[level-membership-for-pediatrics-category]
Chapter 86 Hypoglycemia
Glucose has a central role in fuel economy and is a source of energy storage in the form of glycogen, fat, and protein (Chapter 81). Glucose, an immediate source of energy, provides 38 mol of adenosine triphosphate (ATP) per mol of glucose oxidized. It is essential for cerebral energy metabolism because it is usually the preferred substrate and its utilization accounts for nearly all the oxygen consumption in the brain. Cerebral glucose uptake occurs through a glucose transporter molecule or molecules that are not regulated by insulin. Cerebral transport of glucose is a carrier-mediated, facilitated diffusion process that is dependent on blood glucose concentration. Deficiency of brain glucose transporters can result in seizures because of low cerebral and cerebrospinal fluid (CSF) glucose concentrations (hypoglycorrhachia) despite normal blood glucose levels. To maintain the blood glucose concentration and prevent it from falling precipitously to levels that impair brain function, an elaborate regulatory system has evolved.
Definition
In neonates, there is not always an obvious correlation between blood glucose concentration and the classic clinical manifestations of hypoglycemia. The absence of symptoms does not indicate that glucose concentration is normal and has not fallen to less than some optimal level for maintaining brain metabolism. There is evidence that hypoxemia and ischemia may potentiate the role of hypoglycemia in causing permanent brain damage. Consequently, the lower limit of accepted normality of the blood glucose level in newborn infants with associated illness that already impairs cerebral metabolism has not been determined (Chapter 101). Out of concern for possible neurologic, intellectual, or psychologic sequelae in later life, most authorities recommend that any value of blood glucose <50 mg/dL in neonates be viewed with suspicion and vigorously treated. This is particularly applicable after the initial 2-3 hr of life, when glucose normally has reached its nadir; subsequently, blood glucose levels begin to rise and achieve values of 50 mg/dL or higher after 12-24 hr. In older infants and children, a whole blood glucose concentration of <50 mg/dL (10-15% higher for serum or plasma) represents hypoglycemia.
Substrate, Enzyme, and Hormonal Integration of Glucose Homeostasis
In the Newborn (Chapter 101)
In the early postnatal period, responses of the endocrine pancreas favor glucagon secretion so that blood glucose concentration can be maintained. These adaptive changes in hormone secretion are paralleled by similarly striking adaptive changes in hormone receptors. Key enzymes involved in glucose production also change dramatically in the perinatal period. Thus, there is a rapid fall in glycogen synthase activity and a sharp rise in phosphorylase after delivery. Similarly, the amount of rate-limiting enzyme for gluconeogenesis, phosphoenolpyruvate carboxykinase, rises dramatically after birth, activated in part by the surge in glucagon and the fall in insulin. This framework can explain several causes of neonatal hypoglycemia based on inappropriate changes in hormone secretion and unavailability of adequate reserves of substrates in the form of hepatic glycogen, muscle as a source of amino acids for gluconeogenesis, and lipid stores for the release of fatty acids. In addition, appropriate activities of key enzymes governing glucose homeostasis are required (see Fig. 81-1).
In Older Infants and Children
Hypoglycemia in older infants and children is analogous to that of adults, in whom glucose homeostasis is maintained by glycogenolysis in the immediate postfeeding period and by gluconeogenesis several hours after meals. The liver of a 10 kg child contains 20-25 g of glycogen, which is sufficient to meet normal glucose requirements of 4-6 mg/kg/min for only 6-12 hr. Beyond this period, hepatic gluconeogenesis must be activated. Both glycogenolysis and gluconeogenesis depend on the metabolic pathway summarized in Figure 81-1. Defects in glycogenolysis or gluconeogenesis may not be manifested in infants until the frequent feeding at 3-4 hr intervals ceases and infants sleep through the night, a situation usually present by 3-6 mo of age. The source of gluconeogenic precursors is derived primarily from muscle protein. The muscle bulk of infants and small children is substantially smaller relative to body mass than that of adults, whereas glucose requirements/unit of body mass are greater in children, so the ability to compensate for glucose deprivation by gluconeogenesis is more limited in infants and young children, as is the ability to withstand fasting for prolonged periods. The ability of muscle to generate alanine, the principal gluconeogenic amino acid, may also be limited. Thus, in normal young children, the blood glucose level falls after 24 hr of fasting, insulin concentrations fall appropriately to levels of <5-10 µU/mL, lipolysis and ketogenesis are activated, and ketones may appear in the urine.
The switch from glycogen synthesis during and immediately after meals to glycogen breakdown and later gluconeogenesis is governed by hormones, of which insulin is of central importance. Plasma insulin concentrations increase to peak levels of 50-100 µU/mL after meals, which serve to lower the blood glucose concentration through the activation of glycogen synthesis, enhancement of peripheral glucose uptake, and inhibition of glucose production. In addition, lipogenesis is stimulated, whereas lipolysis and ketogenesis are curtailed. During fasting, plasma insulin concentrations fall to ≤5-10 µU/mL, and together with other hormonal changes, this fall results in activation of gluconeogenic pathways (see Fig. 81-1). Fasting glucose concentrations are maintained through the activation of glycogenolysis and gluconeogenesis, inhibition of glycogen synthesis, and activation of lipolysis and ketogenesis. It should be emphasized that a plasma insulin concentration of >5 µU/mL, in association with a blood glucose concentration of ≤40 mg/dL (2.2 mM), is abnormal, indicating a hyperinsulinemic state and failure of the mechanisms that normally result in suppression of insulin secretion during fasting or hypoglycemia.
Clinical Manifestations (Chapter 101)
Clinical features generally fall into 2 categories. The 1st includes symptoms associated with the activation of the autonomic nervous system and epinephrine release, usually seen with a rapid decline in blood glucose concentration (Table 86-1). The 2nd category includes symptoms due to decreased cerebral glucose utilization, usually associated with a slow decline in blood glucose level or prolonged hypoglycemia (see Table 86-1). Although these classic symptoms occur in older children, the symptoms of hypoglycemia in infants may be subtler and include cyanosis, apnea, hypothermia, hypotonia, poor feeding, lethargy, and seizures. Some of these symptoms may be so mild that they are missed. Occasionally, hypoglycemia may be asymptomatic in the immediate newborn period. Newborns with hyperinsulinemia are often large for gestational age; older infants with hyperinsulinemia may eat excessively because of chronic hypoglycemia and become obese. In childhood, hypoglycemia may present as behavior problems, inattention, ravenous appetite, or seizures. It may be misdiagnosed as epilepsy, inebriation, personality disorders, hysteria, and retardation. A blood glucose determination should always be performed in sick neonates, who should be vigorously treated if concentrations are <50 mg/dL. At any age level, hypoglycemia should be considered a cause of an initial episode of convulsions or a sudden deterioration in psychobehavioral functioning.
Table 86-1 MANIFESTATIONS OF HYPOGLYCEMIA IN CHILDHOOD
FEATURES ASSOCIATED WITH ACTIVATION OF AUTONOMIC NERVOUS SYSTEM AND EPINEPHRINE RELEASE*
FEATURES ASSOCIATED WITH CEREBRAL GLUCOPENIA
* Some of these features will be attenuated if the patient is receiving β-adrenergic blocking agents.
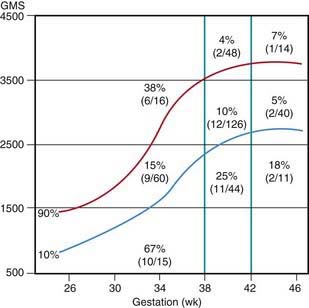
Many neonates have asymptomatic (chemical) hypoglycemia. The incidence of symptomatic hypoglycemia is highest in small for gestational age infants (Fig. 86-1). The exact incidence of symptomatic hypoglycemia has been difficult to establish because many of the symptoms in neonates occur together with other conditions such as infections, especially sepsis and meningitis; central nervous system anomalies, hemorrhage, or edema; hypocalcemia and hypomagnesemia; asphyxia; drug withdrawal; apnea of prematurity; congenital heart disease; or polycythemia.
Classification of Hypoglycemia in Infants and Children
Classification is based on knowledge of the control of glucose homeostasis in infants and children (Table 86-2).
Table 86-2 CLASSIFICATION OF HYPOGLYCEMIA IN INFANTS AND CHILDREN
NEONATAL TRANSIENT HYPOGLYCEMIA
Associated with Inadequate Substrate or Immature Enzyme Function in Otherwise Normal Neonates
Transient Neonatal Hyperinsulinism Also Present in:
NEONATAL, INFANTILE, OR CHILDHOOD PERSISTENT HYPOGLYCEMIAS
Hormonal Disorders
Counter-Regulatory Hormone Deficiency
Glycogenolysis and Gluconeogenesis Disorders
Lipolysis Disorders
Fatty Acid Oxidation Disorders
OTHER ETIOLOGIES
Substrate-Limited
Liver Disease
Amino Acid and Organic Acid Disorders
Systemic Disorders
GSD, glycogen storage disease; HI, hyperinsulinemia; KATP, regulated potassium channel.
Neonatal, Transient, Small for Gestational Age, and Premature Infants (Chapter 101)
The estimated incidence of symptomatic hypoglycemia in newborns is 1-3/1,000 live births. This incidence is increased severalfold in certain high-risk neonatal groups (see Table 86-2 and Fig. 86-1). The premature and small for gestational age (SGA) infants are vulnerable to the development of hypoglycemia. The factors responsible for the high frequency of hypoglycemia in this group, as well as in other groups outlined in Table 86-2, are related to the inadequate stores of liver glycogen, muscle protein, and body fat needed to sustain the substrates required to meet energy needs. These infants are small by virtue of prematurity or impaired placental transfer of nutrients. Their enzyme systems for gluconeogenesis may not be fully developed. Transient hyperinsulinism responsive to diazoxide has also been reported as contributing to hypoglycemia in asphyxiated, SGA, and premature newborn infants. This form of hyperinsulinism associated with perinatal asphyxia, intrauterine growth restriction (IUGR), maternal toxemia and other perinatal stressors, is probably the most common cause of hyperinsulinemic hypoglycemia in neonates and may be quite severe. In most cases, the condition resolves quickly, but it may persist to 7 mo of life or more. A genetic cause of this form of dysregulated insulin secretion has not been established.
Infants Born to Diabetic Mothers (Chapter 101)
Mothers whose diabetes has been well controlled during pregnancy, labor, and delivery generally have infants near normal size who are less likely to develop neonatal hypoglycemia and other complications formerly considered typical of such infants (Chapter 101). In supplying exogenous glucose to these hypoglycemic infants, it is important to avoid hyperglycemia that evokes a prompt exuberant insulin release, which may result in rebound hypoglycemia. When needed, glucose should be provided at continuous infusion rates of 4-8 mg/kg/min, but the appropriate dose for each patient should be individually adjusted. During labor and delivery, maternal hyperglycemia should be avoided because it results in fetal hyperglycemia, which predisposes to hypoglycemia when the glucose supply is interrupted at birth. Hypoglycemia persisting or occurring after 1 wk of life requires an evaluation for the causes listed in Table 86-2.
Persistent or Recurrent Hypoglycemia in Infants and Children
Hyperinsulinism
Most children with hyperinsulinism that causes hypoglycemia present in the neonatal period or later in infancy; hyperinsulinism is the most common cause of persistent hypoglycemia in early infancy. Hyperinsulinemic infants may be macrosomic at birth, reflecting the anabolic effects of insulin in utero. There is no history or biochemical evidence of maternal diabetes. The onset is from birth to 18 mo of age, but occasionally it is 1st evident in older children. Insulin concentrations are inappropriately elevated at the time of documented hypoglycemia; with non-hyperinsulinemic hypoglycemia, plasma insulin concentrations should be <5 µU/mL and no higher than 10 µU/mL. In affected infants, plasma insulin concentrations at the time of hypoglycemia are commonly >5-10 µU/mL. Some authorities set more stringent criteria, arguing that any value of insulin >2 µU/mL with hypoglycemia is abnormal. The insulin (µU/mL):glucose (mg/dL) ratio is commonly >0.4; plasma insulin-like growth factor binding protein-1 (IGFBP-1), ketones, and FFA levels are low with hyperinsulinemia. Macrosomic infants may present with hypoglycemia from the 1st days of life. Infants with lesser degrees of hyperinsulinemia may manifest hypoglycemia after the 1st few weeks to months, when the frequency of feedings has been decreased to permit the infant to sleep through the night and hyperinsulinism prevents the mobilization of endogenous glucose. Increasing appetite and demands for feeding, wilting spells, jitteriness, and frank seizures are the most common presenting features. Additional clues include the rapid development of fasting hypoglycemia within 4-8 hr of food deprivation compared with other causes of hypoglycemia (Tables 86-3 and 86-4); the need for high rates of exogenous glucose infusion to prevent hypoglycemia, often at rates >10-15 mg/kg/min; the absence of ketonemia or acidosis; and elevated C-peptide or proinsulin levels at the time of hypoglycemia. The latter insulin-related products are absent in factitious hypoglycemia from exogenous administration of insulin as a form of child abuse (Munchausen by proxy syndrome) (Chapter 37.2). Hypoglycemia is invariably provoked by withholding feedings for several hours, permitting simultaneous measurement of glucose, insulin, ketones, and FFAs in the same sample at the time of clinically manifested hypoglycemia. This is termed the “critical sample.” The glycemic response to glucagon at the time of hypoglycemia reveals a brisk increment in glucose concentration of at least 40 mg/dL, which implies that glucose mobilization has been restrained by insulin but that glycogenolytic mechanisms are intact (Tables 86-5, 86-6, and 86-7).

Table 86-4 CORRELATION OF CLINICAL FEATURES WITH MOLECULAR DEFECTS IN PERSISTENT HYPERINSULINEMIC HYPOGLYCEMIA IN INFANCY
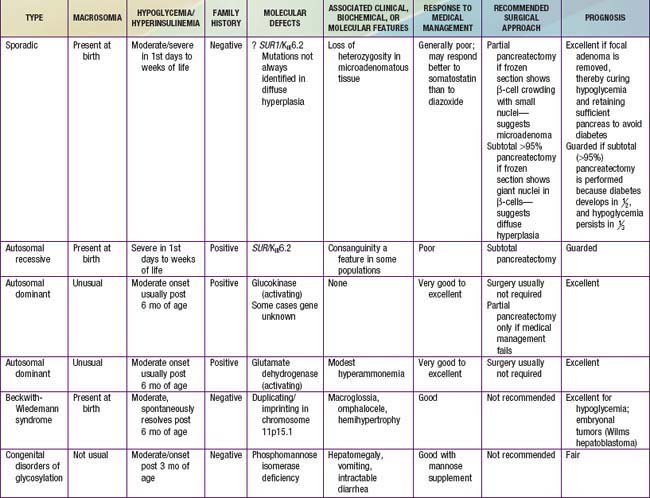
Table 86-5 ANALYSIS OF CRITICAL BLOOD SAMPLE DURING HYPOGLYCEMIA AND 30 MINUTES AFTER GLUCAGON*
SUBSTRATES
HORMONES
IGFBP-1, insulin-like growth factor binding protein-1.
* Glucagon 50 µg/kg with maximum of 1 mg IV or IM.
† Measure once only before or after glucagon administration. Rise in glucose of ≥40 mg/dL after glucagon given at the time of hypoglycemia strongly suggests a hyperinsulinemic state with adequate hepatic glycogen stores and intact glycogenolytic enzymes. If ammonia is elevated to 100-200 µM, consider activating mutation of glutamate dehydrogenase.
Table 86-6 CRITERIA FOR DIAGNOSING HYPERINSULINISM BASED ON “CRITICAL” SAMPLES (DRAWN AT A TIME OF FASTING HYPOGLYCEMIA: PLASMA GLUCOSE <50 MG/DL)
* Depends on sensitivity of insulin assay.
From Stanley CA, Thomson PS, Finegold DN, et al: Hypoglycemia in infants and neonates. In Sperling MA, editor: Pediatric endocrinology, ed 2, Philadelphia, 2002, WB Saunders, pp 135–159.
Table 86-7 DIAGNOSIS OF ACUTE HYPOGLYCEMIA IN INFANTS AND CHILDREN
ACUTE SYMPTOMS PRESENT
HISTORY SUGGESTIVE: ACUTE SYMPTOMS NOT PRESENT
Insulin-secreting macroadenomas are rare in childhood and may be diagnosed preoperatively via CT or MRI. The plasma levels of insulin alone, however, cannot distinguish the aforementioned entities. The diffuse or microadenomatous forms of islet cell hyperplasia represent a variety of genetic defects responsible for abnormalities in the endocrine pancreas characterized by autonomous insulin secretion that is not appropriately reduced when blood glucose declines spontaneously or in response to provocative maneuvers such as fasting (see Tables 86-4, 86-7, and 86-8 and Fig. 86-2). Clinical, biochemical, and molecular genetic approaches now permit classification of congenital hyperinsulinism, formerly termed nesidioblastosis, into distinct entities. Persistent hyperinsulinemic hypoglycemia of infancy (PHHI) may be inherited or sporadic, is severe, and is caused by mutations in the regulation of the potassium channel intimately involved in insulin secretion by the pancreatic β cell (Fig. 86-2). Normally, glucose entry into the β cell is enabled by the non–insulin-responsive glucose transporter GLUT-2. On entry, glucose is phosphorylated to glucose-6-phosphate by the enzyme glucokinase, enabling glucose metabolism to generate ATP. The rise in the molar ratio of ATP relative to adenosine diphosphate (ADP) closes the ATP-sensitive potassium channel in the cell membrane (KATP channel). This channel is composed of two subunits, the KIR 6.2 channel, part of the family of inward-rectifier potassium channels, and a regulatory component in intimate association with KIR 6.2 known as the sulfonylurea receptor (SUR). Together, KIR 6.2 and SUR constitute the potassium-sensitive ATP channel KATP. Normally, the KATP is open, but with the rise in ATP and closure of the channel, potassium accumulates intracellularly, causing depolarization of the membrane, opening of voltage-gated calcium channels, influx of calcium into the cytoplasm, and secretion of insulin via exocytosis. The genes for both SUR and KIR 6.2 are located close together on the short arm of chromosome 11, the site of the insulin gene. Inactivating mutations in the gene for SUR or, less often, KIR 6.2 prevent the potassium channel from opening. It remains essentially variably closed with constant depolarization and, therefore, constant inward flux of calcium; hence, insulin secretion is continuous. A milder autosomal dominant form of these defects is also reported. Likewise, an activating mutation in glucokinase or glutamate dehydrogenase results in closure of the potassium channel through overproduction of ATP and hyperinsulinism. Recently, genetic defects in fatty acid metabolism, in the insulin transcription factor HNF4alpha, and in the uncoupling protein UCP-2 of the mitochondrial gene complex also have been involved in hyperinsulinemic hypoglycemia. Inactivating mutations of the glucokinase gene or activating mutations of the ATP regulated potassium channel which prevent or limit closure of the channel, are responsible for inadequate insulin secretion and form the basis of some forms of maturity-onset diabetes of youth or of neonatal diabetes mellitus (Chapter 583).
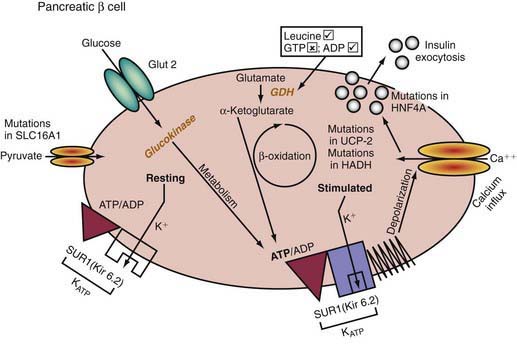
The familial forms of PHHI are more common in certain populations, notably Arabic and Ashkenazi Jewish communities, where it may reach an incidence of about 1/2,500, compared with the sporadic rates in the general population of ≈1/50,000. These autosomal recessive forms of PHHI typically present in the immediate newborn period as macrosomic newborns with a weight >4.0 kg and severe recurrent or persistent hypoglycemia manifesting in the initial hours or days of life. Glucose infusions as high as 15-20 mg/kg/min and frequent feedings fail to maintain euglycemia. Diazoxide, which acts by opening KATP channels (see Fig. 86-2), fails to control hypoglycemia adequately. Somatostatin, which also opens KATP and inhibits calcium flux, may be partially effective in about 50% of patients (see Fig. 86-2). Calcium channel blocking agents have had inconsistent effects. When affected patients are unresponsive to these measures, pancreatectomy is strongly recommended to avoid the long-term neurologic sequelae of hypoglycemia. If surgery is undertaken, preoperative CT or MRI rarely reveals an isolated adenoma, which would then permit local resection. Intraoperative ultrasonography may identify a small impalpable adenoma, permitting local resection. Adenomas often present in late infancy or early childhood.
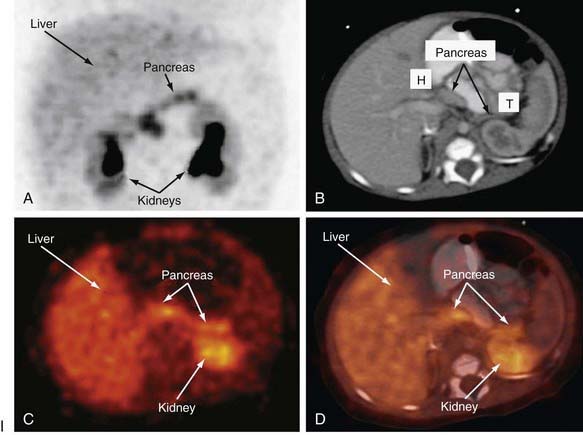
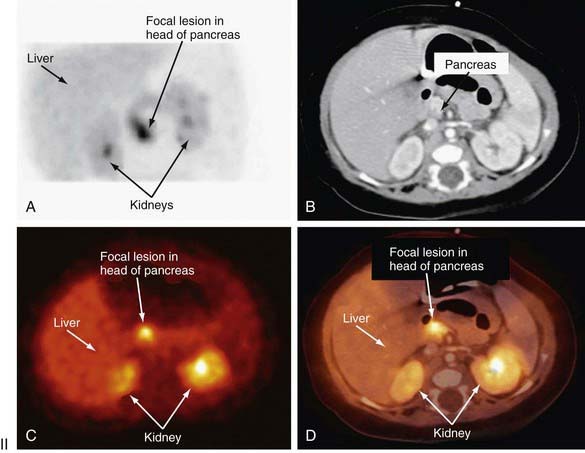
Distinguishing between focal and diffuse cases of persistent hyperinsulinism has been attempted in several ways. Preoperatively, transhepatic portal vein catheterization and selective pancreatic venous sampling to measure insulin may localize a focal lesion from the step-up in insulin concentration at a specific site. Selective catheterization of arterial branches supplying the pancreas, followed by infusion of a secretagogue such as calcium and portal vein sampling for insulin concentration (arterial stimulation-venous sampling) may localize a lesion. Both approaches are highly invasive, restricted to specialized centers, and not uniformly successful in distinguishing the focal from the diffuse forms, hence, these techniques are not recommended. 18F-labeled L-dopa combined with PET scanning is a highly promising means to distinguish the focal from the diffuse lesions of hyperinsulinism unresponsive to medical management (Fig. 86-3). The “gold standard” remains intraoperative histologic characterization. Diffuse hyperinsulinism is characterized by large β cells with abnormally large nuclei, whereas focal adenomatous lesions display small and normal β cell nuclei. Although SUR1 mutations are present in both types, the focal lesions arise by a random loss of a maternally imprinted growth-inhibitory gene on maternal chromosome 11p in association with paternal transmission of a mutated SUR1 or KIR 6.2 paternal chromosome 11p. Thus the focal form represents a double hit-loss of maternal repressor and transmission of a paternal mutation. Local excision of focal adenomatous islet cell hyperplasia results in a cure with little or no recurrence. For the diffuse form, near-total resection of 85-90% of the pancreas is recommended. The near-total pancreatectomy required for the diffuse hyperplastic lesions is, however, often associated with persistent hypoglycemia with the later development of hyperglycemia or frank, insulin-requiring diabetes mellitus.
A 3rd form of persistent PHHI is associated with mild and asymptomatic hyperammonemia, usually as a sporadic occurrence, although dominant inheritance occurs. Presentation is more like the autosomal dominant form than the autosomal recessive form. Diet and diazoxide control symptoms, but pancreatectomy may be necessary in some cases. The association of hyperinsulinism and hyperammonemia is caused by an inherited or de novo gain-of-function mutation in the enzyme glutamate dehydrogenase. The resulting increase in glutamate oxidation in the pancreatic β cell raises the ATP concentration and, hence, the ratio of ATP:ADP, which closes KATP, leading to membrane depolarization, calcium influx, and insulin secretion (see Fig. 86-2). In the liver, the excessive oxidation of glutamate to β-ketoglutarate may generate ammonia and divert glutamate from being processed to N-acetylglutamate, an essential cofactor for removal of ammonia through the urea cycle via activation of the enzyme carbamoyl phosphate synthetase. The hyperammonemia is mild, with concentrations of 100-200 µM/L, and produces no CNS symptoms or consequences, as seen in other hyperammonemic states. Leucine, a potent amino acid for stimulating insulin secretion and implicated in leucine-sensitive hypoglycemia, acts by allosterically stimulating glutamate dehydrogenase. Thus, leucine-sensitive hypoglycemia may be a form of the hyperinsulinemia-hyperammonemia syndrome or a potentiation of mild disorders of the KATP channel; it need not always be associated with a modest increase in serum ammonia.
Hypoglycemia associated with hyperinsulinemia is also seen in about 50% of patients with the Beckwith-Wiedemann syndrome. This syndrome is characterized by omphalocele, gigantism, macroglossia, microcephaly, and visceromegaly (Fig. 86-4). Distinctive lateral earlobe fissures and facial nevus flammeus are present; hemihypertrophy occurs in many of these infants. Diffuse islet cell hyperplasia occurs in infants with hypoglycemia. The diagnostic and therapeutic approaches are the same as those discussed previously, although microcephaly and retarded brain development may occur independently of hypoglycemia. Patients with the Beckwith-Wiedemann syndrome may acquire tumors, including Wilms tumor, hepatoblastoma, adrenal carcinoma, gonadoblastoma, and rhabdomyosarcoma. This overgrowth syndrome is caused by mutations in the chromosome 11p15.5 region close to the genes for insulin, SUR, KIR 6.2, and IGF-2. Duplications in this region and genetic imprinting from a defective or absent copy of the maternally derived gene are involved in the variable features and patterns of transmission. Hypoglycemia may resolve in weeks to months of medical therapy. Pancreatic resection may also be needed.
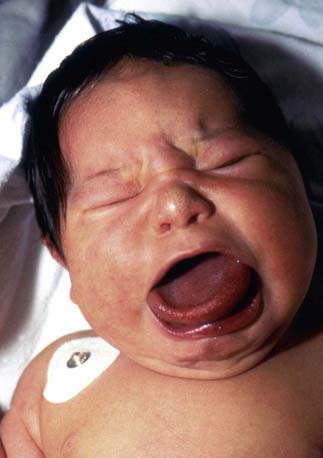
Figure 86-4 Beckwith-Wiedemann syndrome.
(Courtesy of Dr. Michael Cohen, Dalhousie University, Halifax, Nova Scotia. From Jones KL: Smith’s recognizable patterns of human malformation, ed 6, Philadelphia, 2006, Saunders.)
Hyperinsulinemic hypoglycemia in infancy is reported as a manifestation of one form of congenital disorder of glycosylation. Disorders of protein glycosylation usually present with neurologic symptoms but may also include liver dysfunction with hepatomegaly, intractable diarrhea, protein-losing enteropathy, and hypoglycemia (Chapter 81.6). These disorders are often underdiagnosed. One entity associated with hyperinsulinemic hypoglycemia is caused by phosphomannose isomerase deficiency, and clinical improvement followed supplemental treatment with oral mannose at a dose of 0.17 g/kg 6 times per day.
After the 1st 12 mo of life, hyperinsulinemic states are uncommon until islet cell adenomas reappear as a cause after the patient is several years of age. Hyperinsulinemia due to islet cell adenoma should be considered in any child 5 yr or older presenting with hypoglycemia. Islet cell adenomas do not “light up” during scanning with L-dopa labeled with fluorine-18. An islet cell adenoma in a child should arouse suspicion of the possibility of multiple endocrine neoplasia type 1 (Wermer syndrome), which involves mutations in the menin gene and may be associated with hyperparathyroidism and with pituitary tumors. The diagnostic approach is outlined in Tables 86-7 and 86-8. Fasting for up to 24-36 hr usually provokes hypoglycemia; coexisting hyperinsulinemia confirms the diagnosis, provided that factitious administration of insulin by the parents, a form of Munchausen syndrome by proxy, is excluded. Occasionally, provocative tests may be required. Exogenously administered insulin can be distinguished from endogenous insulin by simultaneous measurement of C-peptide concentration. If C-peptide levels are elevated, endogenous insulin secretion is responsible for the hypoglycemia; if C-peptide levels are low but insulin values are high, exogenous insulin has been administered, perhaps as a form of child abuse. Islet cell adenomas at this age are treated by surgical excision. Antibodies to insulin or the insulin receptor (insulin mimetic action) are also rarely associated with hypoglycemia. Some tumors produce insulin-like growth factors, thereby provoking hypoglycemia by interacting with the insulin receptor. The astute clinician must also consider the possibility of deliberate or accidental ingestion of drugs such as a sulfonylurea or related compound that stimulates insulin secretion. In such cases, insulin and C-peptide concentrations in blood will be elevated. Inadvertent substitution of an insulin secretagogue by a dispensing error should be considered in those taking medications who suddenly develop documented hypoglycemia.
Endocrine Deficiency
Hypoglycemia associated with endocrine deficiency is usually caused by adrenal insufficiency with or without associated growth hormone deficiency (Chapters 551 and 569). In panhypopituitarism, isolated adrenocorticotropic hormone (ACTH) or growth hormone deficiency, or combined ACTH deficiency plus growth hormone deficiency, the incidence of hypoglycemia is as high as 20%. In the newborn period, hypoglycemia may be the presenting feature of hypopituitarism; in males, a microphallus may provide a clue to a coexistent deficiency of gonadotropin. Newborns with hypopituitarism often have a form of “hepatitis” associated with cholestatic jaundice and hypoglycemia. The combination of hypoglycemia and cholestatic jaundice requires exclusion of hypopituitarism as a cause, since the jaundice resolves with replacement treatment of growth hormone, cortisol, and thyroid as required. This constellation is often associated with the syndrome of septo-optic dysplasia. When adrenal disease is severe, as in congenital adrenal hyperplasia caused by cortisol synthetic enzyme defects, adrenal hemorrhage, or congenital absence of the adrenal glands, disturbances in serum electrolytes with hyponatremia and hyperkalemia or ambiguous genitals may provide diagnostic clues (Chapter 570). In older children, failure of growth should suggest growth hormone deficiency. Hyperpigmentation may provide the clue to Addison disease with increased ACTH levels or adrenal unresponsiveness to ACTH owing to a defect in the adrenal receptor for ACTH. The frequent association of Addison disease in childhood with hypoparathyroidism (hypocalcemia), chronic mucocutaneous candidiasis, and other endocrinopathies should be considered. Adrenoleukodystrophy should also be considered in the differential diagnosis of primary Addison disease in older male children (Chapter 80.2).
Substrate Limited
Glycogen Storage Disease
Disorders of Gluconeogenesis
Defects in Fatty Acid Oxidation (Chapter 80)
Various congenital enzymatic deficiencies causing defective carnitine or fatty acid metabolism occur. A severe and relatively common form of fasting hypoglycemia with hepatomegaly, cardiomyopathy, and hypotonia occurs with long- and medium-chain fatty acid coenzyme-A dehydrogenase deficiency (LCAD and MCAD). Plasma carnitine levels are low, ketones are not present in urine, but dicarboxylic aciduria is present. Clinically, patients with acyl CoA dehydrogenase deficiency present with a Reye-like syndrome (Chapter 353), recurrent episodes of severe fasting hypoglycemic coma, and cardiorespiratory arrest (sudden infant death syndrome-like events). Severe hypoglycemia and metabolic acidosis without ketosis also occur in patients with multiple acyl CoA dehydrogenase disorders. Hypotonia, seizures, and acrid odor are other clinical clues. Survival depends on whether the defects are severe or mild; diagnosis is established from studies of enzyme activity in liver biopsy tissue or in cultured fibroblasts from affected patients. Tandem mass spectrometry can be employed for blood samples, even those on filter paper, for screening of congenital inborn errors. The frequency of this disorder is at least 1/10,000-15,000 births. Avoidance of fasting and supplementation with carnitine may be lifesaving in these patients who generally present in infancy.
Pyruvate Carboxylase Deficiency (Chapter 81)
Diagnosis and Differential Diagnosis
Table 86-8 and Figure 86-5 list the pertinent clinical and biochemical findings in the common childhood disorders associated with hypoglycemia. A careful and detailed history is essential in every suspected or documented case of hypoglycemia (see Table 86-7). Specific points to be noted include age at onset, temporal relation to meals or caloric deprivation, and a family history of prior infants known to have had hypoglycemia or of unexplained infant deaths. In the 1st wk of life, the majority of infants have the transient form of neonatal hypoglycemia either as a result of prematurity/intrauterine growth restriction or by virtue of being born to diabetic mothers. The absence of a history of maternal diabetes, but the presence of macrosomia and the characteristic large plethoric appearance of an “infant of a diabetic mother” should arouse suspicion of hyperinsulinemic hypoglycemia of infancy probably due to a KATP channel defect that is familial (autosomal recessive) or sporadic; plasma insulin concentrations >5-10 µU/mL in the presence of documented hypoglycemia confirm this diagnosis. The presence of hepatomegaly should arouse suspicion of an enzyme deficiency; if non–glucose-reducing sugar is present in the urine, galactosemia is most likely. In males, the presence of a microphallus suggests the possibility of hypopituitarism, which also may be associated with jaundice in both sexes.
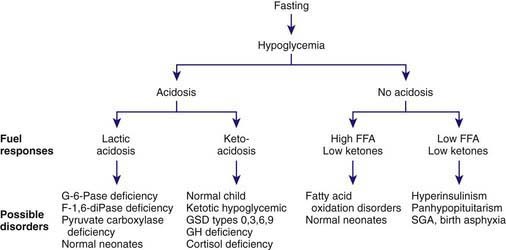
Past the newborn period, clues to the cause of persistent or recurrent hypoglycemia can be obtained through a careful history, physical examination, and initial laboratory findings. The temporal relation of the hypoglycemia to food intake may suggest that the defect is one of gluconeogenesis, if symptoms occur 6 hr or more after meals. If hypoglycemia occurs shortly after meals, galactosemia or fructose intolerance is most likely, and the presence of reducing substances in the urine rapidly distinguishes these possibilities. The autosomal dominant forms of hyperinsulinemic hypoglycemia need to be considered, with measurement of glucose, insulin, and ammonia, and careful history for other affected family members of any age. Measurement of IGFBP-1 may be useful; it is low in hyperinsulinemia states and high in other forms of hypoglycemia. The presence of hepatomegaly suggests one of the enzyme deficiencies in glycogen breakdown or in gluconeogenesis, as outlined in Table 86-8. The absence of ketonemia or ketonuria at the time of initial presentation strongly suggests hyperinsulinemia or a defect in fatty acid oxidation. In most other causes of hypoglycemia, with the exception of galactosemia and fructose intolerance, ketonemia and ketonuria are present at the time of fasting hypoglycemia. At the time of the hypoglycemia, serum should be obtained for determination of hormones and substrates, followed by repeated measurement after an intramuscular or intravenous injection of glucagon, as outlined in Table 86-7. Interpretation of the findings is summarized in Table 86-8. Hypoglycemia with ketonuria in children between ages 18 mo and 5 yr is most likely to be ketotic hypoglycemia, especially if hepatomegaly is absent. The ingestion of a toxin, including alcohol or salicylate, can usually be excluded rapidly by the history. Inadvertent or deliberate drug ingestion and errors in dispensing medicines should also be considered.
When the history is suggestive, but acute symptoms are not present, a 24-36 hr supervised fast can usually provoke hypoglycemia and resolve the question of hyperinsulinemia or other conditions (see Table 86-8). Such a fast is contraindicated if a fatty acid oxidation defect is suspected; other approaches such as mass tandem spectrometry or molecular diagnosis, or both, should be considered. Because adrenal insufficiency may mimic ketotic hypoglycemia, plasma cortisol levels should be determined at the time of documented hypoglycemia; increased buccal or skin pigmentation may provide the clue to primary adrenal insufficiency with elevated ACTH (melanocyte-stimulating hormone) activity. Short stature or a decrease in the growth rate may provide the clue to pituitary insufficiency involving growth hormone as well as ACTH. Definitive tests of pituitary-adrenal function such as the arginine-insulin stimulation test for growth hormone IGF-1, IGFBP-1, and cortisol release may be necessary.
In the presence of hepatomegaly and hypoglycemia, a presumptive diagnosis of the enzyme defect can often be made through the clinical manifestations, presence of hyperlipidemia, acidosis, hyperuricemia, response to glucagon in the fed and fasted states, and response to infusion of various appropriate precursors (see Tables 86-7 and 86-8). These clinical findings and investigative approaches are summarized in Table 86-8. Definitive diagnosis of the glycogen storage disease may require an open liver biopsy (Chapter 81). Occasional patients with all the manifestations of glycogen storage disease are found to have normal enzyme activity. These definitive studies require special expertise available only in certain institutions.
Alkalay AL, Flores-Sarnat L, Sarnat HB, et al. Brain imaging findings in neonatal hypoglycemia: case report and review of 23 cases. Clin Pediatr (Phila). 2005;44:783-790.
Boluyt N, van Kempen A, Offringa M. Neurodevelopment after neonatal hypoglycemia: a systematic review and design of an optimal future study. Pediatrics. 2006;117:2231-2243.
Dalgic N, Ergenekon E, Soysal S, et al. Transient neonatal hypoglycemia—Long-term effects on neurodevelopmental outcome. J Pediatr Endocrinol Metab. 2002;15:319-324.
Daly LP, Osterhoudt KC, Weinzimer SA. Presenting features of idiopathic ketotic hypoglycemia. J Emerg Med. 2003;1:39-43.
Dekelbab BH, Sperling MA. Hypoglycemia in newborns and infants. Adv Pediatr. 2006;53:5-22.
De León DD, Stanley CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;1:57-68.
de Lonlay P, Giurgea I, Touati G, et al. Neonatal hypoglycaemia: aetiologies. Semin Neonatol. 2004;9:49-58.
Giurgea I, Ulinski T, Touati G, et al. Factitious hyperinsulinism leading to pancreatectomy: severe forms of Munchausen syndrome by proxy. Pediatrics. 2005;116:e145-e148.
González-Barroso MM, Giurgea I, Bouillaud F, et al. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One. 2008;3:e3850.
Hardy TO, Hernandez-Pampaloni M, Saffer JR, et al. Accuracy of [18F]fluorodopa positron emission tomography for diagnosing and localizing focal congenital hyperinsulinism. J Clin Endocrinol Metab. 2007;92:4706-4711.
Hardy TO, Hernandez-Pampaloni M, Saffer JR, et al. Diagnosis and localization of focal congenital hyperinsulinism by 18F-fluorodopa PET scan. J Pediatr. 2007;150:140-150.
Inder T. How low can I go? The impact of hypoglycemia on the immature brain. Pediatrics. 2008;122:440-441.
Kapoor RR, James C, Hussain K. Advances in the diagnosis and management of hyperinsulinemic hypoglycemia. Nat Clin Pract Endocrinol Metab. 2008;5:105-112.
Klepper J, Wang D, Fischbarg J, et al. Defective glucose transport across brain tissue barriers: a newly recognized neurological syndrome. Neurochem Res. 1999;24:587-594.
Lupsa BC, Chong AY, Cochran EK, et al. Autoimmune forms of hypoglycemia. Medicine. 2009;88:141-153.
Magge SN, Shyng SL, MacMullen C, et al. Familial leucine-sensitive hypoglycemia of infancy due to a dominant mutation of the beta-cell sulfonylurea receptor. J Clin Endocrinol Metab. 2004;89:4450-4456.
Meissner T, Wendel U, Burgard P, et al. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol. 2003;149:43-51.
Melis D, Parenti G, Della CR, et al. Brain damage in glycogen storage disease type I. J Pediatr. 2004;144:637-642.
Mochel F, Slama A, Touati G, et al. Respiratory chain defects may present only with hypoglycemia. J Clin Endocrinol Metab. 2005;90:3780-3785.
Mohnike K, Blankenstein O, Minn H, et al. [18F]-DOPA positron emission tomography for preoperative localization in congenital hyperinsulinism. Horm Res. 2008;70:65-72.
Neal JM, Han W. Insulin immunoassays in the detection of insulin analogues in factitious hypoglycemia. Endocr Pract. 2008;14:1006-1010.
Ng DD, Ferry RJJr, Kelly A, et al. Acarbose treatment of postprandial hypoglycemia in children after Nissen fundoplication. J Pediatr. 2001;139:877-879.
Njuguna P, Newton C. Management of severe falciparum malaria. J Postgrad Med. 2004;50:45-50.
Osier FH, Berkley JA, Ross A, et al. Abnormal blood glucose concentrations on admission to a rural Kenyan district hospital: prevalence and outcome. Arch Dis Child. 2003;88:621-625.
Otonkoski T, Jiao H, Kaminen-Ahola N, et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am J Hum Genet. 2007;81:467-474.
Palladino AA, Bennett MJ, Stanley CA. Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem. 2008;54:256-263.
Rauchenzauner M, Klepper J, Leiendecker B, et al. The ketogenic diet in children with Glut1 deficiency syndrome and epilepsy. J Pediatr. 2008;153:716-718.
Sperling MA, Menon RK. Differential diagnosis and management of neonatal hypoglycemia. Pediatr Clin North Am. 2004;51:703-723.
Steinkrauss L, Lipman TH, Hendell CD, et al. Effects of hypoglycemia on developmental outcome in children with congenital hyperinsulinism. J Pediatr Nurs. 2005;20:109-118.
Straussman S, Levitsky LL. Neonatal hypoglycemia. Curr Opin Endocrinol Diabetes Obes. 2010;17:20-24.
Sun L, Eklund EA, Chung WK, et al. Congenital disorder of glycosylation is presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J Clin Endocrinol Metab. 2005;90:4371-4375.
Tam EWY, Widjaja E, Blaser SI, et al. Occipital lobe injury and cortical visual outcomes after neonatal hypoglycemia. Pediatrics. 2008;122:507-512.
Wang ML, Dorer DJ, Fleming MP, et al. Clinical outcomes of near-term infants. Pediatrics. 2004;114:372-376.
Weinstein DA, Correia CE, Saunders AC, et al. Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. Mol Genet Metab. 2006;87:284-288.
Wolfsdorf JI. Understanding protein-sensitive hypoglycemia. J Pediatr. 2006;149:6-7.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 86 Hypoglycemia
Glucose has a central role in fuel economy and is a source of energy storage in the form of glycogen, fat, and protein (Chapter 81). Glucose, an immediate source of energy, provides 38 mol of adenosine triphosphate (ATP) per mol of glucose oxidized. It is essential for cerebral energy metabolism because it is usually the preferred substrate and its utilization accounts for nearly all the oxygen consumption in the brain. Cerebral glucose uptake occurs through a glucose transporter molecule or molecules that are not regulated by insulin. Cerebral transport of glucose is a carrier-mediated, facilitated diffusion process that is dependent on blood glucose concentration. Deficiency of brain glucose transporters can result in seizures because of low cerebral and cerebrospinal fluid (CSF) glucose concentrations (hypoglycorrhachia) despite normal blood glucose levels. To maintain the blood glucose concentration and prevent it from falling precipitously to levels that impair brain function, an elaborate regulatory system has evolved.
Definition
In neonates, there is not always an obvious correlation between blood glucose concentration and the classic clinical manifestations of hypoglycemia. The absence of symptoms does not indicate that glucose concentration is normal and has not fallen to less than some optimal level for maintaining brain metabolism. There is evidence that hypoxemia and ischemia may potentiate the role of hypoglycemia in causing permanent brain damage. Consequently, the lower limit of accepted normality of the blood glucose level in newborn infants with associated illness that already impairs cerebral metabolism has not been determined (Chapter 101). Out of concern for possible neurologic, intellectual, or psychologic sequelae in later life, most authorities recommend that any value of blood glucose <50 mg/dL in neonates be viewed with suspicion and vigorously treated. This is particularly applicable after the initial 2-3 hr of life, when glucose normally has reached its nadir; subsequently, blood glucose levels begin to rise and achieve values of 50 mg/dL or higher after 12-24 hr. In older infants and children, a whole blood glucose concentration of <50 mg/dL (10-15% higher for serum or plasma) represents hypoglycemia.
Substrate, Enzyme, and Hormonal Integration of Glucose Homeostasis
In the Newborn (Chapter 101)
In the early postnatal period, responses of the endocrine pancreas favor glucagon secretion so that blood glucose concentration can be maintained. These adaptive changes in hormone secretion are paralleled by similarly striking adaptive changes in hormone receptors. Key enzymes involved in glucose production also change dramatically in the perinatal period. Thus, there is a rapid fall in glycogen synthase activity and a sharp rise in phosphorylase after delivery. Similarly, the amount of rate-limiting enzyme for gluconeogenesis, phosphoenolpyruvate carboxykinase, rises dramatically after birth, activated in part by the surge in glucagon and the fall in insulin. This framework can explain several causes of neonatal hypoglycemia based on inappropriate changes in hormone secretion and unavailability of adequate reserves of substrates in the form of hepatic glycogen, muscle as a source of amino acids for gluconeogenesis, and lipid stores for the release of fatty acids. In addition, appropriate activities of key enzymes governing glucose homeostasis are required (see Fig. 81-1).
In Older Infants and Children
Hypoglycemia in older infants and children is analogous to that of adults, in whom glucose homeostasis is maintained by glycogenolysis in the immediate postfeeding period and by gluconeogenesis several hours after meals. The liver of a 10 kg child contains 20-25 g of glycogen, which is sufficient to meet normal glucose requirements of 4-6 mg/kg/min for only 6-12 hr. Beyond this period, hepatic gluconeogenesis must be activated. Both glycogenolysis and gluconeogenesis depend on the metabolic pathway summarized in Figure 81-1. Defects in glycogenolysis or gluconeogenesis may not be manifested in infants until the frequent feeding at 3-4 hr intervals ceases and infants sleep through the night, a situation usually present by 3-6 mo of age. The source of gluconeogenic precursors is derived primarily from muscle protein. The muscle bulk of infants and small children is substantially smaller relative to body mass than that of adults, whereas glucose requirements/unit of body mass are greater in children, so the ability to compensate for glucose deprivation by gluconeogenesis is more limited in infants and young children, as is the ability to withstand fasting for prolonged periods. The ability of muscle to generate alanine, the principal gluconeogenic amino acid, may also be limited. Thus, in normal young children, the blood glucose level falls after 24 hr of fasting, insulin concentrations fall appropriately to levels of <5-10 µU/mL, lipolysis and ketogenesis are activated, and ketones may appear in the urine.
The switch from glycogen synthesis during and immediately after meals to glycogen breakdown and later gluconeogenesis is governed by hormones, of which insulin is of central importance. Plasma insulin concentrations increase to peak levels of 50-100 µU/mL after meals, which serve to lower the blood glucose concentration through the activation of glycogen synthesis, enhancement of peripheral glucose uptake, and inhibition of glucose production. In addition, lipogenesis is stimulated, whereas lipolysis and ketogenesis are curtailed. During fasting, plasma insulin concentrations fall to ≤5-10 µU/mL, and together with other hormonal changes, this fall results in activation of gluconeogenic pathways (see Fig. 81-1). Fasting glucose concentrations are maintained through the activation of glycogenolysis and gluconeogenesis, inhibition of glycogen synthesis, and activation of lipolysis and ketogenesis. It should be emphasized that a plasma insulin concentration of >5 µU/mL, in association with a blood glucose concentration of ≤40 mg/dL (2.2 mM), is abnormal, indicating a hyperinsulinemic state and failure of the mechanisms that normally result in suppression of insulin secretion during fasting or hypoglycemia.
Clinical Manifestations (Chapter 101)
Clinical features generally fall into 2 categories. The 1st includes symptoms associated with the activation of the autonomic nervous system and epinephrine release, usually seen with a rapid decline in blood glucose concentration (Table 86-1). The 2nd category includes symptoms due to decreased cerebral glucose utilization, usually associated with a slow decline in blood glucose level or prolonged hypoglycemia (see Table 86-1). Although these classic symptoms occur in older children, the symptoms of hypoglycemia in infants may be subtler and include cyanosis, apnea, hypothermia, hypotonia, poor feeding, lethargy, and seizures. Some of these symptoms may be so mild that they are missed. Occasionally, hypoglycemia may be asymptomatic in the immediate newborn period. Newborns with hyperinsulinemia are often large for gestational age; older infants with hyperinsulinemia may eat excessively because of chronic hypoglycemia and become obese. In childhood, hypoglycemia may present as behavior problems, inattention, ravenous appetite, or seizures. It may be misdiagnosed as epilepsy, inebriation, personality disorders, hysteria, and retardation. A blood glucose determination should always be performed in sick neonates, who should be vigorously treated if concentrations are <50 mg/dL. At any age level, hypoglycemia should be considered a cause of an initial episode of convulsions or a sudden deterioration in psychobehavioral functioning.
Table 86-1 MANIFESTATIONS OF HYPOGLYCEMIA IN CHILDHOOD
FEATURES ASSOCIATED WITH ACTIVATION OF AUTONOMIC NERVOUS SYSTEM AND EPINEPHRINE RELEASE*
FEATURES ASSOCIATED WITH CEREBRAL GLUCOPENIA
* Some of these features will be attenuated if the patient is receiving β-adrenergic blocking agents.
Many neonates have asymptomatic (chemical) hypoglycemia. The incidence of symptomatic hypoglycemia is highest in small for gestational age infants (Fig. 86-1). The exact incidence of symptomatic hypoglycemia has been difficult to establish because many of the symptoms in neonates occur together with other conditions such as infections, especially sepsis and meningitis; central nervous system anomalies, hemorrhage, or edema; hypocalcemia and hypomagnesemia; asphyxia; drug withdrawal; apnea of prematurity; congenital heart disease; or polycythemia.
Classification of Hypoglycemia in Infants and Children
Classification is based on knowledge of the control of glucose homeostasis in infants and children (Table 86-2).
Table 86-2 CLASSIFICATION OF HYPOGLYCEMIA IN INFANTS AND CHILDREN
NEONATAL TRANSIENT HYPOGLYCEMIA
Associated with Inadequate Substrate or Immature Enzyme Function in Otherwise Normal Neonates
Transient Neonatal Hyperinsulinism Also Present in:
NEONATAL, INFANTILE, OR CHILDHOOD PERSISTENT HYPOGLYCEMIAS
Hormonal Disorders
Counter-Regulatory Hormone Deficiency
Glycogenolysis and Gluconeogenesis Disorders
Lipolysis Disorders
Fatty Acid Oxidation Disorders
OTHER ETIOLOGIES
Substrate-Limited
Liver Disease
Amino Acid and Organic Acid Disorders
Systemic Disorders
GSD, glycogen storage disease; HI, hyperinsulinemia; KATP, regulated potassium channel.
Neonatal, Transient, Small for Gestational Age, and Premature Infants (Chapter 101)
The estimated incidence of symptomatic hypoglycemia in newborns is 1-3/1,000 live births. This incidence is increased severalfold in certain high-risk neonatal groups (see Table 86-2 and Fig. 86-1). The premature and small for gestational age (SGA) infants are vulnerable to the development of hypoglycemia. The factors responsible for the high frequency of hypoglycemia in this group, as well as in other groups outlined in Table 86-2, are related to the inadequate stores of liver glycogen, muscle protein, and body fat needed to sustain the substrates required to meet energy needs. These infants are small by virtue of prematurity or impaired placental transfer of nutrients. Their enzyme systems for gluconeogenesis may not be fully developed. Transient hyperinsulinism responsive to diazoxide has also been reported as contributing to hypoglycemia in asphyxiated, SGA, and premature newborn infants. This form of hyperinsulinism associated with perinatal asphyxia, intrauterine growth restriction (IUGR), maternal toxemia and other perinatal stressors, is probably the most common cause of hyperinsulinemic hypoglycemia in neonates and may be quite severe. In most cases, the condition resolves quickly, but it may persist to 7 mo of life or more. A genetic cause of this form of dysregulated insulin secretion has not been established.
Infants Born to Diabetic Mothers (Chapter 101)
Mothers whose diabetes has been well controlled during pregnancy, labor, and delivery generally have infants near normal size who are less likely to develop neonatal hypoglycemia and other complications formerly considered typical of such infants (Chapter 101). In supplying exogenous glucose to these hypoglycemic infants, it is important to avoid hyperglycemia that evokes a prompt exuberant insulin release, which may result in rebound hypoglycemia. When needed, glucose should be provided at continuous infusion rates of 4-8 mg/kg/min, but the appropriate dose for each patient should be individually adjusted. During labor and delivery, maternal hyperglycemia should be avoided because it results in fetal hyperglycemia, which predisposes to hypoglycemia when the glucose supply is interrupted at birth. Hypoglycemia persisting or occurring after 1 wk of life requires an evaluation for the causes listed in Table 86-2.
