Chapter 75 Patterns of Genetic Transmission
Family History and Pedigree Notation
A pedigree provides a graphic depiction of a family’s structure and medical history. It is important when taking a pedigree to be systematic and use standard symbols and configurations (Figs. 75-1, 75-2, 75-3, 75-4) so that anyone can read and understand the information. In the pediatric setting, the proband is typically the child or adolescent who is being evaluated. The proband is designated in the pedigree by an arrow.
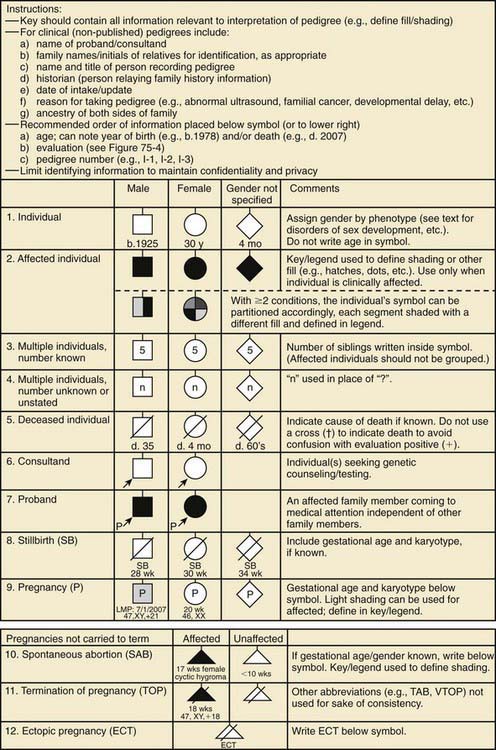
Figure 75-1 Common pedigree symbols, definitions, and abbreviations.
(From Bennett RL, French KS, Resta RG, et al: Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors, J Genet Counsel 17:424–433, 2008.)
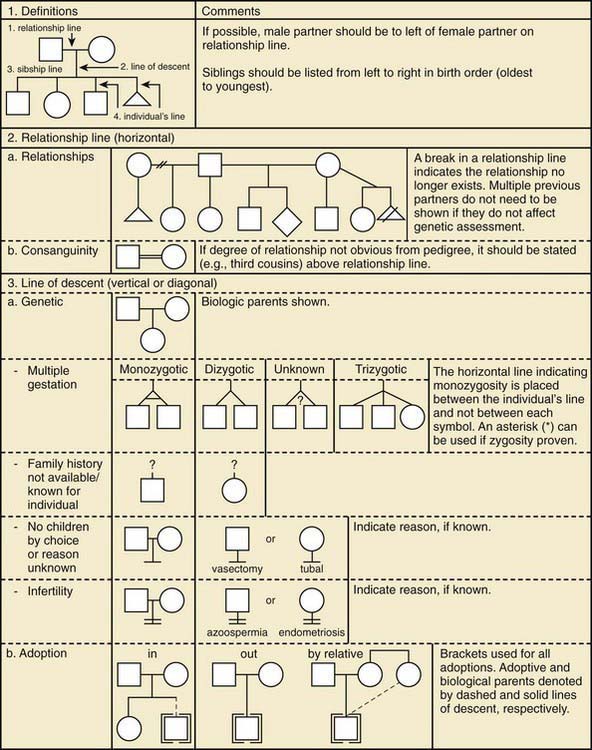
Figure 75-2 Pedigree line definitions.
(From Bennett RL, French KS, Resta RG, et al: Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors, J Genet Counsel 17:424–433, 2008.)
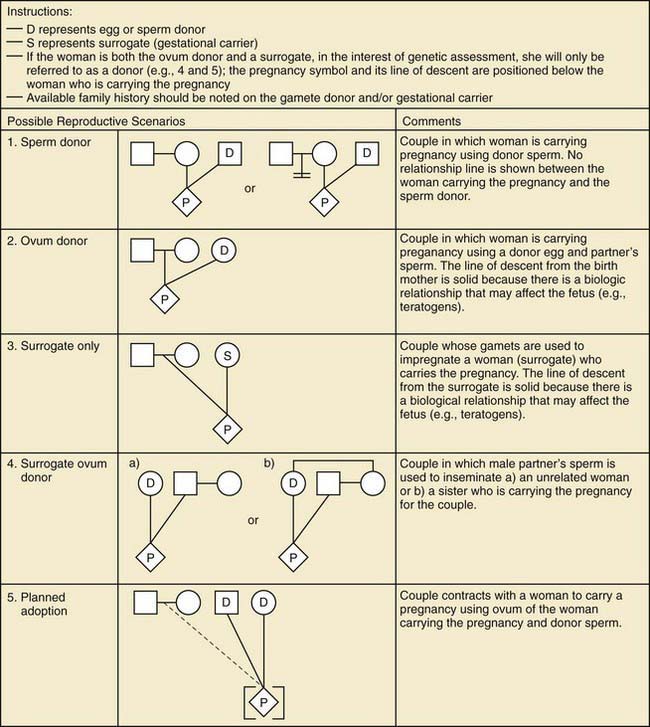
Figure 75-3 Assisted reproductive technology symbols and definitions.
(From Bennett RL, French KS, Resta RG, et al: Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors, J Genet Counsel 17:424–433, 2008.)

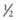 their genetic information on average; 1st cousins share
their genetic information on average; 1st cousins share 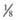 . Sometimes the person providing the family history may mention a distant relative who is affected with a genetic disorder. In such cases a more extensive pedigree may be needed to identify the risk to other family members. For example, a history of a distant maternally related cousin with mental retardation due to fragile X syndrome can still place a male proband at an elevated risk for this disorder.
. Sometimes the person providing the family history may mention a distant relative who is affected with a genetic disorder. In such cases a more extensive pedigree may be needed to identify the risk to other family members. For example, a history of a distant maternally related cousin with mental retardation due to fragile X syndrome can still place a male proband at an elevated risk for this disorder.Mendelian Inheritance
Autosomal Dominant Inheritance
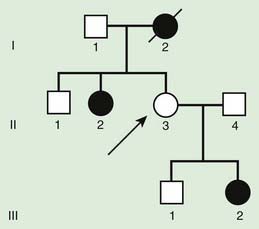
The pedigree for an autosomal dominant disorder (Fig. 75-5) demonstrates certain characteristics. The disorder is transmitted in a vertical (parent to child) pattern and can appear in multiple generations. This is illustrated by individual I.1 (see Fig 75-5) passing on the changed gene to II.2 and II.5. An affected individual has a 50% (1 in 2) chance of passing on the deleterious gene in each pregnancy and, therefore, of having a child affected by the disorder. This is referred to as the recurrence risk for the disorder. Unaffected individuals (family members who do not manifest the trait) do not pass the disorder to their children. Males and females are equally affected. Although not a characteristic per se, the finding of male-to-male transmission essentially confirms autosomal dominant inheritance. Vertical transmission can also be seen with X-linked traits. However, because a father passes on his Y chromosome to a son, male-to-male transmission cannot be seen with an X-linked trait. Therefore, male-to-male transmission eliminates X-linked inheritance as a possible explanation. Although male-to-male transmission can occur with Y-linked genes as well, there are very few Y-linked disorders compared with thousands having the autosomal dominant inheritance pattern.
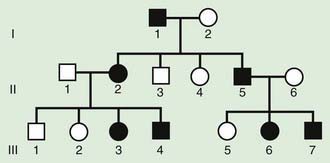
Although parent to child transmission is a characteristic of autosomal dominant inheritance, for many patients with an autosomal dominant disorder there is no history of an affected family member. There are several possible reasons: First, the patient may represent a new mutation that occurred in the DNA of the egg or sperm that came together to form that individual. Second, many autosomal dominant conditions demonstrate incomplete penetrance, meaning that not all individuals who carry the mutation have phenotypic manifestations. In a pedigree this can appear as a skipped generation, in which an unaffected individual links two affected persons (Fig. 75-6). There are many potential reasons that a disorder exhibits incomplete penetrance, including the effect of modifier genes, environmental factors, gender, and age. Third, individuals with the same autosomal dominant mutation can manifest the disorder to different degrees. This is termed variable expression and is a characteristic of many autosomal dominant disorders. Fourth, some spontaneous genetic mutations occur not in the egg or sperm that forms a child but rather in a cell in the developing embryo. Such events are referred to as somatic mutations, and because not all cells are affected, the change is said to be mosaic. The resulting phenotype caused by a somatic mutation can be varied, but it is usually milder than if all cells contain the mutation. In germline mosaicism, the mutation occurs in cells that populate the germline that produce eggs or sperm. A germline mosaic might not have any manifestations of the disorder but might produce multiple eggs or sperm that carry the mutation.
Autosomal Recessive Inheritance
Autosomal recessive inheritance involves mutations in both copies of a gene. Examples of autosomal recessive diseases are cystic fibrosis and sickle cell disease. Characteristics of autosomal recessive traits (Fig. 75-7) include horizontal transmission, the observation of multiple affected members of a kindred in the same generation, but no affected family members in other generations; recurrence risk of 25% for parents with a previous affected child; males and females being equally affected, although some traits exhibit different expression in males and females and increased incidence, particularly for rare traits, in the offspring of consanguineous parents. Consanguinity refers to the existence of a relationship by a common ancestor and increases the chance that both parents carry a gene affected by an identical mutation that they inherited. Consanguinity between parents of a child with a suspected genetic disorder implies (but does not prove) autosomal recessive inheritance. Although consanguineous unions are uncommon in Western society, in other parts of the world (southern India, Japan, and the Middle East) they are common. The risk of a genetic disorder for the offspring of a first-cousin marriage (6-8%) is about double the risk in the general population (3-4%).
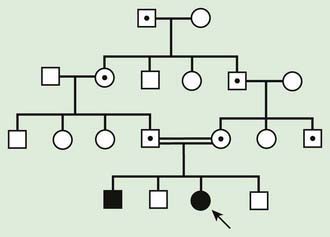
Figure 75-7 Autosomal recessive pedigree with parental consanguinity. Central dot, carriers; black, affected patients.
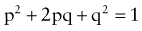
where p is the frequency of one of a pair of alleles and q is the frequency of the other. For example, if the frequency of cystic fibrosis among white Americans is 1 in 2,500 (p2), then the frequency of the heterozygote (2pq) can be calculated: If p2 = 1/2,500, then p = 1/50 and q = 49/50; 2pq = 2 × (1/50) × (49/50) = 98/2500 or 3.92%.
Pseudodominant Inheritance
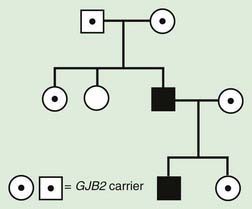
Pseudodominant inheritance refers to the observation of apparent dominant (parent to child) transmission of a known autosomal recessive disorder (Fig. 75-8). This occurs when a homozygous affected individual has a partner who is a heterozygous carrier, and it is most likely to occur for relatively common traits, such as sickle cell anemia or nonsyndromic autosomal recessive hearing loss due to mutations in GJB2, the gene that encodes Connexin 26.
X-Linked Inheritance
Characteristics of X-linked inheritance (Fig. 75-9) include the following:
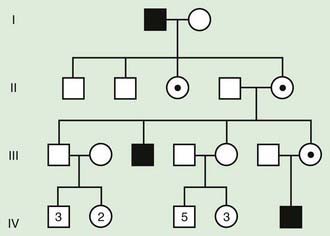
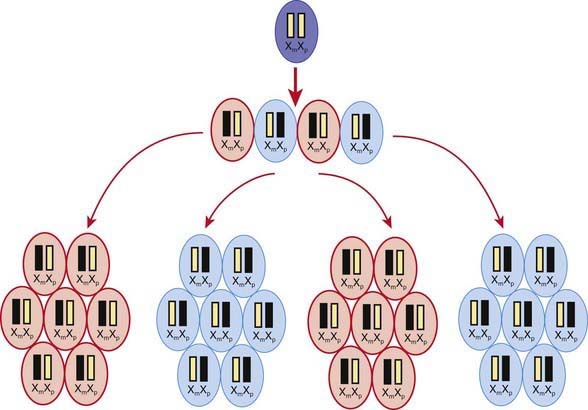
A female occasionally exhibits signs of an X-linked trait similarly to a male. This occurs rarely owing to homozygosity for an X-linked trait or the presence of a sex chromosome abnormality (45,X or 46,XY female) or skewed or nonrandom X-inactivation. X chromosome inactivation occurs early in development and involves random and irreversible inactivation of most genes on one X chromosome in female cells (Fig. 75-10). In some cases, a preponderance of cells inactivates the same X chromosome, resulting in phenotypic expression of an X-linked mutation if it resides on the active chromosome. This can occur owing to chance, selection against cells that have inactivated the X chromosome carrying the normal gene, or X chromosome abnormalities that result in inactivation of the X chromosome carrying the normal gene.
Some X-linked disorders are inherited in an X-linked dominant fashion in which female carriers typically manifest abnormal findings. An affected man will have only affected daughters and unaffected sons, and half of the offspring of an affected woman will be affected (Fig. 75-11). Some X-linked dominant conditions are lethal in a high percentage of males. An example is incontinentia pigmenti (see Chapter 589.7). The pedigree shows only affected females and an overall ratio of 2 : 1 females to males with an increased number of miscarriages (Fig. 75-12).
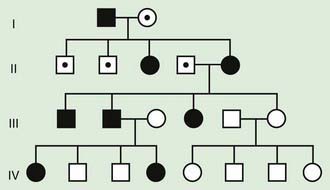
Figure 75-11 Pedigree pattern demonstrating X-linked dominant inheritance. Note there is no father-to-son transmission in this situation, and hemizygosity (i.e., X-linked gene in a male) is not lethal. In some X-linked dominant conditions, X-linked males have a more severe phenotype and might not survive. In that case, only females manifest the disease (see Fig. 75-12).
Y-Linked Inheritance
There are few Y-linked traits. These demonstrate only male-to-male transmission, and only males are affected (Fig. 75-13). Most Y-linked genes are related to male sex determination and reproduction and are associated with infertility. Therefore, it is rare to see familial transmission of a Y-linked disorder. However, advances in assisted reproductive technologies might make it possible to have familial transmission of male infertility.
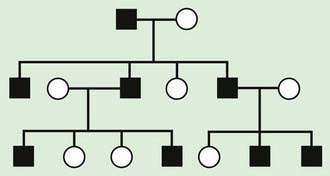
Digenic Inheritance
Digenic inheritance explains the occurrence of retinitis pigmentosa (RP) in children of parents who each carry a mutation in a different RP-associated gene. Both parents have normal vision, as would be expected, but their offspring who are double heterozygotes—having inherited both mutations—develop RP. Digenic pedigrees (Fig. 75-14) can exhibit characteristics of both autosomal dominant (vertical transmission) and autosomal recessive inheritance (1 in 4 recurrence risk). For example, a couple in which the two unaffected partners are carriers for mutation in two different RP-associated genes that show digenic inheritance have a 1 in 4 risk of having an affected child similar to what is seen in autosomal recessive inheritance. However, their affected children, and affected children in subsequent generations, have a 1 in 4 risk of transmitting both mutations to their offspring, who would be affected (vertical transmission).
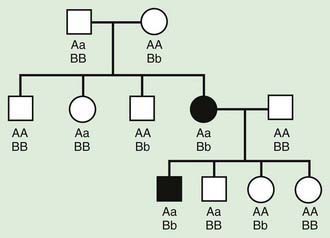
Nontraditional Inheritance
Mitochondrial Inheritance
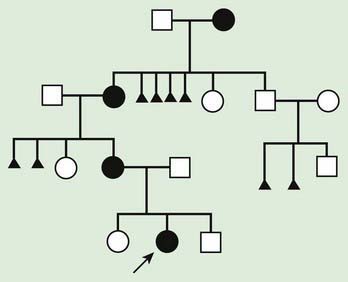
An individual’s mitochondrial genome is entirely derived from the mother because sperm contain few mitochondria, which are typically shed upon fertilization (Fig. 75-15). It follows that mitochondrial disorders exhibit maternal inheritance. A woman with a mitochondrial genetic disorder can have affected offspring of either sex, but an affected father cannot pass on the disease to his offspring (Fig. 75-16). mtDNA mutations are often deletions or point mutations; overall, 1 : 400 people has a maternally inherited pathogenic mtDNA mutation.
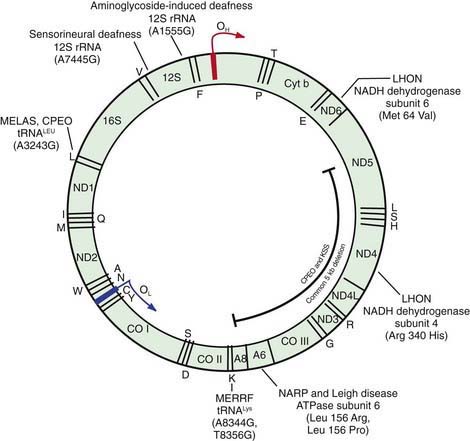
Figure 75-15 The human mitochondrial DNA molecule, showing the location of genes encoding 22 tRNAs, 2 rRNAs, and 13 proteins of the oxidative phosphorylation (OXPHOA) complex. Some of the most common disease-causing substitutions and deletions in the mtDNA genome are also illustrated. OH and OL are the origins of replication of the two DNA strands, respectively; 12S, 12S ribosomal RNA; 16S, 16S ribosomal RNA. The tRNAs are indicated by the single letter code for their corresponding amino acids (e.g., L for leucine, K for lysine). The 13 OXPHOS polypeptides encoded by mtDNA include components of complex I: NADH dehydrogenase (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6); complex III: cytochrome b (Cyt b); complex IV: cytochrome c oxidase I, or Cyt c (COI, COII, COIII); and complex V: ATPase 6 (ATP-6, ATP-8). See Table 75-1 for representative diseases.
(Adapted from Shoffner JM, Wallace DC: Oxidative phosphorylation disease. In Scriver CR, Beaudet AL, Sly WS, et al, editors: The metabolic and molecular basis of inherited disease, ed 7, New York, 1995, McGraw-Hill; and Johns DR: Mitochondrial DNA and disease, N Engl J Med 333:638–644, 1995. From Nussbaum RL, McInnes RR, Willard HF: Thompson & Thompson genetics in medicine, ed 6, Philadelphia, 2001, WB Saunders.)
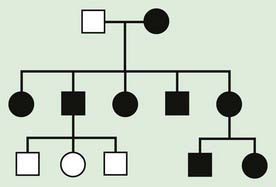
Figure 75-16 Pedigree of a mitochondrial disorder, exhibiting maternal inheritance. Black, affected patient.
In individual families, mitochondrial inheritance may be difficult to distinguish from autosomal dominant or X-linked inheritance, but in many cases paying close attention to the sex of the transmitting and nontransmitting parents can suggests a mitochondrial basis (Table 75-1).
Table 75-1 REPRESENTATIVE EXAMPLES OF DISORDERS CAUSED BY MUTATIONS IN MITOCHONDRIAL DNA AND THEIR INHERITANCE
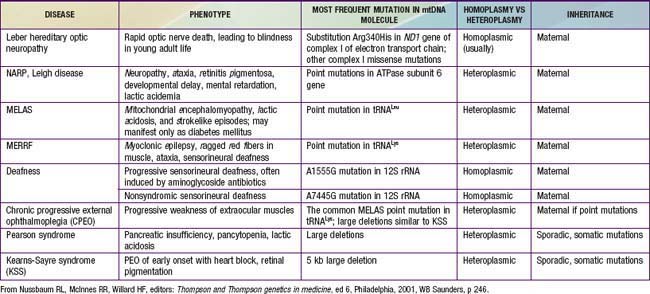
The mitochondria are the cell’s suppliers of energy, and it is not surprising that the organs that are most affected by the presence of abnormal mitochondria are those that have the greatest energy requirements, such as the brain, muscle, heart, and liver (Chapters 81.4, 353, and 591). Common manifestations include developmental delay, seizures, cardiac dysfunction, decreased muscle strength and tone, and hearing and vision problems. Examples of mitochondrial disorders include MELAS (myopathy, encephalopathy, lactic acidosis, and strokelike episodes), MERRF (myoclonic epilepsy associated with ragged red fibers), and Kearns-Sayre syndrome (ophthalmoplegia, pigmentary retinopathy, and cardiomyopathy) (Chapter 591).
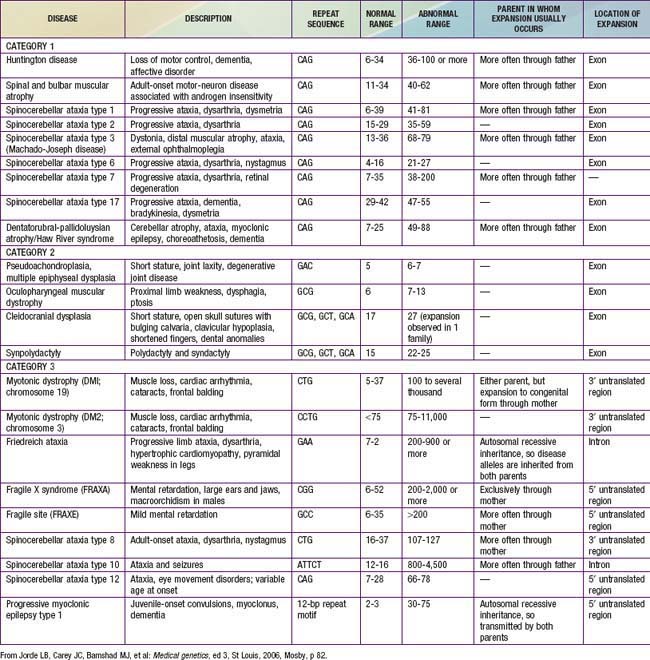
Triplet Repeat Expansion Disorders
Triplet repeat expansion disorders are distinguished by the special dynamic nature of the disease-causing mutation. Triplet repeat expansion disorders include fragile X syndrome, myotonic dystrophy, Huntington disease, spinocerebellar ataxias, and several others (Table 75-2). These disorders are caused by expansion in the number of 3-bp repeats. The fragile X gene, FMR1, normally has 5-40 CGG triplets. An error in replication can result in expansion of that number, to a level in the gray zone between 41 and 58 repeats, or to a level referred to as premutation, which comprises 59-200 repeats. Some male carriers of the premutation develop fragile X–associated tremor/ataxia syndrome (FXTAS) as adults, and female carriers of the permutation are at risk for FMR1-related premature ovarian failure (POF). Persons with a premutation are also at risk for having the gene expand further in subsequent meiosis, hence crossing into the range of full mutation in offspring. In fragile X, the threshold for clinical diagnosis is above 200 repeats. With this number of repeats, the FMR1 gene becomes hypermethylated, and protein production is lost.
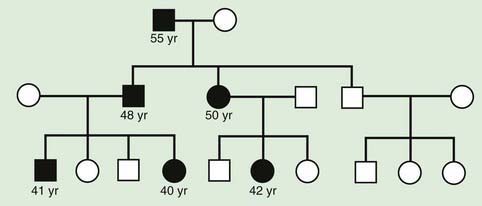
Some triplet expansions associated with other genes can cause disease through a mechanism other than decreased protein production. In Huntington disease, the expansion causes the gene product to have a new, toxic effect on the neurons of the basal ganglia. For most triplet-repeat disorders, there is a clinical correlation to the size of the expansion, with a greater expansion causing more severe symptoms and/or earlier age of onset for the disease. The observation of increasing severity of disease and early age of onset in subsequent generations is termed genetic anticipation and is a defining characteristic of many triplet-repeat expansion disorders (Fig. 75-17).
Genetic Imprinting
The 2 copies of most autosomal genes are functionally equivalent. However, in some cases only 1 copy of a gene is transcribed and the other copy is silenced. This gene silencing is typically associated with methylation of DNA, which is an epigenetic modification, meaning it does not change the nucleotide sequence of the DNA (Fig. 75-18). In imprinting, gene expression depends on the parent of origin of the chromosome (Chapter 76). Imprinting disorders result from an imbalance of active copies of a given gene, which can occur for several reasons. Prader-Willi and Angelman syndromes, two distinct disorders associated with developmental impairment, are illustrative. Both can be caused by microdeletions of chromosome 15q11-12. The microdeletion in Prader-Willi syndrome is always on the paternally derived chromosome 15, whereas in Angelman syndrome it is on the maternal copy. UBE3A is the gene responsible for Angelman syndrome. The paternal copy of UBE3A is transcriptionally silenced in the brain and the maternal copy continues to be transcribed. If an individual has a maternal deletion, an insufficient amount of UBE3A protein is produced in the brain, resulting in the neurologic deficits seen in Angelman syndrome.
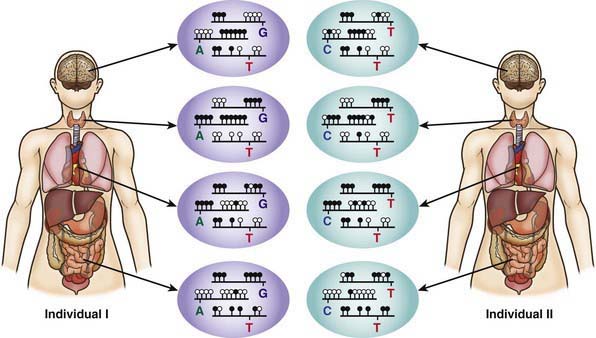
(Redrawn from Brena RM, Huang THM, Plass C: Toward a human epigenome, Nat Genet 38:1359–1360, 2006.)
Uniparental disomy (UPD), the rare occurrence of a child inheriting both copies of a chromosome from the same parent, is another genetic mechanism that can cause Prader-Willi and Angelman syndromes. Inheriting both chromosomes 15 from the mother is functionally the same as deletion of the paternal 15q12 and results in Prader-Willi syndrome. About 30% of cases of Prader-Willi syndrome is caused by paternal UPD15, whereas maternal UPD15 accounts for only 3% of Angelman syndrome (Chapter 76).
Imprinting of a gene can occur during gametogenesis or early embryonic development (reprogramming). Genes can become inactive or active by various mechanisms including DNA methylation or demethylation or histone acetylation or deacytylation, with different patterns of (de)methylation noted on paternal or maternal imprintable chromosome regions. Some genes demonstrate tissue-specific imprinting (see Fig. 75-18). Several studies suggest that there may be a small but significantly increased incidence of imprinting disorders, specifically Beckwith-Wiedemann and Angelman syndrome, associated with assisted reproductive technologies such as in vitro fertilization and intracytoplasmic sperm injection. However, the overall incidence of these disorders in children conceived using assisted reproductive technologies is likely to be <1%.
Multifactorial and Polygenic Inheritance
Multifactorial inheritance refers to traits that are caused by a combination of inherited, environmental, and stochastic factors (Fig. 75-19). Multifactorial traits differ from polygenic inheritance, which refers to traits that result from the additive effects of multiple genes. Multifactorial traits segregate within families but do not exhibit a consistent or recognizable inheritance pattern. Characteristics include the following:
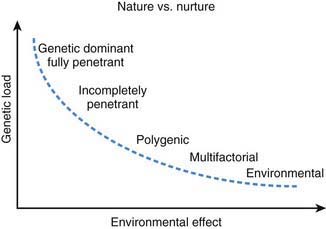
With other multifactorial traits, the distinction between normal and abnormal is based on the presence or absence of a particular trait. Examples include pyloric stenosis, neural tube defects, congenital heart defects, and cleft lip and cleft palate. Such traits follow a threshold model (see Fig. 75-16). A distribution of liability due to genetic and nongenetic factors is postulated in the population. Individuals who exceed a threshold liability develop the trait, and those below the threshold do not.
ACOG Committee on Genetics. ACOG committee opinion. Number 298, August 2004. Prenatal and preconceptional carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol. 2004;104:425-428.
Allegrucci C, Denning C, Priddle H, et al. Stem-cell consequences of embryo epigenetic defects. Lancet. 2004;364:206-208.
Bennett RL, French KS, Resta RG, et al. Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors. J Genet Counsel. 2008;17:424-433.
Bjornsson HT, Sigurdsson MI, Falin MD, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877-2883.
Bowdin S, Allen C, Kirby G, et al. A survey of assisted reproductive technology births and imprinting disorders. Hum Reprod. 2007;22:3237-3240.
Brena RM, Huang THM, Plass C. Toward a human epigenome. Nat Genet. 2006;38:1359-1360.
Choi JK, Kim YJ. Epigenetic regulation and the variability of gene expression. Nat Genet. 2008;40:141-147.
Cree LM, Samuels DC, Chuva de Sousa Lopes S, et al. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249-254.
Gosden RG, Feinberg AP. Genetics and epigenetics—nature’s pen-and-pencil set. N Engl J Med. 2007;356:731-733.
Jacob S, Moley KH. Gametes and embryo epigenetic reprogramming affect developmental outcome: implication for assisted reproductive technologies. Pediatr Res. 2005;58:437-446.
Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X–associated tremor/ataxia syndrome in a permutation carrier population. JAMA. 2004;291:460-468.
Kaminsky ZA, Tang T, Wang SC, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240-245.
Kerkel K, Spadola A, Yuan E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904-908.
Krishnan KJ, Reeve AK, Samuels DC, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275-279.
Maher ER, Brueton LA, Bowdin SC, et al. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). J Med Genet. 2003;40:62-64.
Petrucelli N, Daly MB, Bars Culver JO, et al. BRCA1 and BRCA2 hereditary breast/ovarian cancer (2007) in GeneReviews at GeneTests: Medical Genetics Information Resource (database online). www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=cf#cf. Accessed March 9, 2009
Poulton J, Kennedy S, Oakeshott P, et al. Preventing transmission of maternally inherited mitochondrial DNA diseases. BMJ. 2009;338:b94.
Saul RA, Tarleton J. FMR1-related disorders (2008) in GeneReviews at GeneTests: Medical Genetics Information Resource (database online). www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=cf#cf. Accessed March 9, 2009
Teebi AS, El-Shanti H. Consanguinity: implications for practice, research and policy. Lancet. 2006;367:970-971.