[level-membership-for-pediatrics-category]Chapter 35
Pediatric Brain Neoplasms
Brain tumors are the most common solid pediatric tumors and are the leading cause of death in children from solid tumors.1 The estimated incidence of all childhood primary brain and central nervous system (CNS) tumors is 4.8 cases per 100,000 person-years.2 Approximately 4150 new cases of childhood primary nonmalignant and malignant brain and CNS tumors were diagnosed in the United States in 2011.3 Nearly 50% of brain tumors in children older than 1 year arise in an infratentorial location. However, in neonates, infants, and children up to the age of 3 years, supratentorial tumors are more common.3
Etiology
The etiology of pediatric brain tumors, an area of research beyond the scope of this chapter, requires an understanding of genetic alterations, signaling systems, and molecular genetics and pathways. Although no one risk factor explains more than a small percentage of childhood brain tumors, therapeutic doses of ionizing radiation to the head for brain tumors and radiation for leukemia,4,5 as well as certain genetic syndromes, are known risk factors in the pediatric population. Among the congenital syndromes associated with brain tumors are neurofibromatosis types 1 and 2, Gorlin syndrome (basal cell nevus syndrome), tuberous sclerosis, Turcot syndrome, von Hippel-Lindau syndrome, and Li-Fraumeni syndrome.6
Imaging
Because of its superior soft tissue resolution, multiplanar capability, and lack of ionizing radiation, magnetic resonance imaging (MRI) with contrast is the modality of choice in determining lesion size, location, and characterization. And although contrast enhancement typically reflects disruption of the blood-brain barrier, the degree of contrast enhancement does not always correlate with tumor grade. For example, benign tumors (e.g., choroid plexus papillomas and pilocytic astrocytomas) can enhance avidly, whereas anaplastic astrocytomas may not enhance at all.7
MRI also is used to assess tumor response and progression and monitor treatment effects. Essential to optimal treatment planning is accurate staging of the tumor that confirms whether the tumor has spread through the neural axis. Intraoperative MRI is being used in some centers to guide both conventional and minimally invasive tumor resection. As these systems are refined, they are expected to form the standard of care at many medical centers.8
Diffusion-Weighted Imaging
Contrast on diffusion-weighted images (DWI) reflects the mean distance traveled by free water protons in tissue as a result of Brownian motion.9,10 Diffusion occurs freely in the direction of white matter tract orientation and is restricted in orthogonal planes. DWI can assess the properties of diffusion occurring within a particular voxel, which is expressed as the apparent diffusion coefficient (ADC). A markedly decreased ADC usually correlates well with increased tumor cellularity in brain neoplasms. Vasogenic edema and necrosis show an increased ADC.7,11,12 ADC values are interpreted in conjunction with structural MRI sequences. Diffusion tensor imaging (DTI), an adaptation of DWI, acquires diffusion data in six or more directions to establish the direction and magnitude of water diffusion.
DWI also can be extremely useful in the postoperative period, when low ADC values at the surgical margins or within the resection cavity may be indicative of ischemia or abscess. This technique typically is used in conjunction with conventional MRI, which helps exclude artifact from hematoma.13 Further, DTI aids in identifying patterns of tumor interaction with white matter fiber tracts (i.e., the extent of deviation, edema, infiltration, and destruction),14 and when used in conjunction with volumetric data, it effectively guides surgical resection and predicts possible postoperative deficits resulting from white matter tract damage (e-Fig. 35-1). Reduced fractional anisotropy (FA), which is a measure of the directional diffusivity of water made using DTI, has been found in the white matter of patients with a medulloblastoma, even in the absence of abnormalities on structural sequences.15 Decreased FA values have been shown to correlate with the age at which radiation was administered and with poor academic performance among school-age patients.16 FA thus may be considered a noninvasive biomarker to monitor effects of radiotherapy.16
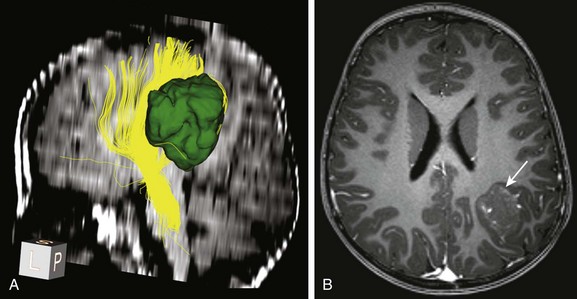
e-Figure 35-1 Presurgical tractography in a glioma lesion.
A, Tractography of a portion of the left corticospinal tract forming the posterior limb of the internal capsule (depicted in yellow) in a patient with left parietal lobe angiocentric glioma (segmented and shown in green). The tract is displaced medially by the tumor but is still intact.
B, An axial postcontrast magnetization-prepared rapid gradient-echo image shows the cortically based minimally enhancing tumor (arrow).
Magnetic Resonance Spectroscopy
Magnetic resonance spectroscopy (MRS) is a noninvasive in vivo technique that provides metabolic information beyond structural imaging sequences. It enables detection and quantification of abnormal metabolites in the brain and can help identify tumor tissue, differentiate tumor types, and separate active tumor from radiation necrosis or scar formation. MRS can be performed with most standard MRI scanners, typically by incorporating either the point resolved spin echo or stimulated echo acquisition mode techniques. Simultaneous acquisition of MRS from multiple voxels increases spatial resolution; this procedure is known as “chemical shift imaging” or MR spectroscopic imaging.17
Most brain tumors are characterized by the presence of increased choline/creatine and decreased NAA/creatine ratios, indicating loss of neuroaxonal integrity and increased cell membrane turnover. The presence of lactate in the tumor suggests an anaerobic process with impaired energy metabolism.18 In general, high-grade tumors have higher choline/creatine and lower NAA/creatine ratios than do low-grade lesions. In rapidly growing malignant tumors, necrotic areas may contain lipid resonances.19 However, in pediatric patients, we frequently (and paradoxically) see elevated levels of choline and lactate in pilocytic astrocytomas, a low-grade tumor.20
The presence of specific metabolites such as alanine (an inverted doublet at 1.44 ppm) in meningiomas (e-Fig. 35-2) and taurine (peak at 3.3 to 3.4 ppm) in primitive neuroectodermal tumors (PNETs) may help narrow the differential diagnosis.21,22 Citrate is a tricarboxylic acid cycle intermediate metabolite that has been described in pediatric brain tumors and is found at particularly high levels in pontine gliomas.23 In one study of grade 2 astrocytomas, citrate was significantly more prominent in tumors that progressed.24
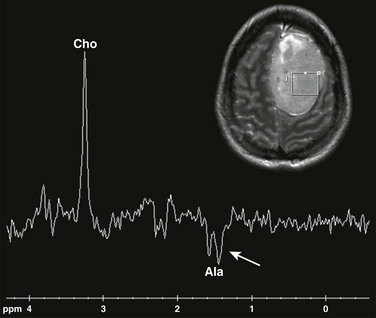
e-Figure 35-2 Magnetic resonance spectroscopy (MRS) in a meningioma tumor.
Single voxel MRS (TE 144 ms) performed over the left frontal meningioma (see inset T2-weighted image) shows a prominent choline peak (Cho) and reduced creatine and N-acetylaspartate peaks. The inverted doublet centered at approximately 1.55 to 1.6 ppm is consistent with an alanine (Ala) peak.
Perfusion-Weighted imaging
Perfusion-weighted imaging measures cerebral hemodynamics at the microcirculation level. Parameters measured by perfusion-weighted imaging include cerebral blood volume (CBV), cerebral blood flow, and mean transit time. Of these, the CBV, defined as the volume of blood in an area of brain tissue expressed in mL/100 g, is the most commonly used parameter in evaluation of brain tumors.25 Lower grade astrocytomas have relatively lower regional CBV than do higher-grade tumors such as anaplastic astrocytomas and glioblastomas26 (e-Fig. 35-3). However, low-grade pediatric pilocytic astrocytomas can have a high relative cerebral blood volume.27
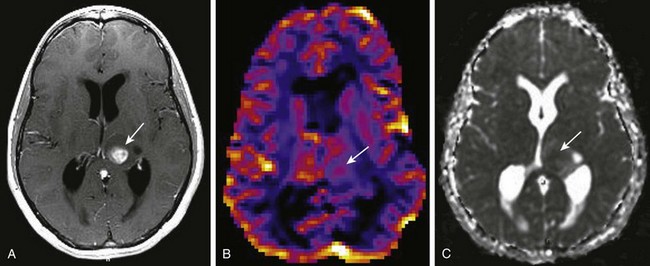
e-Figure 35-3 Perfusion-weighted imaging in a thalamic glioblastoma multiforme tumor.
A, A T1-weighted image shows a rounded centrally enhancing mass lesion in the left thalamus (arrow). B, An axial T2* cerebral blood volume perfusion image shows a focal area of increased perfusion corresponding to the enhancing solid portion of the tumor in the left thalamus (arrow). C, An apparent diffusion coefficient map shows regions of decreased diffusion corresponding to the solid component of the left thalamic tumor (arrow).
The most widely available technique is T2*-weighted dynamic susceptibility contrast imaging, which consists of a rapid bolus of intravenous (IV) paramagnetic contrast agent followed by a rapid acquisition of echo-planar images during the first pass of contrast material through the capillary bed. As the contrast medium is delivered, it goes through the tissues and results in a signal drop proportional to the blood volume during the first pass. Routine use of this technique in children requires the use of rapid contrast medium injection and power injectors, as well as strategies to overcome problems associated with large-bore IV catheter placement, especially in infants.28
ASL uses endogenous blood as a tracer. The two major types of ASL, pulsed and continuous, are now widely available on clinical scanners. A third type, pseudocontinuous ASL, has just recently been introduced for clinical use. ASL has shown promise for hemodynamic evaluation of brain tumors,29,30 but data in children are limited at this time.
Functional MRI
Functional MRI (fMRI) is a technique that essentially relies on two physical principles, namely, that oxyhemoglobin is diamagnetic and deoxyhemoglobin is paramagnetic in nature. Because of the relative increased blood flow and consequent increased utilization of oxygen within the activated portions of the brain, the MR signal, in this case known as the “blood oxygen level dependent signal,” or BOLD, is measurably different compared with other parts of the brain. The primary value of fMRI is in localizing the eloquent areas in the brain controlling language, motor skills, and memory. This information helps provide surgical guidance.31 More detailed information can be found in Chapter 27.
Single PET and PET
The role of PET in the evaluation of pediatric brain tumors is to determine metabolic activity at diagnosis, assess response to therapy, and distinguish treatment effect versus tumor recurrence. Fluorine-18-deoxyglucose (18F-FDG) is the most commonly used isotope for PET studies in children. PET scanning using other labeled agents such as the amino acid analogues [11C] methionine and [11C] tyrosine have shown promise in detecting low-grade tumors in adults, although their diagnostic value in children has not yet been established.32 These amino acid analogues are incorporated via amino acid transport pathways into tumor proteins, and therefore uptake reflects tumor protein synthesis.33,34
Other isotopes still at the investigational stage include cell proliferation agents (e.g., 18F-fluorothymidine) and cell hypoxia imaging agents (e.g., 18F-fluoromisonidazole and 62Cu-labeled diacetyl-bis [N4-methylthiosemicarbazone]).
Specific Tumors
Classification of Pediatric Brain Tumors
The differential diagnosis is effectively limited by classifying tumors by location, describing the appearance of the lesion on conventional MRI, and applying advanced imaging techniques (Box 35-1).
Tumors of the Cerebral Hemispheres
The modified World Health Organization (WHO) classification of CNS tumors divides astrocytomas into low grade (grades I and II) and high grade (grades III and IV).35 Grading of astrocytomas by the WHO criteria is predictive of patient survival.35
Pilocytic astrocytomas are grade I WHO tumors; they account for 20% to 30% of all childhood brain tumors.36 Pilocytic astrocytomas typically arise in the first two decades of life. The most common locations of these tumors are in the optic pathways, hypothalamus, thalamus, basal ganglia, cerebral hemispheres, cerebellum (Fig. 35-4), and brainstem. Patients with neurofibromatosis type 1 (NF1) have an increased risk of the development of pilocytic astrocytomas, including optic pathway tumors. Patients with NF1 who have optic pathway tumors tend to have a better long-term prognosis than do patients without NF1 who have optic pathway tumors.37
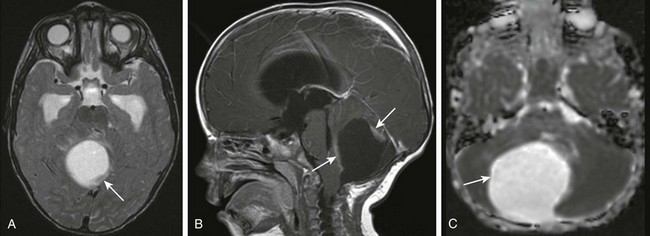
Figure 35-4 A cerebellar astrocytoma.
A, An axial T2-weighted image shows a large cystic mass centered in the right cerebellar hemisphere with small solid components along the posterior and anterior aspects (arrow). B, A postcontrast sagittal T1-weighted image shows the solid components (arrows) along the anterior and posterior margins that enhance after paramagnetic contrast administration. C, An apparent diffusion coefficient map shows increased diffusion within the lesion (arrow) consistent with the relatively low cellularity within the lesion.
Rarely, pilocytic astrocytomas can present with diffuse leptomeningeal spread, which most often is seen in association with the diencephalic syndrome (discussed later in this chapter) or with the pilomyxoid variant of astrocytomas. Pilomyxoid astrocytomas have an indolent course, but their propensity for slow-growing, persistent recurrences makes them difficult to treat.38
Supratentorial High-Grade Gliomas
High-grade gliomas in children are significantly less common than are low-grade lesions, which account for up to 20% of all hemispheric gliomas.39,40
On CT, these lesions demonstrate heterogeneous enhancement and density with edema, occasional hemorrhage, mass effect, and ill-defined margins. On MRI, these lesions have heterogeneous signal intensity (Fig. 35-5). They typically are hypointense on T1-weighted images and hyperintense on T2-weighted images with surrounding white matter edema. They show effect of the mass on surrounding structures and demonstrate irregular enhancement with necrosis and hemorrhage similar to that seen on CT.
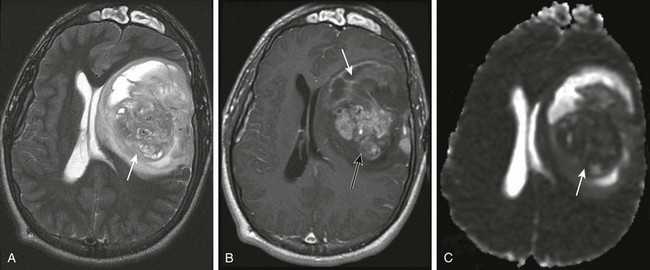
Figure 35-5 A supratentorial high-grade glioma.
A, An axial T2-weighted image shows a large heterogeneous mass (arrow) in the left frontal, temporal, and parietal lobes with marked mass effect, subfalcine herniation, surrounding edema, and rightward midline shift. B, A postcontrast T1-weighted image shows heterogeneous enhancement of the solid components of the tumor (black arrow) and nonenhancing components anteriorly (white arrow), suggestive of necrosis. C, An apparent diffusion coefficient map reveals decreased diffusion within the solid component (arrow), indicating high cellularity.
Aggressive surgical resection with preservation of neurological function, followed by radiotherapy directed at the tumor bed, remains the cornerstone of treatment of pediatric malignant gliomas.41 The addition of chemotherapy has been shown to improve survival compared with surgery and radiotherapy alone.40 The overall prognosis for children with supratentorial malignant gliomas remains poor, however, with 5-year progression-free survival rates of around 30%.40
Supratentorial PNETs
Although supratentorial PNETs are relatively rare, these tumors are more common in the first decade of life, with peak incidence from birth to 5 years of age.42 They account for 5% of all supratentorial tumors in childhood. At presentation they often are large and fairly well defined, occurring either in the cerebral hemispheres or in the lateral ventricles. They may be solid and homogenous or heterogeneous with cyst formation.43 Calcification often is seen on CT. Heterogeneous contrast enhancement is seen along with regions of necrosis.
On MRI, solid areas have restricted diffusion and T2-hypointense areas (Fig. 35-6), reflecting high nuclear-to-cytoplasm ratio, increased cellularity, and increased CBV values. Necrosis and hemorrhage also can occur in these lesions.
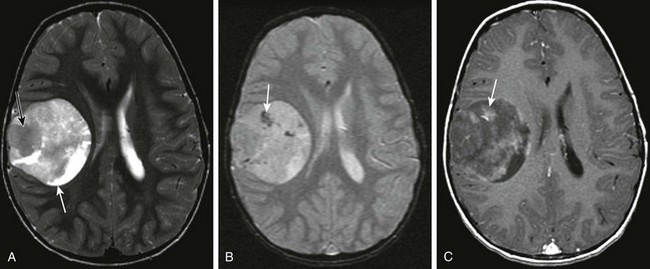
Figure 35-6 A supratentorial primitive neuroectodermal tumor.
A, An axial T2-weighted image shows a well-defined, large, heterogeneous mass lesion in the right cerebral hemisphere with a predominant solid T2-hypointense component (black arrow) and smaller areas of T2 prolongation (white arrow). B, An axial gradient-recall echo image shows scattered foci of susceptibility (arrow) suggestive of calcification or blood products within the lesion. C, An axial postcontrast T1-weighted image shows patchy heterogeneous enhancement within the lesion (arrow).
Supratentorial Ependymoma
Ependymomas constitute approximately 10% of all intracranial tumors in children.44 Of these, supratentorial ependymomas typically occur in children younger than 6 years and account for up to 40% of all ependymomas.45 These tumors are thought to arise from embryonic rests of ependymal tissue trapped in the developing cerebral hemispheres.46 Ependymomas are heterogeneous and often contain calcification and cystic areas. They are hypointense on T1-weighted images and isointense to hyperintense to gray matter on T2-weighted images. Moderate to avid enhancement of the soft tissue components of the tumor is seen, intermixed with poorly enhancing or nonenhancing areas.47
Choroid Plexus Tumors
Choroid plexus tumors account for approximately 3% of pediatric brain tumors.48 Of these, 10% to 20% arise in the first decade of life and 80% occur in the first 2 years of life, including a considerable number of tumors diagnosed in utero.49 Choroid plexus papillomas account for the vast majority of choroid plexus tumors (up to 85%), with the remainder being choroid plexus carcinomas.
These tumors typically occur in the trigone of the lateral ventricles in children, as opposed to adults, in whom they occur in the fourth ventricle. On CT, choroid plexus papillomas are lobulated masses that typically are isodense to hyperdense, may have punctate calcifications, and enhance avidly and homogenously. On MRI, they are homogeneous, enhancing intraventricular masses that are hypointense on T1-weighted images and predominantly hyperintense on T2-weighted images (Fig. 35-7).
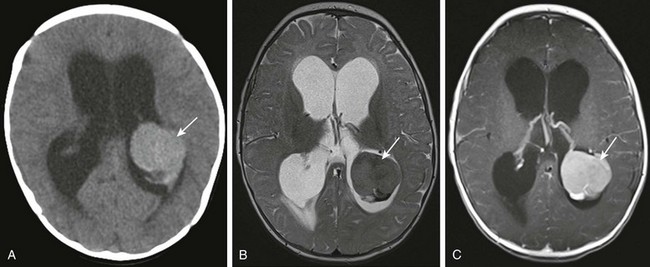
Figure 35-7 A choroid plexus papilloma.
A, Axial noncontrast computed tomography shows a lobulated hyperdense mass in the left lateral ventricle (arrow). B, An axial T2-weighted image shows that the mass in the left lateral ventricle is hypointense compared with the brain parenchyma (arrow). C, An axial postcontrast T1-weighted image shows homogenous enhancement of the lesion (arrow) after administration of contrast material.
Choroid plexus carcinomas may be hyperdense on CT, reflecting increased cellularity. These tumors almost always invade the adjacent brain through the ventricular wall and cause vasogenic edema.50 They are characterized by areas of heterogeneous signal intensity on both T1- and T2-weighted images because of hemorrhage and necrosis. MRS may help distinguish between papillomas and carcinomas. The myoinositol level is significantly lower and the choline level is significantly higher in choroid plexus carcinomas than in choroid plexus papillomas.51
Sellar and Parasellar Tumors
Craniopharyngiomas are slow-growing, benign, nonglial tumors arising in the sellar and parasellar regions. They constitute between 3% and 10% of all pediatric brain tumors.44 Craniopharyngiomas are classified as WHO grade I tumors and arise from ectodermal remnants of the Rathke pouch with a bimodal incidence in the first and fifth decades of life. The adamantinous type is more common in children, whereas the squamous-papillary variant tends to occur in adults.44 Although histologically craniopharyngiomas are benign, they can invade surrounding structures, eliciting a gliotic response that makes resection challenging.
The imaging appearance of craniopharyngiomas reflects their mixed cystic and solid nature, with 90% having calcification and 90% having a cyst formation. On MRI, high signal intensity on both T1- and T2-weighted images is seen in areas with high protein content (Fig. 35-8) or in lesions that show evidence of subacute hemorrhage. Hypointensity on T1-weighted images can be seen reflecting the presence of keratin in some of the cysts. CT often is used to demonstrate calcification, which is important for diagnosis and surgical planning. Surgical treatment remains the mainstay, with radiotherapy having a role in cases that are not amenable to gross total resection. Recurrence-free, 5-year survival is close to 87% but falls to less than 50% with subtotal resection.52 Follow-up imaging is directed toward identifying recurrence, second tumors, and associations with moyamoya syndrome.
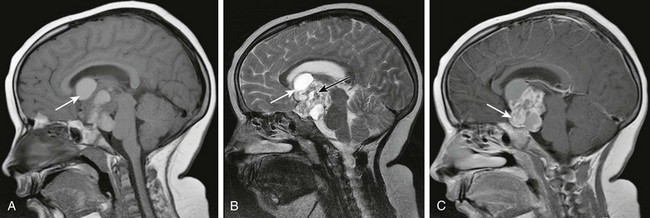
Figure 35-8 A craniopharyngioma.
A, A sagittal T1-weighted image shows a large suprasellar lesion containing solid and cystic components with T1 shortening within the cystic components (arrow) consistent with proteinaceous content. B, A sagittal T2-weighted image shows T2 hyperintensity within the cystic components (white arrow) and scattered hypointensity consistent with foci of calcification (black arrow). C, A sagittal postcontrast T1-weighted image shows avid enhancement of the solid component (arrow).
Chiasmatic/Optic Pathway/Hypothalamic Gliomas
Optic pathway gliomas are low-grade pilocytic astrocytomas (WHO grade I) that represent 15% of supratentorial tumors.53 Although sporadic lesions are not uncommon, a strong association of optic nerve gliomas with NF1 exists; bilateral optic nerve tumors are virtually pathognomonic of NF1.54 Twenty percent to 50% of optic gliomas occur in patients with NF1, whereas the prevalence of optic pathway gliomas in the NF1 population is between 1.5% and 19%.54 The tumors may involve the optic nerves, optic chiasm, optic tracts, lateral geniculate bodies, and/or optic radiations. Tumors in children with NF1 reportedly are less aggressive than those in children without NF1.55
Optic pathway gliomas are usually isointense to hypointense on T1-weighted images (Fig. 35-9). On T2-weighted images, the lesions demonstrate mixed signal intensity; intense enhancement also is common. Use of coronal and axial fat-suppressed thin-section postcontrast T1-weighted images and inversion recovery or T2 images with fat saturation enables optimal visualization of the optic pathways.56
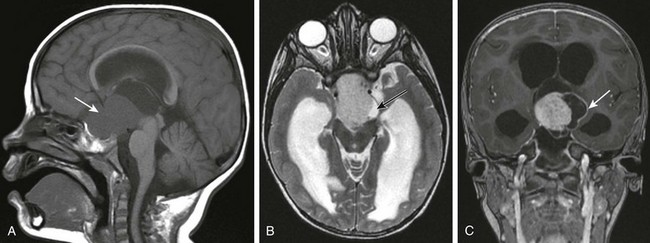
Figure 35-9 A chiasmatic/hypothalamic glioma.
A, A sagittal T1-weighted image shows a large lobulated suprasellar mass (arrow) in the region of the optic chiasm with superior extension into the third ventricle and pontine cistern. B, An axial T2-weighted image shows that the lesion is hyperintense compared with brain parenchyma and that a more hyperintense cystic component is present along its superior and right aspects (black arrow). C, A coronal T1 postcontrast image shows marked enhancement of the central solid component and peripheral enhancement of the cystic components (arrow).
Diencephalic syndrome may be seen in a small percentage of patients with hypothalamic/chiasmatic astrocytomas who present with failure to thrive. These tumors often are larger, occur at a younger age, are more aggressive than others at presentation, and they may seed throughout the cerebrospinal fluid (CSF) pathways.57
Posterior Fossa Tumors
Medulloblastomas are the most common posterior fossa tumors of childhood, accounting for nearly 38% of all posterior fossa tumors and approximately 15% to 20% of all pediatric brain tumors. Medulloblastoma is a heterogeneous disease, with histopathologic and molecular variants that have distinct biological behaviors.58 Medulloblastomas can be separated on the basis of their histopathologic features into the classic type and four variants, including desmoplastic/nodular; medulloblastoma with extensive nodularity; anaplastic medulloblastoma; and large cell medulloblastoma.35 Children who have medulloblastomas with extensive nodularity and desmoplastic/nodular medulloblastomas generally have a better outcome than do children with classic tumors. Patients with large cell and anaplastic medulloblastomas do not fare as well because these tumors behave aggressively and typically are resistant to most therapies.58 Medulloblastomas are characterized by major molecular subgroups that are based on various signaling pathways, including the Shh (sonic hedgehog pathway) variant; Wnt (Wingless); ERBB2 (receptor kinase family); and non-Shh/Wnt subtypes.58,59
Medulloblastomas usually arise in the midline within the vermis and grow into the fourth ventricle, resulting in obstructive hydrocephalus. In older patients and in those with the desmoplastic subtype, they are localized to the cerebellar hemispheres.60 Medulloblastomas usually are hyperdense masses on CT (Fig. 35-10, A); on MRI, they characteristically are T1 and T2 hypointense relative to gray matter with homogeneous enhancement (Fig 35-10, B and C). Elevated taurine content on MRS has been reported in persons with a medulloblastoma.20 The incidence of CSF dissemination at diagnosis is between 20% and 30%.61
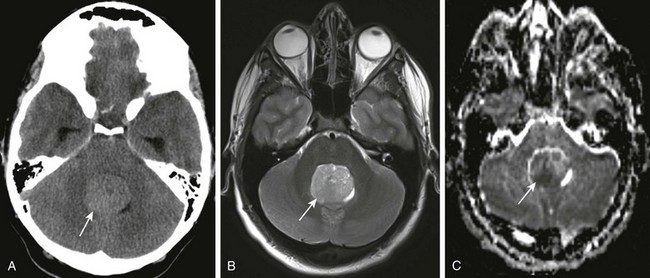
Figure 35-10 A medulloblastoma.
A, Axial noncontrast computed tomography shows a rounded mass (arrow) hyperdense to cerebellar white matter in the fourth ventricle. B, An axial T2-weighted image shows a solid mass centered in the cerebellum close to the midline (arrow). The mass is hypointense on the T2-weighted images. C, An apparent diffusion coefficient map shows restricted diffusion in parts of the mass (arrow), indicating high cellularity.
Treatment of medulloblastomas consists of surgery, radiation therapy, and chemotherapy. Conventional risk stratification is based on the age of the patient, the extent of the tumor at the time of diagnosis, and completeness of surgical resection.62 High-risk features include younger age at diagnosis, incomplete resection or postoperative tumor residuum greater than 1.5 cm2, and metastatic disease.63 More recently, the presence of anaplasia on histopathology, ERBB2 positivity, and classification into the c-Myc and non–Wnt/Shh molecular subgroups have emerged as potential biomarkers of a poor prognosis.59,64
Tectal Gliomas
Patients with tectal gliomas present with symptoms of obstructive hydrocephalus caused by the growth of these lesions adjacent to the aqueduct of Sylvius. Tectal gliomas can be diagnosed on the basis of imaging findings alone. Although imaging appearance is similar to that of pilocytic astrocytomas that appear elsewhere in the cerebral hemispheres, tectal gliomas usually do not enhance after contrast enhancement (Fig. 35-11). They may require CSF diversion procedures to relieve hydrocephalus but usually do not require biopsy or resection. Careful observation suffices for slowly progressing asymptomatic tumors. Rarely, tumors larger than 10 cm may require surgical debulking and/or chemotherapy.65,66
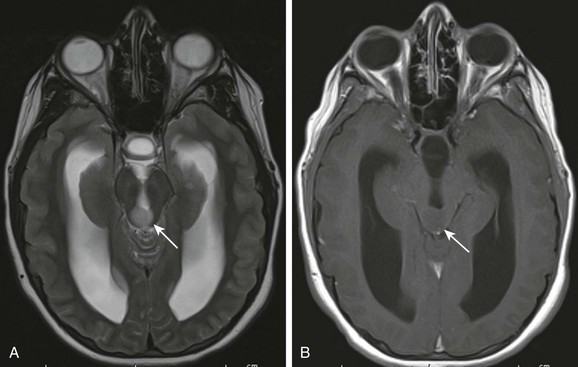
Figure 35-11 A tectal plate glioma.
A, An axial T2-weighted image shows a hyperintense lesion (arrow) involving the tectum that is causing obstruction of the aqueduct. B, An axial postcontrast T1-weighted image shows the mass is T1-isointense to brain parenchyma (arrow) and does not enhance after administration of contrast material.
Brainstem Gliomas
Brainstem tumors account for up to 12% of all brain tumors in children.3 Four types are described on MRI: focal, dorsal exophytic, cervicomedullary, and diffuse intrinsic brainstem glioma.
Diffuse Intrinsic Brain Stem Gliomas (Diffuse Pontine Gliomas): Diffuse intrinsic brainstem gliomas account for up to 85% of all brainstem gliomas. They typically are centered in the pons and hence also are called diffuse pontine gliomas. Because persons with these tumors have a poor long-term survival, they are the focus of numerous clinical trials.67
Because of their location in the brainstem, these lesions previously were considered inoperable. However, with advances in neurosurgical techniques and new molecular analyses using very small amounts of tissue, biopsy of some of these lesions is now being reconsidered.68
On CT, pontine gliomas are hypodense or isodense. On MRI, these tumors are isointense to hypointense on T1-weighted images and hyperintense on T2-weighted images (Fig. 35-12, A). Enhancement is minimal or absent at presentation in most patients (Fig. 35-12, B), but in the later stages of tumor progression, diffuse enhancement and necrosis may be present. Calcification or hemorrhage is rare.

Figure 35-12 A diffuse intrinsic brainstem glioma.
A, An axial T2-weighted image shows a rounded, hyperintense mass centered in the pons (arrow) surrounding the basilar artery and narrowing the pontine cistern. B, A postcontrast sagittal T1 image shows that the lesion does not enhance (arrow) after administration of contrast material. C, An apparent diffusion coefficient map shows increased diffusion within the lesion (arrow), a typical finding at initial presentation.
MR spectroscopy has a potential value in determining tumor treatment response or failure. Decreases in choline : creatine and choline : NAA values are seen within responding tumors after initiating radiotherapy.69 A recent MRS study has shown that increased choline : NAA on single voxel spectroscopy and increased maximum choline : NAA on chemical shift imaging are predictive of a shorter period of survival over time.70
On diffusion images, these tumors have increased ADC values (Fig. 35-12, C) and reduced FA at presentation, with reduced ADC after initiation of radiotherapy.67,71,72 Increased ADC values are thought to be a result of a larger extracellular volume, possibly arising from a combination of vasogenic edema and a lower number of tumor cells.73 Tumor enhancement generally is associated with a shorter survival time, lower tumor diffusion values (and thus increased cellularity), and a smaller drop in diffusion values after radiotherapy.67 Diffusion tensor imaging depicts tracts that initially are infiltrated,74 although not fully disrupted. Improved visualization of white matter tracts is apparent after radiation. As the tumor progresses, complete loss of anisotropy results; this effect may be due to tract infiltration or to possible tract disruption.71
Survival in pediatric patients whose pontine glioma shows 18F-FDG uptake of 50% or more on PET imaging is poorer than in children whose tumor demonstrates less than 50% of 18F-FDG uptake.75 Intense tracer uptake in the tumors, compared with gray matter, also suggests a decreased rate of survival.75 Higher 18F-FDG uptake within the tumor is associated with enhancement on MR images. Increased tumor cellularity, as reflected by restricted MRI diffusion, may be correlated with increased 18F-FDG uniformity throughout the tumor.75
These tumors usually respond initially to radiation therapy, which has improved the median overall survival rate from weeks to months.76 Unfortunately, adjuvant therapies (e.g., radiation sensitizers, differentiation agents, cytotoxic drugs, and molecularly targeted drugs) have not resulted in significantly improved patient outcomes.76,77
Atypical Teratoid Rhabdoid Tumor
Atypical teratoid/rhabdoid tumors (ATRTs) are highly malignant tumors with a peak incidence between birth and 3 years.78 These tumors account for almost 10% of CNS tumors in children and approximately 1% to 2% of all pediatric brain tumors.79 Nearly 60% of these tumors are seen in the posterior fossa at the cerebellopontine angle. However, supratentorial ATRTs also are seen frequently at additional sites in the CNS such as the spine, pineal, and suprasellar regions.80
ATRTs have been identified as being pathologically distinct entities from medulloblastomas and PNETs. This finding is supported by evidence of deletions or loss of material at chromosome 22q11.2, identification of the tumor suppressor gene hSNF5/INI-1, and germline and somatic mutations of INI-1 in approximately 75% of cases of CNS ATRTs.79
Imaging appearances of ATRTs are similar to those of medulloblastomas; namely, they are isointense on T1-weighted images and have hypointense signal intensity on T2-weighted images (e-Fig. 35-13). Cystic areas are common. Because of their high cellularity, T2 hypointensity often is seen in the solid areas and is associated with restricted diffusion. Hemorrhage and calcification are not uncommon (e-Fig. 35-13, A).81 Imaging of the entire neuroaxis is important, because subarachnoid spread throughout the CNS with spinal drop metastases is common, with frequency ranging between 25% and 46%.82,83 ATRT survival rates historically have ranged from 0.5 to 11 months.84 In recent years, the prognosis for these patients has improved with the availability of multimodality treatments. A small number of survivors of relapsed disease have been reported.83,85
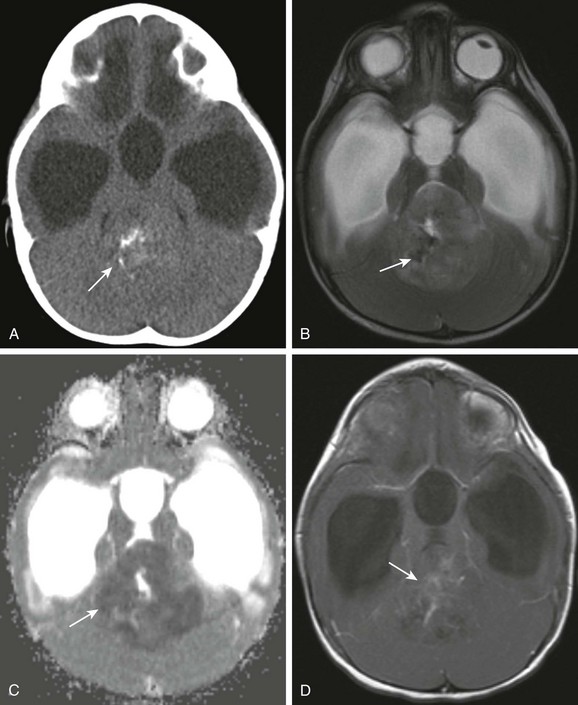
e-Figure 35-13 An atypical teratoid rhabdoid tumor.
A, An axial noncontrast computed tomography (CT) image shows a large mass centered in the fourth ventricle that is isodense to brain parenchyma containing foci of calcification (arrow). B, On the axial T2-weighted image, the predominant solid component is hypointense compared with gray matter. Foci of decreased signal intensity (arrow) correspond to the foci of calcification seen on the CT image. C, An apparent diffusion coefficient map shows decreased diffusion within the lesion (arrow), consistent with increased cellularity. D, A postcontrast axial T1-weighted image shows mild heterogeneous enhancement within the solid component (arrow).
Infratentorial Ependymomas
Infratentorial ependymomas constitute 8% to 15% of posterior fossa tumors in children. They arise from the ventricular ependymal lining and grow out of the fourth ventricle via the foramina of Luschka and Magendie into the cerebellopontine angles and cisternal spaces around the brainstem and cervicomedullary junction. These tumors are hypointense on T1-weighted images and isointense to gray matter on T2-weighted images. Up to 50% contain foci of calcification (Fig. 35-14, A). These lesions demonstrate heterogeneous enhancement on MRI (Fig. 35-14, B and C). Disseminated disease is present in 7% to 15% of patients with ependymomas at diagnosis.86 In the posterior fossa, ependymomas demonstrate significantly higher ADC values than do medulloblastomas and lower ADC values than in astrocytomas.87 These variations in ADC values may help differentiate tumors preoperatively and enable more effective treatment planning.
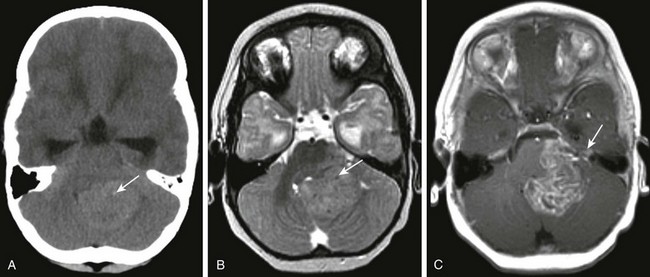
Figure 35-14 An ependymoma.
A, An axial noncontrast computed tomography image shows a hyperdense mass lesion occupying the fourth ventricle and extending through the left foramen of Luschka. Note several tiny punctate areas of calcification (arrow). B, An axial T2-weighted image shows that the lesion (arrow) is isointense compared with gray matter. C, An axial postcontrast T1-weighted image shows heterogeneous enhancement of the lesion and its extension toward the left foramen of Luschka (arrow).
Among all prognostic factors, the extent of surgical resection is the most important. Complete surgical resection followed by other treatments has shown >80% disease-free survival after 3 years of follow-up.88,89 Older age at presentation, along with favorable histologic grading, also may contribute to a better prognosis.90
Pineal Region Tumors
Pineal tumors constitute between 3% and 8% of all pediatric brain tumors.91 Pineal region tumors are divided into four categories: germ cell tumors, pineal cysts, pineal parenchymal tumors, and tumors of tissues supporting the pineal gland or adjacent structures (such as pineal gliomas, dermoids, and epidermoids).
Germ Cell Tumors
The most common tumors of the pineal region are germ cell tumors, of which 65% are pure germinomas.92 Other variants such as nongerminomatous germ cell tumors, mixed germ cell tumors, teratomas and embryonal cell carcinomas, yolk sac tumors, and choriocarcinomas constitute the remainder.
Germinomas are hyperdense on CT and enhance homogenously. On MRI, these lesions have homogenous signal intensity that is isointense to gray matter on all sequences, with intense enhancement (Fig. 35-15). These tumors grow anteriorly into the floor of the third ventricle and may infiltrate the thalami and midbrain. Spinal dissemination is common (in up to 36% of cases).93
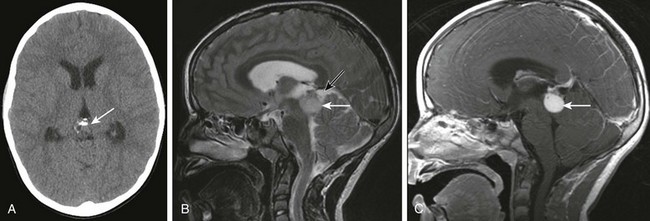
Figure 35-15 A pineal germinoma.
A, An axial unenhanced computed tomography image shows a partially calcified lesion (arrow) in the pineal region. Note the presence of hydrocephalus resulting from third ventricular obstruction. B, A sagittal T2-weighted image shows a rounded mass (white arrow) hyperintense to gray matter centered in the pineal region. Note the presence of a cystic T2-hyperintense component along the superior aspect (black arrow). C, A postcontrast T1-weighted image shows homogeneous enhancement of the solid component (arrow) of the lesion.
Pineal Parenchymal Tumors
Pineocytomas are well-differentiated tumors that retain morphologic features of the pineal parenchymal cells. They are slow-growing tumors that are circumscribed but nonencapsulated. Pineocytomas are more commonly solid tumors, although cystic variants also have been described. Solid tumors are either T1 hypointense or isointense to gray matter and T2 hyperintense. Enhancement is homogeneous, and calcification is common. Cystic variants often are indistinguishable from pineal cysts.94
Pineoblastomas are malignant tumors that resemble PNETs, but they are distinct from PNETs in other locations because of their photosensory differentiation.95 On MRI, these lesions are hypointense to isointense on T1-weighted images and demonstrate variable low, high, or mixed signal on T2-weighted images (e-Fig. 35-16). Pineoblastomas have lobulated contours, enhance homogenously, and calcify less often than pineocytomas. The “exploded pineal pattern” of calcification (characterized by peripheral displacement of pineal gland calcification) is more typical of pineal parenchymal tumors, effectively differentiating them from germ cell tumors.96 Pineoblastomas are resected surgically with adjuvant craniospinal radiation and multiagent chemotherapy. The prognosis is relatively poor.97
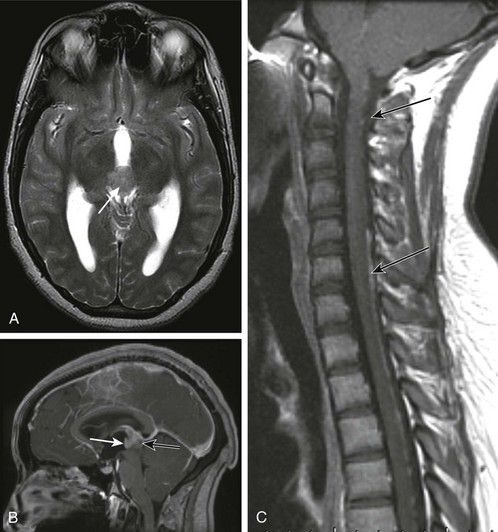
e-Figure 35-16 A pineoblastoma.
A, An axial T2-weighted image shows a heterogeneous lobulated hypointense mass (arrow) in the pineal region causing obstructive hydrocephalus. B, A postcontrast sagittal T1-weighted image shows heterogeneous enhancement of the mass (white arrow) with a small focus of nonenhancement in its posterior aspect (black arrow). C, A sagittal T1-postcontrast image through the cervical and upper thoracic spine shows linear and nodular enhancement (arrows) along the surface of the cervical and upper thoracic spinal cord consistent with leptomeningeal seeding of the tumor.
References
1. Gurney, JG, Smith, MA, Bunin, GR, CNS and miscellaneous intracranial and intraspinal neoplasms. (NIH publication No. 99-4649). Ries LAG, Smith MA, Gurney JG, et al, eds. Cancer incidence and survival among children and adolescents: United States SEER program 1975-1995. Philadelphia: National Cancer Institute, 1999.
2. Kohler, BA, Ward, E, McCarthy, BJ, et al. Annual report to the nation on the status of cancer, 1975-2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst. 2011;103(9):714–736.
3. Central Brain Tumor Registry of the United States. Statistical report: primary brain tumors in the United States, 2000-2004. Hinsdale, IL: Central Brain Tumor Registry of the United States; 2008.
4. Neglia, JP, Meadows, AT, Robison, LL, et al. Second neoplasms after acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;325(19):1330–1336.
5. Vinchon, M, Leblond, P, Caron, S, et al. Radiation-induced tumors in children irradiated for brain tumor: a longitudinal study. Childs Nerv Syst. 2011;27(3):445–453.
6. Bunin, GR. Nongenetic causes of childhood cancers: evidence from international variation, time trends, and risk factor studies. Toxicol Appl Pharmacol. 2004;199(2):91–103.
7. Poussaint, TY, Rodriguez, D. Advanced neuroimaging of pediatric brain tumors: MR diffusion, MR perfusion, and MR spectroscopy. Neuroimaging Clin N Am. 2006;16(1):169–192. [ix].
8. Samdani, A, Jallo, GI. Intraoperative MRI: technology, systems, and application to pediatric brain tumors. Surg Technol Int. 2007;16:236–243.
9. Mukherjee, P, Berman, JI, Chung, SW, et al. Diffusion tensor MR imaging and fiber tractography: theoretic underpinnings. AJNR Am J Neuroradiol. 2008;29(4):632–641.
10. Mukherjee, P, Chung, SW, Berman, JI, et al. Diffusion tensor MR imaging and fiber tractography: technical considerations. AJNR Am J Neuroradiol. 2008;29(5):843–852.
11. Gauvain, KM, McKinstry, RC, Mukherjee, P, et al. Evaluating pediatric brain tumor cellularity with diffusion-tensor imaging. AJR Am J Roentgenol. 2001;177(2):449–454.
12. Cha, S. Update on brain tumor imaging: from anatomy to physiology. AJNR Am J Neuroradiol. 2006;27(3):475–487.
13. Ozturk, A, Oguz, KK, Akalan, N, et al. Evaluation of parenchymal changes at the operation site with early postoperative brain diffusion-weighted magnetic resonance imaging. Diagn Interv Radiol. 2006;12(3):115–120.
14. Jellison, BJ, Field, AS, Medow, J, et al. Diffusion tensor imaging of cerebral white matter: a pictorial review of physics, fiber tract anatomy, and tumor imaging patterns. AJNR Am J Neuroradiol. 2004;25(3):356–369.
15. Khong, PL, Kwong, DL, Chan, GC, et al. Diffusion-tensor imaging for the detection and quantification of treatment-induced white matter injury in children with medulloblastoma: a pilot study. AJNR Am J Neuroradiol. 2003;24(4):734–740.
16. Khong, PL, Leung, LH, Chan, GC, et al. White matter anisotropy in childhood medulloblastoma survivors: association with neurotoxicity risk factors. Radiology. 2005;236(2):647–652.
17. Young, GS, Xia, S, Advanced MR techniques in clinical brain tumor imaging. Handbook of neuro-oncology neuroimaging. Newton, H, Jolesz, FA, eds. Handbook of neuro-oncology neuroimaging, Salt Lake City, Academic Press, 2008;vol 1.
18. Warren, KE. NMR spectroscopy and pediatric brain tumors. Oncologist. 2004;9(3):312–318.
19. Astrakas, LG, Zurakowski, D, Tzika, AA, et al. Noninvasive magnetic resonance spectroscopic imaging biomarkers to predict the clinical grade of pediatric brain tumors. Clin Cancer Res. 2004;10(24):8220–8228.
20. Hwang, JH, Egnaczyk, GF, Ballard, E, et al. Proton MR spectroscopic characteristics of pediatric pilocytic astrocytomas. AJNR Am J Neuroradiol. 1998;19(3):535–540.
21. Cho, YD, Choi, GH, Lee, SP, et al. (1)H-MRS metabolic patterns for distinguishing between meningiomas and other brain tumors. Magn Reson Imaging. 2003;21(6):663–672.
22. Kovanlikaya, A, Panigrahy, A, Krieger, MD, et al. Untreated pediatric primitive neuroectodermal tumor in vivo: quantitation of taurine with MR spectroscopy. Radiology. 2005;236(3):1020–1025.
23. Seymour, ZA, Panigrahy, A, Finlay, JL, et al. Citrate in pediatric CNS tumors? AJNR Am J Neuroradiol. 2008;29(5):1006–1011.
24. Bluml, S, Panigrahy, A, Laskov, M, et al. Elevated citrate in pediatric astrocytomas with malignant progression. Neuro Oncol. 2011;13(10):1107–1117.
25. Rossi, A, Gandolfo, C, Morana, G, et al. New MR sequences (diffusion, perfusion, spectroscopy) in brain tumours. Pediatr Radiol. 2010;40(6):999–1009.
26. Di Costanzo, A, Pollice, S, Trojsi, F, et al. Role of perfusion-weighted imaging at 3 Tesla in the assessment of malignancy of cerebral gliomas. Radiol Med. 2008;113(1):134–143.
27. Ball, WS, Jr., Holland, SK. Perfusion imaging in the pediatric patient. Magn Reson Imaging Clin N Am. 2001;9(1):207–230. [ix].
28. Servaes, S, Epelman, M, Pollock, A, et al. Pediatric malignancies: synopsis of current imaging techniques. In: Blake MA, Kalra MK, eds. Imaging in oncology. New York: Springer, 2008.
29. Lehmann, P, Monet, P, de Marco, G, et al. A comparative study of perfusion measurement in brain tumours at 3 Tesla MR: arterial spin labeling versus dynamic susceptibility contrast-enhanced MRI. Eur Neurol. 2010;64(1):21–26.
30. Noguchi, T, Yoshiura, T, Hiwatashi, A, et al. Perfusion imaging of brain tumors using arterial spin-labeling: correlation with histopathologic vascular density. AJNR Am J Neuroradiol. 2008;29(4):688–693.
31. Gupta, A, Shah, A, Young, RJ, et al. Imaging of brain tumors: functional magnetic resonance imaging and diffusion tensor imaging. Neuroimaging Clin N Am. 2010;20(3):379–400.
32. Jager, PL, Vaalburg, W, Pruim, J, et al. Radiolabeled amino acids: basic aspects and clinical applications in oncology. J Nucl Med. 2001;42(3):432–445.
33. Kubota, K, Yamada, K, Fukada, H, et al. Tumor detection with carbon-11-labelled amino acids. Eur J Nucl Med. 1984;9(3):136–140.
34. Herholz, K, Holzer, T, Bauer, B, et al. 11C-methionine PET for differential diagnosis of low-grade gliomas. Neurology. 1998;50(5):1316–1322.
35. Louis, DN, Ohgaki, H, Wiestler, OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109.
36. Scheithauer, BW, Hawkins, C, Tihan, T. Pilocytic astrocytomas. In: Louis DN, Ohgaki H, Wiestler OD, eds. Classification of tumours of the central nervous system. Geneva: World Heath Organization Press, 2007.
37. Deliganis, AV, Geyer, JR, Berger, MS. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen Disease) in childhood optic glioma. Neurosurgery. 1996;38(6):1114–1118. [discussion 1118-1119].
38. Komotar, RJ, Burger, PC, Carson, BS, et al. Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery. 2004;54(1):72–79. [discussion 79-80].
39. Pollack, IF. Brain tumors in children. N Engl J Med. 1994;331(22):1500–1507.
40. Tamber, MS, Rutka, JT. Pediatric supratentorial high-grade gliomas. Neurosurg Focus. 2003;14(2):e1.
41. Benesch, M, Wagner, S, Berthold, F, et al. Primary dissemination of high-grade gliomas in children: experiences from four studies of the Pediatric Oncology and Hematology Society of the German Language Group (GPOH). J Neurooncol. 2005;72(2):179–183.
42. Kleihues, P, Cavenee, WK, International Agency for Research on Cancer. Pathology and genetics of tumours of the nervous system. Lyon, France: IARC Press; 2000.
43. Figeroa, RE, el Gammal, T, Brooks, BS, et al. MR findings on primitive neuroectodermal tumors. J Comput Assist Tomogr. 1989;13(5):773–778.
44. Kieran, MW, Chi, SN, Samuel, D, et al, Tumors of the brain and spinal cord. Oncology of infancy and childhood. Orkin, SH, Fisher, DE, Look, AT, et al, eds. Oncology of infancy and childhood, Philadelphia, Saunders Elsevier, 2009;vol 1.
45. Spoto, GP, Press, GA, Hesselink, JR, et al. Intracranial ependymoma and subependymoma: MR manifestations. AJR Am J Roentgenol. 1990;154(4):837–845.
46. Koeller, KK, Sandberg, GD. From the archives of the AFIP. Cerebral intraventricular neoplasms: radiologic-pathologic correlation. Radiographics. 2002;22(6):1473–1505.
47. Yuh, EL, Barkovich, AJ, Gupta, N. Imaging of ependymomas: MRI and CT. Childs Nerv Syst. 2009;25(10):1203–1213.
48. Wolff, JE, Sajedi, M, Brant, R, et al. Choroid plexus tumours. Br J Cancer. 2002;87(10):1086–1091.
49. Cavalheiro, S, Moron, AF, Hisaba, W, et al. Fetal brain tumors. Childs Nerv Syst. 2003;19(7-8):529–536.
50. Naeini, RM, Yoo, JH, Hunter, JV. Spectrum of choroid plexus lesions in children. AJR Am J Roentgenol. 2009;192(1):32–40.
51. Horská, A, Ulug, AM, Melhem, ER, et al. Proton magnetic resonance spectroscopy of choroid plexus tumors in children. J Magn Reson Imaging. 2001;14(1):78–82.
52. Di Rocco, C, Caldarelli, M, Tamburrini, G, et al. Surgical management of craniopharyngiomas—experience with a pediatric series. J Pediatr Endocrinol Metab. 2006;19(suppl 1):355–366.
53. Burkhard, C, Di Patre, PL, Schuler, D, et al. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg. 2003;98(6):1170–1174.
54. Chung, EM, Specht, CS, Schroeder, JW. From the archives of the AFIP: Pediatric orbit tumors and tumorlike lesions: neuroepithelial lesions of the ocular globe and optic nerve. Radiographics. 2007;27(4):1159–1186.
55. Janss, AJ, Grundy, R, Cnaan, A, et al. Optic pathway and hypothalamic/chiasmatic gliomas in children younger than age 5 years with a 6-year follow-up. Cancer. 1995;75(4):1051–1059.
56. Simon, J, Szumowski, J, Totterman, S, et al. Fat-suppression MR imaging of the orbit. AJNR Am J Neuroradiol. 1988;9(5):961–968.
57. Poussaint, TY, Barnes, PD, Nichols, K, et al. Diencephalic syndrome: clinical features and imaging findings. AJNR Am J Neuroradiol. 1997;18(8):1499–1505.
58. Ellison, DW. Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathol. 2010;120(3):305–316.
59. Gilbertson, RJ. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5(4):209–218.
60. Bourgouin, PM, Tampieri, D, Grahovac, SZ, et al. CT and MR imaging findings in adults with cerebellar medulloblastoma: comparison with findings in children. AJR Am J Roentgenol. 1992;159(3):609–612.
61. Meyers, SP, Wildenhain, SL, Chang, JK, et al. Postoperative evaluation for disseminated medulloblastoma involving the spine: contrast-enhanced MR findings, CSF cytologic analysis, timing of disease occurrence, and patient outcomes. AJNR Am J Neuroradiol. 2000;21(9):1757–1765.
62. Ray, A, Ho, M, Ma, J, et al. A clinicobiological model predicting survival in medulloblastoma. Clin Cancer Res. 2004;10(22):7613–7620.
63. Helton, KJ, Gajjar, A, Hill, DA, et al. Medulloblastoma metastatic to the suprasellar region at diagnosis: a report of six cases with clinicopathologic correlation. Pediatr Neurosurg. 2002;37(3):111–117.
64. Pollack, IF. Multidisciplinary management of childhood brain tumors: a review of outcomes, recent advances, and challenges. J Neurosurg Pediatr. 2011;8(2):135–148.
65. Bowers, DC, Georgiades, C, Aronson, LJ, et al. Tectal gliomas: natural history of an indolent lesion in pediatric patients. Pediatr Neurosurg. 2000;32(1):24–29.
66. Poussaint, TY, Kowal, JR, Barnes, PD, et al. Tectal tumors of childhood: clinical and imaging follow-up. AJNR Am J Neuroradiol. 1998;19(5):977–983.
67. Poussaint, TY, Kocak, M, Vajapeyam, S, et al. MRI as a central component of clinical trials analysis in brainstem glioma: a report from the Pediatric Brain Tumor Consortium (PBTC). Neuro Oncol. 2011;13(4):417–427.
68. Roujeau, T, Machado, G, Garnett, MR, et al. Stereotactic biopsy of diffuse pontine lesions in children. J Neurosurg. 2007;107(1 suppl):1–4.
69. Laprie, A, Pirzkall, A, Haas-Kogan, DA, et al. Longitudinal multivoxel MR spectroscopy study of pediatric diffuse brainstem gliomas treated with radiotherapy. Int J Radiat Oncol Biol Phys. 2005;62(1):20–31.
70. Hipp, SJ, Steffen-Smith, E, Hammoud, D, et al. Predicting outcome of children with diffuse intrinsic pontine gliomas using multiparametric imaging. Neuro Oncol. 2011;13(8):904–909.
71. Prabhu, SP, Ng, S, Vajapeyam, S, et al. DTI assessment of the brainstem white matter tracts in pediatric BSG before and after therapy: a report from the Pediatric Brain Tumor Consortium. Childs Nerv Syst. 2011;27(1):11–18.
72. Helton, KJ, Edwards, M, Steen, RG, et al. Neuroimaging-detected late transient treatment-induced lesions in pediatric patients with brain tumors. J Neurosurg. 2005;102(2 suppl):179–186.
73. Chen, HJ, Panigrahy, A, Dhall, G, et al. Apparent diffusion and fractional anisotropy of diffuse intrinsic brain stem gliomas. AJNR Am J Neuroradiol. 2010;31(10):1879–1885.
74. Helton, KJ, Phillips, NS, Khan, RB, et al. Diffusion tensor imaging of tract involvement in children with pontine tumors. AJNR Am J Neuroradiol. 2006;27(4):786–793.
75. Zukotynski, KA, Fahey, FH, Kocak, M, et al. Evaluation of 18F-FDG PET and MRI associations in pediatric diffuse intrinsic brain stem glioma: a report from the Pediatric Brain Tumor Consortium. J Nucl Med. 2011;52(2):188–195.
76. Packer, RJ, Boyett, JM, Zimmerman, RA, et al. Hyperfractionated radiation therapy (72 Gy) for children with brain stem gliomas. A Childrens Cancer Group Phase I/II Trial. Cancer. 1993;72(4):1414–1421.
77. Hargrave, D, Bartels, U, Bouffet, E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol. 2006;7(3):241–248.
78. Reddy, AT. Atypical teratoid/rhabdoid tumors of the central nervous system. J Neurooncol. 2005;75(3):309–313.
79. Biegel, JA. Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus. 2006;20(1):E11.
80. Hilden, JM, Meerbaum, S, Burger, P, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22(14):2877–2884.
81. Meyers, SP, Khademian, ZP, Biegel, JA, et al. Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. AJNR Am J Neuroradiol. 2006;27(5):962–971.
82. Parmar, H, Hawkins, C, Bouffet, E, et al. Imaging findings in primary intracranial atypical teratoid/rhabdoid tumors. Pediatr Radiol. 2006;36(2):126–132.
83. Chi, SN, Zimmerman, MA, Yao, X, et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol. 2009;27(3):385–389.
84. Burger, PC, Yu, IT, Tihan, T, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998;22(9):1083–1092.
85. Zimmerman, MA, Goumnerova, LC, Proctor, M, et al. Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol. 2005;72(1):77–84.
86. Robertson, PL, Zeltzer, PM, Boyett, JM, et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s Cancer Group. J Neurosurg. 1998;88(4):695–703.
87. Rumboldt, Z, Camacho, DL, Lake, D, et al. Apparent diffusion coefficients for differentiation of cerebellar tumors in children. AJNR Am J Neuroradiol. 2006;27(6):1362–1369.
88. Timmermann, B, Kortmann, RD, Kuhl, J, et al. Interdisciplinary therapy of childhood ependymomas. Strahlenther Onkol. 2002;178(9):469–479.
89. Timmermann, B, Kortmann, RD, Kuhl, J, et al. Combined postoperative irradiation and chemotherapy for anaplastic ependymomas in childhood: results of the German prospective trials HIT 88/89 and HIT 91. Int J Radiat Oncol Biol Phys. 2000;46(2):287–295.
90. Tihan, T, Zhou, T, Holmes, E, et al. The prognostic value of histological grading of posterior fossa ependymomas in children: a Children’s Oncology Group study and a review of prognostic factors. Mod Pathol. 2008;21(2):165–177.
91. Hoffman, HJ, Yoshida, M, Becker, LE, et al. Pineal region tumors in childhood. Experience at the Hospital for Sick Children. 1983. Pediatr Neurosurg. 1994;21(1):91–103. [discussion 104].
92. Smirniotopoulos, JG, Rushing, EJ, Mena, H. Pineal region masses: differential diagnosis. Radiographics. 1992;12(3):577–596.
93. Maity, A, Shu, HK, Janss, A, et al. Craniospinal radiation in the treatment of biopsy-proven intracranial germinomas: twenty-five years’ experience in a single center. Int J Radiat Oncol Biol Phys. 2004;58(4):1165–1170.
94. Engel, U, Gottschalk, S, Niehaus, L, et al. Cystic lesions of the pineal region—MRI and pathology. Neuroradiology. 2000;42(6):399–402.
95. Hirato, J, Nakazato, Y. Pathology of pineal region tumors. J Neurooncol. 2001;54(3):239–249.
96. Zee, CS, Segall, H, Apuzzo, M, et al. MR imaging of pineal region neoplasms. J Comput Assist Tomogr. 1991;15(1):56–63.
97. Dirks, PB, Harris, L, Hoffman, HJ, et al. Supratentorial primitive neuroectodermal tumors in children. J Neurooncol. 1996;29(1):75–84.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 35
Pediatric Brain Neoplasms
Brain tumors are the most common solid pediatric tumors and are the leading cause of death in children from solid tumors.1 The estimated incidence of all childhood primary brain and central nervous system (CNS) tumors is 4.8 cases per 100,000 person-years.2 Approximately 4150 new cases of childhood primary nonmalignant and malignant brain and CNS tumors were diagnosed in the United States in 2011.3 Nearly 50% of brain tumors in children older than 1 year arise in an infratentorial location. However, in neonates, infants, and children up to the age of 3 years, supratentorial tumors are more common.3
Etiology
The etiology of pediatric brain tumors, an area of research beyond the scope of this chapter, requires an understanding of genetic alterations, signaling systems, and molecular genetics and pathways. Although no one risk factor explains more than a small percentage of childhood brain tumors, therapeutic doses of ionizing radiation to the head for brain tumors and radiation for leukemia,4,5 as well as certain genetic syndromes, are known risk factors in the pediatric population. Among the congenital syndromes associated with brain tumors are neurofibromatosis types 1 and 2, Gorlin syndrome (basal cell nevus syndrome), tuberous sclerosis, Turcot syndrome, von Hippel-Lindau syndrome, and Li-Fraumeni syndrome.6
Imaging
Because of its superior soft tissue resolution, multiplanar capability, and lack of ionizing radiation, magnetic resonance imaging (MRI) with contrast is the modality of choice in determining lesion size, location, and characterization. And although contrast enhancement typically reflects disruption of the blood-brain barrier, the degree of contrast enhancement does not always correlate with tumor grade. For example, benign tumors (e.g., choroid plexus papillomas and pilocytic astrocytomas) can enhance avidly, whereas anaplastic astrocytomas may not enhance at all.7
MRI also is used to assess tumor response and progression and monitor treatment effects. Essential to optimal treatment planning is accurate staging of the tumor that confirms whether the tumor has spread through the neural axis. Intraoperative MRI is being used in some centers to guide both conventional and minimally invasive tumor resection. As these systems are refined, they are expected to form the standard of care at many medical centers.8
Diffusion-Weighted Imaging
Contrast on diffusion-weighted images (DWI) reflects the mean distance traveled by free water protons in tissue as a result of Brownian motion.9,10 Diffusion occurs freely in the direction of white matter tract orientation and is restricted in orthogonal planes. DWI can assess the properties of diffusion occurring within a particular voxel, which is expressed as the apparent diffusion coefficient (ADC). A markedly decreased ADC usually correlates well with increased tumor cellularity in brain neoplasms. Vasogenic edema and necrosis show an increased ADC.7,11,12 ADC values are interpreted in conjunction with structural MRI sequences. Diffusion tensor imaging (DTI), an adaptation of DWI, acquires diffusion data in six or more directions to establish the direction and magnitude of water diffusion.
DWI also can be extremely useful in the postoperative period, when low ADC values at the surgical margins or within the resection cavity may be indicative of ischemia or abscess. This technique typically is used in conjunction with conventional MRI, which helps exclude artifact from hematoma.13 Further, DTI aids in identifying patterns of tumor interaction with white matter fiber tracts (i.e., the extent of deviation, edema, infiltration, and destruction),14 and when used in conjunction with volumetric data, it effectively guides surgical resection and predicts possible postoperative deficits resulting from white matter tract damage (e-Fig. 35-1). Reduced fractional anisotropy (FA), which is a measure of the directional diffusivity of water made using DTI, has been found in the white matter of patients with a medulloblastoma, even in the absence of abnormalities on structural sequences.15 Decreased FA values have been shown to correlate with the age at which radiation was administered and with poor academic performance among school-age patients.16 FA thus may be considered a noninvasive biomarker to monitor effects of radiotherapy.16
e-Figure 35-1 Presurgical tractography in a glioma lesion.
A, Tractography of a portion of the left corticospinal tract forming the posterior limb of the internal capsule (depicted in yellow) in a patient with left parietal lobe angiocentric glioma (segmented and shown in green). The tract is displaced medially by the tumor but is still intact.
B, An axial postcontrast magnetization-prepared rapid gradient-echo image shows the cortically based minimally enhancing tumor (arrow).
Magnetic Resonance Spectroscopy
Magnetic resonance spectroscopy (MRS) is a noninvasive in vivo technique that provides metabolic information beyond structural imaging sequences. It enables detection and quantification of abnormal metabolites in the brain and can help identify tumor tissue, differentiate tumor types, and separate active tumor from radiation necrosis or scar formation. MRS can be performed with most standard MRI scanners, typically by incorporating either the point resolved spin echo or stimulated echo acquisition mode techniques. Simultaneous acquisition of MRS from multiple voxels increases spatial resolution; this procedure is known as “chemical shift imaging” or MR spectroscopic imaging.17
Most brain tumors are characterized by the presence of increased choline/creatine and decreased NAA/creatine ratios, indicating loss of neuroaxonal integrity and increased cell membrane turnover. The presence of lactate in the tumor suggests an anaerobic process with impaired energy metabolism.18 In general, high-grade tumors have higher choline/creatine and lower NAA/creatine ratios than do low-grade lesions. In rapidly growing malignant tumors, necrotic areas may contain lipid resonances.19 However, in pediatric patients, we frequently (and paradoxically) see elevated levels of choline and lactate in pilocytic astrocytomas, a low-grade tumor.20
The presence of specific metabolites such as alanine (an inverted doublet at 1.44 ppm) in meningiomas (e-Fig. 35-2) and taurine (peak at 3.3 to 3.4 ppm) in primitive neuroectodermal tumors (PNETs) may help narrow the differential diagnosis.21,22 Citrate is a tricarboxylic acid cycle intermediate metabolite that has been described in pediatric brain tumors and is found at particularly high levels in pontine gliomas.23 In one study of grade 2 astrocytomas, citrate was significantly more prominent in tumors that progressed.24
e-Figure 35-2 Magnetic resonance spectroscopy (MRS) in a meningioma tumor.
Single voxel MRS (TE 144 ms) performed over the left frontal meningioma (see inset T2-weighted image) shows a prominent choline peak (Cho) and reduced creatine and N-acetylaspartate peaks. The inverted doublet centered at approximately 1.55 to 1.6 ppm is consistent with an alanine (Ala) peak.
Perfusion-Weighted imaging
Perfusion-weighted imaging measures cerebral hemodynamics at the microcirculation level. Parameters measured by perfusion-weighted imaging include cerebral blood volume (CBV), cerebral blood flow, and mean transit time. Of these, the CBV, defined as the volume of blood in an area of brain tissue expressed in mL/100 g, is the most commonly used parameter in evaluation of brain tumors.25 Lower grade astrocytomas have relatively lower regional CBV than do higher-grade tumors such as anaplastic astrocytomas and glioblastomas26 (e-Fig. 35-3). However, low-grade pediatric pilocytic astrocytomas can have a high relative cerebral blood volume.27
e-Figure 35-3 Perfusion-weighted imaging in a thalamic glioblastoma multiforme tumor.
A, A T1-weighted image shows a rounded centrally enhancing mass lesion in the left thalamus (arrow). B, An axial T2* cerebral blood volume perfusion image shows a focal area of increased perfusion corresponding to the enhancing solid portion of the tumor in the left thalamus (arrow). C, An apparent diffusion coefficient map shows regions of decreased diffusion corresponding to the solid component of the left thalamic tumor (arrow).
The most widely available technique is T2*-weighted dynamic susceptibility contrast imaging, which consists of a rapid bolus of intravenous (IV) paramagnetic contrast agent followed by a rapid acquisition of echo-planar images during the first pass of contrast material through the capillary bed. As the contrast medium is delivered, it goes through the tissues and results in a signal drop proportional to the blood volume during the first pass. Routine use of this technique in children requires the use of rapid contrast medium injection and power injectors, as well as strategies to overcome problems associated with large-bore IV catheter placement, especially in infants.28
ASL uses endogenous blood as a tracer. The two major types of ASL, pulsed and continuous, are now widely available on clinical scanners. A third type, pseudocontinuous ASL, has just recently been introduced for clinical use. ASL has shown promise for hemodynamic evaluation of brain tumors,29,30 but data in children are limited at this time.
Functional MRI
Functional MRI (fMRI) is a technique that essentially relies on two physical principles, namely, that oxyhemoglobin is diamagnetic and deoxyhemoglobin is paramagnetic in nature. Because of the relative increased blood flow and consequent increased utilization of oxygen within the activated portions of the brain, the MR signal, in this case known as the “blood oxygen level dependent signal,” or BOLD, is measurably different compared with other parts of the brain. The primary value of fMRI is in localizing the eloquent areas in the brain controlling language, motor skills, and memory. This information helps provide surgical guidance.31 More detailed information can be found in Chapter 27.
Single PET and PET
The role of PET in the evaluation of pediatric brain tumors is to determine metabolic activity at diagnosis, assess response to therapy, and distinguish treatment effect versus tumor recurrence. Fluorine-18-deoxyglucose (18F-FDG) is the most commonly used isotope for PET studies in children. PET scanning using other labeled agents such as the amino acid analogues [11C] methionine and [11C] tyrosine have shown promise in detecting low-grade tumors in adults, although their diagnostic value in children has not yet been established.32 These amino acid analogues are incorporated via amino acid transport pathways into tumor proteins, and therefore uptake reflects tumor protein synthesis.33,34
Other isotopes still at the investigational stage include cell proliferation agents (e.g., 18F-fluorothymidine) and cell hypoxia imaging agents (e.g., 18F-fluoromisonidazole and 62Cu-labeled diacetyl-bis [N4-methylthiosemicarbazone]).
Specific Tumors
Classification of Pediatric Brain Tumors
The differential diagnosis is effectively limited by classifying tumors by location, describing the appearance of the lesion on conventional MRI, and applying advanced imaging techniques (Box 35-1).
Tumors of the Cerebral Hemispheres
The modified World Health Organization (WHO) classification of CNS tumors divides astrocytomas into low grade (grades I and II) and high grade (grades III and IV).35 Grading of astrocytomas by the WHO criteria is predictive of patient survival.35
Pilocytic astrocytomas are grade I WHO tumors; they account for 20% to 30% of all childhood brain tumors.36 Pilocytic astrocytomas typically arise in the first two decades of life. The most common locations of these tumors are in the optic pathways, hypothalamus, thalamus, basal ganglia, cerebral hemispheres, cerebellum (Fig. 35-4), and brainstem. Patients with neurofibromatosis type 1 (NF1) have an increased risk of the development of pilocytic astrocytomas, including optic pathway tumors. Patients with NF1 who have optic pathway tumors tend to have a better long-term prognosis than do patients without NF1 who have optic pathway tumors.37
Figure 35-4 A cerebellar astrocytoma.
A, An axial T2-weighted image shows a large cystic mass centered in the right cerebellar hemisphere with small solid components along the posterior and anterior aspects (arrow). B, A postcontrast sagittal T1-weighted image shows the solid components (arrows) along the anterior and posterior margins that enhance after paramagnetic contrast administration. C, An apparent diffusion coefficient map shows increased diffusion within the lesion (arrow) consistent with the relatively low cellularity within the lesion.
Rarely, pilocytic astrocytomas can present with diffuse leptomeningeal spread, which most often is seen in association with the diencephalic syndrome (discussed later in this chapter) or with the pilomyxoid variant of astrocytomas. Pilomyxoid astrocytomas have an indolent course, but their propensity for slow-growing, persistent recurrences makes them difficult to treat.38
Supratentorial High-Grade Gliomas
High-grade gliomas in children are significantly less common than are low-grade lesions, which account for up to 20% of all hemispheric gliomas.39,40
On CT, these lesions demonstrate heterogeneous enhancement and density with edema, occasional hemorrhage, mass effect, and ill-defined margins. On MRI, these lesions have heterogeneous signal intensity (Fig. 35-5). They typically are hypointense on T1-weighted images and hyperintense on T2-weighted images with surrounding white matter edema. They show effect of the mass on surrounding structures and demonstrate irregular enhancement with necrosis and hemorrhage similar to that seen on CT.
Figure 35-5 A supratentorial high-grade glioma.
A, An axial T2-weighted image shows a large heterogeneous mass (arrow) in the left frontal, temporal, and parietal lobes with marked mass effect, subfalcine herniation, surrounding edema, and rightward midline shift. B, A postcontrast T1-weighted image shows heterogeneous enhancement of the solid components of the tumor (black arrow) and nonenhancing components anteriorly (white arrow), suggestive of necrosis. C, An apparent diffusion coefficient map reveals decreased diffusion within the solid component (arrow), indicating high cellularity.
Aggressive surgical resection with preservation of neurological function, followed by radiotherapy directed at the tumor bed, remains the cornerstone of treatment of pediatric malignant gliomas.41 The addition of chemotherapy has been shown to improve survival compared with surgery and radiotherapy alone.40 The overall prognosis for children with supratentorial malignant gliomas remains poor, however, with 5-year progression-free survival rates of around 30%.40
Supratentorial PNETs
Although supratentorial PNETs are relatively rare, these tumors are more common in the first decade of life, with peak incidence from birth to 5 years of age.42 They account for 5% of all supratentorial tumors in childhood. At presentation they often are large and fairly well defined, occurring either in the cerebral hemispheres or in the lateral ventricles. They may be solid and homogenous or heterogeneous with cyst formation.43 Calcification often is seen on CT. Heterogeneous contrast enhancement is seen along with regions of necrosis.
On MRI, solid areas have restricted diffusion and T2-hypointense areas (Fig. 35-6), reflecting high nuclear-to-cytoplasm ratio, increased cellularity, and increased CBV values. Necrosis and hemorrhage also can occur in these lesions.
Figure 35-6 A supratentorial primitive neuroectodermal tumor.
A,
[/not-level-membership-for-pediatrics-category]
