CHAPTER 7 Patterns of Inheritance
Family Studies
An important reason for studying the pattern of inheritance of disorders within families is to enable advice to be given to members of a family regarding the likelihood of their developing it or passing it on to their children (i.e., genetic counseling; see Chapter 17). Taking a family history can, in itself, provide a diagnosis. For example, a child could come to the attention of a doctor with a fracture after a seemingly trivial injury. A family history of relatives with a similar tendency to fracture and blue sclerae would suggest the diagnosis of osteogenesis imperfecta. In the absence of a positive family history, other diagnoses would have to be considered.
Pedigree Drawing and Terminology
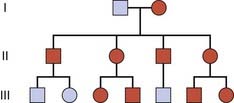
A family tree is a shorthand system of recording the pertinent information about a family. It usually begins with the person through whom the family came to the attention of the investigator. This person is referred to as the index case, proband, or propositus; or, if female, the proposita. The position of the proband in the family tree is indicated by an arrow. Information about the health of the rest of the family is obtained by asking direct questions about brothers, sisters, parents, and maternal and paternal relatives, with the relevant information about the sex of the individual, affection status, and relationship to other individuals being carefully recorded in the pedigree chart (Figure 7.1). Attention to detail can be crucial because patients do not always appreciate the important difference between siblings and half-siblings, or might overlook the fact, for example, that the child of a brother who is at risk of Huntington disease is actually a step-child and not a biological relative.
Mendelian Inheritance
More than 16,000 traits or disorders in humans exhibit single gene unifactorial or mendelian inheritance. However, characteristics such as height, and many common familial disorders, such as diabetes or hypertension, do not usually follow a simple pattern of mendelian inheritance (see Chapter 9).
Autosomal Dominant Inheritance
An autosomal dominant trait is one that manifests in the heterozygous state, that is, in a person possessing both an abnormal or mutant allele and the normal allele. It is often possible to trace a dominantly inherited trait or disorder through many generations of a family (Figure 7.2). In South Africa the vast majority of cases of porphyria variegata can be traced back to one couple in the late seventeenth century. This is a metabolic disorder characterized by skin blistering as a result of increased sensitivity to sunlight (Figure 7.3), and the excretion of urine that becomes ‘port wine’ colored on standing as a result of the presence of porphyrins (p. 179). This pattern of inheritance is sometimes referred to as ‘vertical’ transmission and is confirmed when male–male (i.e., father to son) transmission is observed.
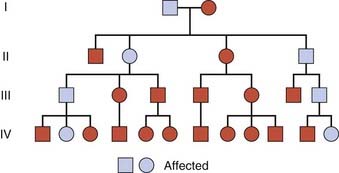
FIGURE 7.2 Family tree of an autosomal dominant trait. Note the presence of male-to-male transmission.
Genetic Risks
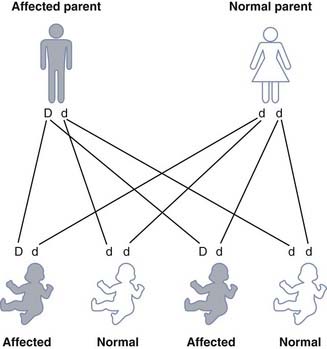
Each gamete from an individual with a dominant trait or disorder will contain either the normal allele or the mutant allele. If we represent the dominant mutant allele as ‘D’ and the normal allele as ‘d’, then the possible combinations of the gametes is seen in Figure 7.4. Any child born to a person affected with a dominant trait or disorder has a 1 in 2 (50%) chance of inheriting it and being similarly affected. These diagrams are often used in the genetic clinic to explain segregation to patients and are more user-friendly than a Punnett square (see Figs. 1.3 and 8.1).
Pleiotropy
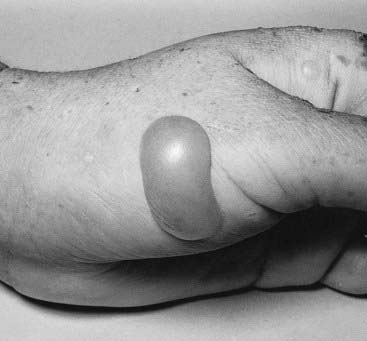
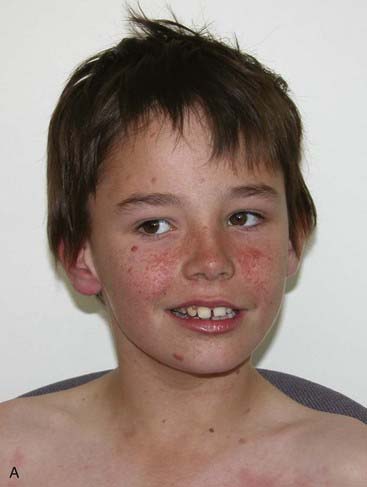
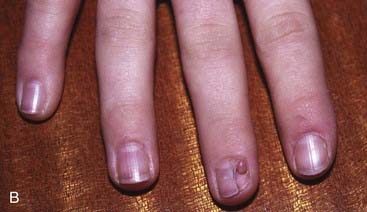
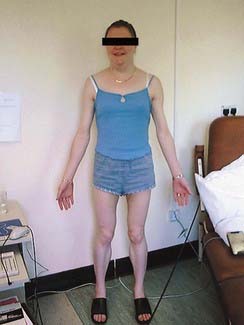
Autosomal dominant traits may involve only one organ or part of the body, for example the eye in congenital cataracts. It is common, however, for autosomal dominant disorders to manifest in different systems of the body in a variety of ways. This is pleiotropy—a single gene that may give rise to two or more apparently unrelated effects. In tuberous sclerosis affected individuals can present with a range of problems including learning difficulties, epilepsy, a facial rash known as adenoma sebaceum (histologically composed of blood vessels and fibrous tissue known as angiokeratoma) or subungual fibromas (Figure 7.5); some affected individuals have all features, whereas others may have almost none. Some discoveries are challenging our conceptual understanding of the term pleiotropy on account of the remarkably diverse syndromes that can result from different mutations in the same gene—for example, the LMNA gene (which encodes lamin A/C) and the X-linked filamin A (FLNA) gene. Mutations in LMNA may cause Emery-Dreifuss muscular dystrophy, a form of limb girdle muscular dystrophy, a form of Charcot-Marie-Tooth disease (p. 305), dilated cardiomyopathy (p. 296) with conduction abnormality, Dunnigan-type familial partial lipodystrophy (Figure 7.6), mandibuloacral dysplasia, and a very rare condition that has always been a great curiosity—Hutchinson-Gilford progeria. These are due to heterozygous mutations, with the exception of the Charcot-Marie-Tooth disease and mandibuloacral dysplasia, which are recessive—affected individuals are therefore homozygous for LMNA mutations. Sometimes an individual with a mutation is entirely normal. Mutations in the filamin A gene have been implicated in the distinct, though overlapping, X-linked dominant dysmorphic conditions oto-palato-digital syndrome, Melnick-Needles syndrome and frontometaphyseal dysplasia. However, it could not have been foreseen that a form of X-linked dominant epilepsy in women, called periventricular nodular heterotopia, is also due to mutations in this gene.
Reduced Penetrance
Reduced penetrance and variable expressivity, together with the pleiotropic effects of a mutant allele, all need to be taken into account when trying to interpret family history information for disorders that follow autosomal dominant inheritance. A good example of a very variable condition for which non-penetrance is frequently seen is Treacher-Collins syndrome. In its most obvious manifestation the facial features are unmistakable (Figure 7.7). However, the mother of the child illustrated is also known to harbor the gene (TCOF1) mutation as she has a number of close relatives with the same condition.

New Mutations
In autosomal dominant disorders an affected person usually has an affected parent. However, this is not always the case and it is not unusual for a trait to appear in an individual when there is no family history of the disorder. A striking example is achondroplasia, a form of short-limbed dwarfism (pp. 93–94), in which the parents usually have normal stature. The sudden unexpected appearance of a condition arising as a result of a mistake occurring in the transmission of a gene is called a new mutation. The dominant mode of inheritance of achondroplasia could be confirmed only by the observation that the offspring of persons with achondroplasia had a 50% chance of having achondroplasia. In less dramatic conditions other explanations for the ‘sudden’ appearance of a disorder must be considered. This includes non-penetrance and variable expression, as mentioned in the previous section. However, the astute clinician also needs to be aware that the family relationships may not be as stated—i.e., there may be undisclosed non-paternity (p. 342) (or, occasionally, non-maternity).
New dominant mutations, in certain instances, have been associated with an increased age of the father. Traditionally, this is believed to be a consequence of the large number of mitotic divisions that male gamete stem cells undergo during a man’s reproductive lifetime (p. 41). However, this may well be a simplistic view. In relation to mutations in FGFR2 (craniosynostosis syndromes), ground-breaking work by Wilkie’s group in Oxford demonstrated that causative gain-of-function mutations confer a selective advantage to spermatogonial stem cells, so that mutated cell lines accumulate in the testis.
Co-Dominance
Co-dominance is the term used for two allelic traits that are both expressed in the heterozygous state. In persons with blood group AB it is possible to demonstrate both A and B blood group substances on the red blood cells, so the A and B blood groups are therefore co-dominant (p. 205).
Homozygosity for Autosomal Dominant Traits
The rarity of most autosomal dominant disorders and diseases means that they usually occur only in the heterozygous state. There are, however, a few reports of children born to couples where both parents are heterozygous for a dominantly inherited disorder. Offspring of such couples are, therefore, at risk of being homozygous. In some instances, affected individuals appear either to be more severely affected, as has been reported with achondroplasia, or to have an earlier age of onset, as in familial hypercholesterolemia (p. 175). The heterozygote with a phenotype intermediate between the homozygotes for the normal and mutant alleles is consistent with a haploinsufficiency loss-of-function mutation (p. 26).
Conversely, with other dominantly inherited disorders, homozygous individuals are not more severely affected than heterozygotes—e.g., Huntington disease (p. 293) and myotonic dystrophy (p. 295).
Autosomal Recessive Inheritance
Recessive traits and disorders are manifest only when the mutant allele is present in a double dose (i.e., homozygosity). Individuals heterozygous for such mutant alleles show no features of the disorder and are perfectly healthy; they are described as carriers. The family tree for recessive traits (Figure 7.8) differs markedly from that seen in autosomal dominant traits. It is not possible to trace an autosomal recessive trait or disorder through the family, as all the affected individuals in a family are usually in a single sibship (i.e., brothers and sisters). This is sometimes referred to as ‘horizontal’ transmission, but this is an inappropriate and misleading term.
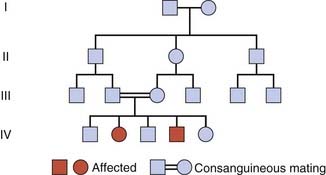
Consanguinity
Enquiry into the family history of individuals affected with rare recessive traits or disorders might reveal that their parents are related (i.e., consanguineous). The rarer a recessive trait or disorder, the greater the frequency of consanguinity among the parents of affected individuals. In cystic fibrosis, the most common ‘serious’ autosomal recessive disorder in western Europeans (p. 1), the frequency of parental consanguinity is only slightly greater than that seen in the general population. By contrast, in alkaptonuria, one of the original inborn errors of metabolism (p. 171), which is an exceedingly rare recessive disorder, Bateson and Garrod, in their original description of the disorder, observed that one-quarter or more of the parents were first cousins. They reasoned that rare alleles for disorders such as alkaptonuria are more likely to ‘meet up’ in the offspring of cousins than in the offspring of parents who are unrelated. In large inbred kindreds an autosomal recessive condition may be present in more than one branch of the family.
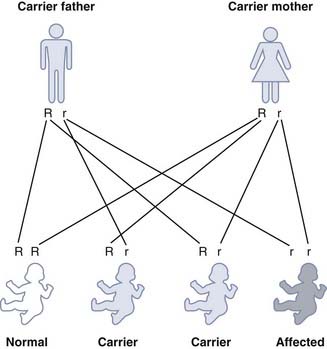
Genetic Risks
If we represent the normal dominant allele as ‘R’ and the recessive mutant allele as ‘r’, then each parental gamete carries either the mutant or the normal allele (Figure 7.9). The various possible combinations of gametes mean that the offspring of two heterozygotes have a 1 in 4 (25%) chance of being homozygous affected, a 1 in 2 (50%) chance of being heterozygous unaffected, and a 1 in 4 (25%) chance of being homozygous unaffected.
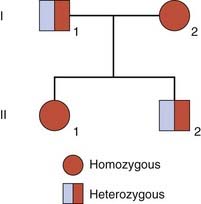
Pseudodominance
If an individual who is homozygous for an autosomal recessive disorder has children with a carrier of the same disorder, their offspring have a 1 in 2 (50%) chance of being affected. Such a pedigree is said to exhibit pseudodominance (Figure 7.10).
Mutational Heterogeneity
Heterogeneity can also occur at the allelic level. In the majority of single-gene disorders (e.g., β-thalassemia) a large number of different mutations have been identified as being responsible (p. 160). There are individuals who have two different mutations at the same locus and are known as compound heterozygotes, constituting what is known as allelic or mutational heterogeneity. Most individuals affected with an autosomal recessive disorder are probably compound heterozygotes rather than true homozygotes, unless their parents are related, when they are likely to be homozygous for the same mutation by descent, having inherited the same mutation from a common ancestor.
Sex-Linked Inheritance
X-Linked Recessive Inheritance
An X-linked recessive trait is one determined by a gene carried on the X chromosome and usually manifests only in males. A male with a mutant allele on his single X chromosome is said to be hemizygous for that allele. Diseases inherited in an X-linked manner are transmitted by healthy heterozygous female carriers to affected males, as well as by affected males to their obligate carrier daughters, with a consequent risk to male grandchildren through these daughters (Figure 7.11). This type of pedigree is sometimes said to show ‘diagonal’ or a ‘knight’s move’ pattern of transmission.
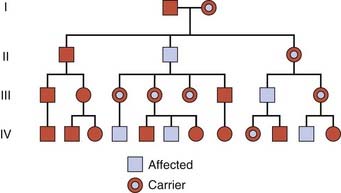
The mode of inheritance whereby only males are affected by a disease that is transmitted by normal females was appreciated by the Jews nearly 2000 years ago. They excused from circumcision the sons of all the sisters of a mother who had sons with the ‘bleeding disease’, in other words, hemophilia (p. 309). The sons of the father’s siblings were not excused. Queen Victoria was a carrier of hemophilia, and her carrier daughters, who were perfectly healthy, introduced the gene into the Russian and Spanish royal families. Fortunately for the British royal family, Queen Victoria’s son, Edward VII, did not inherit the gene and so could not transmit it to his descendants.
Genetic Risks
A male transmits his X chromosome to each of his daughters and his Y chromosome to each of his sons. If a male affected with hemophilia has children with a normal female, then all of his daughters will be obligate carriers but none of his sons will be affected (Figure 7.12). A male cannot transmit an X-linked trait to his son, with the very rare exception of uniparental heterodisomy (p. 121).
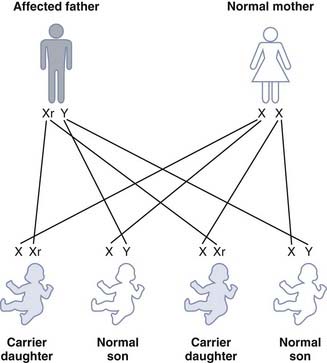
For a carrier female of an X-linked recessive disorder having children with a normal male, each son has a 1 in 2 (50%) chance of being affected and each daughter has a 1 in 2 (50%) chance of being a carrier (Figure 7.13).
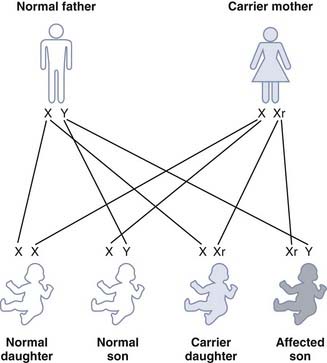
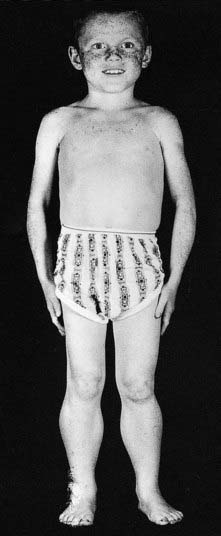
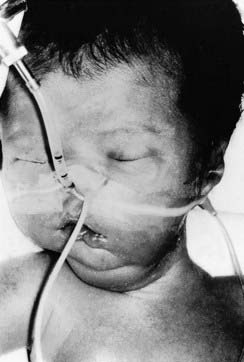
Some X-linked disorders are not compatible with survival to reproductive age and are not, therefore, transmitted by affected males. Duchenne muscular dystrophy is the commonest muscular dystrophy and is a severe disease (p. 307). The first sign is delayed walking followed by a waddling gait, difficulty in climbing stairs unaided, and a tendency to fall easily. By about the age of 10 years affected boys usually need to use a wheelchair. The muscle weakness progresses gradually and affected males ultimately become confined to bed and often die in their late teenage years or early 20s (Figure 7.14). Because affected boys do not usually survive to reproduce, the disease is transmitted by healthy female carriers (Figure 7.15), or may arise as a new mutation.
Variable Expression in Heterozygous Females
In humans, several X-linked disorders are known in which heterozygous females have a mosaic phenotype with a mixture of features of the normal and mutant alleles. In X-linked ocular albinism, the iris and ocular fundus of affected males lack pigment. Careful examination of the ocular fundus in females heterozygous for ocular albinism reveals a mosaic pattern of pigmentation (see Figure 6.25, p. 104). This mosaic pattern of involvement can be explained by the random process of X-inactivation (p. 103). In the pigmented areas, the normal gene is on the active X chromosome, whereas in the depigmented areas the mutant allele is on the active X chromosome.
Females Affected with X-Linked Recessive Disorders
Skewed X-Inactivation
The process of X-inactivation (p. 103) usually occurs randomly, there being an equal chance of either of the two X chromosomes in a heterozygous female being inactivated in any one cell. After X-inactivation in embryogenesis, therefore, in roughly half the cells one of the X chromosomes is active, whereas in the other half it is the other X chromosome that is active. Sometimes this process is not random, allowing for the possibility that the active X chromosome in most of the cells of a heterozygous female carrier is the one bearing the mutant allele. If this happens, a carrier female would exhibit some of the symptoms and signs of the disease and be a so-called manifesting heterozygote or carrier. This has been reported in a number of X-linked disorders, including Duchenne muscular dystrophy and hemophilia A (pp. 307, 309). In addition, there are reports of several X-linked disorders in which there are a number of manifesting carriers in the same family, consistent with the coincidental inheritance of an abnormality of X-inactivation (p. 204).
Numerical X-Chromosome Abnormalities
A female could manifest an X-linked recessive disorder by being a carrier of an X-linked recessive mutation and having only a single X chromosome (i.e., Turner syndrome, see p. 207). Women with Turner syndrome and hemophilia A or Duchenne muscular dystrophy have been reported occasionally.
X-Autosome Translocations
Females with a translocation involving one of the X chromosomes and an autosome can be affected with an X-linked recessive disorder. If the breakpoint of the translocation disrupts a gene on the X chromosome, then a female can be affected. This is because the X chromosome involved in the translocation survives preferentially so as to maintain functional disomy of the autosomal genes (Figure 7.16). The observation of females affected with Duchenne muscular dystrophy with X-autosome translocations involving the same region of the short arm of the X chromosome helped to map the Duchenne muscular dystrophy gene (p. 307). This type of observation has been vital in the positional cloning of a number of genes in humans (p. 75).
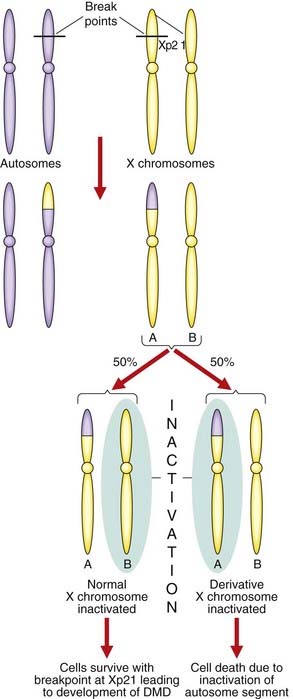
X-Linked Dominant Inheritance
Although uncommon, there are disorders that are manifest in the heterozygous female as well as in the male who has the mutant allele on his single X chromosome. This is known as X-linked dominant inheritance (Figure 7.17). X-linked dominant inheritance superficially resembles that of an autosomal dominant trait because both the daughters and sons of an affected female have a 1 in 2 (50%) chance of being affected. There is, however, an important difference. With an X-linked dominant trait, an affected male transmits the trait to all his daughters but to none of his sons. Therefore, in families with an X-linked dominant disorder there is an excess of affected females and direct male-to-male transmission cannot occur.
A mosaic pattern of involvement can be demonstrated in females heterozygous for some X-linked dominant disorders. An example is the mosaic pattern of abnormal pigmentation of the skin that follows developmental lines seen in females heterozygous for the X-linked dominant disorder incontinentia pigmenti (Figure 7.18). This is also an example of a disorder that is usually lethal for male embryos that inherit the mutated allele. Others include the neurological conditions Rett syndrome and periventricular nodular heterotopia.
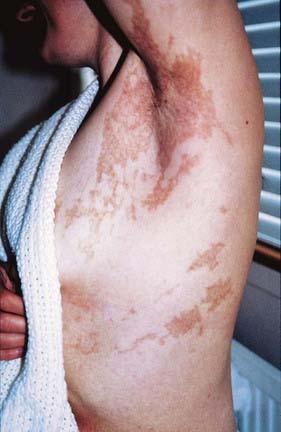
FIGURE 7.18 Mosaic pattern of skin pigmentation in a female with the X-linked dominant disorder, incontinentia pigmenti. The patient has a mutation in a gene on one of her X chromosomes; the pigmented areas indicate tissue in which the normal X chromosome has been inactivated. This developmental pattern follows Blaschko’s lines (see Chapter 18, p. 276).
Y-Linked Inheritance
Y-linked or holandric inheritance implies that only males are affected. An affected male transmits Y-linked traits to all of his sons but to none of his daughters. In the past it has been suggested that bizarre-sounding conditions such as porcupine skin, hairy ears and webbed toes are Y-linked traits. With the possible exception of hairy ears, these claims of holandric inheritance have not stood up to more careful study. Evidence clearly indicates, however, that the H-Y histocompatibility antigen (p. 200) and genes involved in spermatogenesis are carried on the Y chromosome and, therefore, manifest holandric inheritance. The latter, if deleted, leads to infertility from azoospermia (absence of the sperm in semen) in males. The recent advent of techniques of assisted reproduction, particularly the technique of intracytoplasmic sperm injection (ICSI), means that, if a pregnancy with a male conceptus results after the use of this technique, the child will also necessarily be infertile.
Sex Influence
Some autosomal traits are expressed more frequently in one sex than in another—so-called sex influence. Gout and presenile baldness are examples of sex-influenced autosomal dominant traits, males being predominantly affected in both cases. The influence of sex in these two examples is probably through the effect of male hormones. Gout, for example, is very rare in women before the menopause but the frequency increases in later life. Baldness does not occur in males who have been castrated. In hemochromatosis (p. 244), the most common autosomal recessive disorder in Western society, homozygous females are much less likely than homozygous males to develop iron overload and associated symptoms; the explanation usually given is that women have a form of natural blood loss through menstruation.
Establishing the Mode of Inheritance of a Genetic Disorder
In experimental animals it is possible to arrange specific types of mating to establish the mode of inheritance of a trait or disorder. In humans, when a disorder is newly recognized, the geneticist approaches the problem indirectly by fitting likely models of inheritance to the observed outcome in the offspring. Certain features are necessary to support a particular mode of inheritance. Formally establishing the mode of inheritance is not usually possible with a single family and normally requires study of a number of families (Box 7.1).
Box 7.1
Features that Support the Single-Gene or Mendelian Patterns of Inheritance
X-Linked Dominant Inheritance
There are three features necessary to establish X-linked dominant inheritance. First, males and females are affected but affected females are more frequent than affected males. Second, females are usually less severely affected than males. Third, although affected females can transmit the disorder to both male and female offspring, affected males can transmit the disorder only to their daughters (except in partial sex-linkage; see p. 118), all of whom will be affected. In the case of X-linked dominant disorders that are almost invariably lethal in male embryos (e.g., incontinentia pigmenti; see pp. 117–118), only females will be affected and families may show an excess of females over males as well as a number of miscarriages that are the affected male pregnancies.
Multiple Alleles and Complex Traits
So far, each of the traits we have considered has involved only two alleles, the normal, and the mutant. However, some traits and diseases are neither monogenic nor polygenic. Some genes have more than two allelic forms (i.e., multiple alleles). Multiple alleles are the result of a normal gene having mutated to produce various different alleles, some of which can be dominant and others recessive to the normal allele. In the case of the ABO blood group system (p. 205), there are at least four alleles (A1, A2, B, and O). An individual can possess any two of these alleles, which may be the same or different (AO, A2B, OO, and so on). Alleles are carried on homologous chromosomes and therefore a person transmits only one allele for a certain trait to any particular offspring. For example, a person with the genotype AB will transmit to any particular offspring either the A allele or the B allele, but never both or neither (Table 7.1). This relates only to genes located on the autosomes and does not apply to alleles on the X chromosome; in this instance a woman would have two alleles, either of which could be transmitted to offspring, whereas a man only has one allele to transmit.
Table 7.1 Possible Genotypes, Phenotypes, and Gametes Formed from the Four Alleles A1, A2, B, and O at the ABO Locus
| Genotype | Phenotype | Gametes |
|---|---|---|
| A1A1 | A1 | A1 |
| A2A2 | A2 | A2 |
| BB | B | B |
| OO | O | O |
| A1A2 | A1 | A1 or A2 |
| A1B | A1B | A1 or B |
| A1O | A1 | A1 or O |
| A2B | A2B | A2 or B |
| A2O | A2 | A2 or O |
| BO | B | B or O |
The dramatic advances in genome wide scanning using multiple DNA probes has made it possible to begin investigating so-called complex traits (i.e., conditions that are usually much more common than mendelian disorders and likely to be due to the interaction of more than one gene). The effects may be additive, one may be rate limiting over the action of another, or one may enhance or multiply the effect of another; this is considered in more detail in Chapter 15. The possibility of a small number of gene loci being implicated in some disorders has given rise to the concept of oligogenic inheritance, examples of which include the following.
Digenic Inheritance
This refers to the situation where a disorder has been shown to be due to the additive effects of heterozygous mutations at two different gene loci, a concept referred to as digenic inheritance. This is seen in certain transgenic mice. Mice that are homozygotes for rv (rib-vertebrae) or Dll1 (Delta–like-1) manifest abnormal phenotypes, whereas their respective heterozygotes are normal. However, mice that are double heterozygotes for rv and Dll1 show vertebral defects. In humans, one form of retinitis pigmentosa, a disorder of progressive visual impairment, is caused by double heterozygosity for mutations in two unlinked genes, ROM1 and Peripherin, which both encode proteins present in photoreceptors. Individuals with only one of these mutations are not affected. In the field of inherited cardiac arrhythmias and cardiomyopathies (p. 304), it is becoming clear that some cases of arrhythmogenic right ventricular dysplasia exhibit digenic inheritance.
Anticipation
Recent studies, however, have shown that in a number of disorders, including Huntington disease and myotonic dystrophy, anticipation is, in fact, a real biological phenomenon occurring as a result of the expansion of unstable triplet repeat sequences (p. 24). An expansion of the CTG triplet repeat in the 3′ untranslated end of the myotonic dystrophy gene, occurring predominantly in maternal meiosis, appears to be the explanation for the severe neonatal form of myotonic dystrophy that usually only occurs when the gene is transmitted by the mother (Figure 7.19). Fragile X syndrome (CGG repeats) (p. 278) behaves in a similar way, with major instability in the expansion occurring during maternal meiosis. A similar expansion—in this case CAG repeats—in the 5′ end of the Huntington disease gene (Figure 7.20) in paternal meiosis accounts for the increased risk of early onset Huntington disease, occasionally in childhood or adolescence, when the gene is transmitted by the father. The inherited spinocerebellar ataxia group of conditions is another example.
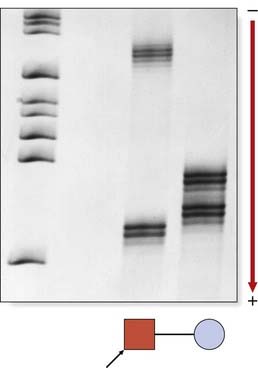
Mosaicism
An individual, or a particular tissue of the body, can consist of more than one cell type or line, through an error occurring during mitosis at any stage after conception. This is known as mosaicism (p. 50). Mosaicism of either somatic tissues or germ cells can account for some instances of unusual patterns of inheritance or phenotypic features in an affected individual.
Somatic Mosaicism
The possibility of somatic mosaicism is suggested by the features of a single-gene disorder being less severe in an individual than is usual, or by being confined to a particular part of the body in a segmental distribution; for example, as occurs occasionally in neurofibromatosis type I (p. 298). The timing of the mutation event in early development may determine whether it is transmitted to the next generation with full expression—this will depend on the mutation being present in all or some of the gonadal tissue, and hence germline cells.
Gonadal Mosaicism
There have been many reports of families with autosomal dominant disorders, such as achondroplasia and osteogenesis imperfecta, and X-linked recessive disorders, such as Duchenne muscular dystrophy and hemophilia, in which the parents are phenotypically normal, and the results of investigations or genetic tests have also all been normal, but in which more than one of their children has been affected. The most favored explanation for these observations is gonadal, or germline, mosaicism in one of the parents; that is, the mutation is present in a proportion of the gonadal or germline cells. An elegant example of this was provided by the demonstration of a mutation in the collagen gene responsible for osteogenesis imperfecta in a proportion of individual sperm from a clinically normal father who had two affected infants with different partners. It is important to keep germline mosaicism in mind when providing recurrence risks in genetic counseling for apparently new autosomal dominant and X-linked recessive mutations (p. 343).
Uniparental Disomy
An individual normally inherits one of a pair of homologous chromosomes from each parent (p. 39). Over the past decade, with the advent of DNA technology, some individuals have been shown to have inherited both homologs of a chromosome pair from only one of their parents, so-called uniparental disomy. If an individual inherits two copies of the same homolog from one parent, through an error in meiosis II (p. 41), this is called uniparental isodisomy (Figure 7.21). If, however, the individual inherits the two different homologs from one parent through an error in meiosis I (p. 39), this is termed uniparental heterodisomy. In either instance, it is presumed that the conceptus would originally be trisomic, with early loss of a chromosome leading to the ‘normal’ disomic state. One-third of such chromosome losses, if they occurred with equal frequency, would result in uniparental disomy. Alternatively, it is postulated that uniparental disomy could arise as a result of a gamete from one parent that does not contain a particular chromosome homolog (i.e., a gamete that is nullisomic), being ‘rescued’ by fertilization with a gamete that, through a second separate chance error in meiosis, is disomic.
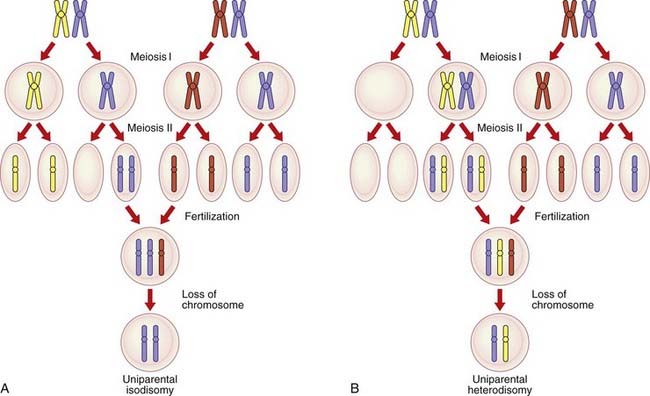
Genomic Imprinting
Genomic imprinting is an epigenetic phenomenon, referred to in Chapter 6 (p. 103). Epigenetics and genomic imprinting give the lie to Thomas Morgan’s quotation at the start of this chapter! Although it was originally thought that genes on homologous chromosomes were expressed equally, it is now recognized that different clinical features can result, depending on whether a gene is inherited from the father or from the mother. This ‘parent of origin’ effect is referred to as genomic imprinting, and methylation of DNA is thought to be the main mechanism by which expression is modified. Methylation is the imprint applied to certain DNA sequences in their passage through gametogenesis, although only a small proportion of the human genome is in fact subject to this process. The differential allele expression (i.e., maternal or paternal) may occur in all somatic cells, or in specific tissues or stages of development. Thus far, at least 80 human genes are known to be imprinted and the regions involved are known as differentially methylated regions (DMRs). These DMRs include imprinting control regions (ICRs) that control gene expression across imprinted domains.
Evidence of genomic imprinting has been observed in two pairs of well known dysmorphic syndromes: Prader-Willi and Angelman syndromes (chromosome 15q), and Beckwith-Wiedemann and Russell-Silver syndromes (chromosome 11p). The mechanisms giving rise to these conditions, although complex, reveal much about imprinting and are therefore now considered in a little detail.
Prader-Willi Syndrome
Prader-Willi syndrome (PWS) (p. 282) occurs in approximately 1 in 20,000 births and is characterized by short stature, obesity, hypogonadism, and learning difficulty (Figure 7.22). Approximately 50% to 60% of individuals with PWS can be shown to have an interstitial deletion of the proximal portion of the long arm of chromosome 15, approximately 2 Mb at 15q11-q13, visible by conventional cytogenetic means, and in a further 15% a submicroscopic deletion can be demonstrated by fluorescent in-situ hybridization (see p. 34) or molecular means. DNA analysis has revealed that the chromosome deleted is almost always the paternally derived homolog. Most of the remaining 25% to 30% of individuals with PWS, without a chromosome deletion, have been shown to have maternal uniparental disomy. Functionally, this is equivalent to a deletion in the paternally derived chromosome 15.
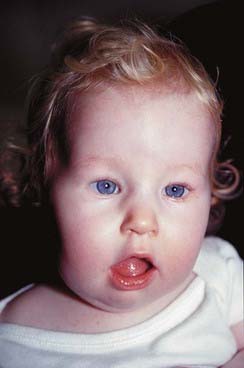
It is now known that only the paternally inherited allele of this critical region of 15q11-q13 is expressed. The molecular organization of the region is shown in Figure 7.23. PWS is a multigene disorder and in the normal situation the small nuclear ribonucleoprotein polypeptide N (SNRPN) and adjacent genes (MKRN3, etc.) are paternally expressed. Expression is under the control of a specific ICR. Analysis of DNA from patients with PWS and various submicroscopic deletions enabled the ICR to be mapped to a segment of about 4 kb, spanning the first exon and promoter of SNRPN and upstream reading frame (SNURF). The 3′ end of the ICR is required for expression of the paternally expressed genes and also the origin of the long SNURF/SNRPN transcript. The maternally expressed genes are not differentially methylated but they are silenced on the paternal allele, probably by an antisense RNA generated from SNURF/SNRPN. In normal cells, the 5′ end of the ICR, needed for maternal expression and involved in Angelman syndrome (see below), is methylated on the maternal allele.
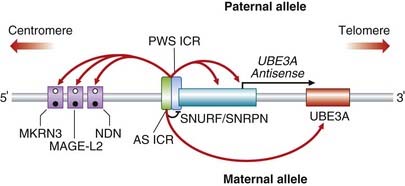
 ) on the paternal allele but methylated (•) on the maternal allele.
) on the paternal allele but methylated (•) on the maternal allele.Angelman Syndrome (AS)
Angelman syndrome (p. 282) occurs in about 1 in 15,000 births and is characterized by epilepsy, severe learning difficulties, an unsteady or ataxic gait, and a happy affect (Figure 7.24). Approximately 70% of individuals with AS have been shown to have an interstitial deletion of the same 15q11-q13 region as is involved in PWS, but in this case on the maternally derived homolog. In a further 5% of individuals with AS, the syndrome can be shown to have arisen through paternal uniparental disomy. Unlike PWS, the features of AS arise through loss of a single gene, UBE3A. In up to 10% of individuals with AS, mutations have been identified in UBE3A, one of the ubiquitin genes, which appears to be preferentially or exclusively expressed from the maternally derived chromosome 15 in brain. How mutations in UBE3A lead to the features seen in persons with AS is not clear, but could involve ubiquitin-mediated destruction of proteins in the central nervous system in development, particularly where UBE3A is expressed most strongly, namely the hippocampus and Purkinje cells of the cerebellum. UBE3A is under control of the AS ICR (see Figure 7.23), which was mapped slightly upstream of SNURF/SNRPN through analysis of patients with AS who had various different microdeletions.
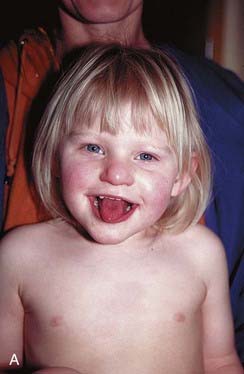
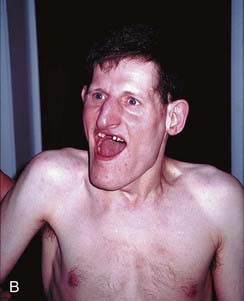
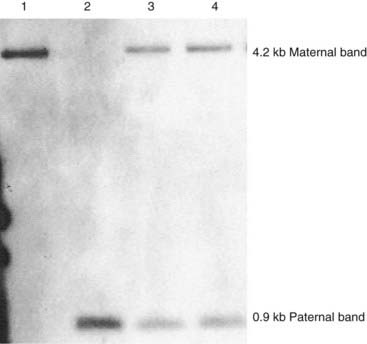
In many genetics service laboratories a simple DNA test is used to diagnose both PWS and AS, exploiting the differential DNA methylation characteristics at the 15q11-q13 locus (Figure 7.25).
Beckwith-Wiedemann Syndrome
Beckwith–Wiedemann syndrome (BWS) is a clinically heterogeneous condition whose main underlying characteristic is overgrowth. First described in 1963 and 1964, the main features are macrosomia (prenatal and/or postnatal overgrowth), macroglossia (large tongue), abdominal wall defect (omphalocele, umbilical hernia, diastasis recti), and neonatal hypoglycemia (Figure 7.26). Hemihyperplasia may be present, as well as visceromegaly, renal abnormalities, ear anomalies (anterior earlobe creases, posterior helical pits) and cleft palate, and there may be embryonal tumors (particularly Wilms tumor).
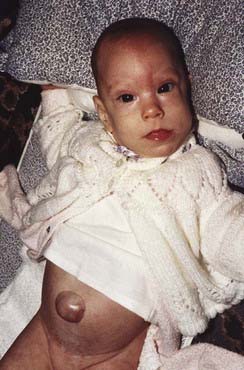
BWS is, in a way, celebrated in medical genetics because of the multiple different (and complex) molecular mechanisms that underlie it. Genomic imprinting, somatic mosaicism, and multiple genes are involved, all within a 1 Mb region at chromosome 11p15 (Figure 7.27). Within this region lie two independently regulated imprinted domains. The more telomeric (differentially methylated region 1 [DMR1] under control of ICR1) contains paternally expressed IGF2 (insulin growth factor 2) and maternally expressed H19. The more centromeric imprinted domain (DMR2, under control of ICR2) contains the maternally expressed KCNQ1 (previously known as KvLQT1) and CDKN1C genes, and the paternally expressed antisense transcript KCNQ1OT1, the promoter for which is located within the KCNQ1 gene.
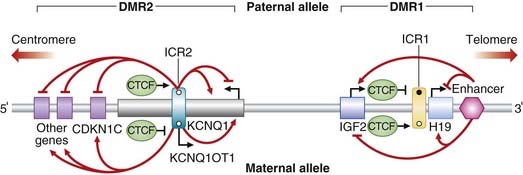
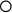 unmethylated). CCCTC-binding factor (CTCF) binds to the unmethylated alleles of both ICRs. In DMR1, coordinated regulation leads to expression of IGF2 only on the paternal allele and H19 expression only on the maternal allele. In DMR2, coordinated regulation leads to maternal expression of KCNQ1 and CDKN1C (plus other genes), and paternal expression of KCNQ1OT1 (a non-coding RNA with antisense transcription to KCNQ1). Angled black arrows show the direction of the transcripts.
unmethylated). CCCTC-binding factor (CTCF) binds to the unmethylated alleles of both ICRs. In DMR1, coordinated regulation leads to expression of IGF2 only on the paternal allele and H19 expression only on the maternal allele. In DMR2, coordinated regulation leads to maternal expression of KCNQ1 and CDKN1C (plus other genes), and paternal expression of KCNQ1OT1 (a non-coding RNA with antisense transcription to KCNQ1). Angled black arrows show the direction of the transcripts.Disruption to the normal regulation of methylation can give rise to altered gene expression dosage and, consequentially, features of BWS. In DMR1, gain of methylation on the maternal allele leads to loss of H19 expression and biallelic IGF2 expression (i.e., effectively two copies of the paternal epigenotype). This occurs in up to 7% of BWS cases and is usually sporadic. In DMR2, loss of methylation results in two copies of the paternal epigenotype and a reduction in expression of CDKN1C; this mechanism is implicated in 50% to 60% of sporadic BWS cases. CDKN1C may be a growth inhibitory gene and mutations have been found in 5% to 10% of cases of BWS. About 15% of BWS cases are familial, and CDKN1C mutations are found in about half of these. In addition to imprinting errors in DMR1 and DMR2, other mechanisms may account for BWS: (1) paternally derived duplications of chromosome 11p5.5 (these cases were the first to identify the BWS locus); (2) paternal uniparental disomy for chromosome 11—invariably present in mosaic form—often associated with neonatal hypoglycemia and hemi-hypertrophy, and associated with the highest risk (about 25%) of embryonal tumors, particularly Wilms tumor; and (3) maternally inherited balanced translocations involving rearrangements of 11p15.
Russell–Silver Syndrome
This well-known condition has ‘opposite’ characteristics to BWS by virtue of marked prenatal and postnatal growth retardation. The head circumference is relatively normal, the face rather small and triangular, giving rise to a ‘pseudohydrocephalic’ appearance (Figure 7.28), and there may be body asymmetry. About 10% of cases appear to be due to maternal uniparental disomy, indicating that this chromosome is subject to imprinting. In contrast to paternally derived duplications of 11p15, which give rise to overgrowth and BWS, maternally derived duplications of this region are associated with growth retardation. Recently it has been shown that about a third of Russell–Silver syndrome (RSS) cases are due to abnormalities of imprinting at the 11p.15.5 locus. Whereas hypermethylation of DMR1 leads to upregulated IGF2 and overgrowth, hypomethylation of H19 leads to downregulated IGF2, the opposite molecular and biochemical consequence, and these patients have features of RSS. Interestingly, in contrast to BWS, there are no cases of RSS with altered methylation of the more centromeric DMR2 region.
Mitochondrial Inheritance
Each cell contains thousands of copies of mitochondrial DNA with more being found in cells that have high energy requirements, such as brain and muscle. Mitochondria, and therefore their DNA, are inherited almost exclusively from the mother through the oocyte (p. 41). Mitochondrial DNA has a higher rate of spontaneous mutation than nuclear DNA, and the accumulation of mutations in mitochondrial DNA has been proposed as being responsible for some of the somatic effects seen with aging.
In humans, cytoplasmic or mitochondrial inheritance has been proposed as a possible explanation for the pattern of inheritance observed in some rare disorders that affect both males and females but are transmitted only through females, so-called maternal or matrilineal inheritance (Figure 7.29).
A number of rare disorders with unusual combinations of neurological and myopathic features, sometimes occurring in association with other conditions such as cardiomyopathy and conduction defects, diabetes, or deafness, have been characterized as being due to mutations in mitochondrial genes (p. 181). Because mitochondria have an important role in cellular metabolism through oxidative phosphorylation, it is not surprising that the organs most susceptible to mitochondrial mutations are the central nervous system, skeletal muscle and heart.
In most persons, the mitochondrial DNA from different mitochondria is identical, or shows what is termed homoplasmy. If a mutation occurs in the mitochondrial DNA of an individual, initially there will be two populations of mitochondrial DNA, so-called heteroplasmy. The proportion of mitochondria with a mutation in their DNA varies between cells and tissues, and this, together with mutational heterogeneity, is a possible explanation for the range of phenotypic severity seen in persons affected with mitochondrial disorders (Figure 7.30).
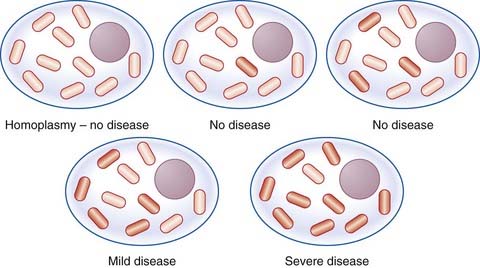
Whilst matrilineal inheritance applies to disorders that are directly because of mutations in mitochondrial DNA, it is also important to be aware that mitochondrial proteins are encoded mainly by nuclear genes. Mutations in these genes can have a devastating impact on respiratory chain functions within mitochondria. Examples include genes encoding proteins within the cytochrome c (COX) system, which follow autosomal recessive inheritance, and the G4.5 (TAZ) gene that is X-linked and causes Barth syndrome (endocardial fibroelastosis) in males (p. 182). There is even a mitochondrial myopathy following autosomal dominant inheritance in which multiple mitochondrial DNA deletions can be detected. Further space is devoted to mitochondrial disorders in Chapter 11 (p. 181).
Bateson W, Saunders ER 1902 Experimental studies in the physiology of heredity, pp 132–134. Royal Society Reports to the Evolution Committee, 1902
Early observations on mendelian inheritance.
Bennet RL, Steinhaus KA, Uhrich SB, et al. Recommendations for standardized human pedigree nomenclature. Am J Hum Genet. 1995;56:745-752.
Goriely A, McVean GAT, Rojmyr M, et al. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301:643-646.
Hall JG. Somatic mosaicism: observations related to clinical genetics. Am J Hum Genet. 1988;43:355-363.
Good review of findings arising from somatic mosaicism in clinical genetics.
Hall JG. Genomic imprinting: review and relevance to human diseases. Am J Hum Genet. 1990;46:857-873.
Extensive review of examples of imprinting in inherited diseases in humans.
Heinig RM. The monk in the garden: the lost and found genius of Gregor Mendel. London: Houghton Mifflin; 2000.
The life and work of Gregor Mendel as the history of the birth of genetics.
Kingston HM. An ABC of clinical genetics, 2nd ed. London: British Medical Association; 1994.
A simple outline primer of the basic principles of clinical genetics.
Reik W, Surami A, editors. Genomic imprinting (frontiers in molecular biology). London: IRL Press, 1997.
Detailed discussion of examples and mechanisms of genomic imprinting.
Vogel F, Motulsky AG. Human genetics, 3rd ed. Berlin: Springer; 1996.
Elements