcification occurs more than 90% of the time.33,34 The next most common tumor in this area is the chiasmatic-hypothalamic glioma, which does not have an intrasellar component and does not commonly calcify.
Endocrine Evaluation
Preoperative
Evaluation of five hormone systems is recommended: thyroid, growth hormone, cortisol, gonadotropins, and prolactin. All children with craniopharyngiomas should be considered ACTH deficient until proved otherwise and should empirically receive stress corticosteroid coverage during surgical procedures. The stress dose of 7 to 15 mg/m2 mis triple the daily maintenance dose of hydrocortisone.25 In a previously operated and treated patient, thyroid deficiency may be profound, and it is critical to ensure that the child has been taking his or her thyroid medication. Of historical interest, it was not until corticosteroid therapy came available in the 1950s that removal of craniopharyngiomas could be accomplished with acceptable morbidity and mortality.46,47
Postoperative
More extensive evaluation of the five hormone systems is necessary after surgery, as the child is followed or goes on to radiation therapy. Thyroid evaluation should include thyroid-stimulating hormone surge and thyrotropin-releasing hormone tests for evaluation of the hypothalamic-pituitary-thyroid axis. Growth hormone evaluation includes the arginine tolerance and levodopa tests for growth hormone secretory capacity and activity. We recommend baseline levels of cortisol, ACTH levels, metapyrone testing for ACTH reserve, and the gonadotropin-releasing hormone stimulation test to determine premature or delayed gonadotropin secretion or response. In addition, serum is obtained for determination of the prolactin level.48 Twenty-four-hour assessment of urine output, serum and urine osmolarity, and electrolytes is important because diabetes insipidus occurs in more than 90% of patients after radical surgery and in 33% to 47% after limited surgery.49 Limited surgical approaches such as stereotactic cyst drainage and biopsy do not result in diabetes insipidus.
Long-term endocrine deficiencies in the five major systems are not much different between the limited surgery and radiotherapy group compared with the radical surgery group. However, there is a difference in time to onset, delayed in the former and immediate in the latter (Table 198-2).35,49,50 In the case of a radiated child, endocrine status must be measured yearly until the baseline nadir is reached.
Management Controversy
Although there is much debate over the proper management of patients with craniopharyngiomas, the treatment must focus on relief of symptoms, cure of disease, and quality of life of the patient. In reaching these goals, the surgeon must choose the treatment strategy that causes the least harm and delivers the most benefit to the patient. The continuum of treatment ranges from radical resection with the avoidance of radiation to limited surgery followed by radiotherapy. The role of other treatment modalities such as intracystic therapy (P-32, bleomycin) or the Gamma Knife is discussed later, but they are usually just adjuncts to one of the major treatment approaches. In expert hands, cure rates using either surgical approach are nearly identical.16,49–52 One certainty is that attempts at gross total resection of large solid tumors have a significantly negative effect on quality of life.17,34,50,51,53–55 As such, the surgeon must look closely at his or her own results in terms of cure of disease and harm from treatment and make the best treatment decision for each patient. A survey of neurosurgeons demonstrated that surgeons who operated on one or fewer craniopharyngiomas per year could not expect the results of radical surgery quoted in the literature with regard to cure rate or complication rate.56
Radical Resection Versus Limited Surgery Plus Radiotherapy
Radical Resection
Radical resection was once considered the “gold standard” for the treatment of craniopharyngioma. The primary goal of radical resection is to obtain a surgical cure of this benign disease, thus sparing the patient the risks associated with radiotherapy or other adjuvant treatment. It can also be useful for decompressing the optic nerve or optic chiasm, thereby improving vision, and for relieving hydrocephalus by decompressing the cerebrospinal fluid pathways and obviating the need for shunting procedures. Although gross total resection has fallen out of favor in some centers, it remains a good option for many patients and should be recommended when it serves the patient better than alternative options. Numerous studies have been published that show excellent surgical results with regard to cure and outcome.10,19,23,24,38,57 Radical resection can be an effective treatment with excellent rates of long-term progression-free survival.23,26
Radical surgery often has an advantage when there is no hypothalamic involvement and resection can be accomplished with minimal complications or side effects. In theses cases, it eliminates the need for further therapies and their associated side effects.49,53 Radical resection is also highly recommended in rare, purely intrasellar tumors. This tumor location allows the potential for gross total resection via the transsphenoidal approach in a teenager with a pneumatized sphenoid sinus.
One must consider a number of factors when deciding whether radical resection is the best option. First and foremost, only experienced surgeons should attempt radical resection in pediatric patients.56 Even among experienced surgeons, the morbidity and mortality associated with adamantinomatous craniopharyngioma in children are higher than in adults.11 Surgeons with less experience should stabilize the patient for transfer or use a less invasive approach for the management of pediatric craniopharyngioma. The worst overall results occur in children in whom an attempt at gross total resection fails, giving them the morbidity associated with that surgery plus the morbidity of radiotherapy.49
Surgical Approach and Technique
Because craniopharyngiomas have such great variability in location, size, and morphology, no one surgical approach should be considered superior to another. The logical approach is the one that allows the best exposure to the tumor and limits the retraction of surrounding structures. The wide variety of approaches is presented in Table 198-3, along with the advantages and disadvantages of each. For the majority of tumors with suprasellar involvement, a unilateral approach from the nondominant side is recommended over dominant hemispheric approaches, thus reducing the risk of vascular or retraction injury to the dominant hemisphere. Bifrontal approaches, which expose large areas of normal brain to potential retraction injury, have largely been abandoned for more limited microscopic approaches. For craniopharyngiomas that extend predominantly into the dominant hemisphere, a dominant-sided approach may be more appropriate. When vision loss is more profound in one eye, approaching the tumor from the side of greater vision loss may prevent surgical injury to the intact optic apparatus, making it the first choice of many surgeons. If the tumor is isolated to the sella, a transsphenoidal approach in a teenager with a pneumatized sella is often the best surgical option.19,58,59 Occasionally, the sellar approach allows resection of an intracranial tumor. This intracranial removal is fraught with risk, however, because it requires traction on nonvisualized structures, and if an arterial injury occurs, rescue is almost impossible. Endoscopic-assisted approaches are becoming more common, adding another tool to the treatment of this disease.59
| APPROACH | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| Subfrontal |
Tumor Removal by Transcranial Approaches
Tumor resection should begin after adequate exposure; addition of the newer skull base approaches is often critical to surgical success. Achieving a surgical approach as inferior as possible facilitates surgical resection with minimal retraction. After the tumor is identified, an attempt to preserve the arachnoid adjacent to the tumor is critical because it allows the identification of the optic nerve and chiasm and the carotid, anterior cerebral, and middle cerebral arteries without troublesome bleeding. Aspiration of cysts is useful early in the procedure, increasing the room available for dissection and often relieving hydrocephalus, thus decreasing the mass effect on adjacent structures. Following aspiration of the cysts, the normal anatomy must be evaluated before debulking of the tumor. As dissection proceeds, it is helpful to preserve the capsular margin and remove it last, thus reducing the chance of leaving residual tumor. Once the tumor margin has been established, tumor removal can begin. During removal, the arterial supply may be coagulated, but care must be taken with vessels in the region of the median eminence because these may supply the optic tract and chiasm. Once the central suprasellar portion is removed, the superior extension of the tumor should descend passively into direct vision. For large tumors, resection must be performed with caution because retracting, pulling, or dissecting hypothalamic adhesions can be quite harmful to the patient postoperatively. Sharp dissection should be used for tumor underlying the hypothalamus, chiasm, and optic nerves whenever possible, rather than blind cyst retraction (historically described surgical technique).16,19
Preservation of the Pituitary Stalk
Preservation of the pituitary stalk is important when attempting gross total resection. Before the introduction of the operating microscope, early attempts to preserve the stalk were seldom successful. Using modern microscopic visualization and illumination, however, stalk preservation is quite possible.38 To attempt stalk preservation, it is necessary to identify the stalk early in the operation. Even with supratentorial tumor displacement, the stalk can be identified as it enters the diaphragma sellae and then followed superiorly. When both the stalk and the pituitary gland are displaced, the stalk can be challenging to locate, but it has a characteristic appearance of alternating striations formed by the portal venous complex that travels its length. It should be noted that even with gross stalk preservation, avoidance of diabetes insipidus or other endocrinopathies is achieved less than 50% of the time.
Complications
For tumors involving the hypothalamus, surgical resection often produces severe morbidity, with a wide range of endocrine and cognitive dysfunction.23,49,60 Damage to the hypothalamus has been associated with changes in personality, apathy, decrease in activity, loss of interest in school and outside activities, and morbid obesity.49,50 Morbid obesity is a common complication of radical surgery and is always associated with the dreaded change in personality.49 Diabetes insipidus occurs in more than 90% of children in spite of preservation of the pituitary stalk.49 This is a major complication in children with athletic pursuits and those who reside in warm climates. Among patients with attempted radical resection, up to 96% have a wide range of permanent endocrinopathies related to the operative intervention (see Table 198-2).61 Diabetes insipidus and other endocrinopathies require lifelong medical management, which has significant socioeconomic and psychological costs.
From the early days of craniopharyngioma surgery to the present, gross total resection—even in highly experienced hands—has been achievable in only about 45% to 75% of cases.38,62,63 As such, a fairly large number of patients have residual disease and go on to receive additional surgery or salvage radiotherapy. Salvage radiotherapy after attempted gross total resection or recurrence has numerous deleterious effects because it combines the side effects of the two modalities.49 Therefore, the surgeon should be confident that he or she can remove the entire tumor before recommending gross total resection. Recurrence after reported gross total resection without radiotherapy is undeniably common, occurring in up to 53% of cases.50,61 Death in patients who undergo attempted radical resection of craniopharyngioma is not uncommon, with a 4% to 9% mortality rate reported in the American Society of Pediatric Neurosurgeons’ survey17 and higher rates in other series.10,64 With these rates of recurrence and residual disease, it behooves the surgeon to follow patients with craniopharyngioma for a lengthy period—at least 5 years—after any surgical intervention.
Other morbidities following attempted gross total resection include epilepsy. Epilepsy occurs in up to 40% of patients managed by attempted radical resection; in contrast, epilepsy is virtually nonexistent in patients receiving limited surgery followed by adjuvant radiotherapy.48 In addition, quality of life is greatly diminished in up to 86% of patients undergoing radical resection.61,65,66 Considering these issues of morbidity, mortality, incomplete resection, high recurrence rates, and diminished quality of life, gross total resection for cure should be attempted only by surgeons with extensive experience at children’s hospitals with high operative volumes.
Limited Surgery and Adjuvant Radiotherapy
Limited surgery is used to improve symptoms, and adjuvant radiotherapy cures the disease. Advocates of radical resection have labeled this approach palliative; however, progressive tumor growth after 5 years is rare.49 The use of limited surgery and adjuvant therapy has been adopted at many centers owing to the improved quality of life and lack of complications.50,67–69 Puget and coworkers50 noted that the degree of hypothalamic involvement was the principal cause of the harmful effects associated with radical surgery and recommended limited surgery when there was extensive involvement (Fig. 198-4). This is our experience as well.49
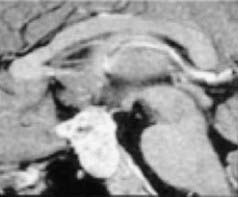
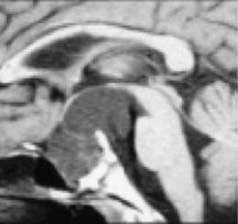
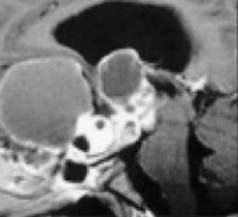
FIGURE 198-4 Grading of hypothalamic involvement on preoperative imaging.
(From Puget S, Garnett M, Sainte-Rose C, et al. Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg. 2007;106[1 suppl]:3-12.)
Figure 198-5 is an example of a solid tumor that we would not attempt to remove because of its extensive involvement with the hypothalamus. The morbidity of resecting a pure cyst from the hypothalamus (see Fig. 198-2D) is less clear and needs to be studied prospectively.
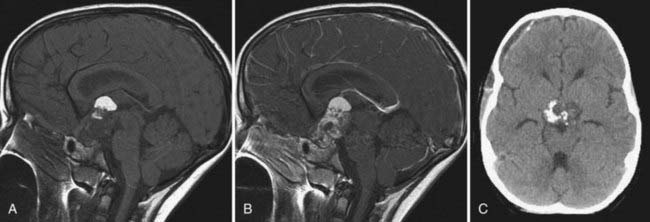
Surgical Strategy
Once a decision has been made to perform limited surgery, the amount of tumor left behind does not affect the cure rate with radiotherapy.49 Thus, the amount of surgery depends on the individual patient. The two principal clinical problems that need to be treated are vision loss and increased intracranial pressure, usually secondary to obstruction of the third ventricle by the tumor, with resultant hydrocephalus. Because the vision loss is chronic, there is usually adequate time to assess acuity, visual field deficit, and the presence of papilledema or optic atrophy preoperatively. Surprisingly, once the hydrocephalus is relieved, vision often improves dramatically. Repeated visual evaluation is necessary after relief of hydrocephalus.
In every case, hydrocephalus must be addressed before radiotherapy begins because of the additional IQ loss and potential vision loss associated with radiating a child with hydrocephalus.49
In terms of operative and perioperative mortality, a number of series have reported no deaths in patients managed with subtotal resection and adjuvant therapy.48,49,69 Visual acuity is preserved or improved in the majority of patients using this treatment regimen, comparable to radical resection attempts.49 This is thought to relate to the treatment of hydrocephalus, which is a common preventable cause of vision loss in children with craniopharyngioma, although direct decompression of the optic nerves and chiasm may be necessary. With limited surgery and radiotherapy, hormone dysfunction occurs, but the onset is later and it is usually less severe. Diabetes insipidus is a major cause of death and morbidity in patients with craniopharyngioma, but it may be avoided if craniotomy is avoided.
In some centers, the use of intracystic bleomycin or other agents has been advocated in young children to delay more aggressive treatment, including to delay radiotherapy.67,70 Alpha interferon is an experimental treatment whose role has yet to be defined in the management of pediatric craniopharyngioma.70 The role of adjuvant chemotherapy will likely expand as newer agents are found that are less toxic and more selective for craniopharyngioma tumor cells.
Radiation
Radiation treatment for patients with craniopharyngioma traditionally involved acquiring lateral radiographs in the treatment position, estimating the location and size of the tumor, and surrounding the estimated tumor with a margin of approximately 2 cm. These conventional methods ensured that the region containing the tumor and a large volume of normal tissue would be encompassed by the prescription dose of radiation. With the advent of newer methods, broadly described as conformal radiation therapy, target volumes can be defined by cross-sectional imaging data using specialized software, and the prescription dose can be shaped to irradiate only the volume at risk, with the goal of sparing normal tissues. A broad range of methods have been developed, each with its own indications for application and relative advantages and disadvantages in terms of dose shaping and normal tissue sparing.71–73 Regardless of the method employed, the most important aspect of radiation treatment planning is defining the treatment volume, which includes the margins surrounding the tumor or tumor bed.
The first study to prospectively define treatment margins for craniopharyngioma was carried out at St. Jude Children’s Research Hospital. The goal of the study was to define a minimal target volume for these patients.49 The gross tumor volume was defined as the residual cystic and solid tumor and the microscopically involved postresection tumor bed. The clinical target volume margin included 10 mm of normal tissue surrounding the gross tumor volume. This volume, meant to account for subclinical microscopic disease, was modified at anatomic barriers to invasion. The clinical target volume was then encompassed by an additional margin of 3 to 5 mm, depending on the method of immobilization, to account for variability in patient positioning. With a median follow-up of 28 months, the 3-year event-free survival was reported as 85% ± 11%. This was comparable to the event-free survival reported for patients treated with conventional radiation therapy. Most important, the correlation of radiation dosimetry with IQ showed a statistically significant relationship between radiation dose and volume, with higher doses having the greatest impact. Based on the state of the art, investigators have proposed treating craniopharyngioma using even smaller margins or with proton beam radiation therapy in an effort to minimize side effects.74
The use of limited surgery and adjuvant therapy has been proved to prolong survival and control disease progression. The reported 10- and 20-year progression-free survival rates were 83% and 79%, respectively, in one large series with extended postradiation follow-up.51 Similar rates of tumor control have been reported in other series.49,50,52 Therefore, there is every reason to believe that long-term tumor control with conformal radiation will be equally efficacious.
Complications
Although it is clear that limited surgery has certain advantages, this treatment paradigm also has some disadvantages that must be carefully weighed. In contrast to radical surgery, short-term complications are rare. However, a number of late morbidities occur as a result of adjuvant radiotherapy. A measurable decrease in IQ, usually less than 10 points, has been noted in patients treated with radiotherapy.49 The decrease in IQ is related to a number of factors, including size of tumor, degree of hydrocephalus, type of previous surgery, and mode of radiotherapy.49 It should be noted that radiation after failed radical surgery increases the risk and severity of a decrease in IQ.49 Following radiation, secondary malignancy is a rare but known complication. In 124 craniopharyngiomas treated over 23 years, we have not encountered a case of secondary malignancy. Secondary malignancy seems to be related to the size of the radiation field and the dosage, so conformal and stereotactic radiation should be safer than conventional radiation.75
In long-term follow-up, we have seen malignant degeneration of the craniopharyngioma itself in two children following radiotherapy, and there are three additional children in the literature.76 Gradual loss of pituitary function has also been noted in long-term follow-up and is an expected sequela rather than a complication.
Radiation-induced moyamoya is an uncommon but challenging complication that can occur in patients receiving radiation to the carotid arteries encased in tumor. Vessel occlusion occurs very slowly over months to years, accompanied by the development of collateral vasculature. The role of direct or indirect revascularization procedures has yet to be defined. We tend to operate only on children with symptoms or small infarcts.77
Tumor growth after radiation requires radical resection for cure. We have found that if craniotomy was avoided in the previous operation, without destruction of the arachnoid planes, surgical removal is no more difficult than it would be at initial surgery. However, if an initial attempt to remove the tumor failed, this tremendously increases the complication rate and decreases the chance of complete resection.78,79
Conclusion
Although no prospective randomized trials have been performed, large cohort studies have closely evaluated quality of life for patients managed by either radical resection or limited surgery and adjuvant radiotherapy. Children managed with limited surgery have better quality of life outcomes compared with those treated by radical resection.49,50,61,66,80 In the Boston series, major disability was found in 33% of radical surgery patients, compared with 15% of those treated with limited surgery and radiotherapy.82,83 In another large series of 173 patients treated with limited surgery and radiotherapy, 52% of patients had no disability and led active lives.51 In contrast, in studies involving radical resection, up to 80% of survivors describe major disability and impairment,23,61 underscoring the importance of extensive surgical experience when gross total resection for cure is attempted. There is no doubt that a craniopharyngioma that does not involve the hypothalamus can be safely removed with a high rate of cure by an experienced neurosurgeon.45,49,50,85,86
Carpentieri SC, Waber RM, Scott LC, et al. Memory deficits among children with craniopharyngiomas. Neurosurgery. 2001;49:1053-1058.
Cavassuti V, Fischer EG, Welch K, et al. Neurological and psychological sequelae following different treatments of craniopharyngioma in children. J Neurosurg. 1983;59:409-417.
Combs SE, Thilmann C, Huber PE, et al. Achievement of long-term local control in patients with craniopharyngiomas using high precision stereotactic radiotherapy. Cancer. 2007;109:2308-2314.
Eldevik OP, Blaivas M, Gabrielsen TO, et al. Craniopharyngioma: radiologic and histologic findings and recurrence. AJNR Am J Neuroradiol. 1996;17:1427-1439.
Epstein FJ, Handler MH. Craniopharyngioma: the answer. Pediatr Neurosurg. 1994;21(suppl 1):1-132.
Fahlbusch RJ, Honegger W, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg. 1999;90:237-250.
Fischer EG, Welch K, Shillito J, et al. Long term effects of conservative surgical procedures combined with radiation therapy. J Neurosurg. 1998;73:534-540.
Harwood-Nash DC. Neuroimaging of childhood craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl-1):2-10.
Hetelekidis S, Da Silva M, Humphres RP, et al. 20 yr. experience in childhood craniopharyngioma. Int J Radiat Oncol Biol Phys. 1993;27:189-195.
Hoffman HJ. Surgical management of craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl 1):44-49.
Hoffman HJ, DeSilva M, Humphreys RP, et al. Aggressive surgical management of craniopharyngiomas in children. J Neurosurg. 1992;76:47-52.
Lapras C, Patet JD, Mottolese C, et al. Craniopharyngiomas in childhood: analysis of 42 cases. Prog Exp Tumor Res. 1987;30:350-358.
Merchant TE, Hua CH, Shukla H, et al. Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr Blood Cancer. 2008;51:110-117.
Merchant TE, Kiehna EN, Sanford RA, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience. Int J Radiat Oncol Biol Phys. 2006;53:533-542.
Merchant TE, Williams T, Sanford RA, et al. Preirradiation endocrinopathies in pediatric brain tumor patients determined by dynamic test of endocrine function. Int J Radiat Oncol Biol Phys. 2002;54:45-50.
Pierre-Kahn A, Recassens C, Pinto G, et al. Social and psycho-intellectual outcome following radical removal of craniopharyngiomas in childhood. Childs Nerv Syst. 2005;21:817-824.
Poretti A, Grotzer MA, Ribi K, et al. Outcome of craniopharyngioma in children: long-term complications and quality of life. Dev Med Child Neurol. 2004;46:220-229.
Puget S, Garnett M, Sainte-Rose C, et al. Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg. 2007;106(1 suppl):3-12.
Raimondi AJ. Parasellar tumors. In: Pediatric Neurosurgery: Theoretical Principles, Art of Surgical Techniques. Berlin: Springer; 1987:276-291.
Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—a long-term result following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1-10.
Regine WF, Kramer S. Pediatric craniopharyngioms: long-term results of combined treatment with surgery and radiation. Int J Radiat Oncol Biol Phys. 1992;24:611-617.
Sami M, Bini W. Surgical treatment of craniopharyngiomas. Zentralbl Neurochir. 1991;52:17-23.
Sands SA, Milner JS, Goldberg J, et al. Quality of life and behavioral follow-up study of pediatric survivors of craniopharyngioma. J Neurosurg. 2005;103:302-311.
Sanford RA, Craniopharyngioma. results of survey of the American Society of Pediatric Neurosurgeons. Pediatr Neurosurg. 1994;21(suppl 1):39-43.
Tomita T, Bowman RM. Craniopharyngiomas in children: surgical experience at Children’s Memorial Hospital. Childs Nerv Syst. 2005;21:729-746.
Tomita T, Mclone DG. Radical resection of childhood craniopharyngiomas. Pediatr Neurosurg. 1993;19:6-14.
Weiner HL, Wisoff JH, Rosenberg ME, et al. Total removal of craniopharyngiomas: a functional outcome. Neurosurgery. 1994;35:1001-1011.
Wisoff JH. Craniopharyngiomas. J Neurosurg Pediatr. 2008;1:124-125.
Yasargil MG, Curic M, Kis M, et al. Total removal of craniopharyngiomas. Approach and long-term results in 144 patients. J Neurosurg. 1990;73:3-11.
1 Cushing H. The craniopharyngioma. In: Thomas CC, editor. Intracranial Tumors. Notes on a Series of Two Thousand Verified Cases with Surgical Mortality Percentages Pertaining Thereto. Springfield, IL: Charles Thomas; 1932:2899-2935.
2 Erdheim J. Uber Hypophysenganggeschwulste and Hirncholesteatome. Sitzungsbericht der Kaiseerlichen Akademie der Wissenchaften. Stzunger Akad Widd Wien. 1904;113:537-726.
3 Halstead AE. Remarks on an operative treatment of tumors of the hypophysis. Surg Gynecol Obstet. 1910;10:494-502.
4 Raimondi AJ. Parasellar tumors. In: Pediatric Neurosurgery: Theoretical Principles, Art of Surgical Techniques. Berlin: Springer; 1987:276-291.
5 Frazier CH. Adamantinoma of the craniopharyngioma duct. Arch Neurol Psychiatry. 1932;26:905-965.
6 Russell DS, Rubinstein LJ. Craniopharyngiomas and suprasellar epidermoid cysts. In: Russell DS, Rubinstein LJ, editors. Pathology of Tumors of the Nervous System. 5th ed. Baltimore: Williams & Wilkins; 1989:695-702.
7 Voelker JL, Campbell RL, Miller J. Clinical radiographic and pathological features of symptomatic Rathke’s cleft cysts. J Neurosurg. 1991;80:535-544.
8 Harrison MJ, Morgello S, Post KD. Epithelial cystic lesions of the sellar and parasellar region: a continuum of ectodermal derivatives. J Neurosurg. 1994;80:1018-1025.
9 Adamson TE, Wiesteler OD, Kleihues P, et al. Correlation of clinical and pathological features in surgically treated craniopharyngiomas. J Neurosurg. 1990;73:12-17.
10 Miller DC. Pathology of craniopharyngiomas: Clinical import of pathological features of symptomatic Rathke’s cleft cysts. J Neurosurg. 1991;74:535-554.
11 Yasargil MG, Curic M, Kis M, et al. Total removal of craniopharyngiomas. Approach and long-term results in 144 patients. J Neurosurg. 1990;73:3-11.
12 Weiner HL, Wisoff JH, Rosenberg ME, et al. Total removal of craniopharyngiomas: a functional outcome. Neurosurgery. 1994;35:1001-1011.
13 Tomita T, Mclone DG. Radical resection of childhood craniopharyngiomas. Pediatr Neurosurg. 1993;19:6-14.
14 Apuzzo ML, Levy ML, Tung H. Surgical strategies and technical methodologies in optimal management of craniopharyngiomas and masses effecting the third ventricular chamber. Acta Neurochir Suppl. 1991;53:77-88.
15 Sweet WH. History of surgery for craniopharyngiomas. Pediatr Neurosurg. 1994;21(suppl 1):28-38.
16 Hoffman HJ. Surgical management of craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl 1):44-49.
17 Sanford RA, Muhlbauer MS. Craniopharyngioma in children. Neurol Clin. 1991;9:453-465.
18 Regine WF, Kramer S. Pediatric craniopharyngioms: long-term results of combined treatment with surgery and radiation. Int J Radiat Oncol Biol Phys. 1992;24:611-617.
19 Carmel PW. Craniopharyngiomas. In: Wilkins RH, Rengachary SS, editors. Neurosurgery, Vol 1. New York: McGraw-Hill; 1985:905-916.
20 Koos WT, Miller MH. Craniopharyngioma. In: Koos WT, Miller MH, editors. Intracranial Tumors of Infancy and Children. St Louis: Mosby; 1971:188-213.
21 Bunin GR, Surawicz TS, Witman PA, et al. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89:547-551.
22 Graham PH, Gattamaneni HR, Birch JM. Paediatric craniopharyngiomas: a regional review. Br J Neurosurg. 1992;76:47-52.
23 Hoffman HJ, DeSilva M, Humphreys RP, et al. Aggressive surgical management of craniopharyngiomas in children. J Neurosurg. 1992;76:47-52.
24 Lapras C, Patet JD, Mottolese C, et al. Craniopharyngiomas in childhood: analysis of 42 case. Prog Exp Tumor Res. 1987;30:350-358.
25 Curtis J, Daneman D, Hoffman HJ. The endocrine outcome after surgical removal of craniopharyngiomas. Pediatr Neurosurg. 1994;21(suppl 1):24-27.
26 Sklar CA. Craniopharyngioma: endocrine abnormalities at presentation. Pediatr Neurosurg. 1994;21(suppl 1):18-20.
27 Rutka JT, Hoffman HJ, Drake JM, et al. Suprasellar and sellar tumors in childhood and adolescence. Neurosurg Clin N Am. 1992;3:803-820.
28 Matson DD, Crigler JFJr. Management of craniopharyngioma in childhood. J Neurol. 1969;30:377-390.
29 Van Effenterre R, Boch AL. Craniopharyngioma in adults and children. J Neurosurg. 2002;97:3-11.
30 Karavitaki N, Cudlip S, Adams CB, et al. Craniopharyngiomas. Endocr Rev. 2006;27:371-397.
31 Luse SA, Kernohan JW. Squamous-cell nests of pituitary gland. Cancer. 1955;8:623-628.
32 Mark RJ, Lutge WR, Shimizu KT, et al. Craniopharyngioma: treatment in the CT and MR imaging era. Radiology. 1995;197:195-198.
33 Cabezudo JM, Vaquero J, Garcia-de-Sola R, et al. Computed tomography with craniopharyngiomas: a review. Surg Neurol. 1981:422-428.
34 Harwood-Nash DC. Neuroimaging of childhood craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl-1):2-10.
35 Sami M, Bini W. Surgical treatment of craniopharyngiomas. Zentralbl Neurochir. 1991;52:17-23.
36 Tsuda M, Takahashi S, Higano S, et al. CT and MR imaging of craniopharyngioma. Eur Radiol. 1997;7:464-469.
37 Eldevik OP, Blaivas M, Gabrielsen TO, et al. Craniopharyngioma: radiologic and histologic findings and recurrence. AJNR Am J Neuroradiol. 1996;17:1427-1439.
38 Fahlbusch RJ, Honegger W, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg. 1999;90:237-250.
39 Young SC, Zimmerman RA, Nowell MA, et al. Giant cystic craniopharyngiomas. Neuroradiology. 1987;29:468-473.
40 Kiran NA, Suri A, Kasliwal MK, et al. Gross total excision of pediatric giant cystic craniopharyngioma with huge retroclival extension to the level of foramen magnum by anterior trans petrous approach: report of two cases and review of literature. Childs Nerv Syst. 2008:385-391.
41 De Vile CJ, Grant DB, Kendall BE, et al. Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J Neurosurg. 1996;85:73-81.
42 Hald JK, Eldevik OP, Skalpe IO. Craniopharyngioma identification by CT and MR imaging at 1.5 T. Acta Radiol. 1995;36:142-147.
43 Ahamadi J, Destian S, Apuzzo ML, et al. Cystic fluid in craniopharyngiomas: MR imaging and quantitative analysis. Radiology. 1992;182:783-785.
44 Barkovich AJ. Craniopharyngiomas: Brain tumors of childhood. In Barkovich AJ, editor: Pediatric Neuroimaging, 2nd ed, New York: Raven Press, 1995.
45 Sutton LN, Gusnard D, Bruce DA, et al. Fusiform dilations of the carotid artery following radical surgery in childhood craniopharyngiomas. J Neurosurg. 1992;74:695-700.
46 Ingraham FD, Matson DD, McLaurin RL. Cortisone and ACTH as an adjunct to the surgery of craniopharyngiomas. N Engl J Med. 1952;246:568-571.
47 Tytus JS, Seltzer HS, Kahn EA. Cortisone as an aid in the surgical treatment of craniopharyngiomas. J Neurosurg. 1952;12:555-564.
48 Merchant TE, Williams T, Sanford RA, et al. Preirradiation endocrinopathies in pediatric brain tumor patients determined by dynamic test of endocrine function. Int J Radiat Oncol Biol Phys. 2002;54:45-50.
49 Merchant TE, Kiehna EN, Sanford RA, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience. Int J Radiat Oncol Biol Phys. 2006;53:533-542.
50 Puget S, Garnett M, Sainte-Rose C, et al. Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg. 2007;106(suppl 1):3-12.
51 Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—a long-term result following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1-10.
52 Ohmori K, Collins J, Fukushima T. Craniopharyngiomas in children. Pediatr Neurosurg. 2007;43:265-278.
53 Carpentieri SC, Waber RM, Scott LC, et al. Memory deficits among children with craniopharyngiomas. Neurosurgery. 2001;49:1053-1058.
54 Cavassuti V, Fischer EG, Welch K, et al. Neurological and psychological sequelae following different treatments of craniopharyngioma in children. J Neurosurg. 1983;59:409-417.
55 Choux M, Lena G, Genitori L. Rapport de la societe francaise de neurochirugie, craniopharyngiomes de l’ enfant. Neurochirurgie. 37(suppl 1), 1991.
56 Sanford RA. Craniopharyngioma: results of survey of the American Society of Pediatric Neurosurgeons. Pediatr Neurosurg. 1994;21(suppl 1):39-43.
57 Epstein FJ, Handler MH. Craniopharyngioma: the answer. Pediatr Neurosurg. 1994;21(suppl 1):1-132.
58 Honegger J, Buchfleder M, Fahlbusch R, et al. Transphenoidal microsurgery for craniopharyngioma. Surg Neurol. 1992;37:189-196.
59 Gardner PA, Prevedello DM, Kassam AB, et al. The evolution of the endonasal approach for craniopharyngiomas. J Neurosurg. 2008;108:1043-1047.
60 Tomita T, Bowman RM. Craniopharyngiomas in children: surgical experience at Children’s Memorial Hospital. Childs Nerv Syst. 2005;21:729-746.
61 Poretti A, Grotzer MA, Ribi K, et al. Outcome of craniopharyngioma in children: long-term complications and quality of life. Dev Med Child Neurol. 2004;46:220-229.
62 Dhellemmes P, Vinchon M. Radical resection for craniopharyngiomas in children: surgical technique and clinical results. J Pediatr Endocrinol Metab. 2006;19(suppl 1):329-335.
63 Zuccaro G. Radical resection of craniopharyngioma. Childs Nerv Syst. 2005;21:679-690.
64 Larijani B, Bastanhagh MH, Pajouhi M, et al. Presentation and outcome of 93 cases of craniopharyngioma. Eur J Cancer Care (Engl). 2004;13:11-15.
65 Pierre-Kahn A, Recassens C, Pinto G, et al. Social and psycho-intellectual outcome following radical removal of craniopharyngiomas in childhood. Childs Nerv Syst. 2005;21:817-824.
66 Sands SA, Milner JS, Goldberg J, et al. Quality of life and behavioral follow-up study of pediatric survivors of craniopharyngioma. J Neurosurg. 2005;103:302-311.
67 Hukin J, Visser J, Sargent M, et al. Childhood craniopharyngioma: Vancouver experience. Childs Nerv Syst. 2005;21:758-765.
68 Mottolese C, Szathmari A, Berlier P, et al. Craniopharyngiomas: our experience in Lyon. Childs Nerv Syst. 2005;21:790-798.
69 Stripp DCH, Maity A, Janss AJ, et al. Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. Int J Radiat Oncol Biol Phys. 2004;58:714-720.
70 Cavalheiro S, Dastoli PA, Silva NS, et al. Use of interferon alpha in intratumoral chemotherapy for cystic craniopharyngioma. Childs Nerv Syst. 2005;21:719-724.
71 Dunbar SF, Tarbell NJ, Kooy HM, et al. Stereotactic radiotherapy for pediatric and adult brain tumors: preliminary report. Int J Radiat Oncol Biol Phys. 1994;30:531-539.
72 Selch MT, DeSalles AA, Wade M, et al. Initial clinical results of stereotactic radiotherapy for the treatment of craniopharyngiomas. Technol Cancer Res Treat. 2002;1:51-59.
73 Combs SE, Thilmann C, Huber PE, et al. Achievement of long-term local control in patients with craniopharyngiomas using high precision stereotactic radiotherapy. Cancer. 2007;109:2308-2314.
74 Merchant TE, Hua CH, Shukla H, et al. Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr Blood Cancer. 2008;51:110-117.
75 Gopalan R, Dassoulas K, Rainey J, et al. Evaluation of the role of Gamma Knife surgery in the treatment of craniopharyngiomas. Neurosurg Focus. 2008;24:E5.
76 Aquilina K, Merchant TE, Rodriguez-Galindo C, et al. Malignant transformation of irradiated craniopharyngioma in children: report of 2 cases. J Neurosurg Pediatr. 2010;5:155-161.
77 Zwienenberg-Lee M, Boop FA, Kun LE, et al. Incidence and management of radiation-induced moya moya in children with optic pathway tumors and craniopharyngiomas: experience at St Jude Children’s Research Hospital (submitted for publication) 2009.
78 Wisoff JH. Surgical management of recurrent craniopharyngiomas. Prdiatr Neurosurg. 1994;21(suppl 1):108-113.
79 Wisoff JH. Craniopharyngiomas. J Neurosurg Pediatr. 2008;1:124-125.
80 Moon SH, Kim IH, Park SW, et al. Early adjuvant radiotherapy toward long-term survival and better quality of life for craniopharyngiomas—a study in single institute. Childs Nerv Syst. 2005;21:799-807.
81 Fischer EG, Welch K, Shillito J, et al. Long term effects of conservative surgical procedures combined with radiation therapy. J Neurosurg. 1998;73:534-540.
82 Hetelekidis S, Da Silva M, Humphres RP, et al. 20 yr. experience in childhood craniopharyngioma. Int J Radiat Oncol Biol Phys. 1993;27:189-195.
83 Sung DI, Chang CH, Carmel PW, et al. Treatment results of craniopharyngioma. Cancer. 1981;47:847-852.
84 Thompson D, Phipps K, Hayward R. Craniopharyngioma in childhood: our evidence-based approach to management. Childs Nerv Syst. 2005;21:660-668.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 198 Pediatric Craniopharyngioma
In 1932 Harvey Cushing stated that craniopharyngiomas were the most baffling problem confronting neurosurgeons.1 To this day, they remain challenging tumors to manage, with a wide range of possible treatment strategies. As the factors influencing treatment-related quality of life are better elucidated, the controversy over the best management of craniopharyngiomas continues.
History
The first pathologic description was from an autopsy performed in 1857, when Zenker reported a suprasellar lesion with cholesterol crystals and squamous epithelial cells. The first accurate pathologic description was by Erdheim2 in 1904. Frazier and Alpers in 1931 and Cushing in 1932 first used the term craniopharyngioma.1 In 1909 Halstead3 was the first to successfully remove a craniopharyngioma using the transsphenoidal route. A child operated on by Cushing in 1923 survived for more than 50 years.4 In 1899 Mont and Barrett suggested that these tumors arise from the hypophysial duct (Rathke’s pouch).5 The hypophysial duct is actually an invagination of the primitive stomodeum, not of the pharynx, making craniopharyngioma a misnomer.6 Rathke’s pouch forms the pars tuberalis in the development of the anterior lobe of the pituitary gland. This embryologic origin becomes important when viewing magnetic resonance imaging (MRI) scans because it is extremely uncommon not to have a portion of the tumor located within the sella turcica.
Rathke’s cleft cysts also arise from Rathke’s pouch, which is normally reduced to a neural cleft by the sixth week of embryonic life. When a Rathke’s cleft cyst persists, it has the potential to become pathologic, progressively enlarging and filling with mucoid material. Rathke’s cleft cysts and craniopharyngiomas may represent a continuum from the simplest form of Rathke’s cleft cyst to a more complex form of craniopharyngioma.7 Other entities found in this area are epithelial, epidermoid, and dermoid cysts.
Histology
There are three histologic types of craniopharyngioma: adenomatous, squamous papillary, and mixed. The adenomatous variety, which is the histologic pattern most common in the pediatric population, resembles tumors of tooth-forming tissue, having a varied histologic pattern of sheet-like epithelium, lobules, anastomosing trabeculae, cysts, and cloverleaves. The distinctive feature is a palisading layer of small cells enclosing a loose stellate reticular zone with alternating, compactly arranged squamous cells that have a tendency to whorl. The epithelium is a wet keratin, differing from the flaky keratin of epidermoid cysts, and is the hallmark of this tumor subtype. Dystrophic calcification, fibrosis, chronic inflammation, and cholesterol clefts are also prominent features.6 The squamous papillary type is rare in children but makes up approximately one third of the adult variety.8 These tumors tend to be solid rather than cystic and may occur within the third ventricle.9 They are less likely to develop calcification and do not form keratin nodules, leading to no evidence of calcification on computed tomography (CT), which is important in the differential diagnosis in adult patients. Mixed tumors contain separate foci with histologic features of both adenomatous and squamous papillary types.9
Adenomatous craniopharyngiomas tend to have finger-like projections that invade the hypothalamus. Therefore, it is common to find small foci of normal brain tissue within resected craniopharyngioma specimens.7–9 This histologic finding is not readily appreciable under the operating microscope, where the tumor capsule may seem to be distinct from the adjacent neural tissue. It is well established that the squamous papillary type, which does not have these microscopic projections, has a better clinical outcome and a lower rate of recurrence with gross total surgical removal than the adenomatous type.8,10 Other authors contend that this histologic difference is not predictive of outcome.9–11 A fibrillary gliotic reaction in the brain immediately adjacent to a craniopharyngioma may create a cleavage plane that actually aids in tumor removal.12–16 Again, there is a difference of opinion; other authors think this dense adherence makes separation from normal brain more difficult.17,18
Epidemiology
Craniopharyngiomas are the most common intracranial pediatric tumors of nonglial origin; they constitute 1.2% to 4% of all brain tumors and 6% to 9% of all pediatric brain tumors.17 There are approximately 0.5 to 2 new cases per million people per year.17 In childhood, approximately 54% of tumors in the sella-chiasmatic region are craniopharyngiomas.20 These tumors can present at any age, but there is a bimodal age distribution, with peak incidence rates in children aged 5 to 15 years and adults aged 40 to 70 years (Fig. 198-1).21 In the United States, 338 cases are expected to occur annually, with 96 patients presenting at an age younger than 14 years. Although classically thought to be more common in males, there has been no obvious gender difference in large population-based studies.21 The peak incidence in the pediatric age group is 5 to 10 years, but craniopharyngioma can occur at any age. There are no known associated genetic abnormalities or risk factors.8
Clinical Presentation
Visual impairment is common, and tumors are occasionally discovered when visual screening or parental observation results in ophthalmologic referral. One should note that very large tumors may cause minimal to no visual defects. Visual defects often go undetected until severe visual impairment and optic atrophy are present.19 Visual disturbance is noted in 37% to 68% of children at presentation.13,21–30 A formal neuro-ophthalmologic examination should be obtained preoperatively whenever possible.
Endocrine dysfunction is very common, occurring in 66% to 90% of new pediatric patients. However, it is rare for a child to be brought to medical attention specifically for an endocrine-related complaint. Short stature (defined as height less than the 3rd percentile), diabetes insipidus (7.5% to 24% of patients), and hypothyroidism (12% to 25% of patients) have been reported.6–9,11,30,31 Endocrine testing reveals deficiencies of growth hormone in up to 75% of patients, luteinizing hormone and follicle-simulating hormone in 40% of patients, adrenocorticotropic hormone (ACTH) in 25%, and thyroid-stimulating hormone in 25%. Less common presenting features are elevation of prolactin (20%), morbid obesity (11% to 18%), diplopia (8% to 11%), and disturbance of mentation.6–8,11,30,31 It should be noted that it is rare for a child with a craniopharyngioma to have clinically significant diabetes insipidus at presentation.
Imaging
Historically, skull radiographs were very important in the diagnosis of craniopharyngioma because they demonstrated sellar enlargement and suprasellar calcification, but they are unnecessary in the modern era. MRI is required in the diagnostic evaluation and is necessary for adequate surgical planning and postoperative evaluation. Because CT scans show calcification, which occurs in 51% to 90% of pediatric craniopharyngiomas,32,33 this modality remains a useful and important part of the preoperative evaluation. CT is also superior in demonstrating the anatomy and size of the sphenoid sinus when the transsphenoidal surgical route is anticipated. The calcifications have a craggy, popcorn-like appearance, with fine eggshell lines (Fig. 198-2A to C).34 Coronal CT can facilitate the detection of intrasellar ossification.
MRI, with and without gadolinium enhancement, is critical for demonstrating the tumor’s relationship to the optic chiasm, infundibulum, hypothalamus, and major vessels (Fig. 198-3C, E, and F). The T1-weighted images should be in the axial, coronal, and sagittal planes. The postcontrast sagittal images are most useful in assessing the relationship of the tumor to the hypothalamus, providing critical information for later decisions regarding treatment options.35,36 If a patient has a cystic, enhancing suprasellar mass with calcifications, it is almost certainly an adamantinomatous type of craniopharyngioma (see Figs. 198-2 and 198-3).33
Craniopharyngiomas are typically 2- to 4-cm lesions in 58% to 76% of cases,37,38 although very large tumors have been reported.39,40 The most common morphologic finding is a predominantly cystic type of tumor in 46% to 64% of cases.30,38,41 The predominantly solid type is seen in 18% to 39% of cases, and mixed solid and cystic tumors are seen in 8% to 36% of cases.29,41 On MRI the solid portion of the tumor typically appears mottled, an indirect effect of signal dampening due to tumor calcification.42 Gadolinium produces heterogeneous enhancement of the solid tumor (see Fig. 198-3B and G). The signal intensity of cystic fluid on T1-weighted images may range from low to high. This variation and degree of intensity is the result of different protein concentrations, free methemoglobin, and cholesterol crystals.43 The walls of the cystic portions of the tumor almost always enhance with contrast (Table 198-1; see Fig. 198-3B).44
TABLE 198-1 Magnetic Resonance Imaging Characteristics of the Cystic and Solid Portions of Craniopharyngioma
| MRI SEQUENCE | TUMOR COMPONENT | DESCRIPTION |
|---|---|---|
| T1 weighted without contrast | Solid | Isointense or hypointense |
| T2 weighted without contrast | Solid | Mixed hypointensity-hyperintensity |
| T1 weighted with contrast | Solid | Homogeneous or heterogeneous enhancement |
| T1 weighted without contrast | Cystic | Hypointense |
| T2 weighted without contrast | Cystic | Hyperintense* |
| T1 weighted with contrast | Cystic | Cystic rim enhancement |
* Acute, subacute, or chronic hemorrhage; protein density; and cholesterol deposition may alter cystic imaging characteristics.
Cerebral angiography provides the best demonstration of the displacement of major vessels by tumor, but it has largely been supplanted by magnetic resonance angiography. When planning a radical surgical removal of recurrent craniopharyngioma, invasive angiography may be useful to delineate constriction of the carotid, middle, or anterior cerebral arteries and facilitate tumor dissection. Fusiform dilation of the carotid artery has been reported after radical tumor resection, and it potentially increases the risk during reoperation.35,45 Because craniopharyngiomas are relatively avascular, even modern angiography may not demonstrate the small nutrient vessels.19
There are many postoperative and intraoperative options for the follow-up of patients with craniopharyngioma. We recommend that MRI be obtained within 48 hours after surgery to document the presence or absence of residual tumor and the size of residual cysts and to minimize confusion when interpreting postsurgical changes (see Fig. 198-2G and H). If complete surgical removal is attempted, an additional CT scan is necessary because a small fleck of calcification may mean that the resection was incomplete, portending recurrent tumor growth in the follow-up period.
Differential Diagnosis
The differential diagnosis of suprasellar-sellar masses includes chiasmatic-hypothalamic glioma, germinoma, Rathke’s cleft cyst, hypothalamic hamartoma, arachnoid cyst, pituitary adenoma, and meningioma; the last two are rare in children.27 The presence of calcification on CT virtually eliminates all these diagnoses except meningioma. In children whose tumors are predominantly adamantinomatous, calcification occurs more than 90% of the time.33,34 The next most common tumor in this area is the chiasmatic-hypothalamic glioma, which does not have an intrasellar component and does not commonly calcify.
Endocrine Evaluation
Preoperative
Evaluation of five hormone systems is recommended: thyroid, growth hormone, cortisol, gonadotropins, and prolactin. All children with craniopharyngiomas should be considered ACTH deficient until proved otherwise and should empirically receive stress corticosteroid coverage during surgical procedures. The stress dose of 7 to 15 mg/m2 mis triple the daily maintenance dose of hydrocortisone.25 In a previously operated and treated patient, thyroid deficiency may be profound, and it is critical to ensure that the child has been taking his or her thyroid medication. Of historical interest, it was not until corticosteroid therapy came available in the 1950s that removal of craniopharyngiomas could be accomplished with acceptable morbidity and mortality.46,47
Postoperative
More extensive evaluation of the five hormone systems is necessary after surgery, as the child is followed or goes on to radiation therapy. Thyroid evaluation should include thyroid-stimulating hormone surge and thyrotropin-releasing hormone tests for evaluation of the hypothalamic-pituitary-thyroid axis. Growth hormone evaluation includes the arginine tolerance and levodopa tests for growth hormone secretory capacity and activity. We recommend baseline levels of cortisol, ACTH levels, metapyrone testing for ACTH reserve, and the gonadotropin-releasing hormone stimulation test to determine premature or delayed gonadotropin secretion or response. In addition, serum is obtained for determination of the prolactin level.48 Twenty-four-hour assessment of urine output, serum and urine osmolarity, and electrolytes is important because diabetes insipidus occurs in more than 90% of patients after radical surgery and in 33% to 47% after limited surgery.49 Limited surgical approaches such as stereotactic cyst drainage and biopsy do not result in diabetes insipidus.
Long-term endocrine deficiencies in the five major systems are not much different between the limited surgery and radiotherapy group compared with the radical surgery group. However, there is a difference in time to onset, delayed in the former and immediate in the latter (Table 198-2).35,49,50 In the case of a radiated child, endocrine status must be measured yearly until the baseline nadir is reached.
Management Controversy
Although there is much debate over the proper management of patients with craniopharyngiomas, the treatment must focus on relief of symptoms, cure of disease, and quality of life of the patient. In reaching these goals, the surgeon must choose the treatment strategy that causes the least harm and delivers the most benefit to the patient. The continuum of treatment ranges from radical resection with the avoidance of radiation to limited surgery followed by radiotherapy. The role of other treatment modalities such as intracystic therapy (P-32, bleomycin) or the Gamma Knife is discussed later, but they are usually just adjuncts to one of the major treatment approaches. In expert hands, cure rates using either surgical approach are nearly identical.16,49–52 One certainty is that attempts at gross total resection of large solid tumors have a significantly negative effect on quality of life.17,34,50,51,53–55 As such, the surgeon must look closely at his or her own results in terms of cure of disease and harm from treatment and make the best treatment decision for each patient. A survey of neurosurgeons demonstrated that surgeons who operated on one or fewer craniopharyngiomas per year could not expect the results of radical surgery quoted in the literature with regard to cure rate or complication rate.56
Radical Resection Versus Limited Surgery Plus Radiotherapy
Radical Resection
Radical resection was once considered the “gold standard” for the treatment of craniopharyngioma. The primary goal of radical resection is to obtain a surgical cure of this benign disease, thus sparing the patient the risks associated with radiotherapy or other adjuvant treatment. It can also be useful for decompressing the optic nerve or optic chiasm, thereby improving vision, and for relieving hydrocephalus by decompressing the cerebrospinal fluid pathways and obviating the need for shunting procedures. Although gross total resection has fallen out of favor in some centers, it remains a good option for many patients and should be recommended when it serves the patient better than alternative options. Numerous studies have been published that show excellent surgical results with regard to cure and outcome.10,19,23,24,38,57 Radical resection can be an effective treatment with excellent rates of long-term progression-free survival.23,26
Radical surgery often has an advantage when there is no hypothalamic involvement and resection can be accomplished with minimal complications or side effects. In theses cases, it eliminates the need for further therapies and their associated side effects.49,53
[/not-level-membership-for-neurosurgery-category]
